

Abstract
Mammalian neocortex has a laminated structure that develops in a birth-date-dependent “inside-out” pattern. This layered structure is established by neuronal migration with sequential changes of the migratory mode regulated by several signaling cascades, including the Reelin–Disabled homolog 1 (Dab1) pathway. Although the importance of “locomotion,” the major migratory mode, has been well established, the physiological significance of the mode change from locomotion to “terminal translocation,” the final migratory mode, is unknown. In this study, we found that the outermost region of the mouse cortical plate has several histologically distinct features and named this region the primitive cortical zone (PCZ). Time-lapse analyses revealed that “locomoting” neurons paused transiently just beneath the PCZ before migrating into it by “terminal translocation.” Furthermore, whereas Dab1–knockdown (KD) neurons could reach beneath the PCZ, they failed to enter the PCZ, suggesting that the Dab1-dependent terminal translocation is necessary for entry of the neurons into the PCZ. Importantly, sequential in utero electroporation experiments directly revealed that failure of the Dab1-dependent terminal translocation resulted in disruption of the inside-out alignment within the PCZ and that this disrupted pattern was still preserved in the mature cortex. Conversely, Dab1–KD locomoting neurons could pass by both wild-type and Dab1–KD predecessors beneath the PCZ. Our data indicate that the PCZ is a unique environment, passage of neurons through which involves molecularly and behaviorally different migratory mechanisms, and that the migratory mode change from locomotion to terminal translocation just beneath the PCZ is critical for the Dab1-dependent inside-out lamination in the mature cortex.
Introduction
Mammalian cerebral cortex has a six-layered structure that is formed in a birth-date-dependent “inside-out” manner; migrating excitatory neurons invariably pass by their predecessors and settle at the top of the cortical plate (CP), just beneath the marginal zone (MZ). Because this six-layered structure is not observed in other vertebrates, it is thought that the mechanisms that regulate the formation of this inside-out pattern has been acquired during the course of mammalian evolution (Molnár et al., 2006).
The extracellular protein Reelin, which is secreted from the Cajal-Retzius cells in the MZ and activates Disabled homolog 1 (Dab1) in migrating neurons, plays indispensable roles in this inside-out neuronal positioning (Caviness and Sidman, 1973; D'Arcangelo et al., 1995; Ogawa et al., 1995; Rice and Curran, 2001; Tissir and Goffinet, 2003). Previous studies have proposed several abnormalities that could affect the positioning of the neurons in the reeler cortex, which lacks reelin, or that of the Dab1-suppressed neurons (Pinto-Lord et al., 1982; Tabata and Nakajima, 2002; Hartfuss et al., 2003; Luque et al., 2003; Olson et al., 2006). However, it is controversial which developmental abnormality is the primary failure that causes entire “inversion” of the reeler cortex, because it is difficult to exclude secondary effects that affect neuronal migration in the disrupted mutant cortex and to reproduce the whole process of laminar formation in vitro.
In the intermediate zone (IMZ) and CP, postmitotic neurons with one leading process migrate long distances along the radial fibers (locomotion mode) (Rakic, 1972; Nadarajah et al., 2001). In contrast, when the leading process reaches the MZ, only the soma moves rapidly short distances toward the top of the CP, whereas the tip of the process remains attached to the MZ (“terminal translocation” mode) (Nadarajah et al., 2001). Although the importance of “locomotion” has been studied extensively (Kawauchi and Hoshino, 2008), the physiological significance of the migratory mode change from locomotion to terminal translocation remains unknown.
In this study, we characterized the outermost region of the CP as the primitive cortical zone (PCZ), because the PCZ shows histologically distinct features, being composed of densely packed “immature” neurons, and because “locomoting” neurons transiently pause just beneath the PCZ before their migratory mode change to terminal translocation. In addition, Dab1–knockdown (KD) neurons could migrate past the “mature” predecessors beneath the PCZ but could not migrate past the immature predecessors within the PCZ, suggesting that Dab1 is essential for entry of the neurons into the PCZ by terminal translocation. Furthermore, we established sequential in utero electroporation experiments to directly analyze the process of the inside-out layer formation in the wild-type cortex and found that Dab1-dependent process for the final inside-out alignment was mainly regulated by the terminal translocation within the PCZ rather than by the locomotion below the PCZ. Our results suggest that the PCZ is the pivotal region for the Dab1-dependent eventual inside-out lamination and that switching of the migratory mode from locomotion to terminal translocation plays critical roles for the neuronal entry into this unique region.
Materials and Methods
Animals.
Pregnant ICR mice were purchased from Japan SLC. The colony of reeler mice (B6CFe a/a-Relinrl/J) obtained from the The Jackson Laboratory was maintained by allowing heterozygous females to mate with homozygous males. The day of vaginal plug detection was considered to be embryonic day 0 (E0). For example, “E14” was used to denote experiments performed in the morning of embryonic day 14, and “E14.5” was used for the experiments performed in the afternoon of embryonic day 14. All animal experiments were performed according to the Guidelines for the Care and Use of Laboratory Animals of Keio University School of Medicine.
Construction of plasmids.
pCAGGS–GFP or pCAGGS–mCherry was constructed by inserting the cDNAs of enhanced green fluorescent protein (GFP) or mCherry (Clontech) into the pCAGGS vector (Niwa et al., 1991).
The pSilencer–control vector and the Dab1–knockdown vector (pSilencer–Dab396) has been described previously (Kubo et al., 2010a). An expression vector for Dab1–HA fusion protein was constructed using the expression vector for Dab1–EGFP fusion protein (pCAGGS–Dab1–EGFP) (Honda and Nakajima, 2006). After excision of the EGFP site of pCAGGS–Dab1–EGFP, the oligonucleotides of the HA tag containing an XhoI site and an NotI site at the 5′ and 3′ ends, respectively (sense, 5′-TCGAGtacccatacgatgttccagattacgcttgaGC-3′; antisense, 5′ GGCCGCtcaagcgtaacttggaacatcgtatgggtaC-3′), were inserted to generate pCAGGS–Dab1–HA. To generate a mutant Dab1, Dab1–3MT, which was resistant to pSilencer–Dab396, three-point mutations were introduced into Dab1 by PCR (Zheng et al., 2004). The primers used were as follows: sense primer, GGAGGGTAACCACAGATTTGTGGCCATCAAACAGCC; antisense primer, GTGGTTACCCTCCTTCCCGCAAACGTATCCG (underlines are the points of the mutations). The Tα1 expression vector described previously (Gloster et al., 1994) was used with some modification to its multicloning site. The Dab1–HA and Dab1–3MT–HA fragments were inserted into this vector.
In utero electroporation.
Pregnant mice were deeply anesthetized with pentobarbital sodium (Nembutal), and their intrauterine embryos were surgically manipulated as described previously (Nakajima et al., 1997). In utero electroporation was performed as described previously (Tabata and Nakajima, 2001). Approximately 1–2 μl of the plasmid solution (3 mg/ml for KD vectors, 1 mg/ml for the GFP or mCherry expression vectors, and 0.5 mg/ml for the Tα1–Dab1–3MT–HA vectors) containing 0.01% fast green solution was injected into the lateral ventricle of the intrauterine embryos and electronic pulses (31 V, 50 ms, four times) were then applied using an electroporator (CUY-21 or CUY-21PE; NEPA GENE) with a forceps-type electrode (CUY650P5). For the sequential electroporation experiments, plasmids were introduced into the same cortex at E14.5 and then after a 24 h interval at E15.5.
Time-lapse imaging.
Time-lapse imaging was performed as described previously (Tabata and Nakajima, 2003). Briefly, the brains were embedded in 3% low-melting agarose, and coronal brain slices (350 μm thick) from the anterior one-third of the cortex were placed on a Millicell-CM membrane (pore size, 0.4 μm; Millipore). Slices were cultured in Neurobasal medium containing B27 (Invitrogen). The dishes were then mounted in a CO2 incubator chamber fitted onto a confocal microscope (FV300 or FV1000; Olympus). Approximately 7–10 optical Z-section images were acquired every 5 min, and all focal planes (50–70 μm) were merged. The migrating neurons were tracked and analyzed using NIH ImageJ software.
Preparation of frozen sections.
The embryos and neonates were placed on ice for anesthesia and perfused transcardially with 4% paraformaldehyde (PFA) in a 0.1 m sodium phosphate buffer, pH 7.4. The embryonic brains were removed and postfixed in the same fixative for 2–4 h at 4°C. After the brains had been replaced in a 30% sucrose solution with PBS, they were embedded in OCT compound (Sakura) and frozen in liquid nitrogen. The frozen sections were cut with a cryostat (CM1900 or CM3050 S; Leica) into 20-μm-thick sections.
Immunohistochemistry.
The sections were blocked with 10% normal donkey serum in PBS containing 0.01% Triton X-100 (Sigma) (PBS-Tx) at room temperature (RT) for 1 h and then incubated with the primary antibodies at 4°C overnight. The sections were then washed with PBS-Tx and incubated with TRITC-, Cy5-, Dylight 488-, Dylight 549-, or Dylight 649-conjugated secondary antibodies at RT for 1 h (Jackson ImmunoResearch). For nuclear staining, TO-PRO3 (0.1 μm quinolinium, 4-[3-(3-methyl-2(3H)-benzothiazolylidene)-1-propenyl]-1-[3-(trimethylammonio) propyl]-diiodide) or DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) (Invitrogen) was used. The fluorescence images were captured by confocal microscopy (FV300 or FV1000; Olympus). The primary antibodies used were mouse anti-NeuN (1:100, clone A60; Millipore), mouse anti-Nestin (1:200, Rat401; BD Biosciences Pharmingen), rabbit anti-GFP (1:1000; MBL International), rat anti-Ctip2 (1:500, 25B6; Abcam), rabbit anti-Cux1 (1:100, M-222; Santa Cruz Biotechnology), and goat anti-Brn2 (1:100, C-20; Santa Cruz Biotechnology). To detect NeuN, sections were incubated directly with the primary antibody or incubated at 85°C in 0.01 m citrate buffer, pH 6.0, for 10 min before incubation with the primary antibody. The fluorescences of GFP and mCherry were mostly captured directly. After the citrate buffer treatment, GFP was detected with an anti-GFP antibody.
Nissl staining.
Thin slices of PFA-fixed paraffin sections (1 μm) were prepared using a microtome. Nissl staining was performed using 0.1% Toluidine blue according to the standard protocol.
Cell culture and transfection.
Neuro2a cells were cultured in DMEM containing 100 U/ml penicillin and 100 μg/ml streptomycin supplemented with 10% fetal bovine serum. The cells were transfected with FuGene HD (Roche Diagnostics) and cultured for 2 d.
Western blot analysis.
Neuro2a cells were collected with a cell scraper and centrifuged, and the cell pellets were lysed using a cell lysis buffer [1% Triton X-100, 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, protease inhibitor mix (Roche Diagnostics), 50 mm NaF, 1 mm orthovanadate, and 10 mm β-glycerophosphate] and incubated on ice for 1 h. The lysates were sonicated and centrifuged for 5 min at 5000 rpm. The supernatants were solubilized with a sample buffer (50 mm Tris-HCl, pH 6.8, 2% SDS, 0.005% bromophenol blue, 10% glycerol, and 100 mm DTT). The solubilized materials were boiled for 3 min at 100°C, subjected to SDS-PAGE (8% acrylamide), and then electrotransferred onto a PVDF membrane using an iBlot gel transfer system (Invitrogen). The blot was treated with a blocking buffer, 5% skimmed milk in PBS containing 0.05% Tween 20, for 1 h at RT, incubated overnight at 4°C with mouse anti-HA antibody (1:2000, HA.11; Covance), washed three times, incubated for 1 h at RT with HRP-labeled goat anti-mouse IgG (1:2000; Dako), and washed again three times. After the final wash, the blot was treated with ECL Plus Western blotting detection reagents (GE Healthcare). The signals were detected and measured using a cooled charge-coupled device camera (LAS-4000mini; Fujifilm). As the loading control, mouse anti-β-tubulin antibody (1:2000, tub2.1; Sigma) was used.
Results
Locomoting neurons tend to pause transiently and change their migratory mode to terminal translocation near the outermost region of the CP
To analyze the physiological importance of the migratory mode change from locomotion to terminal translocation, we first performed intensive live monitoring of radial migration in slice cultures after in utero electroporation (Tabata and Nakajima, 2001, 2003). We introduced GFP expression plasmids into migrating cells at E14.5 and obtained cortical slices 3 d after electroporation. Although previous time-lapse analyses of terminal translocation have been performed every 20–30 min (Nadarajah et al., 2001), we obtained images every 5 min for ∼24 h, because terminal translocation is reportedly completed within ∼60 min (Nadarajah et al., 2001). Under this intensive monitoring, we observed that many cells migrated toward the surface of the CP in a saltatory manner in the deeper parts of the CP. However, they tended to stop transiently near the top of the CP (Fig. 1A, 2:30–3:00, B, Cell 1). After this brief pause, the cells changed their migration speed along with a shortening of the leading process, whose tip remained attached to the MZ (Fig. 1A, 3:00, arrowheads). When the shortening of the leading process was completed, the cells stopped migrating. When we tracked the behaviors of the migrating neurons through the final phase of radial migration, the cells appeared to stop transiently and change their migration speed at similar position (Fig. 1B, Cell 1 to Cell 3, arrows; C, 40 of 55 cells, 5 independent slices obtained from different embryos). The average ± SEM distance from the position of this speed change to the endpoint of migration was 50.9 ± 2.8 μm. Because this transient pause and change in the soma speed were often observed in association with a shortening of the leading process, it was suggested that this unique migratory behavior was correlated with the change in the migratory mode from locomotion to terminal translocation. The transient pause of the locomoting neurons in the outermost 40–50 μm region of the CP may be necessary for the change of the migratory behavior of the neurons.
Figure 1.

Time-lapse imaging of the terminal translocation. A, Time-lapse images of the terminal translocation of neurons were obtained from a cortical slice prepared at E17.5 after electroporation of GFP expression vectors at E14.5. Note that the cell soma (arrow) tended to stop near the top of the CP (2:30–3:00). After this pause, the soma alone moved rapidly toward the top of the CP, with a shortening of the leading process (3:00, arrowheads). Scale bar, 20 μm. The time indicates the time (hours and minutes) elapsed since the start of the observation period. B, Tracking graph of the terminal translocation. The somal movement of cell 1 (shown in A) was tracked using NIH ImageJ software. Note that the speed of the somal movement changed at 3:00 (arrow). The migration distance after this time point until the end of migration was 54 μm. Representative graphs of the terminal translocation for cell 2 and cell 3 are also shown. C, Average terminal translocation distance. The distances from the point at which the migratory speed changed until the end of migration were plotted. The average ± SEM distance was 50.9 ± 2.8 μm (n = 40, 5 independent slices obtained from different embryos).
The outermost region of the cortical plate is composed of densely packed immature neurons
To characterize the relationship between the change in the migratory behavior of the neurons and the outermost region of the CP, we first examined the histological features of this region by Nissl staining. We noticed dense accumulation of darkly stained neurons in the outermost region of the CP at E18.5 (Fig. 2A,A′, white arrows), whereas neurons in the other CP regions were more lightly stained and less densely packed. This distinct staining pattern was also observed at E16.5 but not at E15.5 (Fig. 2A″, white arrows). Because immature neurons are known to show dark staining of the nuclei and small somata (Rakic, 1972), it seemed that the outermost region of the CP was composed of immature neurons. To further confirm this possibility, we performed immunohistochemistry using a marker of mature neurons, NeuN, at P0.5 (Mullen et al., 1992; Sarnat et al., 1998). The results showed that the outermost part of the CP was almost negative (or only weakly positive) for NeuN (Fig. 2B, arrows), whereas the other CP regions contained an abundance of strongly NeuN-positive neurons, indicating that the superficial part of the CP was composed of immature neurons. The thickness of this NeuN-negative zone was ∼40–50 μm at P0.5, almost the same as the distance of the terminal translocation observed in the time-lapse analyses. The NeuN-negative zone was also observed in other stages of cortical development (Fig. 2C, white arrows). At E15.5, whereas the subplate cells were positive for NeuN, the cells in the CP showed weak or negative staining for NeuN. In contrast, at E16.5, some cells in the middle part of the CP became NeuN positive, whereas other cells at the top of the CP were still NeuN negative, although the neurons in the deeper part of the CP also showed weak NeuN staining during the developmental stages. These staining patterns were also consistent with the results of Nissl staining. Subsequently, whereas the thickness of this zone gradually decreased after P2.5, a NeuN-negative zone at the top of the CP was discernible until P3.5 but, by P4.5, all the neurons became NeuN positive. Because the former NeuN-negative predecessors at the top of the CP became NeuN positive after the later-born migrating neurons arrived at the top of the CP, the NeuN-negative zone was formed one after another from the inside to the outside. We also observed that this dense accumulation of the immature cells was not present in the reeler cortex at P0 (Fig. 2D), which showed scattered and mixed distribution of NeuN-positive and -negative cells in the CP (Fig. 2E). Together, these findings suggest that this zone is a unique structure that is observed in the developing wild-type CP but not in the reeler CP.
Figure 2.
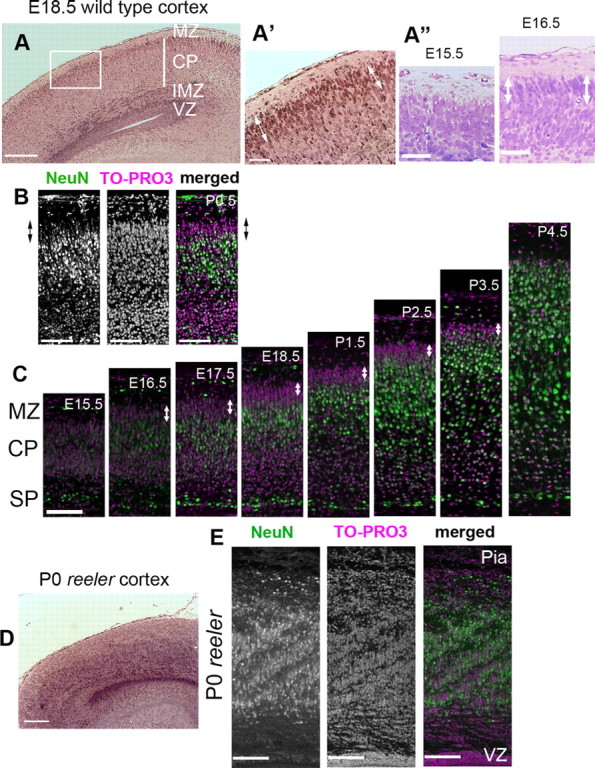
The outermost region of the developing CP was composed of immature neurons. A–A″, Nissl staining of the wild-type developing cortex. A, At E18.5. Note that the staining pattern of the outermost region of the CP differed from that of the other parts of the CP. A higher magnification of the square is shown in A′. Note the accumulation of the darkly stained neurons around the superficial part of the CP (white arrows). Accumulation of these cells was also identified at E16.5 but not at E15.5 (A″, white arrows). VZ, Ventricular zone. Scale bars: A, 200 μm; A′, A″, 50 μm. B, NeuN staining at P0.5; the outermost part of the CP showed weak or almost negative staining for NeuN. The arrows indicate the NeuN-negative zone. Scale bars, 100 μm. C, Developmental course of the NeuN-negative zone (white arrows). At E15.5, the subplate cells (SP) were NeuN positive (green), whereas the other cells in the CP were NeuN negative. Magenta is the nuclear staining. At E16.5, some cells in the CP became NeuN positive. From E17.5 to P1.5, the thicknesses of the NeuN-negative zone at the outermost part of the CP remained almost the same. Thereafter, the thickness gradually decreased, and, at P4.5 the NeuN-negative zone entirely disappeared. Note that the cells in the deep part of the CP showed weak or negative staining for NeuN until approximately P1.5. Scale bar, 100 μm. D, Nissl staining of the reeler cortex at P0. Note that no obvious accumulation of darkly stained neurons was visible. Scale bar, 200 μm. E, NeuN staining of the reeler cortex at P0. An admixture of NeuN-positive cells and NeuN-negative cells was observed in the deep part of the mutant cortex. Most of the cells at the superficial part of the reeler cortex were NeuN negative, corresponding to the deep part of the wild-type CP. Scale bars, 100 μm.
Dab1–KD prevents the entry of neurons into the PCZ, the NeuN-negative zone of the CP
Because this NeuN-negative zone is adjacent to the Reelin-positive MZ, we next examined whether Reelin–Dab1 signaling might regulate the entry of neurons into this NeuN-negative zone. We introduced a control or Dab1–KD vector (Kubo et al., 2010a) (Fig. 3A) with a GFP expression vector at E14.5 by in utero electroporation and analyzed the positions of the labeled neurons at E17.5, E18.5, and P0.5 (Fig. 3B–B″,C–C″,D–D″). Because in utero electroporation labels cells attached to the ventricular surface, most labeled cells shared the same birth date (Hatanaka et al., 2004; Tabata et al., 2009). Although the labeled cells were scattered throughout the CP and IMZ at E17.5 both in the control and Dab1–KD cortices (Fig. 3B–D), the control cells were arranged again at the NeuN-negative zone of the CP according to their birth date at the end of their migration (Fig. 3B′,B″,D′,D″), consistent with a previous report (Ajioka and Nakajima, 2005). In the case of the Dab1–KD cells, conversely, some of the cells were abnormally localized in the IMZ at E18.5 (Fig. 3C′,D′, *), but these cells migrated into the CP only 1 d later (Fig. 3C″,D″). Because the migration modes below the CP are multipolar migration (Tabata and Nakajima, 2003) and locomotion, these data suggest that, although the Dab1–KD neurons showed moderately delayed migration before entering the CP, they still had the ability to migrate into the CP. However, even by E18.5 or P0.5, the soma of the Dab1–KD neurons had not entered the NeuN-negative zone of the CP, although they were positioned in the upper part of the CP (Fig. 3C′,C″,D′,D″, arrows). The distances from the top of the CP to the soma at P0.5 were 26.5 ± 0.5 μm (SEM) in the control brains and 55.7 ± 0.7 μm in the Dab1–KD brains (Fig. 3L), the latter of which was similar to the distance of the terminal translocation in our time-lapse observation (Fig. 1C). Because the tip of the leading process of Dab1–KD neurons remained attached to the MZ, these results indicate that Dab1–KD resulted in terminal translocation failure, consistent with a previous report (Olson et al., 2006). Furthermore, although many control neurons were located within the NeuN-negative zone at P0.5 (Fig. 3E–E″, arrow), most of the Dab1–KD neurons had not entered the NeuN-negative zone (Fig. 3F–F″, arrow) (the percentages ± SEM of GFP-positive cells within the NeuN-negative zone were 69.0 ± 4.1% for control neurons and 10.3 ± 1.0% for Dab1–KD neurons; n = 4 brains each, >200 neurons were counted; p=0.0209, Mann–Whitney's test), indicating that Dab1 is required for entry of the neurons by terminal translocation into the NeuN-negative zone of the CP.
Figure 3.
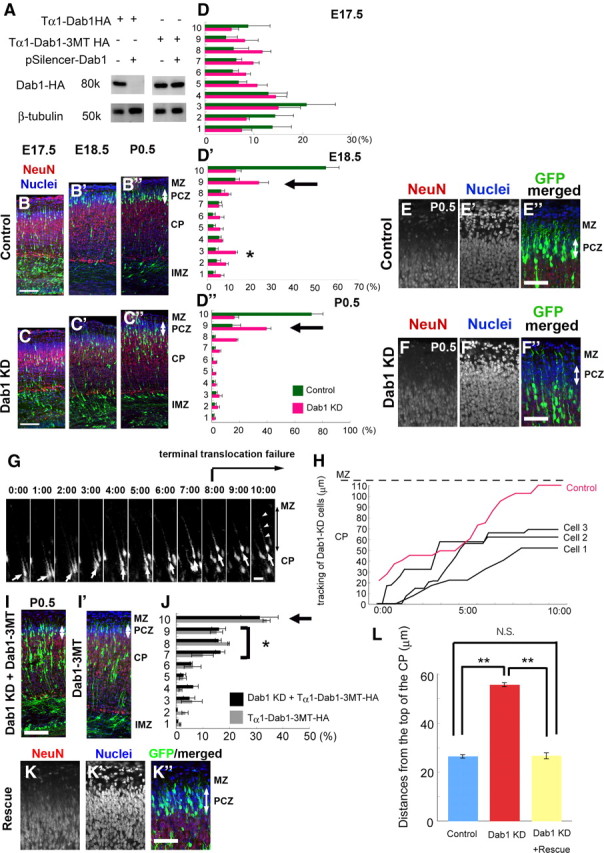
Dab1 is required for neuronal entry into the NeuN-negative zone or PCZ. A, In vitro analysis of the Dab1–KD vectors. Wild-type or mutant Dab1-expressing vectors were transfected with or without Dab1–KD vectors into Neuro2A cells. The cell lysates were analyzed by Western blotting. Dab1–KD vectors effectively suppressed the expression of wild-type Dab1 but not the expression of mutant Dab1, which was designed to be resistant to this Dab1–KD vector. B–B″, C–C″, D–D″, Effects of Dab1–KD on neuronal migration. Electroporation was performed at E14.5, and the brains were examined at E17.5, E18.5, and P0.5. The area from the outer margin of the CP to the inner margin of the IMZ was divided into 10 bins. B–B″, Control case. C–D″, The distribution of the Dab1–KD neurons was almost the same as that of the control neurons at E17.5 (C, D), but, even by E18.5 (C′, D′, black arrow) or P0.5 (C″, D″, black arrow), most of these neurons had not entered the NeuN-negative PCZ (white arrows). Note that some cells were abnormally observed in the IMZ (C′) and the third bin of the Dab1–KD experiments at E18.5 (D′, *), but most of these neurons had migrated to the CP by P0.5. n = 3 brains each. Scale bars, 100 μm. E–E″, F–F″, Dab1 is required for neuronal entry into the NeuN-negative PCZ. E–E″, Control case at P0.5. GFP-expressing cells had entered the top of the CP. Note that this zone was negative or weak for NeuN (red) (arrows). The nuclei were showed by DAPI (blue). F–F″, Dab1–KD neurons (green) were located beneath the NeuN-negative zone. Scale bars, 50 μm. G, Time-lapse images of the Dab1–KD neurons. The Dab1–KD neuron (arrow) migrated toward the surface of the CP but stopped migrating at 8:00 and did not move anymore. Note that the long leading process (arrowheads) is attached to the MZ. Scale bar, 20 μm. The time indicates the time (hours and minutes) elapsed since the start of the observation period. H, Three tracking graphs of the Dab1–KD neurons. Cell 1 is the tracking graph of the cell shown in G. The red line is the tracking graph of the pSilencer–control vector-transfected neuron. I–I′, Rescue experiments of the Dab1–KD phenotype. Electroporation was performed at E14.5, and the brains were examined at P0.5. In the rescue experiments (cotransfection of a Dab1–KD vector and a Tα1–Dab1–3MT–HA vector), many cells were successfully located in the NeuN-negative PCZ (white arrow). Note some neuronal accumulation around the middle of the CP; such accumulation was also observed when both a pSilencer–control vector and a Tα1–Dab1–3MT–HA vector were cotransfected (I′) but not observed in the Dab1–KD experiments (C″). Scale bar, 100 μm. J, Bin analysis of the rescue experiment. The area from the outer margin of the CP to the inner margin of the IMZ was divided into 10 bins. Note the accumulation of cells in the seventh to ninth bins (*). n = 5 brains for rescue experiments (black bar) and n = 3 brains for Dab1 overexpression experiments (gray bar). K–K″, Higher-magnification views of the rescue experiments around the PCZ. Note that the Dab1–KD phenotype was rescued by cotransfection of a Tα1–Dab1–3MT–HA vector and located within the NeuN-negative PCZ (arrow). Scale bar, 50 μm. L, Statistical analysis of the distances from the outer margin of the CP to the soma of the neurons. Only neurons located above the middle of the CP were counted. The distance was 26.5 ± 0.5 μm for the control neurons (n = 4 brains, >500 neurons were counted), 55.7 ± 0.7 μm for the Dab1–KD neurons (n = 4 brains, >500 neurons were counted), and 26.7 ± 1.1 μm for the Dab1–KD+Rescue neurons (n = 5 brains, >250 neurons were counted). **p < 0.01, Scheffé's F test.
To directly analyze the role of Dab1 in the migratory behaviors of the cells, we performed time-lapse analyses of Dab1–KD neurons in slice cultures (Fig. 3G,H). Similar to the control neurons, many Dab1–KD neurons smoothly migrated in a saltatory manner in the deeper part of the CP. Conversely, even after the leading processes of the Dab1–KD neurons became attached to the MZ, the shortening of the leading processes and the additional somal movement were not observed in most cases (Fig. 3G, arrowheads). Although some GFP weakly positive cells escaped this migration failure and were aligned more superficially, these GFP weakly positive cells were also observed in the NeuN-negative zone in the in utero electroporation experiments (10.3 ± 1.0%). Therefore, we speculated that the effect of the Dab1–KD vector was not sufficient in these GFP weakly positive cells and that they were aligned at the border between the CP and MZ, like the control cells. Because the “cell-dense zone” in the outermost region of the CP was not discernible in the slice cultures, we estimated the distances of the terminal translocation failure by measuring the distances from these GFP weakly positive cells to the GFP strongly positive Dab1–KD cells. The mean ± SEM distance was 49.1 ± 2.6 μm (25 cells in three independent slices obtained from different embryos), which was similar to the terminal translocation distance of the control neurons and the width of the NeuN-negative zone of the CP.
This terminal translocation failure caused by transfection of the Dab1–KD vector was rescued by cotransfection with an HA-tagged mutant-Dab1 expression vector driven by the Tα1 promoter (Tα1–Dab1–3MT–HA, a form of Dab1 resistant to the Dab1–KD vector) (Fig. 3A,I,J). Because the Tα1 promoter is not expressed in the radial glial cells (Gal et al., 2006), these data indicate that the terminal translocation failure was caused primarily by Dab1–KD in the migrating neurons rather than by its KD in the radial glial cells (Fig. 3I,J, arrow, K–K″,L). Although we observed that some cells remained around the middle of the CP (Fig. 3I,J, *), this abnormal positioning was also observed after cotransfection of a Tα1–Dab1–3MT–HA vector and a pSilencer–control vector (Fig. 3I′,J). An abnormally high amount of exogenous Dab1–HA was found to be expressed in these cells that remained in the middle of the CP (data not shown), suggesting that overexpressed Dab1 may have adverse effects on locomotion, as was also suggested in a previous report (Simó et al., 2010).
The above-mentioned results collectively indicate that the outermost 40–50 μm zone of the CP is histologically distinct, filled with densely packed immature neurons, compared with the mature neurons in the other part of the CP. Furthermore, locomoting neurons tend to pause transiently beneath this outermost zone and then move quickly through this zone by Dab1-dependent terminal translocation, suggesting that the passage through this zone requires a molecularly and behaviorally distinct mechanism from that in the other part of the CP. Thus, we named this zone the primitive cortical zone (PCZ).
Dab1-dependent somal/terminal translocation is required for neuronal entry into the outermost region of the CP or the PCZ throughout the period of neocortical development
To investigate whether Dab1 was required to enter the PCZ during other stages of development, we performed a series of Dab1–KD experiments during the development of the neocortex. Because the PCZ is histologically discernible until P3.5 (Fig. 2C), we first introduced a Dab1–KD vector at E16 to label the last cohort of neurons born in the ventricular zone of the lateral cortex (Takahashi et al., 1999) and examined the brains at P3.5 (Fig. 4A–A″). Similar to the results obtained after electroporation at E14.5 (Fig. 3E–E″,F–F″), most of the Dab1–KD neurons could not enter the NeuN-negative PCZ, whereas control neurons were settled within the PCZ (the percentages ± SEM of GFP-positive cells within the PCZ were 68.5 ± 2.8% for the control neurons and 14.2 ± 1.3% for the Dab1–KD neurons; n = 3 brains each, >120 neurons were counted; p=0.0495, Mann–Whitney's test).
Figure 4.
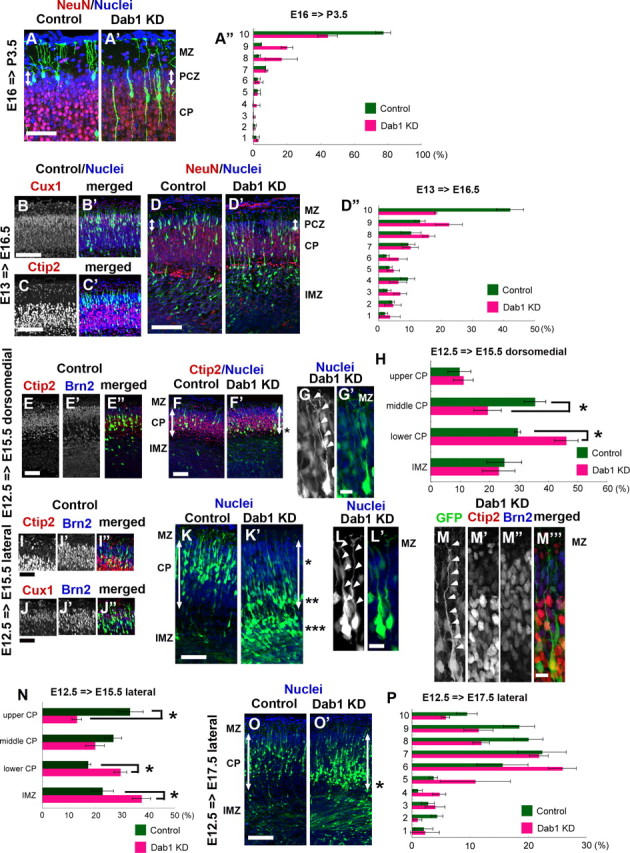
Dab1 is required for neuronal entry to the outermost region of the CP during all developmental stages. A–A″, Control or Dab1–KD vector was introduced at E16 and the brains were examined at P3.5. Control neurons were positioned within the NeuN-negative PCZ (A, arrow), whereas the Dab1–KD neurons could not enter this zone (A′, arrow). A″, Bin analysis. The thickness of the CP was divided into 10 bins (n = 3 brains each, >120 neurons were counted.) Scale bar, 50 μm. B–B′, C–C′, D–D″, Control or Dab1–KD vector was introduced at E13 and the brains were examined at E16.5. Many control neurons were positioned in the NeuN-negative PCZ (D, arrow), and these neurons were positive for Cux1 (B–B′) but negative for Ctip2 (C–C′). The Dab1–KD neurons, conversely, could not enter the PCZ (D′, arrow). D″, Bin analysis. The distance between the outer margin of the CP and the inner margin of the IMZ was divided into 10 bins. Note that the abnormal accumulation of the Dab1–KD neurons in the ninth bin, but the distribution pattern in the deeper parts of the cortex was similar to that in the control (n = 4 brains each, >400 neurons were counted.) Scale bars, 100 μm. E–E″, F–F′, G–G′, H, Control or Dab1–KD vector was introduced into the dorsomedial cortex at E12.5 and the brains were examined at E15.5. Control neurons were positive for Ctip2 but negative for Brn2 (E–E″), and they were positioned in the middle one-third of the CP (F). Many Dab1–KD neurons, conversely, were located at the border between the CP and the IMZ (F′, *). Arrows indicate the thickness of the CP. G–G′, Higher-magnification view of the Dab1–KD neurons located at the border between the CP and the IMZ. Note that their leading processes are attached to the MZ (arrowheads). H, Bin analysis. The cortex was divided into upper CP, middle CP, lower CP, and IMZ. Note that Dab1–KD neurons were abnormally accumulated in the lower CP (*p = 0.0495, Mann–Whitney's test, n = 3 brains each, >200 neurons were counted.) Scale bars: E-E″, F–F′, 50 μm; G–G′, 10 μm. I–I″, J–J″, K–K′, L–L′, M–M‴, N, Control or Dab1–KD vector was introduced into the lateral cortex at E12.5 and the brains were examined at E15.5. Control neurons were positioned in the outermost region of the CP (K), and many of them were double-positive for Ctip2 and Brn2 (I–I″) or Cux1 and Brn2 (J–J″). Dab1–KD neurons, conversely, were accumulated in three different areas (*, **, ***) (K′). * denotes the neuronal accumulation around the border between the upper one-third of the CP and the middle one-third of the CP. ** denotes the neuronal accumulation around the border between the CP and the IMZ. *** denotes the neuronal accumulation below the CP. Arrows indicate the thickness of the CP. L–L′, Higher-magnification view of Dab1–KD neurons located at the border between the upper CP and the middle CP (representative images at *). Note that their leading processes reach the MZ (arrowheads). M–M‴, Higher-magnification view of Dab1–KD neurons located at the border between the CP and the IMZ (representative images at **). Note that their leading processes also reach the MZ (arrowheads). Red represents Ctip2-positive cells, and blue represents Brn2-positive cells. N, Bin analysis. The cortex was divided into the upper CP, middle CP, lower CP, and IMZ (*p = 0.0209, Mann–Whitney's test; n = 4 brains each, >700 neurons were counted.) Scale bars: I–I″, J–J″, K–K′, 50 μm; L–L′, M–M‴, 10 μm. O–O′, P, The control or Dab1–KD vector was introduced into the lateral cortex at E12.5 and the brains were examined at E17.5. O, Control case. O′, Most of the Dab1–KD neurons were located in the CP, but they were settled in a slightly deeper place than the control neurons. Note the positioning of some Dab1–KD neurons at the border between the CP and the IMZ (*). Arrows indicate the thickness of the CP. P, Bin analysis. The distance between the outer margin of the CP and the inner margin of the IMZ was divided into 10 bins (n = 3 brains each, >400 neurons were counted.). Scale bar, 100 μm.
Next, we introduced a Dab1–KD vector at E13 and examined the brains at E16.5 because the PCZ became discernible at E16.5 (Figs. 2C, 4B–B′,C–C′,D–D″). Many control neurons were positioned in the NeuN-negative PCZ (Fig. 4D). Conversely, most of the Dab1–KD neurons could not enter the PCZ (the percentages ± SEM of GFP-positive cells within the PCZ were 38.0 ± 3.4% for the control neurons and 14.0 ± 4.5% for the Dab1–KD neurons; n = 4 brains each, >400 neurons were counted; p=0.0105, Mann–Whitney's test) (Fig. 4D′–D″), which was consistent with the findings obtained during the later developmental stages after electroporation at E14.5 or E16. In addition, the distribution pattern of the Dab1–KD neurons in the deeper parts of the cortex was similar to that of the control neurons (Fig. 4D″), suggesting that Dab1–KD mainly affected terminal translocation rather than locomotion. These data indicate that Dab1-dependent terminal translocation is necessary for the neuronal entry into the PCZ throughout the time period when the PCZ is discernible (from E16.5 to P3.5). The labeled cells at E13 were upper-layer neurons, because they were mostly positive for Cux1 (layer II/III/IV marker) (Fig. 4B–B′) but negative for Ctip2 (layer V/VI marker) (Fig. 4C–C′) (Molyneaux et al., 2007), suggesting that the upper-layer neurons exhibit Dab1-dependent terminal translocation to enter the PCZ.
We next investigated whether Dab1 was also required for the deep-layer neurons (layer V/VI) to migrate into the top of the CP, by introducing a Dab1–KD vector at E12.5 and examining the brains at E15.5. It was reported that the layer VI neurons do not use locomotion but directly change their migratory mode from multipolar migration to somal translocation (Nadarajah et al., 2001; Hatanaka et al., 2004). In addition, it was also reported that electroporation into the lateral cortex at E12.5 can label the neurons that mainly use locomotion, whereas electroporation into the dorsomedial cortex labels the neurons that use somal translocation at this stage, because there is a neurogenic gradient between the lateral cortex and the dorsomedial cortex (i.e., the lateral cortex is older than the dorsomedial cortex) (Hatanaka et al., 2004). Control neurons in the dorsomedial cortex were located in the middle of the CP at E15.5 (Fig. 4F,H). These cells were positive for Ctip2 and negative for Brn2 (layer II/III/V marker, with moderate labeling of layer IV neurons, too, at this stage), suggesting that these labeled cells included the layer VI neurons and some of the layer V neurons (Fig. 4E–E″). Interestingly, many Dab1–KD neurons were located near the border between the CP and the IMZ (Fig. 4F′, *, H). The leading process of the Dab1–KD neurons reached the MZ (Fig. 4G–G′), suggesting the possibility that Dab1–KD affected the somal translocation of the layer VI neurons.
When electroporation was performed into the lateral cortex at E12.5 and the brains were examined at E15.5, many control neurons were positioned in the superficial region of the CP; these neurons were double positive for Ctip2 and Brn2 or Cux1 and Brn2 (Fig. 4I–I″,J–J″,K,N). Some cells in the deep part of the CP were single positive for Ctip2. These staining patterns suggest that the labeled cells were the admixture of the layer IV neurons, the layer V neurons, and the layer VI neurons, consistent with a previous report (Takahashi et al., 1999). Although we could not discern the PCZ-like structure by Nissl staining or NeuN staining at E15.5, most of the Dab1–KD neurons labeled at E12.5 in the lateral cortex could not reach the top of the CP (Fig. 4K′,N). Interestingly, there were some cells reached at the outer one-third of the CP (40–50 μm below the border between the MZ and the CP) (Fig. 4K′, *, L–L′), from where they extended their leading processes to the MZ. These results suggest that these Dab1–KD neurons could reach the superficial part of the CP by locomotion but could not enter the outermost region of the CP because of the terminal translocation failure, similar to the Dab1–KD neurons in the subsequent stages. We also observed accumulation of Dab1–KD neurons around the border between the CP and the IMZ (Fig. 4K′, **, N) or below the CP (Fig. 4K′, ***, N), but these neurons migrated into the CP by E17.5 (Fig. 4O–O′,P), suggesting that Dab1–KD only somehow affected the early phase of neuronal migration (multipolar migration or locomotion) before the cells entered the CP, similar to the phenotype of the Dab1–KD at a later stage [electroporation at E14.5 (Fig. 3C′,D′)]. Interestingly, some cells around the border between the CP and the IMZ extended the abnormally long leading processes to the MZ, and they were Ctip2-positive but Brn2-negative neurons (Fig. 4K′, **, M–M‴). The accumulation of Dab1–KD neurons at the border between the CP and the IMZ was also observed at E17.5 (Fig. 4O′, *). Therefore, it is possible that the accumulation of Dab1–KD cells around this border was partly caused by somal translocation failure of the layer VI neurons or terminal translocation failure of the layer V neurons, similar to the case for the Dab1–KD cells at the dorsomedial cortex. Collectively, these series of data suggest that Dab1 is mainly required for the layer VI neurons and for the subsequent-layer neurons to enter the outermost region of the CP by somal translocation and terminal translocation, respectively.
Neuronal entry into the PCZ by terminal translocation is required for the Dab1-dependent final inside-out neuronal alignment
Although Dab1-mutant mice showed a disrupted inside-out neuronal layering (Howell et al., 1997; Sheldon et al., 1997; Ware et al., 1997; Yoneshima et al., 1997; Kojima et al., 2000), it remains unknown whether the entry of the neurons into the PCZ by Dab1-dependent terminal translocation is critical for the layer formation. To address this issue directly, we established sequential in utero electroporation experiments to analyze the process of the inside-out layer formation in the developing wild-type cortex. We first introduced a GFP expression vector at E14.5 to label the earlier-born neurons; then, 24 h later, we introduced an mCherry expression vector to label the later-born neurons (control–control case). At E18.5, most of the GFP-labeled earlier-born neurons were observed in the superficial part of the CP, whereas the mCherry-labeled later-born neurons were still undergoing locomotion (Fig. 5A–A‴). In contrast, at P1.5, most of the mCherry-labeled later-born neurons had completed migration and were located at the top of the CP, just above the GFP-labeled earlier-born neurons (Fig. 5B–B‴). Furthermore, at P7, at which time all the neurons had settled in the layered structure, the GFP-labeled earlier-born neurons and the mCherry-labeled later-born neurons were arranged in a highly segregated inside-out manner (Fig. 5C–C‴). These data indicate that the sequential in utero electroporation successfully visualized the process of the inside-out layer formation in vivo.
Figure 5.
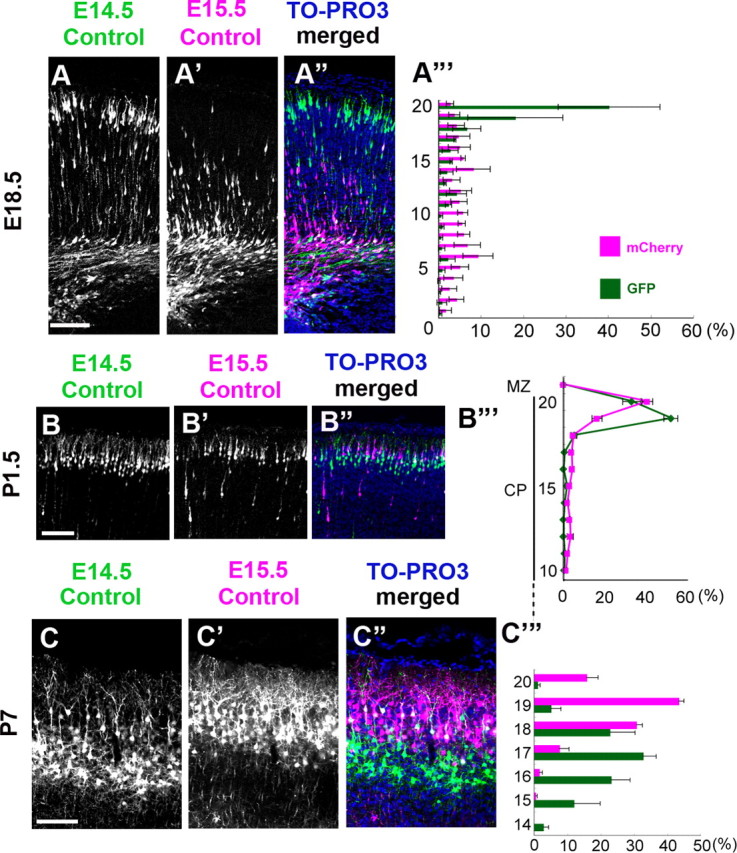
Sequential electroporation can visualize the process of the inside-out alignment of neurons in the wild-type cortex. A–A‴, The GFP expression vector was introduced at E14.5, and the mCherry expression vector was electroporated at E15.5. At E18.5, most of the GFP-labeled earlier-born control neurons were seen in the superficial part of the CP, whereas the mCherry-labeled later-born control neurons were mainly located in the IMZ and the deep part of the CP. Scale bar, 100 μm. A‴, Bin analysis. The distance between the outer margin of the CP and the inner margin of the IMZ was divided into 20 bins. n = 3 brains. B–B‴, At P1.5, the mCherry-labeled later-born neurons were located more superficially than the GFP-labeled earlier-born neurons in the outermost region of the CP. B‴, Bin analysis. The distance between the outer margin of the CP and the inner margin of the IMZ was divided into 20 bins. Many mCherry-labeled later-born neurons were located in the top 20th bin, whereas the GFP-labeled earlier-born neurons were seen in the 19th bin. Scale bar, 100 μm. n = 4 brains. C–C‴, At P7. Note that the mCherry-labeled later-born neurons and the GFP-labeled earlier-born neurons exhibited a highly segregated inside-out pattern. C‴, Bin analysis. The thickness of the CP (from the outer border of the layer II/III to the inner border of the layer VI) was divided into 20 bins. The mean ± SEM distances from the top of the CP were 117.4 ± 22.7 μm for the GFP-labeled neurons and 59.8 ± 7.3 μm for the mCherry-labeled neurons, respectively (Mann–Whitney's test, p=0.0495, n = 3 brains, >700 neurons were counted). Scale bar, 100 μm.
We then introduced a Dab1–KD vector and an mCherry expression vector at E15.5 and examined the positional relationships relative to the GFP-labeled earlier-born control cells labeled at E14.5 (control–Dab1–KD case). At E18.5, the mCherry-labeled Dab1–KD later-born neurons were still undergoing locomotion, similar to the control–control case (Fig. 6A–A‴). Importantly, however, the Dab1–KD later-born neurons were unable to reach the top of the CP and stopped just beneath the PCZ because of the terminal translocation failure at P1.5, and these neurons were located at almost the same position as the GFP-labeled earlier-born control neurons (Fig. 6B–B‴). Furthermore, at P7, the mCherry-labeled Dab1–KD later-born neurons were located at almost the same position as the GFP-labeled earlier-born control neurons, similar to the phenotypes observed at P1.5 (Fig. 6C–C‴), in dicating that the terminal translocation failure resulted in a disruption of the final inside-out pattern of neuronal alignment. These data suggest that the final inside-out pattern is originally established at the top of the CP (PCZ) when the neuronal migration is completed and that the neuronal entry into the PCZ by terminal translocation plays a crucial role in the eventual Dab1-dependent inside-out alignment of neurons in the mature cortex.
Figure 6.
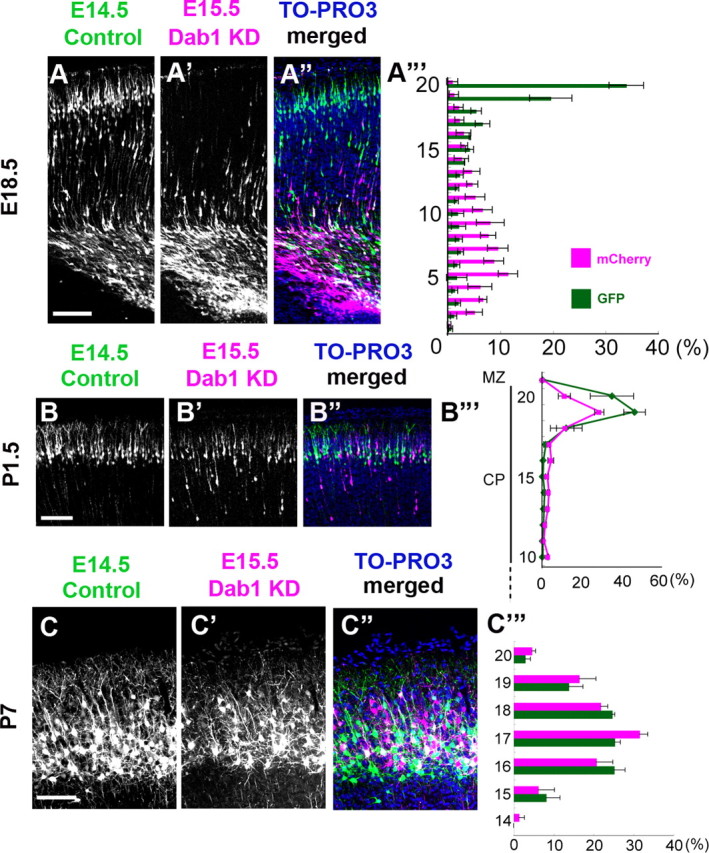
Dab1 is required for the eventual inside-out alignment of the mature cortex. A–A‴, Control-Dab1–KD case at E18.5. The distributions of the GFP-labeled earlier-born control neurons and the mCherry-labeled later-born Dab1–KD neurons were almost the same as those in the control–control case (Fig. 5A-A‴). A‴, Bin analysis. The distance between the outer margin of the CP and the inner margin of the IMZ was divided into 20 bins. Scale bar, 100 μm. n = 3 brains. B–B‴, Control–Dab1–KD case at P1.5. The mCherry-labeled later-born Dab1–KD neurons were seen at almost the same position as the GFP-labeled earlier-born control neurons. B‴, Bin analysis. The distance between the outer margin of the CP and the inner margin of the IMZ was divided into 20 bins. The later-born Dab1–KD neurons were seen in the 19th bin, which was almost similar to the case of the earlier-born control neurons. Scale bar, 100 μm. n = 4 brains. C–C‴, Control–Dab1–KD case at P7. The mCherry-labeled later-born Dab1–KD neurons were located at almost the same position as the GFP-labeled earlier-born control neurons, showing disruption of the inside-out alignment pattern. C‴, Bin analysis. The thickness of the CP was divided into 20 bins. The distribution of the later-born Dab1–KD neurons was similar to that of the earlier-born control neurons. The mean ± SEM distances from the top of the CP were 132.9 ± 16.1 μm for the GFP-labeled neurons and 124.4 ± 10.3 μm for the mCherry-labeled neurons, respectively (p = 0.8273, Mann–Whitney's test, n = 3 brains, >800 neurons were counted.). Scale bar, 100 μm.
Dab1–KD neurons can migrate past their Dab1–KD predecessors until they reach beneath the PCZ
Several abnormal factors that could affect the neuronal migration were demonstrated in the reeler or Dab1-null cortex (Pinto-Lord et al., 1982; Tabata and Nakajima, 2002; Sanada et al., 2004). To investigate whether the earlier-born Dab1–KD neurons might affect the subsequent migration of the later-born Dab1–KD neurons even in the absence of these factors, we performed sequential Dab1–KD–Dab1–KD electroporation experiments in the wild-type cortex. As a result, many mCherry-labeled Dab1–KD later-born neurons were located above the GFP-labeled Dab1–KD earlier-born neurons at P7 (Fig. 7A,A′). This pattern was also observed at P1.5 (Fig. 7B,B′), and the morphology of the radial fibers seemed unaffected in the sequential Dab1–KD experiments (Fig. 7C,C′,D,D′,E,E′). These data suggest that the Dab1–KD later-born neurons could migrate past the Dab1–KD predecessors in the wild-type cortex and that the alignment pattern at P7 was originally established when the neuronal migration was completed.
Figure 7.
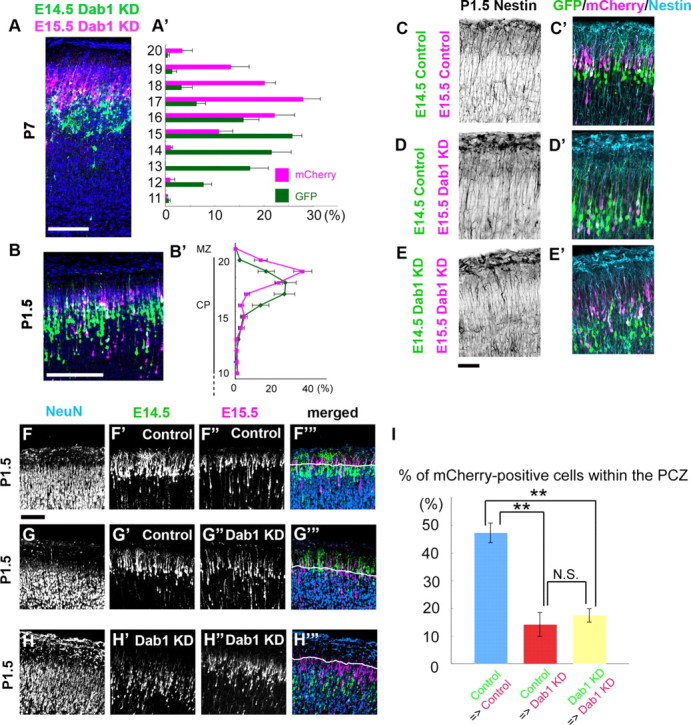
Dab1–KD neurons can go past the Dab1–KD predecessors until they reach the region just beneath the PCZ. A–A′, Sequential electroporation of Dab1–KD neurons was performed at E14.5 and E15.5. The brains were then examined at P7. Blue is the nuclear staining. A′, Bin analysis. The thickness of the CP was divided into 20 bins. Note that the later-born neurons were located above the earlier-born neurons (the mean ± SEM distances from the top of the CP to the soma were 236.3 ± 10.9 μm for the earlier-born neurons and 138.9 ± 6.9 μm for the later-born neurons, respectively; n = 3 brains, >700 neurons were counted; p = 0.0495, Mann–Whitney's test). Scale bar, 100 μm. B–B′, The brains were examined at P1.5. B′, Bin analysis. The distance between the outer margin of the CP and the inner margin of the IMZ was divided into 20 bins. n = 4 brains. Scale bar, 100 μm. C–C′, D–D′, E–E′, Staining for nestin (cyan), a marker for radial fibers, was unaffected in these sequential electroporation experiments at P1.5. C, C′, Control–control case. D, D′, Control–Dab1–KD case. E, E′, Dab1–KD–Dab1–KD case. Scale bar, 50 μm. F–F‴, G–G‴, H–H‴, NeuN staining at P1.5 in the series of sequential electroporation. F–F‴, In the control–control case, the GFP-labeled earlier-born neurons were seen below the NeuN-negative zone (F′), whereas the mCherry-labeled later-born neurons were seen within the NeuN-negative zone (F″). Cyan blue staining showed the NeuN-positive nuclei, and the white line is the lower border of the NeuN-negative zone (F‴). Scale bar, 100 μm. G–G‴, In the control–Dab1–KD case, mCherry-labeled Dab1–KD later-born neurons were located below the PCZ. H–H‴, In the Dab1–KD–Dab1–KD case, both earlier-born neurons and later-born neurons were located below the PCZ. I, Statistical analysis of the percentages of the mCherry-labeled later-born neurons within the PCZ. Control–control case; the percentage ± SEM of the mCherry-positive cells within the PCZ was 47.4 ± 3.5% (n = 4 brains, >600 neurons were counted). Control–Dab1–KD case; the percentage of the mCherry-positive cells within the PCZ was 14.2 ± 4.3% (n = 3 brains, >180 neurons were counted). Dab1–KD–Dab1–KD case; the percentage of the mCherry-positive cells within the PCZ was 17.6 ± 2.2% (n = 3 brains, >250 neurons were counted). **p < 0.01, ANOVA followed by the Scheffé's F test.
We observed some interesting features of this Dab1-independent neuronal alignment in the wild-type cortex. First, when we measured the distance from the top of the CP to the mCherry-labeled Dab1–KD neurons in the Dab1–KD–Dab1–KD case at P7, we found that the distance was significantly longer than the distance to the mCherry-labeled control neurons in the control–control case [the mean distances to the mCherry-labeled neurons from the top of the CP were 138.9 ± 6.9 μm in the Dab1–KD–Dab1–KD case (n = 3 brains) and 59.8 ± 7.3 μm in the control–control case (n = 3 brains); p=0.0495, Mann–Whitney's test]. These results indicate that the mCherry-labeled Dab1–KD neurons in the Dab1–KD–Dab1–KD case were located deeper in the CP than the mCherry-labeled control neurons in the control–control case at P7. Considering that Dab1-“dependent” passage was established within the PCZ in the control–control case, the Dab1-“independent” passage was likely to be accomplished in a different part of the developing CP. In fact, NeuN staining at P1.5 revealed that only a small number of the mCherry-labeled Dab1–KD neurons were located within the NeuN-negative PCZ in the Dab1–KD–Dab1–KD case, whereas many of the mCherry-labeled control neurons had entered the PCZ in the control–control case (Fig. 7F–F‴,H–H‴,I); these findings indicated that the Dab1–KD migrating neurons could pass by their Dab1–KD predecessors, but their migration stopped aberrantly beneath the PCZ. Because the PCZ is a transient structure formed from the inside to the outside (Fig. 2C), the Dab1-independent passage below the PCZ was considered to be accomplished as follows; the earlier-born Dab1–KD neurons stopped migrating to just beneath the PCZ, the new PCZ was formed above the old one by nonlabeled cells, the later-born Dab1–KD neurons migrated past the earlier-born Dab1–KD neurons, and the later-born Dab1–KD neurons completed their migration just beneath the new PCZ (Fig. 8). Because the mutant cortex seemed to lack a PCZ-like distinct structure in the CP (Fig. 2D,E), this Dab1-independent passage of predecessors below the PCZ could be observed only in the wild-type CP and not in the mutant CP.
Figure 8.
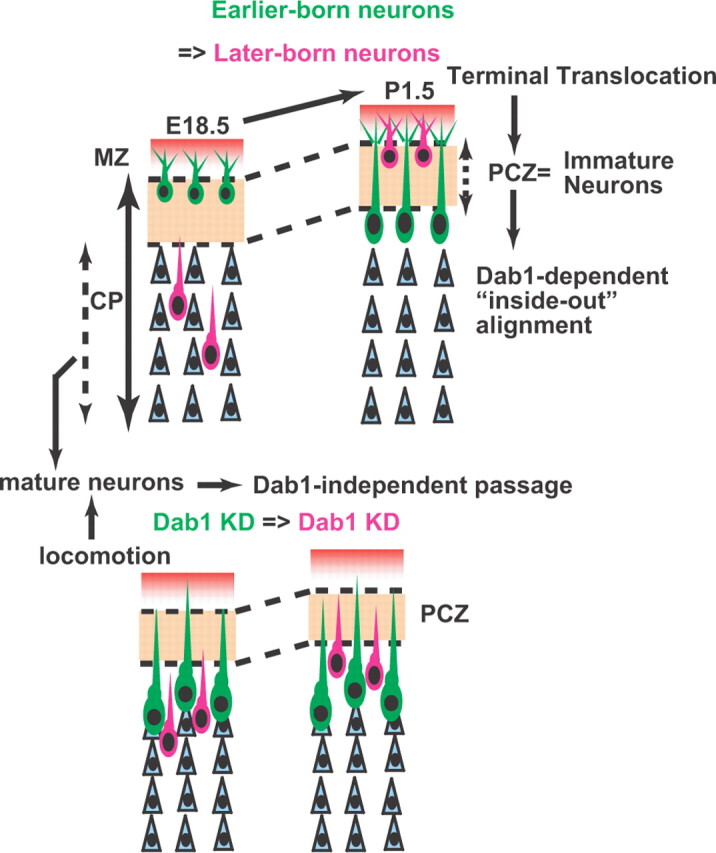
The Dab1-dependent terminal translocation forms the inside-out neuronal alignment within the PCZ. In the developing CP, there are two distinct predecessors: the immature predecessors within the PCZ and the mature predecessors below the PCZ. Terminal translocation is necessary for both neuronal entry into the PCZ and establishment of the Dab1-dependent inside-out alignment of neurons. Conversely, locomoting neurons can pass through the mature predecessors independent of Dab1 until they reach the region just beneath the PCZ. Note that the PCZ is a transient structure that is formed from the inside to the outside and that Dab1–KD neurons cannot enter the PCZ.
Second, the alignment of the Dab1–KD–Dab1–KD neurons showed a more loosely scattered pattern (Fig. 7A) compared with the packed and clearly segregated pattern of the control–control neurons (Fig. 5C). When the distances from the top of the CP to the labeled cells were measured at P7, the statistical variances, the square value of the SDs, were significantly higher in the Dab1–KD–Dab1–KD case than in the control–control case, indicative of impairment of the birth-date-dependent alignment in the former case. The variances were as follows: GFP-positive neurons in the control–control case, 2181 μm2 (3 brains, 351 neurons); GFP-positive neurons in the Dab1–KD–Dab1–KD case, 4200 μm2 (3 brains, 362 neurons) (p < 0.001, F test); mCherry-positive neurons in the control–control case, 954 μm2 (3 brains, 376 neurons); mCherry-positive neurons in the Dab1–KD–Dab1–KD case, 3436 μm2 (3 brains, 381 neurons) (p < 0.001, F test). Because the birth-date-dependent alignment was originally established within the PCZ (Fig. 3B,E), these data suggest that not only the simple migration past the predecessors by locomotion but also entry into the PCZ by terminal translocation in a Dab1-dependent manner is required for the proper birth-date-dependent laminar alignment of neurons in the mature cortex.
Discussion
It is known that migrating neurons need to move past “all” their predecessors for establishment of the inside-out neuronal alignment. In this study, we found that there were two types of predecessors in the CP: the immature predecessors within the PCZ, the outermost region of the CP, and the mature predecessors below the PCZ (Fig. 8). The Dab1-dependent terminal translocation was required for the entry of the neurons into the PCZ and the birth-date-dependent alignment of neurons within this zone. Our sequential in utero electroporation experiments in the wild-type cortex revealed that locomoting Dab1–KD neurons could migrate past the mature predecessors located beneath the PCZ but could not go past the immature predecessors within the PCZ and that the Dab1-dependent eventual inside-out neuronal alignment in the wild-type cortex is primarily established within the PCZ by the “local” migratory mode, terminal translocation.
Although migrating neurons sharing the same birth date show scattered distribution throughout the CP and IMZ when they are in the locomotion mode, these neurons rearrange within the PCZ according to their birth date after the completion of terminal translocation (Fig. 3B′,B″). These data suggest that the PCZ is a unique and transient structure in which the neurons are arranged and segregated according to their birth dates. In the case of sequential Dab1–KD–Dab1–KD electroporation (Fig. 7H–H‴), the later-born Dab1–KD neurons were located above the earlier-born Dab1–KD neurons but still remained beneath the normal PCZ, suggesting that the earlier-born Dab1–KD neurons were prevented from becoming a part of the PCZ. Furthermore, ectopically overexpressed Reelin induces neuronal aggregation similar to that in the PCZ (Kubo et al., 2010a). Thus, it is possible that Reelin–Dab1 signaling may be involved in the formation of the PCZ itself.
It is important to identify the specific molecular mechanisms involved in each specific mode of migration. In regard to Dab1, a moderate delay of migration of the Dab1–KD neurons in the IMZ was observed (Fig. 3C′,D′) (Yano et al., 2010). This delay seemed to be consistent with the previous reports that suggested the presence of Reelin other than from the MZ and the functional Reelin receptors in the deeper part of the cortex (Yoshida et al., 2006; Uchida et al., 2009). However, it seemed that this delay had only a minor impact on the final neuronal alignment in the control–Dab1–KD case (Fig. 6), because, for the most part, the KD cells could go past their predecessors in the CP until they reached beneath the PCZ. In addition, if the delay of locomotion of the Dab1–KD cells were to affect the final positioning, the cells would be observed more superficially than their normal positions in the mature cortex (Kubo et al., 2010b). However, no such superficial shift was observed in our sequential electroporation experiments. Rather, we and other groups (Olson et al., 2006; Franco et al., 2011) revealed that Dab1 was mainly involved in terminal translocation. We recently found that the KD of the Reelin receptors also affected terminal translocation (Kubo et al., 2010a). Thus, Reelin–Dab1 signaling likely regulates the terminal translocation into the PCZ, which eventually shows an inside-out neuronal alignment. Considering that recent studies have also reported several cellular cascades regulated by Reelin–Dab1 signaling (Feng et al., 2007; Hashimoto-Torii et al., 2008; Park and Curran, 2008; Voss et al., 2008; Chai et al., 2009), future studies will be needed to uncover how Reelin–Dab1 signaling might orchestrate the molecular cascades to regulate the terminal translocation into the PCZ and the eventual inside-out neuronal alignment.
Terminal translocation is known to involve several steps, such as detachment of the cells from radial fibers, anchorage of the leading process to the MZ, and somal movement (Nadarajah et al., 2001). It was reported previously that neurons adhered abnormally to the radial fibers even after the completion of migration in the reeler cortex. This abnormal adherence to the radial fibers was hypothesized to cause a “traffic jam” for the later-born neurons (Pinto-Lord et al., 1982). If a similar situation was considered for the Dab1–KD neurons in our experiments, a simple interpretation of the pattern of neuronal alignment in the Dab1–KD–Dab1–KD case would be that many of the radial fibers might be unoccupied by the Dab1–KD earlier-born neurons, allowing the later-born neurons to migrate along the unoccupied fibers. Alternatively, another interpretation would be that the Dab1-null neurons might become detached from the radial fibers but not translocated. This “translocation failure” hypothesis has also been proposed previously (Cooper, 2008; Feng and Cooper, 2009), based on the findings that the average distance between radial fibers was almost the same as the reported distance between the postmigratory Dab1-null neurons and their parental radial fibers (Sanada et al., 2004). Lending support to the latter possibility, the introduction of Dab1 into a Dab1-null cortex at an appropriate stage was capable of rescuing the migration failure (Morimura and Ogawa, 2009; Simó et al., 2010), and, in Dab1 chimeras, wild-type neurons could migrate past the Dab1-null neurons and establish the super cortex above the Dab1-null cortex (Hammond et al., 2001); these findings would be inconsistent with the notion that many of the postmigratory early-born neurons adhere to the radial fibers. Collectively, our data can be interpreted as indicating that the Dab1–KD neurons also detach from the radial fibers once they reach beneath the already formed PCZ in the wild-type cortex and that the later-born Dab1–KD neurons might migrate past the earlier-born neurons using locomotion until they reach beneath the newly formed PCZ (Fig. 8).
During cortical development, the earliest-born neurons form the preplate. The next cohort migrates into the preplate and causes the preplate splitting. Invasion into the preplate has been suggested to be accomplished by somal translocation of neurons regulated by Reelin–Dab1 signaling (Caviness and Sidman, 1973; Goffinet, 1979; Nadarajah et al., 2001). Because our data showed that Dab1 was required for both somal translocation and terminal translocation, the primary defect in the Dab1-null or reeler cortex may be migration failure into the respective regions of the cortex, i.e., the preplate or PCZ through translocation. In addition, it was reported that several non-cell-autonomous factors were involved in the defective neuronal migration in the mutant cortex such as the internal plexiform zone (IPZ) (Tabata and Nakajima, 2002) and that the migration failure in the Dab1-null cortex was rescued by the introduction of Dab1 at an appropriate stage (Morimura and Ogawa, 2009; Simó et al., 2010). These studies may be interpreted as suggesting that the introduction of Dab1 rescues the ability of the mutant neurons to migrate past several distinct regions of the mutant CP, including IPZ, probably by adopting the unique migratory mode, translocation. Furthermore, considering that the preplate and the IPZ are cell-sparse and fiber-rich structures (Marín-Padilla, 1998; Tabata and Nakajima, 2002) unlike the cell-dense PCZ, it is possible that not only the cell density, but also other factors, such as the extracellular matrix or cell–cell interactions that exist preferentially around the PCZ (Tachikawa et al., 2008), might affect the migratory behavior of neurons.
From an evolutionary standpoint, the expression levels of Reelin have been noted to be the highest in mammals, although the protein is also expressed in other vertebrates (Bar et al., 2000). Preplate neurons are also known to be the oldest neurons phylogenetically and to exist in other vertebrates as well (Marín Padilla, 2001). The migration and arrangement of these neurons and layer VI neurons are thought to be regulated through somal translocation, which in turn is thought to be a relatively early mode of migration from an evolutionary perspective (Meyer et al., 1998; Nadarajah et al., 2001). Conversely, considering that the neurons in the deeper part of the CP are more differentiated than those located more superficially (i.e., the “inside-out gradient of maturation”) (Caviness and Rakic, 1978), radial-fiber-guided locomotion may have developed in the mammalian neocortex to enable the cells to migrate long distances past mature predecessors that had arrived earlier in the expanding cortex (Rakic, 1972). However, locomotion alone is not sufficient to establish the proper inside-out layered structure, because locomoting neurons cannot migrate past the immature predecessors within the PCZ. Therefore, we hypothesize that locomotion is an evolutionarily acquired mode that enables the movement of neurons toward the surface of the brain, whereas terminal translocation is a phylogenetically conserved migratory mode that allows the neurons to be arranged locally in a birth-date-dependent manner. We also hypothesize that the switch from locomotion to terminal translocation is regulated by the high amount of Reelin in the mammalian cortex to establish the inside-out neuronal alignment.
In conclusion, the present study provides the evidence to indicate that the PCZ has a distinct role in the establishment of the final inside-out alignment of the neurons. The change of the migratory mode of neurons just beneath the PCZ by Reelin–Dab1 signaling and the sequence of the birth-date-dependent alignment of neurons within the PCZ are likely to be important for the establishment of the proper layered structure of the mammalian neocortex.
Footnotes
This project was supported by the Strategic Research Program for Brain Sciences (“Understanding of Molecular and Environmental Bases for Brain Health”), the Ministry of Education, Culture, Sports, and Science and Technology of Japan, the Keio Gijuku Academic Development Funds, and the Promotion and Mutual Aid Corporation for Private Schools of Japan. We thank Drs. J. Cooper, F. Miller, and J. Miyazaki for providing the plasmids, Dr. H. Tabata for assisting the time-lapse analyses, and all the members of the Nakajima laboratory for their valuable discussions. K.S. is a research fellow of the Japan Society for the Promotion of Science.
The authors declares no competing financial interests.
References
- Ajioka I, Nakajima K. Birth-date-dependent segregation of the mouse cerebral cortical neurons in reaggregation cultures. Eur J Neurosci. 2005;22:331–342. doi: 10.1111/j.1460-9568.2005.04214.x. [DOI] [PubMed] [Google Scholar]
- Bar I, Lambert de Rouvroit C, Goffinet AM. The evolution of cortical development. An hypothesis based on the role of the Reelin signaling pathway. Trends Neurosci. 2000;23:633–638. doi: 10.1016/s0166-2236(00)01675-1. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Rakic P. Mechanisms of cortical development: a view from mutations in mice. Annu Rev Neurosci. 1978;1:297–326. doi: 10.1146/annurev.ne.01.030178.001501. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Sidman RL. Time of origin or corresponding cell classes in the cerebral cortex of normal and reeler mutant mice: an autoradiographic analysis. J Comp Neurol. 1973;148:141–151. doi: 10.1002/cne.901480202. [DOI] [PubMed] [Google Scholar]
- Chai X, Förster E, Zhao S, Bock HH, Frotscher M. Reelin stabilizes the actin cytoskeleton of neuronal processes by inducing n-cofilin phosphorylation at serine3. J Neurosci. 2009;29:288–299. doi: 10.1523/JNEUROSCI.2934-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA. A mechanism for inside-out lamination in the neocortex. Trends Neurosci. 2008;31:113–119. doi: 10.1016/j.tins.2007.12.003. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- Feng L, Cooper JA. Dual functions of Dab1 during brain development. Mol Cell Biol. 2009;29:324–332. doi: 10.1128/MCB.00663-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Allen NS, Simo S, Cooper JA. Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev. 2007;21:2717–2730. doi: 10.1101/gad.1604207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco SJ, Martinez-Garay I, Gil-Sanz C, Harkins-Perry SR, Müller U. Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron. 2011;69:482–497. doi: 10.1016/j.neuron.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal JS, Morozov YM, Ayoub AE, Chatterjee M, Rakic P, Haydar TF. Molecular and morphological heterogeneity of neural precursors in the mouse neocortical proliferative zones. J Neurosci. 2006;26:1045–1056. doi: 10.1523/JNEUROSCI.4499-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloster A, Wu W, Speelman A, Weiss S, Causing C, Pozniak C, Reynolds B, Chang E, Toma JG, Miller FD. The Tα1α-tubulin promoter specifies gene expression as a function of neuronal growth and regeneration in transgenic mice. J Neurosci. 1994;14:7319–7330. doi: 10.1523/JNEUROSCI.14-12-07319.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goffinet AM. An early development defect in the cerebral cortex of the reeler mouse. A morphological study leading to a hypothesis concerning the action of the mutant gene. Anat Embryol (Berl) 1979;157:205–216. doi: 10.1007/BF00305160. [DOI] [PubMed] [Google Scholar]
- Hammond V, Howell B, Godinho L, Tan SS. disabled-1 functions cell autonomously during radial migration and cortical layering of pyramidal neurons. J Neurosci. 2001;21:8798–8808. doi: 10.1523/JNEUROSCI.21-22-08798.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartfuss E, Förster E, Bock HH, Hack MA, Leprince P, Luque JM, Herz J, Frotscher M, Götz M. Reelin signaling directly affects radial glia morphology and biochemical maturation. Development. 2003;130:4597–4609. doi: 10.1242/dev.00654. [DOI] [PubMed] [Google Scholar]
- Hashimoto-Torii K, Torii M, Sarkisian MR, Bartley CM, Shen J, Radtke F, Gridley T, Sestan N, Rakic P. Interaction between Reelin and Notch signaling regulates neuronal migration in the cerebral cortex. Neuron. 2008;60:273–284. doi: 10.1016/j.neuron.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatanaka Y, Hisanaga S, Heizmann CW, Murakami F. Distinct migratory behavior of early- and late-born neurons derived from the cortical ventricular zone. J Comp Neurol. 2004;479:1–14. doi: 10.1002/cne.20256. [DOI] [PubMed] [Google Scholar]
- Honda T, Nakajima K. Mouse Disabled1 (DAB1) is a nucleocytoplasmic shuttling protein. J Biol Chem. 2006;281:38951–38965. doi: 10.1074/jbc.M609061200. [DOI] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- Kawauchi T, Hoshino M. Molecular pathways regulating cytoskeletal organization and morphological changes in migrating neurons. Dev Neurosci. 2008;30:36–46. doi: 10.1159/000109850. [DOI] [PubMed] [Google Scholar]
- Kojima T, Nakajima K, Mikoshiba K. The disabled 1 gene is disrupted by a replacement with L1 fragment in yotari mice. Brain Res Mol Brain Res. 2000;75:121–127. doi: 10.1016/s0169-328x(99)00313-7. [DOI] [PubMed] [Google Scholar]
- Kubo K, Honda T, Tomita K, Sekine K, Ishii K, Uto A, Kobayashi K, Tabata H, Nakajima K. Ectopic Reelin induces neuronal aggregation with a normal birthdate-dependent “inside-out” alignment in the developing neocortex. J Neurosci. 2010a;30:10953–10966. doi: 10.1523/JNEUROSCI.0486-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo K, Tomita K, Uto A, Kuroda K, Seshadri S, Cohen J, Kaibuchi K, Kamiya A, Nakajima K. Migration defects by DISC1 knockdown in C57BL/6, 129X1/SvJ, and ICR strains via in utero gene transfer and virus-mediated RNAi. Biochem Biophys Res Commun. 2010b;400:631–637. doi: 10.1016/j.bbrc.2010.08.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luque JM, Morante-Oria J, Fairén A. Localization of ApoER2, VLDLR and Dab1 in radial glia: groundwork for a new model of reelin action during cortical development. Brain Res Dev Brain Res. 2003;140:195–203. doi: 10.1016/s0165-3806(02)00604-1. [DOI] [PubMed] [Google Scholar]
- Marín-Padilla M. Cajal-Retzius cells and the development of the neocortex. Trends Neurosci. 1998;21:64–71. doi: 10.1016/s0166-2236(97)01164-8. [DOI] [PubMed] [Google Scholar]
- Marín Padilla M. The evolution of the structure of the neocortex in mammals: a new theory of cytoarchitecture (in Spanish) Rev Neurol. 2001;33:843–853. [PubMed] [Google Scholar]
- Meyer G, Soria JM, Martínez-Galán JR, Martín-Clemente B, Fairén A. Different origins and developmental histories of transient neurons in the marginal zone of the fetal and neonatal rat cortex. J Comp Neurol. 1998;397:493–518. [PubMed] [Google Scholar]
- Molnár Z, Métin C, Stoykova A, Tarabykin V, Price DJ, Francis F, Meyer G, Dehay C, Kennedy H. Comparative aspects of cerebral cortical development. Eur J Neurosci. 2006;23:921–934. doi: 10.1111/j.1460-9568.2006.04611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Morimura T, Ogawa M. Relative importance of the tyrosine phosphorylation sites of Disabled-1 to the transmission of Reelin signaling. Brain Res. 2009;1304:26–37. doi: 10.1016/j.brainres.2009.09.087. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Brunstrom JE, Grutzendler J, Wong RO, Pearlman AL. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Mikoshiba K, Miyata T, Kudo C, Ogawa M. Disruption of hippocampal development in vivo by CR-50 mAb against reelin. Proc Natl Acad Sci U S A. 1997;94:8196–8201. doi: 10.1073/pnas.94.15.8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Olson EC, Kim S, Walsh CA. Impaired neuronal positioning and dendritogenesis in the neocortex after cell-autonomous Dab1 suppression. J Neurosci. 2006;26:1767–1775. doi: 10.1523/JNEUROSCI.3000-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TJ, Curran T. Crk and Crk-like play essential overlapping roles downstream of disabled-1 in the Reelin pathway. J Neurosci. 2008;28:13551–13562. doi: 10.1523/JNEUROSCI.4323-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto-Lord MC, Evrard P, Caviness VS., Jr Obstructed neuronal migration along radial glial fibers in the neocortex of the reeler mouse: a Golgi-EM analysis. Brain Res. 1982;256:379–393. doi: 10.1016/0165-3806(82)90181-x. [DOI] [PubMed] [Google Scholar]
- Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145:61–83. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- Sanada K, Gupta A, Tsai LH. Disabled-1-regulated adhesion of migrating neurons to radial glial fiber contributes to neuronal positioning during early corticogenesis. Neuron. 2004;42:197–211. doi: 10.1016/s0896-6273(04)00222-3. [DOI] [PubMed] [Google Scholar]
- Sarnat HB, Nochlin D, Born DE. Neuronal nuclear antigen (NeuN): a marker of neuronal maturation in early human fetal nervous system. Brain Dev. 1998;20:88–94. doi: 10.1016/s0387-7604(97)00111-3. [DOI] [PubMed] [Google Scholar]
- Sheldon M, Rice DS, D'Arcangelo G, Yoneshima H, Nakajima K, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389:730–733. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- Simó S, Jossin Y, Cooper JA. Cullin 5 regulates cortical layering by modulating the speed and duration of Dab1-dependent neuronal migration. J Neurosci. 2010;30:5668–5676. doi: 10.1523/JNEUROSCI.0035-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: visualization of neuronal migration in the developing cortex. Neuroscience. 2001;103:865–872. doi: 10.1016/s0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Neurons tend to stop migration and differentiate along the cortical internal plexiform zones in the Reelin signal-deficient mice. J Neurosci Res. 2002;69:723–730. doi: 10.1002/jnr.10345. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Multipolar migration: the third mode of radial neuronal migration in the developing cerebral cortex. J Neurosci. 2003;23:9996–10001. doi: 10.1523/JNEUROSCI.23-31-09996.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata H, Kanatani S, Nakajima K. Differences of migratory behavior between direct progeny of apical progenitors and basal progenitors in the developing cerebral cortex. Cereb Cortex. 2009;19:2092–2105. doi: 10.1093/cercor/bhn227. [DOI] [PubMed] [Google Scholar]
- Tachikawa K, Sasaki S, Maeda T, Nakajima K. Identification of molecules preferentially expressed beneath the marginal zone in the developing cerebral cortex. Neurosci Res. 2008;60:135–146. doi: 10.1016/j.neures.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Goto T, Miyama S, Nowakowski RS, Caviness VS., Jr Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J Neurosci. 1999;19:10357–10371. doi: 10.1523/JNEUROSCI.19-23-10357.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissir F, Goffinet AM. Reelin and brain development. Nat Rev Neurosci. 2003;4:496–505. doi: 10.1038/nrn1113. [DOI] [PubMed] [Google Scholar]
- Uchida T, Baba A, Pérez-Martínez FJ, Hibi T, Miyata T, Luque JM, Nakajima K, Hattori M. Downregulation of functional Reelin receptors in projection neurons implies that primary Reelin action occurs at early/premigratory stages. J Neurosci. 2009;29:10653–10662. doi: 10.1523/JNEUROSCI.0345-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss AK, Britto JM, Dixon MP, Sheikh BN, Collin C, Tan SS, Thomas T. C3G regulates cortical neuron migration, preplate splitting and radial glial cell attachment. Development. 2008;135:2139–2149. doi: 10.1242/dev.016725. [DOI] [PubMed] [Google Scholar]
- Ware ML, Fox JW, González JL, Davis NM, Lambert de Rouvroit C, Russo CJ, Chua SC, Jr, Goffinet AM, Walsh CA. Aberrant splicing of a mouse disabled homolog, mdab1, in the scrambler mouse. Neuron. 1997;19:239–249. doi: 10.1016/s0896-6273(00)80936-8. [DOI] [PubMed] [Google Scholar]
- Yano M, Hayakawa-Yano Y, Mele A, Darnell RB. Nova2 regulates neuronal migration through an RNA switch in disabled-1 signaling. Neuron. 2010;66:848–858. doi: 10.1016/j.neuron.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneshima H, Nagata E, Matsumoto M, Yamada M, Nakajima K, Miyata T, Ogawa M, Mikoshiba K. A novel neurological mutant mouse, yotari, which exhibits reeler-like phenotype but expresses CR-50 antigen/reelin. Neurosci Res. 1997;29:217–223. doi: 10.1016/s0168-0102(97)00088-6. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Assimacopoulos S, Jones KR, Grove EA. Massive loss of Cajal-Retzius cells does not disrupt neocortical layer order. Development. 2006;133:537–545. doi: 10.1242/dev.02209. [DOI] [PubMed] [Google Scholar]
- Zheng L, Baumann U, Reymond JL. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 2004;32:e115. doi: 10.1093/nar/gnh110. [DOI] [PMC free article] [PubMed] [Google Scholar]