

Abstract
Group I metabotropic glutamate receptors (mGluRs) stimulation activates translation-dependent epileptogenesis in the hippocampus. This translation is regulated by repressors, including BC1 RNA and fragile X mental retardation protein (FMRP). Recent data indicate that group I mGluR stimulation exerts bidirectional control over FMRP level by activating translation and ubiquitin-proteasome system (UPS)-dependent proteolysis for the up- and downregulation of the protein, respectively. At present, the temporal relationship of translation and proteolysis on FMRP and their interplay for group I mGluR-mediated translation and epileptogenesis are unknown. We addressed these issues by using mouse hippocampal slices. Agonist [(S)-3,5-dihydroxyphenylglycine (DHPG)] stimulation of group I mGluRs caused a biphasic change in FMRP level. An initial decrease (within 10 min) was followed by an increase at 30 min. When slices were pretreated with translation inhibitor (anisomycin or cycloheximide), group I mGluRs elicited a sustained decrease in FMRP. This decrease was prevented by a proteasome inhibitor [Z-Leu-Leu-Leu-CHO (MG-132)]. When slices were pretreated with MG-132 alone, DHPG no longer elicited any change in FMRP. MG-132 also suppressed increase in other proteins, including postsynaptic density-95and α-calcium/calmodulin-dependent protein kinase II, normally elicited by group I mGluR stimulation. Physiological experiments showed that proteasome inhibitor suppressed group I mGluR-induced prolonged synchronized discharges. However, proteasome inhibitor did not affect group I mGluR-induced prolonged synchronized discharges in Fmr1−/− preparations, where functional FMRP is absent. The results suggest that constitutive FMRP in hippocampal cells acts as a brake on group I mGluR-mediated translation and epileptogenesis. FMRP downregulation via UPS removes this brake enabling group I mGluR-mediated translation and epileptogenesis.
Introduction
Group I metabotropic glutamate receptor (mGluR) responses in the hippocampus is a useful model for the study of epileptogenesis for three reasons: (1) stimulation of the receptor elicits population discharges (prolonged synchronized discharges) in the CA3 pyramidal cells in the hippocampus in a pattern and duration that can be compared with ictal events recorded in electrographic seizures (Zifkin and Gracco, 1990; D'Ambrosio et al., 2009); (2) the ictal-like prolonged synchronized discharges, once initiated, persist even when the stimulating agonist is washed away, suggesting that an epileptogenic process has occurred, permanently transforming the network (Merlin and Wong, 1997); and (3) group I mGluR-mediated epileptogenesis has clinical relevance because it may constitute a mechanism underlying epilepsy in subjects with fragile X syndrome (Chuang et al., 2005; Yan et al., 2005; Bianchi et al., 2009). Group I mGluR-induced epileptogenesis depends on activation of mRNA translation (Merlin et al., 1998) and is independent of transcription (Chuang et al., 2005). Recent studies indicate that the translation underlying epileptogenesis is regulated by a number of repressors including fragile X mental retardation protein (FMRP) (Chuang et al., 2005) and BC1 RNA (Zhong et al., 2009). These repressors safeguard normal subjects from group I mGluR-mediated epileptogenesis. In instances where repressor function is compromised, as in the case of fragile X syndrome, susceptibility to group I mGluR-mediated epileptogenesis is enhanced (Chuang et al., 2005; Yan et al., 2005; Bianchi et al., 2009).
FMRP level and function are affected by group I mGluR stimulation in various ways: (1) dephosphorylation and phosphorylation, (2) proteolysis via a ubiquitin-proteasome system (UPS), and (3) protein synthesis. The phosphorylated form of FMRP is reported to be the active form in translation repression (Ceman et al., 2003). Stimulation of group I mGluRs elicits rapid FMRP dephosphorylation enabling a permissive state for protein synthesis (Narayanan et al., 2007). In addition, stimulation of group I mGluRs facilitates FMRP downregulation via UPS (Hou et al., 2006). Conversely, group I mGluR stimulation also upregulates FMRP by activating its synthesis (Weiler et al., 1997; Todd et al., 2003a,b; Hou et al., 2006; Zhong et al., 2009).
We evaluated the interplay between group I mGluR-mediated FMRP proteolysis and translation in epileptogenesis by studying the two processes independently and by examining the temporal relationship between the two processes following group I mGluR stimulation. Our results indicate: (1) that the constitutive level of FMRP is sufficient to arrest group I mGluR-induced translation, including that associated with epileptogenesis, and (2) that downregulation of FMRP, via ubiquitin-proteasome system, is an obligatory step preceding the onset of group I mGluR-mediated translation and epileptogenesis.
Materials and Methods
Animals.
Fmr1 knock-out (FVB.129P2-Fmr1tm1Cgr/J) and control (FVB.129P2-Pde6b+Tyrc-ch/AntJ) mice were purchased from The Jackson Laboratory. Four- to 6-week-old male mice were used.
Slice preparation.
Transverse hippocampal slices (400 μm thick) were prepared as described previously (Chuang et al., 2002). During slicing, the hippocampi were submerged in an ice-cold low-Ca2+/high-Mg2+ buffer, consisting of the following (in mm): 124 NaCl, 26 NaHCO3, 2.5 KCl, 8 MgCl2, 0.5 CaCl2, and 10 d-glucose, bubbled with 95% O2 and 5% CO2. For electrophysiology, slices were transferred to an interface chamber (Fine Science Tools) perfused with normal solution [artificial CSF (ACSF)] consisting of the following (in mm): 124 NaCl, 26 NaHCO3, 5 KCl, 1.6 MgCl2, 2 CaCl2, and 10 d-glucose bubbled with 95% O2 and 5% CO2; pH 7.4. Slices were maintained at 34.5°C for at least 60 min before recording. For Western blot experiments, slices were maintained at 32.5°C in a slice holding chamber filled with ACSF for at least 90 min before drug application. To test whether the DHPG stimulation paradigm used in Western blot experiments was effective in inducing epileptiform discharges, slices were exposed to DHPG for 30 min at 32.5°C in the holding chamber, then briefly washed in ACSF and transferred to the interface chamber perfused with ACSF at 34.5°C. Electrophysiological recordings were performed after 20 min of perfusion in the interface chamber.
Electrophysiology.
Microelectrodes were pulled from glass tubes (1 mm outer diameter with glass filament inside; World Precision Instruments) using a micropipette puller (Sutter Instruments). Microelectrodes were filled with 2 m potassium acetate and the resistances were ∼70 MΩ. Signals were amplified (Axoclamp 2B; Molecular Devices) and recorded simultaneously in a computer running pClamp6 (Molecular Devices) and on a chart recorder (Gould).
Drugs.
For electrophysiology, pharmacological agents were delivered to the hippocampal slices through continuous perfusion. Depending on the flow rate, the final concentration was reached within 15 min. For Western blot experiments, agents were directly applied to the holding chamber. Pretreatment of slices with various agents lasted for 45 min. (S)-3,5-Dihydroxyphenylglycine (DHPG), (2R,3S,4S)-2-[(4-methoxyphenyl)methyl]-3,4-pyrrolidinediol 3-acetate (anisomycin), 4-[2-(3,5-dimethyl-2-oxo-cyclohexyl)-2-hydroxyethyl]-2,6-piperidinedione (cycloheximide), and 1–4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene (U0126) were purchased from Tocris Bioscience. Z-Leu-Leu-Leu-CHO (MG-132) and lactacystin were from Calbiochem. Antibodies for FMRP, postsynaptic density-95(PSD-95), α-calcium/calmodulin-dependent protein kinase II (α-CaMKII), and α-tubulin were purchased from Millipore. Antibody for phospho-ERK1/2 and secondary antibodies were from Cell Signaling Technology. Other chemicals were from Sigma. MG-132, lactacystin, anisomycin, cycloheximide, and U0126 were prepared in DMSO stock solutions. Before drug application, stock solutions were diluted 500 (0.2% DMSO) or 1000 (0.1% DMSO) times in normal ACSF with strong agitation to ensure complete solubility. Stock solutions were aliquoted and stored at −20°C.
Sample preparation.
For time course studies, hippocampal slices were collected at 0, 10, and 30 min after DHPG exposure from the slice-holding chamber and snap frozen on dry ice. Each sample containing four to six slices was stored at −80°C until biochemical analysis. In some experiments, the CA1 region was micro-dissected for biochemical analysis. Each sample was sonicated in ice-cold homogenization buffer containing the following (in mm): 320 sucrose, 10 HEPES pH 7.4, 2 EDTA, 1 EGTA, and protease inhibitors (20 μg/ml aprotinin, 20 μg/ml leupeptin, 20 μg/ml Pepstatin A, 1 mm PMSF) and phosphatase inhibitors (1 NaF, 1 Na3VO4, 2.5 Na4P2O7, and 1 β-glycerophosphate). The homogenate was then mixed with detergents (final: 0.1% SDS, 0.5% DOC, and 1% NP-40) and left on ice for 30 min. The suspension was cleared by centrifugation. Lysates were aliquoted and stored at −80°C. One aliquot of lysate was used to determine protein concentration with BCA method (Fisher Scientific). For Western blots, samples were mixed with SDS sample buffer (10% glycerol, 5% β-mercaptoethanol, and 2% SDS, in 62 mm Tris-HCl, pH 6.8) and boiled for 5 min.
Western blots.
Equal amount of samples were subjected to electrophoresis in 10% SDS-polyacrylamide gels (1.0 mm Novex Tris-Glycine; Invitrogen). Gels were then transferred to PVDF membranes for 1 h at a constant voltage of 100 V (mini Trans-Blot Cell; Bio-Rad). The membrane was stained with Ponceau S briefly to verify the quality of transfer. The membrane was blocked in 5% nonfat milk dissolved in TBS-Tween 20 (TBST) buffer (containing the following: 20 mm Tris-HCl, pH 7.6, 15 mm NaCl, and 0.1% Tween 20) for 1 h at room temperature or overnight at 4°C. After blocking, the membrane was probed with primary antibody for 2 h at room temperature or overnight at 4°C. After incubation, the membrane was washed three times for 5 min each with TBST. The membrane was then incubated with HRP-conjugated secondary antibody in TBST for 1 h. The membrane was washed again three times for 5 min each with TBST. And then the membrane was treated with LumiGLO (Cell Signaling Technology) and developed. For total protein stain (Ehlers, 2003), equal amount of proteins (20 μg) were resolved in 4–20% SDS-polyacrylamide gel. The protein amount of each band was measured by the enclosed area underneath the corresponding peak revealed in line scan analysis.
Immunoprecipitation.
Hippocampal slices were maintained in a slice holding chamber at 32.5°C. After drug application, slices were snap-frozen on dry ice and stored at −80°C. Pooled equal number of hippocampal slices were briefly sonicated in ice-cold homogenization buffer containing the following (in mm): 50 Tris-HCl, pH 7.4, 10 EDTA, 25 NaF, protease inhibitor cocktail, tyrosine phosphatase inhibitor cocktail, 50 MG-132, and 1 ubiquitin aldehyde. All further manipulations were performed at 4°C. The homogenate was then adjusted to contain 150 mm NaCl and was cleared at 20,000 × g for 20 min. The resulting hippocampal lysate was used for immunoprecipitation (IP). The IP experiments using Protein G Dynabeads (Invitrogen) were performed following the manufacturer's protocol with minor modifications. Briefly, Dynabeads were first incubated with anti-FMRP (7G1, Developmental Studies Hybridoma Bank) diluted in antibody binding and washing buffer for 1 h. After binding, the Dynabeads bound with anti-FMRP were pelleted with a magnet, the supernatant was discarded, and the pellet was washed once with antibody binding and washing buffer to further remove any unbounded antibody. After removing the washing buffer by pelleting again, the Dynabeads–antibody (Ab) complex was resuspended in hippocampal lysate (prepared as described above) and incubated with rotation for 1 h. After incubation, the Dynabeads–Ab–agonist (Ag) complex was separated from the hippocampal lysate with a magnet. Aliquots from hippocampal lysate before and after the IP procedure were also saved for later analysis. The Dynabeads–Ab–Ag complex pellet was washed three times using a washing buffer before transfer to a clean tube. Following separation with the magnet, the supernatant was discarded, and the Dynabeads–Ab–Ag complex was resuspended in a SDS sample buffer. The mixture was boiled for 5 min and pelleted with a magnet, and the resulting supernatant was used for 8% SDS-polyacrylamide gel loading. After transferring, membrane was blotted with another FMRP antibody (Cell Signaling Technology) or ubiquitin antibody (Millipore). For experiments examining the action of MG-132 on proteasome activity in hippocampal slices, the monoclonal ubiquitin antibody FK2 (Enzo Life Sciences) was used for IP, followed by detection with another ubiquitin antibody (Millipore).
Data analysis.
The duration of an individual synchronized discharge was measured from the beginning of the first action potential of the burst to the repolarization of the last action potential of the burst. Burst durations of all synchronized bursts in a 5 min period for each slice in the various experimental conditions were averaged. Synchronized discharges with duration longer than 1.5 s are referred to as prolonged synchronized discharges. Immunoreactive bands in Western blots were scanned with a desktop scanner (Epson Expression 1680) and quantified with Scion Image software. The optical densities (ODs) of bands of interest were normalized to the OD value of α-tubulin bands. Summary data are reported as means ± SEM. Student's t test was used for the statistical comparison of two sets of data. For analysis of data among groups, one- or two-way ANOVA followed by the Newman–Keuls test for repeated measures was applied. In all cases, the level of significant difference was p < 0.05.
Results
UPS activity is required for the induction of prolonged synchronized discharges
Application of the group I mGluR agonist DHPG (50 μm) induced short synchronized discharges (<1 s) in hippocampal slices (Fig. 1Aa(i)). After ∼10 min following onset of short synchronized discharges rhythmic prolonged synchronized discharges of longer duration (>2 s) appeared and persisted for the duration of DHPG treatment (Fig. 1Aa(ii)). The emergence of prolonged synchronized discharges requires de novo protein synthesis (Merlin et al., 1998), while that of initial short synchronized discharges does not (Zhao et al., 2004). When DHPG was applied for 10 min and then washed out, short synchronized discharges with stable duration were recorded for up to 120 min after DHPG washout (10 min: 271 ± 13 ms; 120 min: 286 ± 19 ms; n = 6; p = 0.37).
Figure 1.
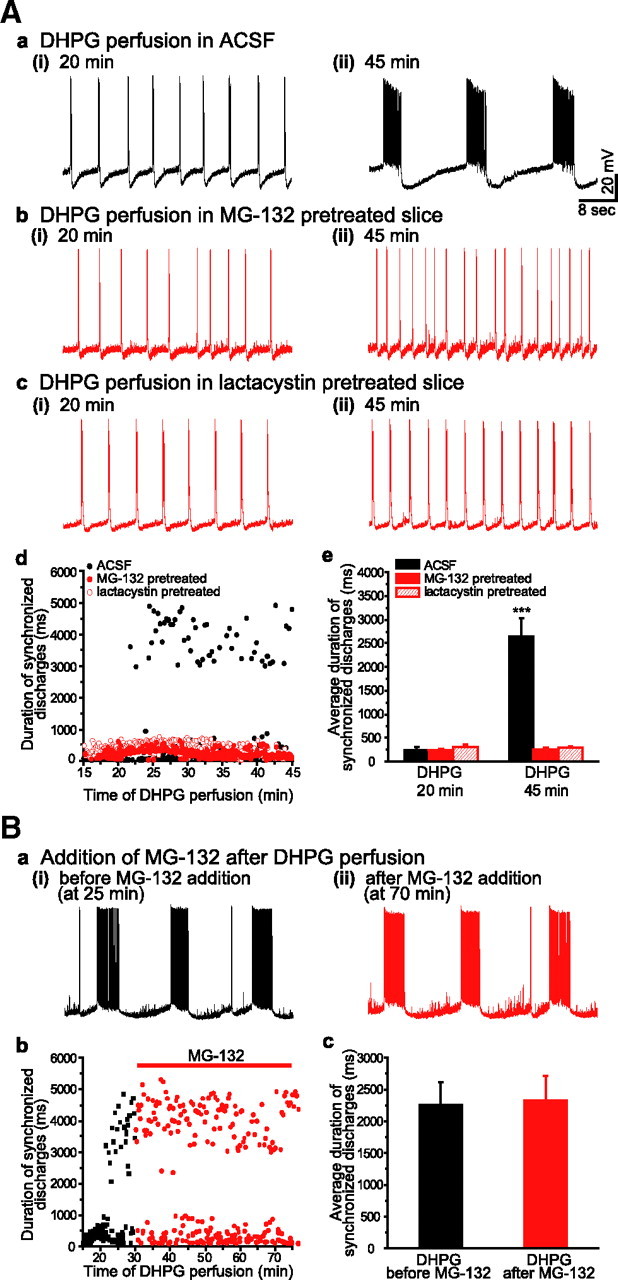
Suppression of UPS activity by proteasome inhibitors blocked the induction of prolonged synchronized discharges in response to group I mGluR stimulation. Aa–c, Intracellular recordings from CA3 pyramidal cell at early (i) and late (ii) time points after DHPG (50 μm) application. In ACSF (Aa), DHPG elicited short synchronized discharges (Aa(i)) followed by prolonged synchronized discharges (Aa(ii)). In slices pretreated with a proteasome inhibitor, MG-132 (1 μm, Ab) or lactacystin (5 μm, Ac), extended perfusion of DHPG failed to induce prolonged synchronized discharges. Ad, The duration time course of DHPG-induced synchronized discharges in ACSF (black filled circles), MG-132 (red filled circles), and lactacystin (red empty circles). Pretreatment of proteasome inhibitors prevented the occurrence of prolonged synchronized discharges. Ae, Synchronized discharge durations averaged over 5 min periods ending at 20 min and at 45 min of DHPG perfusion. In ACSF, at 20 min, the average discharge duration was 246 ± 56 ms (mean ± SEM; short synchronized discharges) and significantly increased to 2639 ± 383 ms at 45 min (prolonged synchronized discharges; n = 12, ***p < 0.001 compared with that at 20 min). Under the condition of proteasome inhibitors, there were no significant change in average discharge durations between at 20 min and at 45 min and only short synchronized discharges were recorded (MG-132: at 20 min, 227 ± 33 ms; at 45 min, 222 ± 20 ms; n = 7; p = 0.89; lactacystin: at 20 min, 325 ± 58 ms; at 45 min, 290 ± 30 ms; n = 5; p = 0.60). Ba, Intracellular recordings of DHPG-induced prolonged synchronized discharges before (at 25 min) and after (at 70 min) the addition of MG-132. Bb, the duration time course of DHPG-induced synchronized discharges in response to the addition of MG-132 (black squares, before MG-132; red circles, during MG-132). Bc, Average durations of synchronized discharges duration before and after the addition of MG-132. Average duration of prolonged was not affected by MG-132 addition (before MG-132: 2254 ± 363 ms; after MG-132 addition: 2319 ± 390 ms; n = 5; p = 0.91). Red label indicates the presence of proteasome inhibitor.
The involvement of UPS in the induction of prolonged synchronized discharges was examined using MG-132, a proteasome inhibitor (Lee and Goldberg, 1996). Slices were pretreated with MG-132 (1 μm) for 45 min before application of DHPG. DHPG perfusion elicited short synchronized discharges in a manner similar to that observed in ACSF (Fig. 1Ab(i)). However, the presence of MG-132 prevented the subsequent development of the prolonged synchronized discharges observed in control conditions (Fig. 1Ab(ii)).
The involvement of UPS in the induction of prolonged synchronized discharges was further examined using another proteasome inhibitor, lactacystin (5 μm) (Fenteany et al., 1995). Pretreatment with lactacystin for 45 min was also effective in preventing the occurrence of DHPG-induced prolonged synchronized discharges (Fig. 1Ac(ii)). The results indicate that UPS is required for the activation of prolonged synchronized discharges.
Following an extended period of DHPG exposure, when prolonged synchronized discharges were elicited, application of MG-132 to the perfusate did not significantly affect the duration or the frequency of ongoing prolonged synchronized discharges (Fig. 1B). Thus, MG-132 prevents prolonged synchronized discharge activation but does not affect the properties of these discharges once they are expressed.
DHPG elicits downregulation of FMRP via UPS
Results in Figure 1 demonstrate that UPS activity appears to play a role in the induction but not in the maintenance of the prolonged synchronized discharges. UPS also regulates group I mGluR-mediated LTD in hippocampal slices (Hou et al., 2006; Citri et al., 2009). Proteolysis of FMRP has been implicated in this regulation (Hou et al., 2006).
The time course of FMRP expression in hippocampal slices in response to group I mGluR stimulation was studied using Western blots. Slices were collected at 0, 10, and 30 min after DHPG (100 μm) application. DHPG elicited a biphasic change in FMRP (Fig. 2A). A decrease was observed 10 min following DHPG application. At 30 min, an increase of FMRP over control was observed. These results suggest that group I mGluRs exert a bidirectional control on FMRP levels, possibly eliciting an initial degradation and subsequent synthesis.
Figure 2.

UPS-dependent FMRP downregulation elicited by group I mGluR stimulation. Hippocampal slices were incubated in a slice holding chamber at 32.5°C. After DHPG (D; 100 μm) exposure, slices were collected at 0 (Con), 10 min (D10min), and 30 min (D30min). Pooled slices were subjected to Western blot analysis of FMRP expression level. Representative Western blot results are shown in the right panel and the summary data in the left panel. A, In ACSF, stimulation of group I mGluR elicited a transient decrease in FMRP at 10 min followed by the increase in FMRP at 30 min (at 10 min: 85 ± 2.1%, n = 8, ***p < 0.001; at 30 min: 121.4 ± 3.7%, n = 8, ***p < 0.001; compared with control). The DHPG-induced FMRP decrease was further evaluated in the presence of protein synthesis inhibitors. B, In slices pretreated with anisomycin (Aniso; 20 μm), DHPG also induced decrease (84.8 ± 2%, n = 8, p < 0.001; compared with control) in FMRP at 10 min, comparable to that observed in ACSF. In the presence of anisomycin, the decrease in FMRP was sustained (at 30 min: 81 ± 2.6%, n = 8, ***p < 0.001; compared with control). The involvement of UPS was examined using a proteasome inhibitor MG-132. In slices pretreated with anisomycin and MG-132, application of DHPG no longer elicited decrease in FMRP at either 10 min (99.5 ± 1.7%, n = 8, p = 0.8; compared with control) or 30 min (97.9 ± 2.2%, n = 8, p = 0.76; compared with control) time point. C, Similarly, DHPG-induced FMRP decrease was sustained in slices pretreated with another protein synthesis inhibitor cycloheximide (CHX) (100 μm) (at 10 min: 72.7 ± 4.1%, n = 8, ***p < 0.001; at 30 min: 76.8 ± 3.5%, n = 8, ***p < 0.001; compared with control). Such decrease was prevented by the co-pretreatment of MG-132 (at 10 min: 98.3 ± 5%, n = 8, p = 0.74; at 30 min: 101 ± 4%, n = 8, p = 0.93; compared with control). FMRP immunoresponse was normalized to that of α-tubulin (α-Tub).
Since we assumed that the conditions for biochemical studies were adequate for the activation of DHPG-induced synchronized discharges, we transferred the same slices that were preincubated under conditions for biochemical studies into the recording chamber for electrophysiological studies. Slices were first treated with 100 μm DHPG at 32.5°C in the incubation chamber previously used for biochemical studies. Afterward, slices were briefly washed with control solution and immediately transferred to an interface recording chamber perfused with control (i.e., no DHPG) solution. Recordings were performed after 20 min at 34.5°C in the interface chamber. Prolonged epileptiform discharges of similar properties as those observed in electrophysiological studies were recorded in 6 of 6 slices tested (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Additional data show that the duration of prolonged epileptiform discharges were similar when slices were treated with 50 or 100 μm DHPG at 32.5°C in the incubation chamber (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). These results indicate that the conditions for DHPG exposure in the biochemical studies reflect those required for induction of prolonged epileptiform discharges.
To examine the degradation process in isolation (independent of synthesis), Western blot experiments were performed in the presence of protein synthesis inhibitors (anisomycin or cycloheximide). The data show that at 10 min of DHPG exposure the FMRP level decreased below baseline (Fig. 2B,C, 10 min, gray columns), similar to that observed in the absence of protein synthesis inhibitors. At 30 min of DHPG exposure, the FMRP level remained below baseline (Fig. 2B,C, 30 min, gray columns). This result differs from that observed in control (i.e., in the absence of protein synthesis inhibitors), where FMRP level was increased (Fig. 2A, 30 min). Thus, protein synthesis inhibitors do not affect initial FMRP decrease but do prevent its subsequent increase and uncovered a decrease at 30 min.
To evaluate the role of UPS in FMRP decrease, slices were incubated in a solution containing both a protein synthesis inhibitor and the proteasome inhibitor MG-132. Under this condition, a stable FMRP level was measured following DHPG at 10 and 30 min (Fig. 2B,C, black columns). These data indicate that the FMRP decrease is caused by the UPS proteolytic pathway.
Proteins destined for degradation via the UPS are processed by two concerted enzymatic actions: the attachment of chains of the polypeptide cofactor, ubiquitin, and subsequent recognition and cleavage by the proteasome. MG-132 blocks the proteolytic activity of proteasome. Thus, the presence of MG-132 would be expected to cause accumulation of proteins linked with multiple repeats of ubiquitin. To test the effectiveness of MG-132 at the dose and duration that we used, the ubiquitinated proteins in hippocampal slices treated with MG-132 (1 μm for 45 min) were immunoprecipitated with the mouse FK2 anti-ubiquitin antibody, and subsequently subjected to Western blot analysis with a different ubiquitin antibody. We found that ubiquitinated proteins were concentrated by immunoprecipitation (supplemental Fig. S2A, IP: ubiquitin, available at www.jneurosci.org as supplemental material) and that MG-132 exposure enhanced the amount of ubiquitinated proteins with molecular weight greater than ∼98 kDa (supplemental Fig. S2A,B, MG, available at www.jneurosci.org as supplemental material).
Data shown in Figure 2, B and C, suggest that the UPS is involved in DHPG-induced decrease of FMRP levels. We examined whether FMRP is a direct target of the UPS in response to group I mGluR stimulation. FMRP was successfully immunoprecipitated using the mouse 7G1 anti-FMRP antibody (supplemental Fig. S3A, IP: FMRP, WT, available at www.jneurosci.org as supplemental material). Following the same IP procedure, the ubiquitination status of FMRP in slices treated with or without DHPG in the presence of proteasome inhibitors (MG-132 or lactacystin) was examined using a ubiquitin antibody (supplemental Fig. S3Ba, available at www.jneurosci.org as supplemental material). Ubiquitin conjugates of FMRP (supplemental Fig. S3Ba, band indicated by arrowhead and higher MW bands, available at www.jneurosci.org as supplemental material) were detected in ACSF and DHPG-treated preparations. In the presence of proteasome inhibitors (MG-132 or lactacystin), DHPG increased FMRP ubiquitin conjugates (supplemental Fig. S3Ba,c, available at www.jneurosci.org as supplemental material). Probing the IP lysate with an FMRP antibody revealed, in addition to the concentrated FMRP bands, higher molecular weight bands that had the same mobility as those detected with the ubiquitin antibody (supplemental Fig. S3Bb, available at www.jneurosci.org as supplemental material).
MG-132 did not prevent induction of prolonged synchronized discharges in Fmr1−/− preparations
One function of FMRP is to repress mRNA translation (Laggerbauer et al., 2001; Li et al., 2001; Bear et al., 2004). A transient decrease in FMRP (Fig. 2A, DHPG 10 min) may unleash translation and allow for DHPG-induced epileptogenesis. MG-132, by blocking this decrease, may thereby prevent translation and epileptogenesis. If baseline FMRP levels were sufficiently low and a transient decrease were no longer necessary for translation to occur, blocking this transient decrease by MG-132 should not arrest on-going translation and associated epileptogenesis. We examined MG-132 action in Fmr1−/− preparations, where FMRP is functionally absent (The Dutch-Belgian Fragile X Consortium, 1994).
After DHPG perfusion, stimulation of group I mGluRs sequentially elicited short and prolonged synchronized discharges in Fmr1−/− slice (Fig. 3Aa), as observed in wild-type slices (Fig. 1Aa). In a parallel set of experiments, slices from Fmr1−/− animals were pretreated with MG-132 for 45 min. DHPG application induced typical prolonged synchronized discharges with duration and frequency comparable to those observed in the absence of MG-132 (Fig. 3Ab). The results demonstrate that MG-132 did not affect the induction or maintenance of prolonged synchronized discharges in the Fmr1−/− preparation.
Figure 3.

UPS activity is no longer required for the induction of prolonged synchronized discharges in Fmr1−/− preparations. Aa–d, Effect of MG-132 on agonist induced prolonged synchronized discharges. Intracellular recordings from Fmr1−/− mice showing the development of synchronized discharges at early (i) and late (ii) time points after DHPG application under the condition of ACSF (Aa) and MG-132 pretreatment (Ab). Stimulation of group I mGluRs in Fmr1−/− preparation elicited prolonged synchronized discharges (Aa(ii)) with comparable duration to that of observed in WT animal. In slices pretreated with MG-132, DHPG perfusion also induced prolonged synchronized discharges (Ab(ii)) indistinguishable from those recorded in ACSF (Aa(ii)). Ac, The duration time course of synchronized discharges from Fmr1−/− preparations in response to DHPG perfusion under the condition of ACSF (black filled circles) and MG-132 pretreatment (red filled circles). Ad, Synchronized discharge durations averaged over 5 min periods ending at 20 min (264 ± 32 ms) and at 45 min of DHPG perfusion (2316 ± 319 ms; n = 6; **p < 0.01 compared with that at 20 min). In the Fmr1−/− preparations, prolonged synchronized discharges also appeared in MG-132 pretreated slices (2232 ± 263 ms; n = 6; **p < 0.01, compared with DHPG 20 min in MG-132) and no significant difference in their duration was detected between records in ACSF and in MG-132 pretreatment (at 45 min: ACSF, 2316 ± 319 ms; MG-132, 2232 ± 263 ms; n = 6; p = 0.60). Ba–d, Effect of MG-132 on the induction of prolonged synchronized discharges elicited by synaptically released glutamate. Ba, b, Recordings from Fmr1−/− preparations. Synchronized discharges at 45 min (i) and at 75 min (ii) in the presence of bicuculline (50 μm). Rhythmic short synchronized discharges was induced in CA3 hippocampus with duration similar to that recorded in WT animal (at 45 min: WT, 327 ± 26 ms; Fmr1−/−, 313 ± 20 ms; n = 8; p = 0.68). However, extended perfusion of bicuculline brought out the occurrence of prolonged synchronized discharges in Fmr1−/− slices. Pretreated with MG-132 did not affect prolonged synchronized discharges (ACSF: 2652 ± 382 ms; n = 8; MG-132 pretreatment: 2589 ± 649 ms; n = 6; p = 0.93). Bc, The time course of synaptically induced prolonged synchronized discharges under the condition of ACSF (black filled circles) and MG-132 pretreatment (red filled circles). Bd, Synchronized discharge durations averaged over 5 min periods ending at 45 min and at 75 min of bicuculline (Bic) perfusion. The average durations at 75 min were significantly longer than those at 45 min under the condition of both ACSF and MG-132 pretreatment (ACSF: at 45 min, 313 ± 20 ms; at 75 min: 2652 ± 382 ms; n = 8; ***p < 0.001; MG-132: at 45 min, 356 ± 27 ms; at 75 min: 2589 ± 649 ms; n = 6; **p < 0.01).
Group I mGluR-mediated prolonged synchronized discharges can also be induced synaptically in Fmr1−/− preparations (Chuang et al., 2005). The effects of MG-132 on synaptically induced prolonged synchronized discharges were also examined. Similar to agonist-elicited events, synaptically activated prolonged discharges persisted in the presence of MG-132 (Fig. 3Bb–d).
MG-132 prevents group I mGluR-mediated protein synthesis
The data above indicate that MG-132 prevents DHPG-induced transient FMRP decrease (Fig. 2) and the associated prolonged synchronized discharges (Fig. 1). We examined the effects of MG-132 on group I mGluR-mediated protein synthesis.
A subset of three proteins—FMRP, PSD-95, and α-CaMKII—whose syntheses are known to be stimulated by group I mGluR stimulation (Weiler et al., 1997; Todd et al., 2003a; Hou et al., 2006; Muddashetty et al., 2007) were monitored using Western blots. Their expression levels were measured at 10 min and 30 min after DHPG. At the 10 min time point, group I mGluR stimulation elicited a decrease in FMRP level, while the levels of PSD-95 and α-CaMKII were not significantly altered (Fig. 4A, DHPG 10 min). At the 30 min time point, significant increases in FMRP, PSD-95, and α-CaMKII were detected (Fig. 4A, 30 min).
Figure 4.

Control of FMRP downregulation on group I mGluR-mediated protein synthesis. In response to DHPG (D) exposure, the changes in expression of FMRP, PSD-95, and α-CaMKII at 10 min (D10min) and 30 min (D30min) time points were examined using Western blot analysis. A, In ACSF, stimulation of group I mGluRs elicited a biphasic changes in FMRP expression level: a transient decrease at 10 min (85 ± 2.1%, n = 8, **p < 0.01) followed by an increase at 30 min (121.4 ± 3.7%, n = 8, **p < 0.01). For PSD-95 and α-CaMKII, the expression levels were stable at 10 min DHPG application, at 30 min exposure significant increases in their expression levels were detected (PSD-95: at 10 min, 97.9 ± 4.1%, n = 8, p = 0.62; at 30 min, 118.1 ± 3.4%, n = 8, **p < 0.01; α-CaMKII: at 10 min, 98.2 ± 2.6%, n = 8, p = 0.52; at 30 min, 117 ± 4.4%, n = 8, **p < 0.01). B, Effect of anisomycin. DHPG exposure no longer affected PSD-95 and α-CaMKII expression levels (PSD-95: at 10 min, 99.8 ± 5.3%, n = 8, p = 0.96; at 30 min, 100.8 ± 3.3%, n = 8, p = 0.83; α-CaMKII: at 10 min, 101 ± 1.9%, n = 8, p = 0.95; at 30 min, 97.8 ± 4.0%, n = 8, p = 0.60). For FMRP, the late increase (at 30 min) in response to DHPG exposure was blocked unmasking a sustained decrease comparable to that detected at 10 min (FMRP: at 10 min, 84.8 ± 2%, n = 8, ***p < 0.001; at 30 min, 81 ± 2.6%, n = 8, ***p < 0.001). C, The early (at 10 min) and late (30 min) decreases of FMRP in the presence of anisomycin was prevented in slices co-pretreated with MG-132, a proteasome inhibitor (FMRP: at 10 min, 99.5 ± 1.7%, n = 8, p = 0.80; at 30 min, 97.9 ± 2.2%, n = 8, p = 0.40). D, Effect of MG-132 alone on group I mGluR-mediated protein synthesis. In slices pretreated with MG-132, 10 min DHPG exposure no longer caused any change in FMRP expression level (at 10 min: 98 ± 4.1%, n = 8, p = 0.67; at 30 min: 99 ± 1.7%, n = 8, p = 0.80). MG-132 also suppressed the late increases (at 30 min) in PSD-95 and α-CaMKII levels (PSD-95: at 10 min, 98.8 ± 4.4%, n = 8, p = 0.80; at 30 min, 100.4 ± 5.1%, n = 8, p = 0.94; α-CaMKII: at 10 min, 100 ± 3.6%, n = 8, p = 0.97; at 30 min, 98 ± 2.5%, n = 8, p = 0.42). FMRP, PSD-95, and α-CaMKII immunoresponses were normalized to that of α-tubulin (α-Tub).
In slices pretreated with the translation inhibitor anisomycin, the protein level profile at 10 min after DHPG resembled that observed in the absence of anisomycin (control), namely, FMRP decreased below baseline whereas PSD-95 and α-CaMKII levels were maintained at baseline (Fig. 4B, DHPG 10 min). After 30 min of DHPG in anisomycin, protein levels differed significantly from those expressed in control conditions. Instead of an overall increase, PSD-95 and α-CaMKII remained at baseline levels (Fig. 4B, DHPG 30 min). FMRP, instead of an increase, showed a significant decrease below baseline (Fig. 4B, DHPG 30 min), as described before in Figure 2.
MG-132 applied in the presence of anisomycin eliminated the DHPG-induced decrease of FMRP at the 10 and 30 min time points. Furthermore, the levels of all three proteins monitored under these conditions remained stable following DHPG application (Fig. 4C).
The isolated effects of MG-132 were then tested on DHPG-mediated protein synthesis under the control condition (i.e., in the absence of anisomycin). Figure 4D shows that in the presence of MG-132 alone application of DHPG no longer elicited measurable changes in the level of all three proteins at 10 and 30 min. Additional experiments show that lactacystin also prevented changes in FMRP levels during DHPG (supplemental Fig. S5B, available at www.jneurosci.org as supplemental material).
ERK1/2 phosphorylation in MG-132
Previously, we showed that activation of group I mGluR-mediated, translation-dependent, responses including prolonged synchronized discharges requires ERK1/2 phosphorylation (Gallagher et al., 2004; Zhao et al., 2004; Chuang et al., 2005). Thus, the action of the ERK1/2 inhibitor U0126 (20 μm) on DHPG-stimulated changes in FMRP (Fig. 5A) is similar to that caused by protein synthesis inhibitors (Fig. 4B). In slices pretreated with U0126, 30 min DHPG exposure failed to elicit changes in PSD-95 and α-CaMKII levels, whereas FMRP downregulation was not affected (Fig. 5A).
Figure 5.

Group I mGluR-mediated parallel and independent controls of ERK1/2 activation and FMRP downregulation. A, In slices pretreated with the ERK1/2 inhibitor U0126 (20 μm), 30 min DHPG exposure (D30min) failed to elicit group I mGluR-mediated protein synthesis of PSD-95 and α-CaMKII (PSD95: 99.4 ± 5.0%, n = 8, p = 0.91; α-CaMKII: 98.6 ± 3.5%, n = 8, p = 0.70). FMRP downregulation was not affect by U0126 pretreatment (U0126: 85.4 ± 4.5%, n = 8, **p < 0.01). B, MG-132 effects on DHPG-induced ERK1/2 phosphorylation in wild-type (a) and Fmr1−/− (b) preparations. Ba, In WT, DHPG increased ERK1/2 phosphorylation at 10 min and 30 min in ACSF (gray bars: DHPG 10 min, 179 ± 4%, n = 8; DHPG 30 min, 149 ± 3%, n = 8; **p < 0.01 compared with control). MG-132 did not affect the basal level of ERK1/2 phosphorylation (black bars, Control, 103 ± 3%, n = 8; p = 0.28 compared with control in ACSF) and did not block the DHPG-induced ERK1/2 phosphorylation (black bars: DHPG 10 min, 219 ± 8%, n = 8; DHPG 30 min, 198 ± 7%, n = 8; **p < 0.01 compared with control). In fact, DHPG-induced ERK1/2 phosphorylation in slices pretreated with MG-132 was maintained at higher levels at both time points than those in ACSF (**p < 0.01). Bb, In Fmr1−/− preparations, DHPG still induced ERK1/2 phosphorylation (gray bars: DHPG 10 min, 156 ± 7%, n = 8; DHPG 30 min, 118 ± 3%, n = 8; **p < 0.01 compared with control). Compared with WT, the ERK1/2 phosphorylation occurred at a suppressed level (DHPG 10 min: WT vs Fmr1−/−, p < 0.05; DHPG 30 min: WT vs Fmr1−/−, p < 0.001). In Fmr1−/−, DHPG induced ERK1/2 phosphorylation also after MG-132 treatment (black bars: DHPG 10 min, 169 ± 4%, n = 8; DHPG 30 min, 147 ± 3%, n = 8; **p < 0.01 compared with control), with a pattern similar to that observed in WT.
We then examined (1) ERK1/2 activation at both 10 and 30 min time points in ACSF, (2) the effects of MG-132 on ERK1/2 activation at 10 and 30 min, and (iii) ERK1/2 activation in the presence or absence of MG-132 in Fmr1−/− preparations. (1) Consistent with previous results (Zhao et al., 2004), the data show that DHPG increased ERK1/2 activation at both time points (10 and 30 min) in ACSF. The increase peaked at 10 min and then declined at 30 min, yet remaining at levels higher than in control conditions (Fig. 5Ba, gray bars), likely as a result of increased neuronal activity (Zhao et al., 2004). (2) DHPG-induced ERK1/2 activation also occurred in the presence of MG-132 (Fig. 5Ba, black bars). ERK1/2 activation was enhanced at both time points in the presence of MG-132, which blocks UPS activity, consistent with previous data from Citri et al. (2009). (3) Also, the patterns of DHPG-induced ERK1/2 activation observed in Fmr1−/− preparations perfused in ACSF or with added MG-132 were similar to those observed in wild type, but at suppressed levels (Fig. 5Bb).
Discussion
Our data show that the UPS plays a critical role in regulating group I mGluR-mediated epileptogenesis. This action of UPS is mediated by proteolysis of FMRP. By targeting FMRP, UPS presents a permissive state for group I mGluR to activate protein synthesis that underlies epileptogenesis.
FMRP as a target of UPS proteolysis
The first indication of UPS involvement in group I mGluR-mediated epileptogenesis is the arrest of group I mGluR-mediated prolonged synchronized discharges when UPS action is blocked by MG-132 (Fig. 1Ab,e) or lactacystin (Fig. 1Ac,e). Using Western blots we searched for targets of UPS proteolysis and detected an early transient decrease in FMRP level following agonist stimulation. This transient decrease is eliminated by UPS blocker MG-132 suggesting that FMRP is a target of UPS proteolysis, a finding that is further supported by data shown in supplemental Fig. S3, available at www.jneurosci.org as supplemental material. Figure 2A demonstrates that decreased FMRP level is followed by an increase elicited by de novo protein synthesis. In the presence of a translation inhibitor, a sustained decrease in FMRP is uncovered (Fig. 2B,C). The data suggest that group I mGluR stimulation elicits two parallel processes that cause the proteolysis and synthesis of FMRP, respectively.
Mechanism of UPS regulation of group I mGluR-mediated epileptogenesis
We monitored three proteins—PSD-95, α-CaMKII, and FMRP—whose syntheses are known to be stimulated by group I mGluR activation (Weiler et al., 1997; Todd et al., 2003a; Hou et al., 2006; Muddashetty et al., 2007), and we detected their increases following receptor stimulation (Fig. 4A). Western blot data show that the syntheses of these proteins are arrested when UPS activity is blocked by MG-132 (Fig. 4D). This observation can be explained by an action of UPS on FMRP. One known function of FMRP is repression of translation (Laggerbauer et al., 2001; Li et al., 2001; Bear et al., 2004). Presumably, constitutive FMRP is present at a sufficient level to repress group I mGluR-mediated translation. Proteolysis of FMRP by UPS relieves translation repression and enables protein synthesis and the generation of group I mGluR-mediated synchronized discharges. The above hypothesis, emphasizing the importance of UPS action on FMRP as a gating mechanism for epileptogenesis, is depicted in Figure 6.
Figure 6.

Group I mGluR activates UPS-mediated proteolysis of FMRP to enable translation and epileptogenesis. A, FMRP is an inhibitor of protein synthesis in normal conditions (Control). B, Group I mGluR-mediated translation is relieved from FMRP repression, via UPS-dependent proteolysis of FMRP, and proceeds to activate epileptogenesis. C, MG132, by blocking proteasome action, inhibits proteolysis of FMRP by UPS. FMRP is now present at a sufficient level to repress group I mGluR-mediated translation and epileptogenesis. D, in Fmr1−/− preparations, FMRP is absent; proteolysis of FMRP is no longer necessary for protein synthesis and epileptogenesis.
We monitored protein synthesis in our study by using Western blots and not directly by 35S-Met incorporation. This leaves open at least two alternative explanations other than changes in synthesis: (1) that fluctuations in basal protein synthesis may be sufficiently large to obscure DHPG-induced responses or (2) that the increase in protein level by DHPG is due to downregulation of protein degradation. We addressed these issue by measuring protein profiles under three conditions: in control (ACSF), in MG-132 (45 min treatment), and in anisomycin (45 min treatment). No significant differences were detected in the baseline protein levels measured (supplemental Fig. S4, available at www.jneurosci.org as supplemental material), suggesting that on-going protein synthesis (in ACSF) and degradation (in MG-132 and in anisomycin) are unlikely to contribute significantly to the DHPG-stimulated changes observed in the time window of our experiments.
The hypothesis depicted in Figure 6 is faced with two challenges. First, since many proteins are targets of UPS and group I mGluR stimulation can downregulate these proteins (Citri et al., 2009), a unique action of UPS on FMRP cannot be the only conclusion. Second, the hypothesis assumes that the action of MG-132 is specifically on UPS and that MG-132 does not have nonspecific effects on translation. The Fmr1−/− preparation allowed for an assessment of these possibilities. The single difference between the Fmr1−/− preparation and the wild-type preparation is the absence of functional FMRP in the former (The Dutch-Belgian Fragile X Consortium, 1994). Figure 3B shows that, in the absence of FMRP, MG-132 no longer suppressed group I mGluR-mediated synchronized discharges in the Fmr1−/− preparation, in distinct contrast to the wild-type preparation. These data suggest that the target of MG-132 is FMRP; in the absence of FMRP, MG-132 is no longer effective. Data obtained in Fmr1−/− preparations also indicate that the permissive action of UPS on the generation of prolonged synchronized discharges is specifically on the proteolysis of FMRP.
Group I mGluR-mediated protein synthesis in the Fmr1−/− preparation
We have previously shown that group I mGluR-mediated synaptic induction of epileptogenesis in Fmr1−/− mice is dependent on protein synthesis (Chuang et al. 2005). Specific protein(s) involved have not been identified, but the data indicate that synthesis of the involved protein(s) is regulated by the translation repressor FMRP (Chuang et al. 2005). The synthesis of a number of other proteins, including PSD-95 (Todd et al., 2003a; Muddashetty et al., 2007), α-CaMKII (Grossman et al., 2006), and protein(s) involved in LTD induction (as yet to be identified) (Nosyreva and Huber, 2006), is also stimulated by group I mGluRs and regulated by FMRP. We monitored this group of proteins in correlation with FMRP level as indicators of the status of the translation process involved in epileptogenesis. Biochemical studies of PSD-95 and α-CaMKII show that, while mGluR stimulation elevates their levels in the wild-type preparations, their levels are no longer stimulated by group I mGluRs in Fmr1−/− preparations (Todd et al., 2003a; Hou et al., 2006; Muddashetty et al., 2007). This finding seems to contradict data indicating that FMRP represses the synthesis of these proteins. A possible explanation is that baseline protein synthesis is enhanced in Fmr1−/− (Todd et al., 2003a; Qin et al., 2005; Muddashetty et al., 2007) so that additional stimulated synthesis is occluded in two ways. First, the elevated baseline may decrease signal-to-noise ratio for biochemical detection. Second, the elevated baseline translation may decrease the translational capacity for additional stimulated synthesis. Thus, group I mGluR-stimulated protein synthesis may still occur in Fmr1−/− preparations but has yet to be demonstrated biochemically.
Group I mGluR-dependent epileptiform discharges can be elicited synaptically in Fmr1−/− preparations but not in the wild type (Chuang et al., 2005; Bianchi et al., 2009). In the wild type, such epileptiform discharges can only be elicited by extended periods of group I mGluR agonist stimulation (Merlin et al., 1998) whereupon proteolysis of FMRP occurs (Figs. 1, 2). This induction protocol may be relevant to pathological conditions, as in stroke, where prolonged release of glutamate occurs (Rothman and Olney, 1986) providing a mechanism for the group I mGluR-mediated epileptogenesis.
Group I mGluR modulation of FMRP
Hou et al. (2006) reported a lingering effect of UPS action on FMRP after group I mGluR stimulation. Our study emphasizes an earlier action of UPS that plays a critical permissive role in the activation of group I mGluR-mediated translation and prolonged synchronized discharges (Fig. 6). The study by Hou et al. (2006) shows an increase in FMRP at 10 min DHPG, whereas we demonstrated a decrease at this time point (Fig. 2A). We note that the former study is performed in the CA1 region of the slice whereas we used the whole hippocampal slice. We performed additional experiments in the CA1 region for a more direct comparison. The data showed a decrease in FMRP at 10 min after DHPG application and an increase at 30 min in the CA1 preparation (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material), as observed in the whole slice preparation (Fig. 4). In addition—different from Hou et al. (2006)—in the presence of MG-132, DHPG no longer affected FMRP levels (i.e., the FMRP level was stable) (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material). At present the reason for the discrepancy in the findings is not immediately apparent. Variations in age, sex or strain may be contributing factors. In addition to proteolysis and translation, group I mGluR stimulation also elicits FMRP phosphorylation (Narayanan et al., 2007) or trafficking in dendrites and synapses (Antar et al., 2004). These modifications regulate the effectiveness of FMRP as a translation repressor. Our study does not evaluate the overall effect of these mechanisms on group I mGluR-mediated responses but does point out that proteolysis of FMRP is a necessary step for the initiation of group I mGluR-dependent translation and prolonged synchronized discharges.
Footnotes
This work has been supported by the FRAXA Research Foundation and National Institutes of Health Grant NS35481. The FMRP 7G1-1 monoclonal antibody developed by Stephen T. Warren was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by The University of Iowa, Department of Biology, Iowa City, Iowa 52242.
References
- Antar LN, Afroz R, Dictenberg JB, Carroll RC, Bassell GJ. Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J Neurosci. 2004;24:2648–2655. doi: 10.1523/JNEUROSCI.0099-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Bianchi R, Chuang SC, Zhao W, Young SR, Wong RK. Cellular plasticity for group I mGluR-mediated epileptogenesis. J Neurosci. 2009;29:3497–3507. doi: 10.1523/JNEUROSCI.5447-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceman S, O'Donnell WT, Reed M, Patton S, Pohl J, Warren ST. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet. 2003;12:3295–3305. doi: 10.1093/hmg/ddg350. [DOI] [PubMed] [Google Scholar]
- Chuang SC, Zhao W, Young SR, Conquet F, Bianchi R, Wong RK. Activation of group I mGluRs elicits different responses in murine CA1 and CA3 pyramidal cells. J Physiol. 2002;541:113–121. doi: 10.1113/jphysiol.2001.013309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci. 2005;25:8048–8055. doi: 10.1523/JNEUROSCI.1777-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Soler-Llavina G, Bhattacharyya S, Malenka RC. N-Methyl-d-aspartate receptor- and metabotropic glutamate receptor-dependent long-term depression are differentially regulated by the ubiquitin-proteasome system. Eur J Neurosci. 2009;30:1443–1450. doi: 10.1111/j.1460-9568.2009.06950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Ambrosio R, Hakimian S, Stewart T, Verley DR, Fender JS, Eastman CL, Sheerin AH, Gupta P, Diaz-Arrastia R, Ojemann J, Miller JW. Functional definition of seizure provides new insight into post-traumatic epileptogenesis. Brain. 2009;132:2805–2821. doi: 10.1093/brain/awp217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Fenteany G, Standaert RF, Lane WS, Choi S, Corey EJ, Schreiber SL. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science. 1995;268:726–731. doi: 10.1126/science.7732382. [DOI] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, Bear MF, Huber KM. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci. 2004;24:4859–4864. doi: 10.1523/JNEUROSCI.5407-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman AW, Aldridge GM, Weiler IJ, Greenough WT. Local protein synthesis and spine morphogenesis: Fragile X syndrome and beyond. J Neurosci. 2006;26:7151–7155. doi: 10.1523/JNEUROSCI.1790-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron. 2006;51:441–454. doi: 10.1016/j.neuron.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet. 2001;10:329–338. doi: 10.1093/hmg/10.4.329. [DOI] [PubMed] [Google Scholar]
- Lee DH, Goldberg AL. Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J Biol Chem. 1996;271:27280–27284. doi: 10.1074/jbc.271.44.27280. [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29:2276–2283. doi: 10.1093/nar/29.11.2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin LR, Wong RK. Role of group I metabotropic glutamate receptors in the patterning of epileptiform activities in vitro. J Neurophysiol. 1997;78:539–544. doi: 10.1152/jn.1997.78.1.539. [DOI] [PubMed] [Google Scholar]
- Merlin LR, Bergold PJ, Wong RK. Requirement of protein synthesis for group I mGluR-mediated induction of epileptiform discharges. J Neurophysiol. 1998;80:989–993. doi: 10.1152/jn.1998.80.2.989. [DOI] [PubMed] [Google Scholar]
- Muddashetty RS, Kelić S, Gross C, Xu M, Bassell GJ. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci. 2007;27:5338–5348. doi: 10.1523/JNEUROSCI.0937-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan U, Nalavadi V, Nakamoto M, Pallas DC, Ceman S, Bassell GJ, Warren ST. FMRP phosphorylation reveals an immediate-early signaling pathway triggered by group I mGluR and mediated by PP2A. J Neurosci. 2007;27:14349–14357. doi: 10.1523/JNEUROSCI.2969-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Metabotropic receptor-dependent long-term depression persists in the absence of protein synthesis in the mouse model of fragile X syndrome. J Neurophysiol. 2006;95:3291–3295. doi: 10.1152/jn.01316.2005. [DOI] [PubMed] [Google Scholar]
- Qin M, Kang J, Burlin TV, Jiang C, Smith CB. Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J Neurosci. 2005;25:5087–5095. doi: 10.1523/JNEUROSCI.0093-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Olney JW. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann Neurol. 1986;19:105–111. doi: 10.1002/ana.410190202. [DOI] [PubMed] [Google Scholar]
- The Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Todd PK, Mack KJ, Malter JS. The fragile X mental retardation protein is required for type-I metabotropic glutamate receptor-dependent translation of PSD-95. Proc Natl Acad Sci U S A. 2003a;100:14374–14378. doi: 10.1073/pnas.2336265100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Malter JS, Mack KJ. Whisker stimulation-dependent translation of FMRP in the barrel cortex requires activation of type I metabotropic glutamate receptors. Brain Res Mol Brain Res. 2003b;110:267–278. doi: 10.1016/s0169-328x(02)00657-5. [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT. Fragile X mental retardation protein is translated near synapses in response to neurotransmitter activation. Proc Natl Acad Sci U S A. 1997;94:5395–5400. doi: 10.1073/pnas.94.10.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Zhao W, Bianchi R, Wang M, Wong RK. Extracellular signal-regulated kinase 1/2 is required for the induction of group I metabotropic glutamate receptor-mediated epileptiform discharges. J Neurosci. 2004;24:76–84. doi: 10.1523/JNEUROSCI.4515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Chuang SC, Bianchi R, Zhao W, Lee H, Fenton AA, Wong RK, Tiedge H. BC1 regulation of metabotropic glutamate receptor-mediated neuronal excitability. J Neurosci. 2009;29:9977–9986. doi: 10.1523/JNEUROSCI.3893-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zifkin BG, Gracco RQ. An orderly approach to the abnormal EEG. In: Daly DD, Pedley TA, editors. Current practice of clinical electroencephalograph. New York: Raven; 1990. pp. 253–267. [Google Scholar]