

Abstract
A major event in the maturation of CNS GABAergic transmission is the qualitative change in GABAA-mediated responses from depolarizing to hyperpolarizing. In cortical regions, this is attributed to the increased expression of potassium chloride cotransporter 2b (KCC2b), the main isoform of the neuron-specific K-Cl cotransporter KCC2. We have previously shown that transcription factor early growth response 4 (Egr4) can activate the KCC2b promoter. Here we demonstrate that in immature hippocampal neurons BDNF robustly induces ERK1/2 (extracellular signal-regulated kinase 1/2)-dependent Egr4 expression and rapid Egr4-dependent activation of the KCC2b promoter. The subsequent increase in KCC2b mRNA contributes to the expression of total KCC2 protein levels. These results indicate that Egr4 is an important component in the mechanism of BDNF-dependent KCC2 gene regulation via the ERK1/2 pathway in immature neurons.
Introduction
A major event during maturation of GABAergic neurotransmission is the developmental shift in GABAA-mediated responses from depolarizing to hyperpolarizing (for review, see Blaesse et al., 2009). This is caused by a decrease in intracellular chloride concentration (Berglund et al., 2006), attributed to the developmental upregulation and functional activation of the neuronal potassium chloride cotransporter 2 (KCC2) (Rivera et al., 1999; Hübner et al., 2001; Kelsch et al., 2001; Khirug et al., 2005). The shift in polarity of GABAA responses occurs in rodent hippocampus during the first 2 postnatal weeks (Farrant and Kaila, 2007). We have shown that the KCC2 gene generates two isoforms, KCC2a and KCC2b, by an alternative promoter and first exon usage (Uvarov et al., 2007). During development, expression levels of KCC2a mRNA in hippocampus are relatively constant, whereas KCC2b mRNA increases significantly. This indicates that the developmental increase in the expression of KCC2 mRNA and protein is mainly due to the KCC2b isoform (Uvarov et al., 2007, 2009).
Previous studies suggest that KCC2 expression is regulated by BDNF through a complex signaling cascade that is not yet fully understood. Increased KCC2 mRNA levels were found in embryos overexpressing BDNF (Aguado et al., 2003). In line with this observation, it was found that KCC2 expression decreases in early postnatal knock-out mice lacking TrkB (tyrosine receptor kinase B) receptor (Carmona et al., 2006). In contrast, in mature neurons the BDNF/TrkB signaling mediates activity-dependent decrease in KCC2 expression. This requires the simultaneous binding of Shc (Src homology 2 domain-containing transforming protein) and PLCγ (phospholipase C γ) adaptor proteins to TrkB and downstream activation of the two signal transduction pathways (Rivera et al., 2002, 2004; Payne et al., 2003). Interestingly, when only the Shc pathway is active and PLCγ binding is disrupted, BDNF increases the expression of KCC2 in adult neurons (Rivera et al., 2004). Elucidation of the molecular mechanisms and pathways regulating KCC2 gene expression is important for understanding the changes occurring in KCC2 expression during normal development and under various pathological conditions. (Coull et al., 2005; Boulenguez et al., 2010).
Neurotrophins robustly induce expression of several immediate early genes including early growth response (Egr) transcription factors (Crosby et al., 1991; Harada et al., 2001; Roberts et al., 2006; Calella et al., 2007). Egr4 (or NGFI-C), which displays a neural-specific pattern of expression, is rapidly upregulated by nerve growth factor and neuronal activity (Crosby et al., 1991, 1992; Honkaniemi and Sharp, 1999; for review, see Beckmann and Wilce, 1997). Recently, we have demonstrated that Egr4 contributes directly to the transcriptional upregulation of KCC2 through the Egr4 binding site present in the proximal promoter of the KCC2b isoform (Uvarov et al., 2006).
In the present work, we show that brief application of BDNF induces a dramatic extracellular signal-regulated kinase 1/2 (ERK1/2)-dependent upregulation of Egr4 transcription factor and increases KCC2b mRNA and protein levels in immature cultured hippocampal neurons. Using luciferase reporter constructs controlled by the proximal KCC2b promoter, we demonstrate that intact Egr4 binding site is required for BDNF-mediated KCC2b promoter activation. Our results provide evidence for a significant role of Egr4 in BDNF-dependent regulation of KCC2b expression in immature neurons.
Materials and Methods
Primary culture of hippocampal neurons.
All animal experiments were approved by the local ethics committee for animal research at the University of Helsinki. Standard dissociated hippocampal cultures were prepared from embryonic day 17 mice as described by Banker and Goslin (1998), with slight modifications. Briefly, a pregnant mouse was anesthetized in a CO2 chamber and killed by cervical dislocation, embryos were removed, and hippocampi were dissected. Cells were dissociated by enzymatic treatment (0.25% trypsin for 15 min at 37°C) and plated on poly-dl-ornithine-coated cover glasses (50,000 cells/cm2) in Neurobasal medium containing B27 supplement (Invitrogen). Before plating, the medium was incubated on astroglial culture for 24 h. Neuronal cultures were fed once a week by changing half of the medium. Astroglial cultures were prepared according to Banker and Goslin (1998) and maintained in DMEM supplemented with 10% of fetal calf serum, penicillin (100 U/ml), and streptomycin (100 μg/ml).
Pharmacological treatments of neuronal cultures.
BDNF (R&D Systems), K252a, U0126, and tetrodotoxin (TTX) (Tocris Bioscience) treatments were conducted by replacing the original culture medium with the drug diluted in fresh glia-conditioned medium. No further medium change was performed until the time of analysis (5 min–3 d later). In treatments requiring a combination of drugs, K252a, U0126, and TTX were always added to the medium 30 min before the application of BDNF. The final concentrations of the drugs are specified in the Results section.
Western blotting.
Neuronal cultures were rinsed in PBS, scraped, and homogenized in ice-cold RIPA lysis buffer (NaCl 150 mm; Triton X-100 1%; Doc 0.5%; SDS 0.1%; TrisHCl 50 mm, pH 8.0) with Complete Protease Inhibitor Cocktail (Roche). Protein concentrations were determined using DC Protein Assay kit (Bio-Rad). Samples were separated using 7.5% SDS-PAGE and transferred onto Hybond ECL nitrocellulose membrane (GE Healthcare/Pharmacia Biotech). Membranes were probed with various antibodies at appropriate dilution as indicated in supplemental Table 1 (available at www.jneurosci.org as supplemental material), developed with ECL-plus (GE Healthcare/Pharmacia Biotech), visualized with a luminescent image analyzer LAS-3000 (Fujifilm), and analyzed with AIDA imaging software (Raytest).
Real-time PCR analysis.
Total RNA was isolated with RNeasy Micro kit (Qiagen). Typically, 1 μg of total RNA was reverse transcribed using the SuperScript III Reverse Transcriptase (Invitrogen) and random primers according to the manufacturer's protocol. The cDNA samples were amplified in triplicate using the iQ SYBR Green Supermix (Bio-Rad) and detected via the CFX96 Real Time Detection System (Bio-Rad). Primers for the genes of interest were designed with the Express v2.0 software (Applied Biosystems) or manually, and contained, when possible, intronic regions in between the sequences. Primer sequences are indicated in supplemental Table 2 (available at www.jneurosci.org as supplemental material). Glyceraldehyde-3-phosphate dehydrogenase or phosphoglycerate kinase-1 were used for normalization (Willems et al., 2006).
Transfection and luciferase assay.
Cultured neurons were transfected with the luciferase reporter constructs using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. To avoid cytotoxicity, low amounts of the luciferase constructs (0.5 μg per 1.6 cm2 well) were used, and the culture medium was changed 4 h after transfection. Two days after transfection, cultures were briefly washed with PBS and lysed in Passive Lysis Buffer (Promega). Renila and Firefly luciferase activities were measured with a Dual-Luciferase Reporter Assay System according to the manufacturer's protocol.
Immunocytochemistry.
Neurons were fixed with 4% paraformaldehyde in PBS at room temperature and permeabilized with 0.5% Triton X-100. Cells were blocked with 10% sheep serum and 0.2% Triton X-100 in PBS at room temperature, and then were incubated at 4°C overnight in 2% sheep serum containing primary antibody: mouse monoclonal anti-pERK (Santa Cruz Biotechnology) (1:500) or rabbit polyclonal anti-KCC2 (Ludwig et al., 2003) (1:1000) (supplemental Table 1, available at www.jneurosci.org as supplemental material). Species-specific donkey anti-mouse Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 568 (Invitrogen) secondary antibodies were used at 1:1000 dilution. Cover glasses were mounted in Vectashield mounting medium (Vector Laboratories). Cells were visualized with Leica TCS SP2 AOBS confocal system. The specificity of the KCC2 antibody was controlled on KCC2-deficient mice (Ludwig et al., 2003) and by omitting primary antibody in some of the samples.
Results
BDNF can induce fast increase in endogenous KCC2b mRNA and protein levels in immature dissociated hippocampal neurons
Consistent with the previously published data (Aguado et al., 2003; Carmona et al., 2006), Western blot analysis showed that KCC2 protein levels were increased in neuronal cultures treated with BDNF for 3 d (Fig. 1A). The BDNF effect was larger in immature cultures [day in vitro (DIV) 4, 142 ± 12%; DIV 11, 126 ± 12% (with respect to control values)] compared with relatively more mature (DIV 18) hippocampal neurons (92 ± 5% of control) (Fig. 1A). The absence of BDNF-induced KCC2 upregulation in DIV 18 neurons is in accordance with our previous data (Rivera et al., 2004) and indicates that BDNF loses the ability to stimulate KCC2 expression with neuronal development. In immature (DIV 6–8) cultures KCC2 protein expression already increased 24 h after BDNF application as shown by a significantly increased KCC2 immunoreactivity (120 ± 2% of control) (Fig. 1B). The KCC2 antibody used in this study recognizes an epitope present in the C-terminal domain of both KCC2a and KCC2b isoforms (Ludwig et al., 2003). Thus, the analysis of KCC2 protein expression accounts for both KCC2a and KCC2b isoforms.
Figure 1.
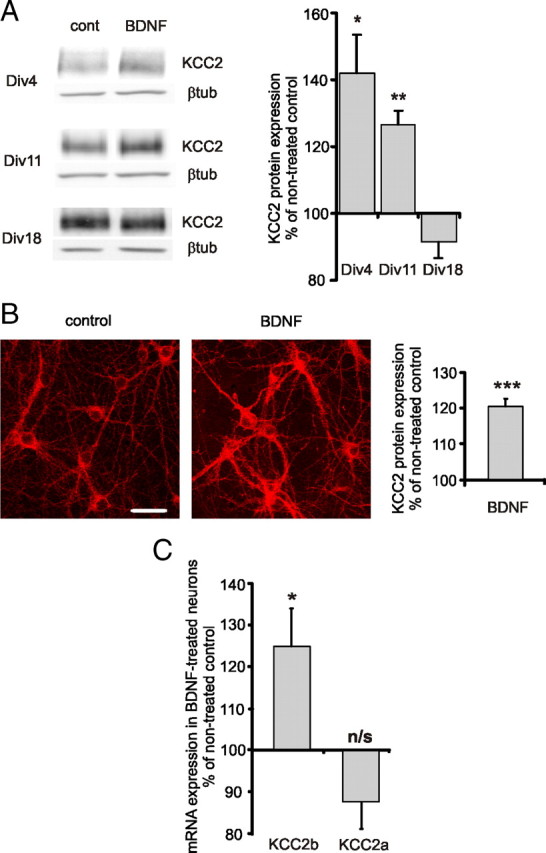
BDNF increases KCC2 expression in immature hippocampal cultures. A, Representative Western blot analysis and quantification of KCC2 expression in dissociated hippocampal cultures treated with BDNF (50 ng/ml) (n = 3–7). Dissociated cultures were treated with BDNF at DIV 1, DIV 8, and DIV 15, and analyzed 3 d after the treatment. Data are normalized with respect to the average in nontreated controls of corresponding age. *p < 0.05, **p < 0.01, one-sample t test. Error bars represent SEM. βtub, β-tubulin. B, Representative immunofluorescent staining and quantification of KCC2 expression in dissociated hippocampal cultures treated with BDNF (50 ng/ml) (n = 119–125). Neurons were treated with BDNF at DIV 6–8 and analyzed 24 h later. Data are normalized to the average in nontreated controls. ***p < 0.001, one-sample t test. Error bars represent SEM. Scale bar, 40 μm. C, KCC2b and KCC2a mRNA levels in DIV 6–8 cultured neurons 1–2 h after BDNF (50 ng/ml) application as detected by real-time PCR (n = 10). Values of corresponding nontreated controls were set to 100%. *p < 0.05, not significant (n/s); p > 0.05, one-sample t test. Error bars represent SEM.
Next, we studied whether BDNF application can activate endogenous KCC2 expression at a short time scale. Immature cultured hippocampal neurons (DIV 6–8) were treated with BDNF (50 ng/ml) and analyzed 1–4 h after BDNF exposure. We found that 1–2 h after BDNF application the levels of KCC2b mRNA increased rapidly and significantly to 125 ± 9% above control (Fig. 1C). The rise in transcript expression was followed by an increase (140 ± 20% above control) in KCC2 protein expression 4 h after BDNF application. The effect displayed some variability between the analyzed samples. Interestingly, the variability was greatly reduced upon silencing of network activity with the sodium channel blocker TTX (1 μm), which resulted in a significant upregulation of KCC2 by BDNF at this time point (142 ± 9% of TTX-treated control; p < 0.05, one-sample t test).
Thus, endogenous KCC2b mRNA and protein levels can be quickly upregulated in cultured hippocampal neurons after BDNF treatment. The effect of BDNF is more pronounced in immature DIV 4–11 neurons compared with more mature DIV 18 cultures.
BDNF significantly increases Egr4 expression via ERK1/2 signaling pathway
Previous studies have shown that BDNF induces a relatively rapid increase in expression of Egr genes Egr1, Egr2, and Egr3 (Harada et al., 2001; Roberts et al., 2006; Calella et al., 2007). To determine whether Egr4 expression is regulated in a similar way, cultured hippocampal neurons (DIV 5–6) were treated with BDNF (50 ng/ml). Already 5 min after BDNF application, ERK1/2 phosphorylation was increased sevenfold (Fig. 2A,B). This increase was significantly attenuated when neurons were pretreated with U0126 (20 μm), a specific inhibitor of the MAP/ERK kinase (MEK) (Favata et al., 1998). U0126 did not inhibit BDNF-induced phosphorylation of cellular homolog of the viral oncogene v-Akt (AKT) supporting the specificity of this compound in our culture conditions (data not shown). Within 2 h after BDNF application, Egr4 mRNA levels increased substantially (22 ± 7-fold) as detected by real-time PCR (Fig. 2C). This BDNF-induced increase in Egr4 mRNA expression was abolished in the presence of MEK inhibitor U0126.
Figure 2.
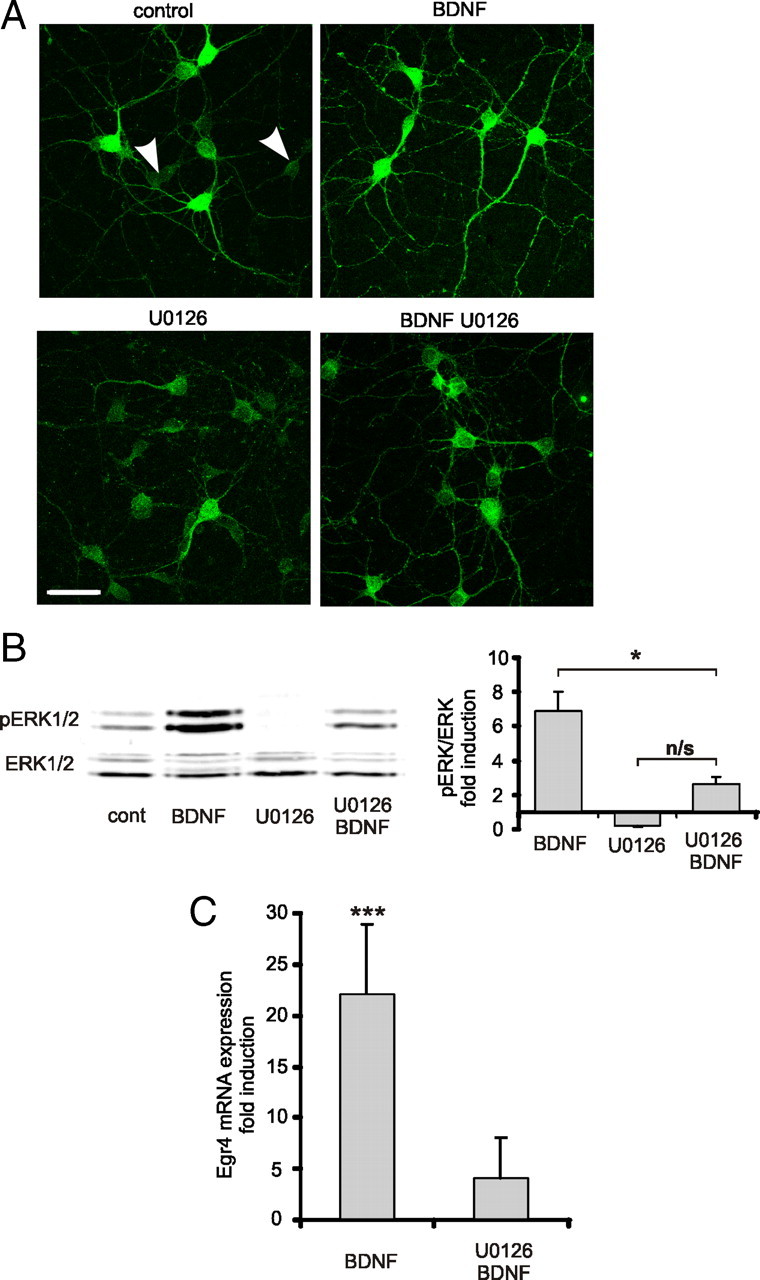
BDNF-induced Egr4 expression is dependent on ERK1/2 phosphorylation. A, Representative immunofluorescent staining of phosphorylated ERK1/2 (pERK1/2) in dissociated DIV 5–6 hippocampal neurons treated for 5 min with BDNF (50 ng/ml). Some of the cultures were pretreated with MEK inhibitor U0126 (20 μm) 30 min before BDNF treatment. Note that in nontreated control cultures there are neurons with very low levels of pERK1/2 (white arrowheads). In BDNF-treated cultures, all neurons exhibit high levels of pERK1/2 immunostaining intensity. Scale bar, 50 μm. B, Representative Western blot analysis and quantification of BDNF-induced ERK1/2 phosphorylation (n = 4). Cell lysates were collected 5 min after BDNF (50 ng/ml) application. Some cultures were pretreated with MEK inhibitor U0126 (20 μm) 30 min before BDNF application (n = 3). Data are normalized to the density of nonphosphorylated ERK1/2 bands and to a value in nontreated controls. *p < 0.05, not significant (n/s); p > 0.05; one-way ANOVA with subsequent Bonferroni's multiple-comparison test. Error bars represent SEM. C, Egr4 mRNA levels in dissociated DIV 5–6 hippocampal neurons 1–2 h after BDNF (50 ng/ml) application as detected by real-time PCR (n = 12). In some cases, cultures were pretreated with MEK inhibitor U0126 (20 μm) 30 min before BDNF application (n = 4). Nontreated control value set to 1. ***p < 0.001, one-sample t test. Error bars represent SEM.
Altogether, these results suggest that BDNF rapidly and significantly increases Egr4 mRNA expression in immature cultured hippocampal neurons via an MEK/ERK signal transduction pathway.
BDNF activation of KCC2b promoter requires an intact Egr4 transcription factor binding site
In a previous study, we demonstrated that the transcription factor Egr4 increases KCC2 expression in immature neurons through the Egr4 binding site present in the KCC2b proximal promoter region (Uvarov et al., 2006). To assess whether the Egr4 induced by BDNF is able to activate KCC2 gene transcription, we used a previously characterized luciferase reporter construct KCC2(−309/+42). In this construct, luciferase was placed under control of the proximal KCC2b promoter bearing the Egr4 binding site (Fig. 3A). Details of this construct were described in a previous publication (Uvarov et al., 2006). Egr4 regulates KCC2(−309/+42) reporter similarly to reporters driven by longer (up to 1.4 kb) KCC2b promoter sequences (Uvarov et al., 2006).
Figure 3.

BDNF positively regulates KCC2b promoter activity through Egr4. A, Scheme of KCC2(−309/+42) and KCC2(−309/+42)Egr4mut constructs used to study the activity of KCC2b promoter. The constructs include a 309 bp fragment of KCC2b promoter sequence controlling luciferase reporter expression and containing Egr4 binding site. Egr4 binding site was mutated in the KCC2(−309/+42)Egr4mut construct to prevent Egr4 binding to the promoter (Uvarov et al., 2006). B, Luciferase (LUC) activity in cultures transfected with KCC2(−309/+42) construct and treated with varying concentrations of BDNF (n = 8–31). Luciferase activity was measured 2–4 h after BDNF application. Nontreated control value is set to 100%. ***p < 0.001, one-sample t test. Error bars represent SEM. C, Luciferase activity in cultures transfected with either KCC2(−309/+42) or KCC2(−309/+42)Egr4mut constructs, and treated with BDNF (50 ng/ml) (n = 12–31). Luciferase activity was measured 2–4 h after BDNF application. Values are normalized to the activity of KCC2(−309/+42) construct without BDNF treatment. ***p < 0.001, not significant (n/s); p > 0.05; one-way ANOVA with subsequent Bonferroni's multiple-comparison test. Error bars represent SEM.
Dissociated hippocampal neurons were transfected with the KCC2(−309/+42) reporter construct at DIV 4, treated with 10 and 50 ng/ml BDNF 2 d after transfection, and lysed 2–4 h after BDNF application. Luciferase activity increased significantly (130 ± 5% of nontreated controls) only in neurons treated with 50 ng/ml BDNF (Fig. 3B). BDNF-induced activation of KCC2b promoter was abolished by the TrkB inhibitor K252A (200 nm) or the MEK inhibitor U0126 (20 μm) (K252A, 88 ± 4%, vs K252A + BDNF, 88 ± 10%; p > 0.05; U0126, 81 ± 6%, vs U0126 + BDNF, 89 ± 10%; p > 0.05, one-way ANOVA with subsequent Bonferroni's multiple-comparison test). These results show that KCC2b promoter activation by BDNF is mediated via TrkB/mitogen-activated protein kinase (MAPK) signaling.
To rule out the possibility that BDNF-induced network activity (Lang et al., 2007) participated in the observed BDNF effect on KCC2b promoter, cultures were treated with TTX (1 μm) 30 min before and during BDNF application. TTX did not prevent BDNF-induced KCC2 promoter activation (130 ± 6% of TTX-treated control; p < 0.01, one-sample t test).
To investigate whether BDNF-induced activation of KCC2b promoter is Egr4 dependent and occurs exclusively through the Egr4 binding site present in the proximal KCC2b promoter, we used KCC2(−309/+42)Egr4mut construct (Fig. 3A). This construct is identical to KCC2(−309/+42) reporter, except for the two point mutations introduced into the Egr4 binding site. These mutations hinder Egr4 binding and render it unable to conduct Egr4-mediated activation of the KCC2b promoter (Uvarov et al., 2006). The reporter activity of the KCC2(−309/+42)Egr4mut construct was significantly lower compared with the nonmutated KCC2(−309/+42) construct, reflecting the influence of endogenous Egr4 (Fig. 3C). BDNF treatment (50 ng/ml, 2–4 h) increased the luciferase activity of the KCC2(−309/+42) construct bearing the intact Egr4 binding site (132 ± 4%), but had no effect on the KCC2(−309/+42)Egr4mut construct (control, 84 ± 2%, vs BDNF, 80 ± 5%; p > 0.05; one-way ANOVA with subsequent Bonferroni's multiple-comparison test).
In summary, these data demonstrate that a brief application of BDNF can activate KCC2b transcription via the TrkB/MAPK pathway. Furthermore, BDNF-induced KCC2b promoter activation requires the intact Egr4 binding site in the proximal promoter region.
Discussion
Uncovering the mechanisms of developmental KCC2 expression is fundamental for understanding the functional maturation of GABAA-mediated neurotransmission during development. In the present study, we demonstrate a conspicuous effect of the neurotrophic factor BDNF on KCC2 expression in immature neurons. The key point in the pathway is the activation of transcription factor Egr4 downstream of the MAPK signaling cascade. We provide evidence that Egr4 binding to KCC2b promoter is essential for a fast BDNF-induced KCC2b promoter activation (Fig. 4).
Figure 4.

Simplified schematic diagram of the signal transduction cascade involved in BDNF-induced KCC2 expression.
BDNF-mediated upregulation of KCC2
Previous studies demonstrated that KCC2 expression increased in BDNF-overexpressing mice (Aguado et al., 2003) and decreased in TrkB−/− transgenic mice (Carmona et al., 2006). However, a direct link between BDNF and KCC2 regulation was not established in either of the above cases. BDNF/TrkB signaling has been shown to play an important role in cell survival, differentiation, and plasticity (e.g., by inducing changes in the frequency and synchronization of spontaneous network events in the developing cortex) (Aguado et al., 2003, Carmona et al., 2006). Taking this into consideration, it is plausible that long-term changes in BDNF signaling could indirectly affect KCC2 expression. The results presented here show that BDNF can induce KCC2 expression in a short time window. In particular, when monitoring changes in KCC2b mRNA, the effect could be observed as fast as 1–2 h after BDNF application.
Identifying the intracellular cascades involved in KCC2 regulation
BDNF can activate several intracellular cascades including Shc/Frs2 (FGF receptor substrate 2) that in turn triggers MAPK and AKT signaling (Kotzbauer et al., 1996; Huang and Reichardt, 2003). The MAPK pathway is known to be involved in differentiation and maturation, whereas AKT is implicated in cell growth and survival. In the present study, we used a luciferase reporter controlled by proximal KCC2b promoter sequence to monitor BDNF-induced KCC2b promoter activation. Suppression of MAPK signaling by the specific MEK inhibitor U0126 resulted in a significant attenuation of BDNF-induced KCC2b promoter activation. We have previously shown that in adult neurons activation of mutant TrkB receptor, in which only Shc/Frs2 docking site was preserved, resulted in increase of KCC2 levels (Rivera et al., 2004). The present results are in agreement with this and confirm that the intracellular Shc/Frs2 and MAPK cascades are required for BDNF-induced KCC2 upregulation.
Egr1, Egr2, and Egr3 are activated by BDNF in a MAPK-dependent manner (Harada et al., 2001; Roberts et al., 2006; Calella et al., 2007). We have recently demonstrated that Egr4 can induce KCC2 transcription in developing neurons (Uvarov et al., 2006). Here we show that BDNF can increase Egr4 mRNA levels and that this effect can be blocked by a specific MEK inhibitor U0126. The question arises as to whether Egr4 is involved in the trophic factor-induced KCC2 upregulation. An important role of Egr4 in this process was demonstrated by the experiment where the effect of BDNF on KCC2 reporter construct was completely abolished by mutations introduced into the Egr4 binding site in the KCC2b promoter sequence.
BDNF differently regulates KCC2 expression in developing and adult neurons
We and others have reported that BDNF decreases KCC2 mRNA and protein expression in mature neurons (Rivera et al., 2002, 2004; Wardle and Poo, 2003; Coull et al., 2005; Wake et al., 2007). In the present study, we show that BDNF increases KCC2 expression in developing hippocampal neurons in culture. Interestingly, BDNF ability to induce KCC2 expression declines during culture maturation. Assuming that similar responses are found in vivo, our results suggest a developmental shift in the effect of BDNF on KCC2 expression. This shift is not likely to be attributed to changes in expression of the BDNF cognate receptor TrkB as PCR results showed that this receptor was expressed at all developmental time points analyzed (data not shown) (Ivanova and Beyer, 2001).
Although in the present study we focused on the components underlying the upregulation of KCC2 in immature embryonic neurons in culture, it is tempting to suggest that KCC2 is regulated by at least two competing mechanisms. In support of this idea, our previous data showed that even in adult neurons BDNF is able to induce upregulation of KCC2 via the Shc pathway in the absence of the PLCγ activation (Rivera et al., 2004). In cultured neurons, BDNF induced a rise in KCC2b mRNA levels in the majority but not in all of the cultures analyzed. Some cultures reacted to BDNF application with a rapid decrease in KCC2b mRNA levels despite a substantial increase in Egr4 mRNA expression. This suggests that the currently unknown mechanism suppressing KCC2b expression may be able to overcome the Egr4-induced KCC2 gene activation and may work through one of the multiple additional factors identified in the KCC2b promoter (Uvarov et al., 2006; Yeo et al., 2009).
In conclusion, we show that in developing embryonic neurons in culture, the trophic factor BDNF produces a rapid increase in KCC2 expression. BDNF induces the expression of the immediate early gene Egr4 expression via activation of the MAPK pathway, which in turn stimulates KCC2b promoter activity. As in vivo the MAPK pathway is used by various signaling mechanism, it remains for future studies to elucidate whether and how other MAPK signaling mechanisms can synergize to cause the developmental upregulation of KCC2 during embryonic and early postnatal development.
Footnotes
This work was supported by grants from the Finnish Academy and the Juselius Foundation. M.S.A. and C.R. are members of the Finnish Center of Excellence in Molecular and Integrative Neuroscience. We acknowledge Miika Palviainen for excellent technical assistance.
References
- Aguado F, Carmona MA, Pozas E, Aguiló A, Martínez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibañez CF, Soriano E. BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl- co-transporter KCC2. Development. 2003;130:1267–1280. doi: 10.1242/dev.00351. [DOI] [PubMed] [Google Scholar]
- Banker G, Goslin K. Ed 2. Cambridge, MA: MIT Press; 1998. Culturing nerve cells; pp. 339–370. [Google Scholar]
- Beckmann AM, Wilce PA. Egr transcription factors in the nervous system. Neurochem Int. 1997;31:477–510. doi: 10.1016/s0197-0186(96)00136-2. [DOI] [PubMed] [Google Scholar]
- Berglund K, Schleich W, Krieger P, Loo LS, Wang D, Cant NB, Feng G, Augustine GJ, Kuner T. Imaging synaptic inhibition in transgenic mice expressing the chloride indicator, Clomeleon. Brain Cell Biol. 2006;35:207–228. doi: 10.1007/s11068-008-9019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;6:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med. 2010;16:302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- Calella AM, Nerlov C, Lopez RG, Sciarretta C, von Bohlen und Halbach O, Bereshchenko O, Minichiello L. Neurotrophin/Trk receptor signaling mediates C/EBPalpha, -beta and NeuroD recruitment to immediate-early gene promoters in neuronal cells and requires C/EBPs to induce immediate-early gene transcription. Neural Dev. 2007;2:4. doi: 10.1186/1749-8104-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona MA, Pozas E, Martínez A, Espinosa-Parrilla JF, Soriano E, Aguado F. Age-dependent spontaneous hyperexcitability and impairment of GABAergic function in the hippocampus of mice lacking trkB. Cereb Cortex. 2006;16:47–63. doi: 10.1093/cercor/bhi083. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Crosby SD, Puetz JJ, Simburger KS, Fahrner TJ, Milbrandt J. The early response gene NGFI-C encodes a zinc finger transcriptional activator and is a member of the GCGGGGGCG (GSG) element-binding protein family. Mol Cell Biol. 1991;11:3835–3841. doi: 10.1128/mcb.11.8.3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosby SD, Veile RA, Donis-Keller H, Baraban JM, Bhat RV, Simburger KS, Milbrandt J. Neural-specific expression, genomic structure, and chromosomal localization of the gene encoding the zinc-finger transcription factor NGFI-C. Proc Natl Acad Sci U S A. 1992;89:4739–4743. doi: 10.1073/pnas.89.10.4739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Kaila K. The cellular, molecular and ionic basis of GABA(A) receptor signalling. Prog Brain Res. 2007;160:59–87. doi: 10.1016/S0079-6123(06)60005-8. [DOI] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Harada T, Morooka T, Ogawa S, Nishida E. ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat Cell Biol. 2001;3:453–459. doi: 10.1038/35074516. [DOI] [PubMed] [Google Scholar]
- Honkaniemi J, Sharp FR. Prolonged expression of zinc finger immediate-early gene mRNAs and decreased protein synthesis following kainic acid induced seizures. Eur J Neurosci. 1999;11:10–17. doi: 10.1046/j.1460-9568.1999.00401.x. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Hübner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30:515–524. doi: 10.1016/s0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- Ivanova T, Beyer C. Pre- and postnatal expression of brain-derived neurotrophic factor mRNA/protein and tyrosine protein kinase receptor B mRNA in the mouse hippocampus. Neurosci Lett. 2001;307:21–24. doi: 10.1016/s0304-3940(01)01905-x. [DOI] [PubMed] [Google Scholar]
- Kelsch W, Hormuzdi S, Straube E, Lewen A, Monyer H, Misgeld U. Insulin-like growth factor 1 and a cytosolic tyrosine kinase activate chloride outward transport during maturation of hippocampal neurons. J Neurosci. 2001;21:8339–8347. doi: 10.1523/JNEUROSCI.21-21-08339.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khirug S, Huttu K, Ludwig A, Smirnov S, Voipio J, Rivera C, Kaila K, Khiroug L. Distinct properties of functional KCC2 expression in immature mouse hippocampal neurons in culture and in acute slices. Eur J Neurosci. 2005;21:899–904. doi: 10.1111/j.1460-9568.2005.03886.x. [DOI] [PubMed] [Google Scholar]
- Kotzbauer PT, Lampe PA, Heuckeroth RO, Golden JP, Creedon DJ, Johnson EM, Jr, Milbrandt J. Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature. 1996;384:467–470. doi: 10.1038/384467a0. [DOI] [PubMed] [Google Scholar]
- Lang SB, Stein V, Bonhoeffer T, Lohmann C. Endogenous brain-derived neurotrophic factor triggers fast calcium transients at synapses in developing dendrites. J Neurosci. 2007;27:1097–1105. doi: 10.1523/JNEUROSCI.3590-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Li H, Saarma M, Kaila K, Rivera C. Developmental up-regulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur J Neurosci. 2003;18:3199–3206. doi: 10.1111/j.1460-9568.2003.03069.x. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K. Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 2003;26:199–206. doi: 10.1016/S0166-2236(03)00068-7. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl-cotransporter KCC2 and impairs neuronal Cl- extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipilä S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DS, Hu Y, Lund IV, Brooks-Kayal AR, Russek SJ. Brain-derived neurotrophic factor (BDNF)-induced synthesis of early growth response factor 3 (Egr3) controls the levels of type A GABA receptor α 4 subunits in hippocampal neurons. J Biol Chem. 2006;281:29431–29435. doi: 10.1074/jbc.C600167200. [DOI] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Rivera C, Airaksinen MS. Upregulation of the neuron-specific K+/Cl− cotransporter expression by transcription factor early growth response 4. J Neurosci. 2006;26:13463–13473. doi: 10.1523/JNEUROSCI.4731-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Pruunsild P, Kaila K, Delpire E, Timmusk T, Rivera C, Airaksinen MS. A novel N-terminal isoform of the neuron-specific K-Cl cotransporter KCC2. J Biol Chem. 2007;282:30570–30576. doi: 10.1074/jbc.M705095200. [DOI] [PubMed] [Google Scholar]
- Uvarov P, Ludwig A, Markkanen M, Soni S, Hübner CA, Rivera C, Airaksinen MS. Coexpression and heteromerization of two neuronal K-Cl cotransporter isoforms in neonatal brain. J Biol Chem. 2009;284:13696–13704. doi: 10.1074/jbc.M807366200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Watanabe M, Moorhouse AJ, Kanematsu T, Horibe S, Matsukawa N, Asai K, Ojika K, Hirata M, Nabekura J. Early changes in KCC2 phosphorylation in response to neuronal stress result in functional downregulation. J Neurosci. 2007;27:1642–1650. doi: 10.1523/JNEUROSCI.3104-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle RA, Poo MM. Brain-derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. J Neurosci. 2003;23:8722–8732. doi: 10.1523/JNEUROSCI.23-25-08722.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems E, Mateizel I, Kemp C, Cauffman G, Sermon K, Leyns L. Selection of reference genes in mouse embryos and in differentiating human and mouse ES cells. Int J Dev Biol. 2006;50:627–635. doi: 10.1387/ijdb.052130ew. [DOI] [PubMed] [Google Scholar]
- Yeo M, Berglund K, Augustine G, Liedtke W. Novel repression of Kcc2 transcription by REST-RE-1 controls developmental switch in neuronal chloride. J Neurosci. 2009;29:14652–14662. doi: 10.1523/JNEUROSCI.2934-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]