

Abstract
Neurons recruit numerous mechanisms to facilitate the development of synaptic connections. However, little is known about activity-dependent mechanisms that control the timing and fidelity of this process. Here we describe a novel pathway used by neurons to regulate glutamate receptors at maturing central synapses. This pathway relies on communication between neurons and astrocytes and the ability of astrocytes to release the factor SPARC (secreted protein, acidic and rich in cysteine). SPARC expression is dynamically regulated and plays a critical role in determining the level of synaptic AMPARs. SPARC ablation in mice increases excitatory synapse function, causes an abnormal accumulation of surface AMPARs at synapses, and impairs synaptic plasticity during development. We further demonstrate that SPARC inhibits the properties of neuronal β3-integrin complexes, which are intimately coupled to AMPAR stabilization at synapses. Thus neuron–glial signals control glutamate receptor levels at developing synapses to enable activity-driven modifications of synaptic strength.
Introduction
Synapses are key sites of information exchange in the CNS, and perturbation of their formation and maintenance is correlated with cognitive impairments found in patients with neurodevelopmental disorders and neurodegenerative diseases (Fiala et al., 2002). Neurons employ an assortment of mechanisms to promote synapse formation. Proteins including those of the SynCAM, NGL (Netrin-G ligand), neuroligin, neurexin, LAR (leukocyte common antigen-related), LRRTM (leucine-rich repeat transmembrane neuronal), and Eph receptor families are required for proper formation and maintenance of synapses (Waites et al., 2005; Linhoff et al., 2009). Many of these molecules function in transsynaptic signaling complexes that coordinate the development of presynaptic and postsynaptic specializations. Following the initial stages of synaptogenesis, neurons enlist additional activity-dependent processes to fine-tune the maturation and pattern of synaptic connections (Katz and Shatz, 1996; Cline and Haas, 2008; Kerschensteiner et al., 2009). Several proteins, including the growth factor BDNF (Kuczewski et al., 2009), are implicated in these processes, but in general, the molecular cues that govern this phase of synapse development remain unclear.
Neurons also rely on glial cells for synapse formation (Barres, 2008). Remarkably, culturing neurons with media conditioned by astrocytes controls the number and effectiveness of synapses (Pfrieger and Barres, 1997; Nägler et al., 2001; Ullian et al., 2001), indicating that soluble factors secreted from astrocytes play an important role in synapse formation. It is thought that astrocyte-secreted factors function during discrete phases of synapse development (Barres, 2008). Indeed, Christopherson et al. (2005) identified thrombospondins as necessary components of glial-conditioned medium for the initial steps of synaptogenesis. Glial cells also release factors such as cholesterol to generally increase the functionality of synapses (Mauch et al., 2001) and other cues that promote their elimination (Stevens et al., 2007). However, the importance of glial-secreted factors in controlling the process of synapse maturation remains unresolved.
Here we demonstrate that bidirectional signals between neurons and astrocytes play a pivotal role in controlling the level of postsynaptic glutamate receptors during synaptic development. We found that astrocytes secrete the matricellular protein known as SPARC (secreted protein, acidic and rich in cysteine) to regulate the level of postsynaptic AMPA receptors (AMPARs) at maturing synapses. SPARC ablation in mice enhances synaptic function and impairs long-term potentiation (LTP) and the ability of neurons to scale up their synapses during periods of activity deprivation. These changes coincide with an abnormal accumulation of surface AMPARs at developing synapses. We show that SPARC regulates surface AMPARs through β3-integrins, receptors that have been recently shown to stabilize GluR2 subunits at synapses (Cingolani et al., 2008). This study reveals a novel cellular and molecular mechanism for determining glutamate receptor levels at maturing synapses to control synaptic development and plasticity in the CNS.
Materials and Methods
Animals.
For electrophysiological experiments, littermates of SPARC knock-out (KO) mice (The Jackson Laboratory) and wild-type (WT) mice of C57BL/6J/129SVJ (both male and female) were used for analysis. The only exception was the use of age-matched mice of each genotype for the de-depression analysis. WT C57BL/6J mice were used for all other experiments. Animal procedures were all performed in accordance with the guidelines of the Canadian Council for Animal Care and the Montreal General Hospital Facility Animal Care Committee.
Antibodies and recombinant proteins.
The following antibodies were used in this study: mouse SPARC AF942 (goat polyclonal, R and D Systems), Hevin/SC1 (rat polyclonal, R and D Systems), GFAP (mouse monoclonal, Sigma), GFAP (rabbit polyclonal, Millipore), glutamine synthetase (mouse monoclonal, Millipore), IBA-1 (rabbit polyclonal, Wako), GluR1 N-terminal (rabbit polyclonal, Calbiochem), GluR1 C-terminal CT3 (rabbit monoclonal, Millipore), GluR1 N-terminal RH95 (mouse monoclonal, Millipore), GluR2 N-terminal (mouse monoclonal, Millipore), β3-integrin (rabbit polyclonal AB1932, Millipore), NeuN (mouse monoclonal, Millipore), MAP2 clone HM-2 (mouse monoclonal, Sigma), PSD-95 (mouse monoclonal, NeuroMab/Antibodies), Synapsin (rabbit polyclonal, Synaptic Systems), NR1 (mouse monoclonal clone 54.1, Millipore), GAPDH (Abcam), hamster anti-mouse β3-integrin clone 2C9.G2 (BD Biosciences), hamster IgG1 κ isotype control (BD Biosciences), and anti-goat and rat HRP (GE Healthcare). Recombinant mouse SPARC protein was purchased from R and D Systems. Peptides were synthesized by New England Peptide. Peptide 2.3 was previously described (Lane and Sage, 1990). The sequence of scrambled Peptide 2.3 was KLTEKTGLGPLHGDKIHY.
Hippocampal slice preparation.
Organotypic hippocampal slices were prepared as described previously (Zhou et al., 2007). Briefly, 300 μm hippocampal slices were prepared from postnatal day 6 (P6) pups and transferred onto semiporous tissue culture inserts (0.4 μm pore size; Millipore). Culture media consisted of 50% Minimum Essential Medium, 25% horse serum, 25% HBSS, 6.5 mg/ml d-glucose, and 0.5% penicillin/streptomycin (Invitrogen). Medium was replaced three times per week. For immunohistochemistry, slices were fixed in 4% paraformaldehyde/0.1 m phosphate buffer for 30 min, washed with TBS, and blocked for 1 h at room temperature in 10% donkey serum (Jackson ImmunoResearch Laboratories) in TBS (Tris-buffered saline) containing 0.2% Triton X-100 (TBS-T) and then incubated with primary antibodies GFAP (1:300) and SPARC (1:300) overnight. The next day, slices were washed four times with TBS-T before incubation with donkey anti-goat Alexa-568, anti-mouse Alexa-488, and/or anti-rabbit Alexa 647 (Invitrogen). Slices were then washed four times with TBS-T and imaged with confocal microscopy. For immunohistochemistry of hippocampal tissue sections, mice (WT and KO littermates at P14 or P27) were perfused with 4% paraformaldehyde/0.1 m phosphate buffer and brains were postfixed overnight in the same solution followed by 30% (w/v) sucrose/0.1 m phosphate buffer. Thirty-micrometer-thick coronal sections were prepared by a cryostat (Leica, CM 3050 S) and processed free-floating for immunohistochemistry. After 1 h incubation in blocking solution (10% donkey serum, TBS-T), sections were incubated overnight at 4°C with the following primary antibodies: SPARC (1:300), glutamine synthetase (1:200), IBA-1 (1:500), NeuN (1:400), and MAP-2 (1:300). After three washes in TBS, primary antibodies were revealed with a 2 h incubation with Alexa Fluor secondary antibodies (1:300, 10% donkey serum, TBS-T) and confocal microscopy.
Spine analysis.
To visualize dendritic spine morphology, we made use of Semliki Forest viruses (SFVs) expressing membrane-targeted mCherry protein, a variant of RFP. Preparation of plasmid constructs and virus were described previously (Zhou et al., 2007). At 11 d in vitro (11DIV), SFV particles were injected into hippocampal slices with a picospritzer (General Valve). Sixteen to twenty hours after infection, slices were fixed and mounted for confocal imaging. Confocal stacks of dendrites were collected with a Yokogawa spinning disk confocal system (PerkinElmer) using a 60× oil-immersion objective connected to a Nikon Eclipse TE2000. Z-stacks were collected using MetaMorph imaging software (Molecular Devices). For analysis of CA1 spines from SPARC KO slices, at least 24 dendritic segments per genotype were analyzed from at least four independent experiments (753 WT spines and 730 knock-out spines). Measurements of dendritic spine density and morphology were acquired using Photoshop and Reconstruct software according to previously published methods (Zhou et al., 2007). Differences between samples were assessed using either a Student's t test or Mann–Whitney U test as indicated in the figure legend. All analysis was performed by an investigator blind to the genotype or experimental manipulation.
Dissociated hippocampal cultures.
Dissociated hippocampal neurons were grown suspended above astrocyte feeder cultures using a modified method previously published (Kaech and Banker, 2006). An astrocyte feeder layer was prepared by plating mouse astrocytes at 20,000 cells/cm2 onto 12-well dishes coated with poly-d-lysine (0.1 mg/ml) (Sigma) in Minimal Essential Medium containing Earle's salts and l-glutamine supplemented with 10% horse serum, 0.6% glucose, and 1% penicillin/streptomycin (Invitrogen). Media was replaced with Neurobasal-A medium supplemented with 2% B27, 1 mm GlutaMax, and 1% penicillin/streptomycin (Invitrogen) 24 h before neuron dissection. After 5 d of growth of astrocyte cultures, hippocampal neurons were isolated from P0 pups. Briefly, hippocampi were dissociated by treatment with papain (0.1% papain, 0.02% BSA in Neurobasal-A medium, 15 min at 37°C) followed by trituration with a fire-polished glass pipette in Neurobasal-A media containing trypsin inhibitor (1%) (Sigma) and BSA (1%). Neurons were plated onto poly-l-lysine-coated coverslips (0.1 mg/ml; Sigma) at a density of 20,000 cells/cm2 and were allowed to attach for 3 h before transfer to dishes containing the astrocyte feeder layers. Coverslips were suspended above the feeder layer on wax dots adhered to the bottom of the culture dish well.
Immunohistochemistry of dissociated neurons.
For visualization of surface GluR1, GluR2, and β3-integrin levels, we used a protocol described previously (Stellwagen et al., 2005). Briefly, 14-d-old cultures were chilled on ice, washed with ice-cold PBS, and fixed with 4% paraformaldehyde/0.1 m phosphate buffer for 10 min. Nonpermeabilized cells were blocked in 5% BSA and incubated with an antibody against the N terminus of GluR1 (1:30), GluR2 (1:200), or β3-integrin (1:200) for 1 h. Immunolabeling with an antibody against MAP2 (1:200) as well as Synapsin (1:300) and PSD-95 (1:100) was used to confirm the lack of cell permeabilization (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). For other immunohistochemistry, cells were permeabilized following fixation in PBS containing 0.2% Triton X-100 and immunolabeled with antibodies against the C terminus of GluR1 (1:100), PSD-95 (1:100), and Synapsin (1:300) as indicated. For analysis of surface GluR1 levels, images were thresholded blindly using ImageJ (NIH) to exclude background noise; values were held constant across each experiment. For each neuron, a 50 μm region of three dendrites (one body distance away) was measured. The average pixel intensity was recorded for each dendrite (>70 measured per condition for each experiment) and used to compare surface GluR1 levels across conditions. This method was also used to assess total GluR1 levels. Significance was determined by ANOVA and a post hoc Holm–Sidak test. For measurement of synaptic punctum density, RGB overlays of red (Synapsin) and green (PSD-95) images (channels were separately thresholded to exclude background noise) were used. Colocalization masks were generated using the “RG2B colocalization” ImageJ plugin (using default parameters). Synaptic density per 50 μm was calculated by counting puncta automatically using ImageJ.
For analysis of silent synapses, cells were fixed in cold methanol and immunolabeled with antibodies against the N terminus of NR1 (1:200) and GluR1 (1:30). Total NR1 and colocalized GluR1/NR1 punctum density were measured using ImageJ using the method described above.
Immunoblot analysis.
To examine the temporal expression of SPARC during development, hippocampi from mice of P2–P60 were dissected and homogenized in Triton lysis buffer (20 mm Tris, pH 7.4, 137 mm NaCl, 25 mm β-glycerophosphate, 2 mm EDTA, 1% Triton X-100, 10% glycerol, and 0.1% SDS supplemented with protease inhibitors and sodium orthovanadate) using a Dounce homogenizer and lysed on ice for 15 min. Lysates were centrifuged at 13,000 rpm for 10 min at 4°C to pellet cell debris. Supernatants were diluted with 3× sample buffer, resolved by SDS-PAGE, and analyzed by immunoblotting with anti-SPARC (1:1000) and GAPDH (1:2000) as a control for protein levels. For other immunoblotting, SC1 (1:2000), GluR1 RH95 (1:1000), GluR2 (1:1000), and NR1 (1:2000) were used.
Cell surface biotinylation.
Neurons were gently washed twice with ice-cold PBS (containing Ca2+ and Mg2+) and then incubated with Sulfo-NHS-SS-Biotin/PBS (0.2 mg/ml) for 30 min at 4°C. The biotin reaction was quenched by washing two times with PBS containing 100 mm glycine. Neurons were lysed on ice with PBS/0.1% Triton X-100/0.1% SDS supplemented with protease inhibitors and sodium orthovanadate. An aliquot of lysate was kept for analysis of total receptor levels. Biotinylated receptors were precipitated with streptavidin beads (Sigma) for 2 h at 4°C on a rotating platform and were eluted with 3× sample buffer. Samples (biotinylated complexes and lysates) were heated to 75°C for 10 min and then resolved by SDS-PAGE and analyzed by immunoblotting with GluR1, GluR2, and NR1 antibodies. As a control for protein levels, membranes were stripped and reprobed with an antibody against NR1 (1:2000). Densitometric quantification was carried using ImageJ (NIH). Cell surface receptor values were adjusted relative to total receptor levels.
Cell attachment assay.
HEK 293T cells were transfected with either pcDNA3 (empty vector control) or pcDNA3-β3-integrin (kindly provided by Dr P. Holland, McGill University, Montreal, QC, Canada) using Lipofectamine 2000 (Invitrogen). After 48 h, cells were trypsinized and counted. Before cell attachment, cell suspensions (2.5 × 105 cells/ml) were incubated with or without SPARC (0.5 μg/ml) for 10 min at room temperature. Cells were then allowed to attach to vitronectin-coated (5 μg/ml, Calbiochem) cover glasses for 1 h at 37°C, washed twice with PBS, and fixed in 4% paraformaldehyde/0.1 m phosphate buffer for 30 min. Following fixation, attached cells were permeabilized and labeled by incubation with 1 μm TOTO-3 iodide (Invitrogen) before mounting and imaging (10×). Imaging and analysis were performed blindly. Four independent experiments were carried out, with cells from 10 image fields counted per condition.
mEPSC measurements.
Miniature EPSCs (mEPSCs) were recorded by whole-cell patch recordings made on CA1 pyramidal cells from organotypic hippocampal slices after 11–13DIV. Recordings were made with an Axopatch 200B (Molecular Devices) using low-resistance pipettes (2–5 MΩ) containing 140 mm K-gluconate, 5 mm NaCl, 2 mm MgCl2, 0.1 mm CaCl2, 1.1 mm EGTA, 7 mm Na2-phosphocreatine, 10 mm HEPES, 4 mm Mg-ATP, and 0.4 mm Na3-GTP (∼285 mOsm). The extracellular solution was carbogenated (5% CO2/95% O2) and consisted of 130 mm NaCl, 3 mm KCl, 26 mm NaHCO3, 2 mm CaCl2, 1.48 mm MgCl2, 1.23 mm NaH2PO4, 10 mm glucose, and 100 μm lidocaine (∼312 mOsm). Cells were held at −65 mV and 50 μm picrotoxin was added to the extracellular solution to eliminate IPSPs. mEPSCs were inhibited by adding the AMPAR antagonist CNQX (10 μm) in the extracellular medium (data not shown). Membrane currents were monitored in voltage-clamp mode using pClamp9 software (Molecular Devices), filtered at 1 kHz, sampled at 5 kHz, and recorded at 35°C. Series resistance (Rs) was compensated and checked before and after every recording period; cells containing >15% Rs change, unstable holding current, and low membrane capacitance (Cm < 60 pF) were excluded from the analysis. mEPSCs were collected over a 5–10 min period from blinded WT or SPARC KO hippocampal slices. Events smaller than −4 pA were considered as noise and discarded. To obtain the mean mEPSC amplitude from each cell, all events were aligned and the peak amplitude of each event was measured with Clampfit. An average amplitude was then calculated. The mean mEPSC amplitude and frequency for WT and SPARC KO were obtained from the averaged values of each neuron.
LTP/long-term depression and NMDAR/AMPAR ratios in hippocampal slices.
Hippocampal slices (350 μm thickness) prepared from control or SPARC KO mice were placed in an incubating chamber with carbogenated artificial CSF (ACSF) consisting of 125 mm NaCl, 2.5 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 25 mm glucose (∼310 mOsm). ACSF containing bicuculline methobromide (5 μm) to block GABAA receptor-mediated inhibitory synaptic currents was used to perfuse slices in all recordings. Postsynaptic responses, which were evoked by stimulating the Schaeffer collateral–commissural pathway via constant current pulses (0.08 ms) delivered through a tungsten bipolar electrode (FHC), were recorded from the hippocampal CA1 region, amplified by a Multiclamp 700B (Molecular Devices), and stored in a PC for offline analysis using Clampfit (Molecular Devices). All recordings were performed at room temperature. Field EPSPs (fEPSPs) were evoked at 0.05 Hz and detected by an ACSF-filled glass electrode that was placed in the stratum radiatum of the hippocampal CA1 region. In another set of experiments, long-term plasticity of fEPSPs such as LTP and long-term depression (LTD) were induced by high-frequency (100 Hz, 100 pulses) and low-frequency (1 Hz, 900 pulses) tetanus, respectively. Initial fEPSP amplitude (i.e., before LTP induction) was similar between control (0.35 ± 0.02 mV) and SPARC KO (0.37 ± 0.02 mV) mice (two-tailed t test, p = 0.364). In addition, the strength of stimulation used in these experiments was quite similar for each genotype (150.3 ± 17.9 μA in control vs 164.0 ± 26.6 μA in SPARC KO; two-tailed t test, p = 0.689). For de-depression analysis, fEPSPs were evoked at 0.05 Hz and LTD was first induced as before. De-depression was then elicited by the LTP-inducing stimulus mentioned above. For assessment of paired-pulse facilitation, paired constant-current stimuli at varying interpulse intervals (25, 50, 100, and 200 ms) were applied to the Schaeffer collaterals. Paired-pulse ratio was calculated as the slope of the second fEPSP divided by the slope of the first fEPSP. For determining the ratio of NMDAR- and AMPAR-mediated postsynaptic currents, whole-cell recordings of evoked EPSCs were recorded from CA1 neurons. Recording pipettes were filled with solution containing 110 mm Cs-gluconate, 17.5 mm CsCl, 2 mm MgCl2, 0.5 mm EGTA, 10 mm HEPES, 4 mm ATP, and 5 mm QX-314 (∼290 mOsm) for experiments performed in acute slices or with 130 mm CsCl, 10 mm NaCl, 10 mm HEPES, 4 mm ATP, 0.4 mm GTP, and 5 mm QX-314 in organotypic slices AMPAR-mediated and AMPAR+NMDAR-mediated EPSCs were evoked at 0.05 Hz via voltage clamping the recorded cell at −60 mV and +40 mV, respectively. The NMDAR-mediated component of evoked EPSCs was represented by the EPSC amplitude at +40 mV recordings at 200 ms or 100 ms after stimulation in acute and organotypic slices, respectively, when the AMPAR-mediated component has decayed back to baseline.
Statistical analysis.
All data are presented as mean ± SEM, and comparisons were made using a two-tailed Student's t test, Mann–Whitney U, or ANOVA with post hoc Holm–Sidak test as specified. Differences were considered to be statistically significant for p < 0.05.
Results
SPARC is expressed by hippocampal astrocytes during development and is regulated by neural activity
SPARC is a secreted matricellular protein that is incorporated into the extracellular matrix and modifies a variety of cell signaling pathways in non-neuronal tissues (Arnold and Brekken, 2009). It was previously reported that SPARC is expressed by glial cells in the developing and adult CNS (Mendis et al., 1995; Lein et al., 2007; Vincent et al., 2008). To more precisely define the localization pattern of SPARC protein in the developing hippocampus, we performed colabeling studies at postnatal day 14 (P14) with an antibody that specifically detects SPARC by immunofluorescence (Fig. 1A; supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Based upon Western blot analysis of hippocampal lysates, SPARC expression is near its peak around P14 during hippocampal development (Fig. 1B) before being downregulated in adulthood. SPARC was mainly expressed by glutamine synthetase-positive astrocytes. In more rare instances, SPARC was detected in cells positive for IBA-1, a marker for microglia. In contrast, SPARC was not detected in NeuN-positive neurons. SPARC was also localized in astrocytes in organotypic hippocampal slices and in preparations of dissociated astrocytes (supplemental Figs. 1C,D, available at www.jneurosci.org as supplemental material). Furthermore, SPARC was secreted by dissociated astrocytes and was not detectable in media recovered from cultured hippocampal neurons (Fig. 1D; supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Thus SPARC is developmentally regulated and is highly produced by astrocytes during the period of intense synapse formation and reorganization in the hippocampus (De Simoni et al., 2003).
Figure 1.
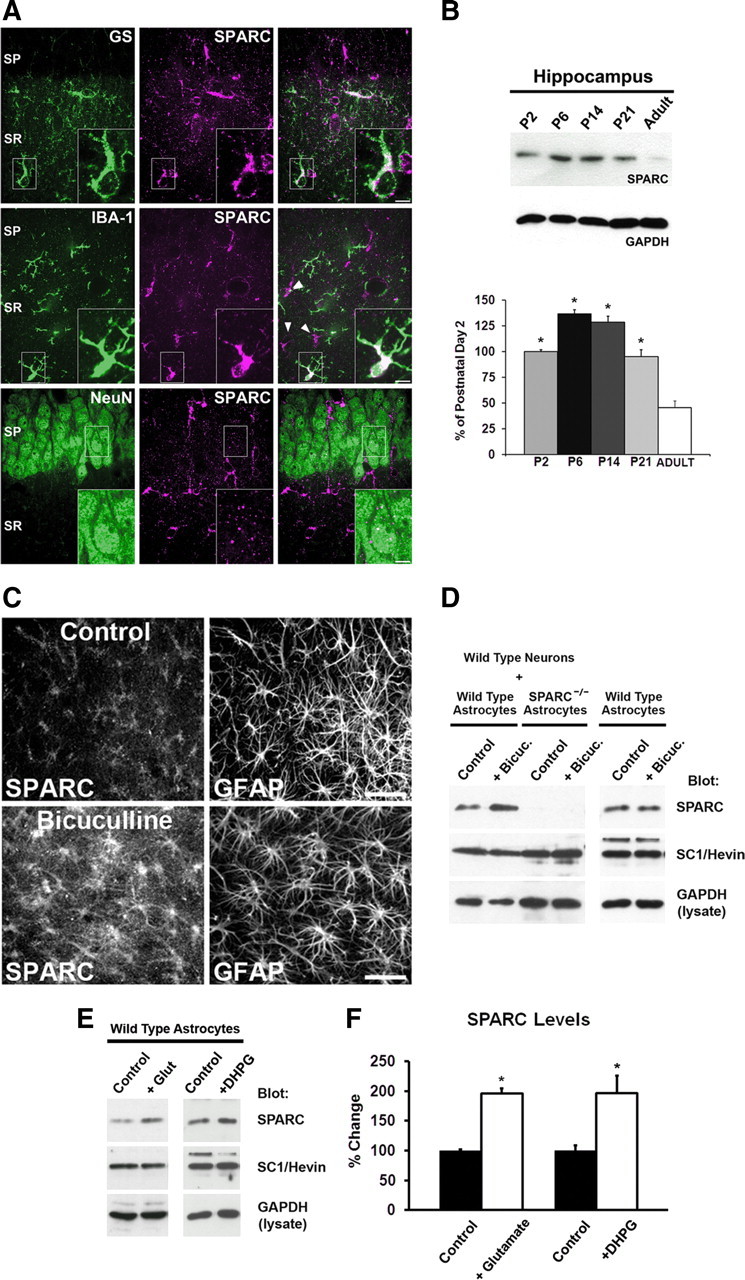
SPARC is expressed by hippocampal astrocytes during development and is regulated by neural activity. A, Immunolabeling for SPARC and either GS, IBA-1, or NeuN (P14 hippocampus) demonstrates that SPARC is expressed in astrocytes, some microglia but not neurons. SP, Stratum pyramidale; SR, stratum radiatum. B, Developmental time course for the expression of SPARC in the hippocampus revealed by immunoblot. SPARC levels peak during the first couple of postnatal weeks (ANOVA with post hoc Holm–Sidak test vs adult time point, *p < 0.05). C, Treatment of organotypic hippocampal slices with the GABAA receptor antagonist bicuculline (20 μm, 24 h) to elevate activity results in significantly increased SPARC expression [3.58-fold increase with bicuculline as compared to control; 2-tailed t test, *p = 0.0054, (n = 4)]. GFAP is shown as a control for relative astrocyte number. D, Increased SPARC protein can be detected in media taken from bicuculline-treated (20 μm, 24 h) neuron–astrocyte feeder cultures (see Fig. 4A), but not in bicuculline-treated astrocyte cultures. Immunoblot analysis of SC1/Hevin and GAPDH was performed as a control for protein level. E, F, Treatment of dissociated astrocytes with glutamate (20 μm, 6 h) or DHPG (10 μm, 6 h) leads to a significant increase in SPARC expression [2-tailed t test, *p = 0.0057 for glutamate, *p = 0.0297 for DHPG (n = 3)]. Error bars indicate SEM. Scale bars: A, 5 μm; C, 20 μm.
Given that synaptic activity modulates the formation of synapses and neuronal networks (Katz and Shatz, 1996; Cline and Haas, 2008), we examined whether changes in activity could affect SPARC expression. To test this, cultured hippocampal slices were treated with bicuculline to increase neuronal firing by blocking GABAA receptors. Elevating neural activity resulted in a robust increase in SPARC expression by astrocytes (3.58-fold increase with bicuculline as compared to control; two-tailed t test, p = 0.0054) (Fig. 1C). To determine whether the upregulation of SPARC required communication between neurons and astrocytes, we compared the effect of bicuculline on SPARC expression in cultures of astrocytes alone versus astrocytes in the presence of neurons (Figs. 1D, 4A). The bicuculline-induced increase in SPARC expression in astrocytes only occurred when neurons were present with astrocytes, demonstrating a requirement for neuron–astrocyte communication to mediate this activity-dependent effect. In contrast, probing for the SPARC-related family member Hevin/SC1 (SPARC Like-1) did not reveal any change in its expression upon bicuculline treatment. These results indicate that astrocytes respond to increases in neural activity through the upregulation of SPARC.
Figure 4.
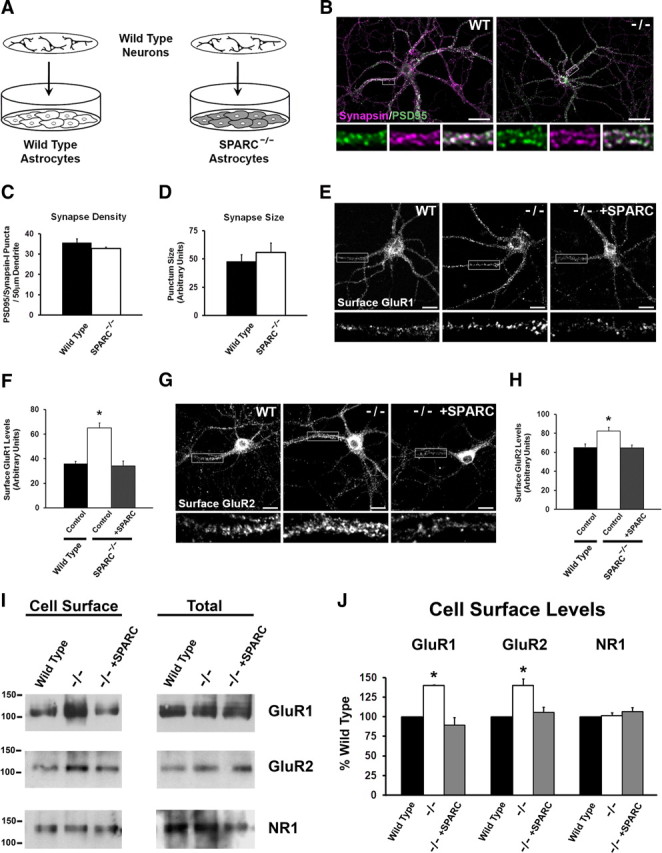
Neurons grown with SPARC KO astrocytes develop normally but have increased GluR1 and GluR2 surface expression. A, Diagram illustrating the culturing of WT neurons above a feeder layer of WT or KO astrocytes. B, Immunostaining showing the colocalization of presynaptic synapsin (magenta) and postsynaptic PSD-95 (green) to reveal synaptic puncta in WT and SPARC-deficient cultures. C, D, Quantification of synaptic punctum density and size. There were no detectable differences between WT and KO cultures [2-tailed t test, p = 0.0912 for density, p = 0.1563 for size (n = 4)]. E, F, Representative images of surface GluR1 expression showing increased intensity of puncta in SPARC-deficient cultures compared to WT cultures. The boxed region is magnified below each image. Recombinant SPARC application (+SPARC; 0.5 μg/ml, 48 h) rescues surface GluR1 levels in SPARC-deficient cultures [ANOVA with post hoc Holm–Sidak test, *p = 0.00025 (n = 3)]. G, H, Representative images of surface GluR2 expression showing increased intensity of puncta in SPARC-deficient cultures compared to WT cultures. The boxed region is magnified below each image. Application of recombinant SPARC (0.5 μg/ml, 48 h) restores surface GluR2 levels to WT levels [ANOVA with post hoc Holm–Sidak test, *p = 0.00246 (n = 4)]. I, J, Representative immunoblots showing cell surface (left panel) and total GluR1, GluR2, and NR1 (right panel) levels in WT, SPARC-deficient, and SPARC-deficient cultures treated with SPARC (0.5 μg/ml, 48 h). Cell surface receptor levels were determined by cell surface biotinylation. Neurons cultured with SPARC KO astrocytes had significantly higher surface GluR1 [ANOVA with post hoc Holm–Sidak test (n = 3), *p = 0.003] and GluR2 (*p = 0.008) but not NR1 (p = 0.472) levels without an increase in total receptor levels. Abnormal levels of GluR1 and GluR2 in the SPARC KO condition could be completely rescued with recombinant SPARC. Scale bars: B, 40 μm; E, G, 20 μm.
Astrocytes are known to respond to synaptic release of neurotransmitter. Glutamate, in particular, is well known to trigger intracellular signals in astrocytes (Fiacco and McCarthy, 2006). To determine whether SPARC expression could be influenced by glutamate, we treated dissociated hippocampal astrocytes with glutamate (20 μm for 6 h) and assessed the level of SPARC protein. We found that glutamate readily increased the levels of SPARC in astrocytes, whereas it had no effect on the expression of the closely related family member Hevin/SC1 (Fig. 1E,F). Consistent with a role of a glutamate pathway in increasing SPARC in astrocytes, application of dihydroxyphenylglycine (DHPG; an antagonist of group I metabotropic glutamate receptors) significantly increased SPARC levels (Fig. 1E,F). This suggests that signaling downstream of astrocytic mGluR1/5 receptors is involved in sensing neural activity and increasing SPARC expression.
SPARC KO mice show increases in excitatory synapse function, altered NMDAR/AMPAR ratios, and impairments in long-term plasticity
SPARC KO mice are viable and fertile but have reduced bone density, impaired wound healing, and increased fat stores and develop cataracts in adulthood (Framson and Sage, 2004). However, the role of SPARC in the development of the CNS is unknown. Given that SPARC is enriched during hippocampal development, we examined whether SPARC KO mice had normal hippocampal anatomy and synapse function. We found the gross hippocampal architecture and dendritic organization of SPARC KO mice to be similar to that of WT mice (Fig. 2A). Furthermore, SPARC KO mice did not display differences in dendritic spine density (Fig. 2B,C). Astrocyte morphology and distribution were also not overtly affected by loss of SPARC (Fig. 1A; supplemental Fig. 1A,C,D, available at www.jneurosci.org as supplemental material). We next investigated whether synaptic function was impaired in SPARC KO mice. Comparing presynaptic fiber volley versus the slope of fEPSPs in acute slices did not reveal significant differences in general evoked synaptic transmission between genotypes (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). To perform a more detailed analysis of individual synaptic events, we recorded AMPAR-mediated mEPSCs (miniature EPSCs) from CA1 neurons in WT and SPARC KO slices. Strikingly, mEPSCs in SPARC KO mice were significantly greater in both amplitude (Fig. 2D–F) and frequency (Fig. 2D,G), indicating that synapses in the KO mice are altered and show increased synaptic strength.
Figure 2.

SPARC KO mice show enhanced synaptic strength and alterations in NMDAR/AMPAR ratios. A, NeuN and MAP2 immunolabeling (P27 hippocampus) indicating that hippocampal architecture and dendritic structure is similar in WT and KO mice. B, Representative images of CA1 apical dendrites expressing membrane-targeted mCherry in WT and KO organotypic slices. C, Quantification of spine density between WT and SPARC KO neurons (Mann–Whitney U, p = 0.5362) D, Sample traces of mEPSC recordings from hippocampal pyramidal neurons of WT (top) and SPARC KO (bottom) organotypic hippocampal slices. E, Graph of average mEPSC amplitudes showing significant increases in amplitudes in SPARC KO neurons (2-tailed t test, *p = 0.0073). F, Cumulative distribution of mEPSC amplitudes from cells shown in the histogram (K–S test, p < 0.001). G, Graph of average mEPSC frequencies showing a significant increase in SPARC KO neurons (2-tailed t test, *p = 0.0375). H, Representative current traces of AMPAR-mediated inward currents (−60 mV) and mixed NMDAR+AMPAR-mediated outward currents (+40 mV) recorded from hippocampal neurons in acute WT and SPARC KO slices. I, SPARC KO mice show a significant reduction in the ratio of NMDAR/AMPAR currents (2-tailed t test, *p = 0.0154). Scale bars: A, top panels, 100 μm; bottom panels, 20 μm; B, 3 μm.
Since synapses from SPARC KO mice show elevated AMPAR-mediated mEPSC amplitudes when compared to WT controls, we were interested in determining whether the ratio of NMDARs to AMPARs at synapses was changed. In both WT and SPARC KO acute slices, AMPAR-mediated and AMPAR+NMDAR-mediated EPSCs were evoked while voltage clamping CA1 cells at −60 mV and +40 mV, respectively. Interestingly, neurons from SPARC KO mice showed a significant reduction in the NMDAR/AMPAR ratio when compared to WT neurons (Fig. 2H,I). A reduction in this ratio is in line with the mEPSC analysis, which found that AMPAR-mediated currents are significantly greater in SPARC KO mice.
We next investigated whether SPARC KO mice show deficits in synaptic plasticity. We first examined whether SPARC KO mice have defects in presynaptic plasticity by measuring paired-pulse facilitation, a form of short-term plasticity that is known to rely on changes in presynaptic release. This analysis did not reveal differences among genotypes (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). We then examined forms of long-term plasticity including LTP and LTD at CA1 synapses. Slices were prepared from WT and SPARC KO mice at P25–P30 and subjected to different stimulation paradigms while recording fEPSPs. We first tested for LTP by stimulating Schaeffer collaterals with 100 pulses at 100 Hz. WT slices displayed persistent LTP that lasted for >55–60 min after induction. In contrast, slices from SPARC KO mice showed impaired LTP (Fig. 3A). We also tested whether KO mice have defects in LTD. We stimulated Schaeffer collaterals in hippocampal slices of both genotypes at P25–P30 with 900 pulses at 1 Hz. However, in contrast to LTP, SPARC KO mice showed a level of LTD similar to that of the WT controls (Fig. 3B). Thus, synapses in SPARC KO mice have a selective reduction of LTP in the developing hippocampus.
Figure 3.

SPARC KO mice show defects in long-term plasticity. A, fEPSPs were recorded from the stratum radiatum of CA1 in acute hippocampal slices from WT and KO mice. Representative averaged traces of fEPSPs recorded before (1) and 55 min after (2) high-frequency stimulation (bottom left). Note that significant LTP, as shown in the graph (bottom right), was only expressed in slices from WT mice at 55–60 min after high-frequency stimulation (2-tailed t test, *p = 0.0127). B, LTD was induced by low-frequency stimulation (LFS). Representative averaged traces of fEPSP recorded before (1) and at 45 min after (2) LFS (bottom left). The graph shows the percentage depression of fEPSPs at 45–50 min after LFS. No differences between genotypes were found (2-tailed t test, p = 0.7564).
Loss of SPARC increases surface AMPAR levels at synapses
We next determined how SPARC regulates the properties of developing synapses. WT hippocampal neurons were grown with a feeder layer of either WT or SPARC KO astrocytes (Fig. 4A). This system allowed us to test the effect of soluble, astrocyte-derived SPARC on synapses in the absence of direct contact between neurons and astrocytes. At 14 d in vitro, a time point when synapses are actively recruiting AMPARs (Mammen et al., 1997), neurons grown with SPARC KO astrocytes had a similar number of synapses to cultures with WT astrocytes as determined by the number of colabeled synapsin and PSD-95 puncta (Fig. 4B,C). The size of individual colabeled puncta was also the same between conditions (Fig. 4D). We next evaluated the levels of AMPARs at the cell surface by labeling for GluR1 and GluR2 under nonpermeabilizing conditions. To exclude the possibility that synaptic sites are being selectively permeabilized with formaldehyde and may complicate the analysis, we performed immunostaining for the presynaptic protein synapsin and postsynaptic protein PSD-95 under nonpermeabilizing conditions (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). This showed that nonspecific permeabilization was limited to the soma and initial dendritic segment, which were excluded from our analysis (see Materials and Methods). Strikingly, neurons grown with SPARC KO astrocytes had significantly higher levels of surface GluR1 and GluR2 at synapses than neurons cultured with WT astrocytes (Fig. 4E–H). These abnormalities were completely rescued by the addition of exogenous recombinant SPARC for 48 h (Fig. 4E,G). Importantly, the defects found at synapses in the SPARC KO condition occurred without an increase in overall GluR1 or GluR2 content in neurons (Fig. 4I). To further support the findings that cell surface AMPAR levels are regulated by SPARC, we performed surface biotinylation experiments. The results showed the same effect as cell surface immunolabeling. In addition, we also found that cell surface NMDA receptor subunit 1 (NR1) is not altered by loss of SPARC. Thus astrocyte-derived SPARC controls the level of surface AMPARs in maturing neurons. This result is consistent with the electrophysiological recordings, which found an increase in AMPAR function in neurons from SPARC KO mice (Fig. 2B–G).
During development, a greater proportion of synapses have NMDARs but lack functional AMPARs and are referred to as electrophysiologically “silent” (Kerchner and Nicoll, 2008). We tested whether SPARC influences the proportion of synapses that contain both NMDARs and AMPARs by colabeling for NR1 and GluR1 at 7 d in vitro, a time point when there is a greater number of silent synapses in culture (Liao et al., 1999). However, this analysis did not reveal a difference in the density of NR1 puncta, or the fraction of NR1 puncta colocalized with GluR1 puncta between conditions (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These results suggest that SPARC does not play a role in maintaining silent synapses during development, but rather constrains the level of AMPARs at synapses as they mature.
SPARC regulates the ability of developing synapses to show plasticity
Knowing that synapses in the SPARC KO condition have significantly more AMPA receptors, we next asked whether neurons cultured with SPARC KO astrocytes retained the ability to modify their synapses with activity. We first tested this by subjecting neurons cultured with WT or KO astrocytes to activity deprivation for 48 h to induce homeostatic synaptic scaling (Turrigiano, 2008). In response to activity blockade with tetrodotoxin (TTX), neurons compensate by recruiting AMPARs to increase synaptic strength (Turrigiano, 2008). Consistent with previous studies, treating cultures with TTX increased surface GluR1 levels in control WT cultures after 48 h (O'Brien et al., 1998; Stellwagen and Malenka, 2006). In contrast, surface GluR1 levels in neurons cultured with SPARC KO astrocytes were unchanged by TTX, remaining at a significantly higher level than in the control condition (Fig. 5A,B). One explanation could be that SPARC maintains surface GluR1 levels below a saturation point and synapses in the KO condition are unable to increase their GluR1 content further upon activity deprivation. Another possibility is that SPARC participates in homeostatic regulation of AMPARs at synapses. To distinguish between these possibilities, we first added recombinant SPARC to KO cultures to rescue the surface GluR1 levels and then subjected the neurons to TTX for 48 h. We found that neurons in the KO condition that were rescued with recombinant SPARC before TTX administration showed higher levels of surface GluR1 on neurons than those treated with recombinant SPARC alone (Fig. 5C). This effect occurred regardless of whether SPARC was present (Fig. 5C) or absent (data not shown) during TTX treatment. This indicates that neurons in the SPARC KO condition maintain the capacity to increase their surface AMPARs in response to chronic activity deprivation. We also tested whether loss of SPARC prevented a homeostatic reduction in surface GluR1 levels in response to chronic increases in activity (O'Brien et al., 1998). However, neurons grown in the absence of SPARC responded to bicuculline with a significant decrease in surface GluR1 levels (Fig. 5D). Therefore, neurons cultured in the absence of SPARC retain the potential to show homeostatic increases in AMPAR levels at synapses but are unable to do so because their AMPAR content is abnormally high.
Figure 5.

Application of SPARC protein or prior synaptic depression to the KO condition restores synapses to a permissible range for synaptic plasticity. A, B, Synaptic strength in SPARC-deficient cultures is not increased further by chronic activity blockade. WT cultures display significant increases in surface GluR1 levels in response to TTX treatment (1 μm, 48 h). In contrast, SPARC-deficient cultures do not exhibit any changes in surface GluR1 levels [ANOVA with post hoc Holm–Sidak test, *p = 0.0055, for KO cultures p = 0.865 (n = 5)]. C, Rescue of KO cultures with recombinant SPARC allows TTX-induced increases in synaptic strength to occur [ANOVA with post hoc Holms–Sidak test versus SPARC treatment, *p < 0.05 (n = 4)]. SPARC protein (0.5 μg/ml) was applied for 24 h and then incubated with TTX (1 μm) for a subsequent 48 h. D, Synaptic strength in SPARC-deficient cultures is decreased by bicuculline application [2-tailed t test, *p = 0.00283 (n = 5)] (20 μm, 48 h) Error bars indicate SEM. Scale bars, 20 μm. E, F, Synapses in acute slices from SPARC KO mice can show potentiation when given a prior LTD-inducing stimulus. The graph shows the average percentage change in fEPSPs at 55–60 min after high-frequency stimulation (HFS) (2) compared to 25–30 min after LFS (1) in both WT [2-tailed t test, p = 0.0388 (n = 6)] and KO slices [2-tailed t test, *p = 0.042 (n = 5)]. Post-HFS potentiation was not significantly different between WT and KO slices (2-tailed t test, p = 0.29). Scale bars, 20 μm.
To further investigate how the overabundance of AMPARs at maturing synapses in SPARC KO mice impairs synaptic plasticity during development, we took advantage of the bidirectionality of long-term synaptic plasticity (Lee et al., 2000). The rationale was to reduce AMPARs and synaptic strength in SPARC KO slices before applying a potentiating stimulus. To accomplish this, we first provided an LTD-inducing stimulus to the Schaeffer collateral pathway in acute slices and followed this with 100 Hz stimulation to induce de-depression (Lee et al., 2000). Remarkably, synapses in the SPARC KO were able to potentiate to the same extent as WT synapses when synaptic efficacy was first lowered by LTD-inducing stimuli (Fig. 5E,F). These results, along with those presented above, indicate that SPARC maintains synapses within a useful “working range” that enables them to show activity-dependent postsynaptic modifications during development.
SPARC interacts with β-integrins to control AMPAR levels at maturing synapses
SPARC has been shown to interact with several growth factors (i.e., VEGF, PDGF, and TGFβ) and modulate their downstream signaling cascades (Brekken and Sage, 2001; Arnold and Brekken, 2009). In addition, SPARC was found to bind integrin subunits and reduce cell-substrate adhesion of non-neuronal cells (Said et al., 2007; Nie et al., 2008; Weaver et al., 2008). Recently, β3-integrins have been shown to stabilize surface GluR2 to regulate synaptic efficacy (Cingolani et al., 2008). We followed up on the putative relationship between SPARC and β-integrin complexes in the brain and the possibility that this interaction is important for regulating synaptic AMPARs. We investigated whether SPARC affects β-integrins in neurons by examining the cell surface levels of β-integrins on hippocampal neurons cultured with SPARC KO astrocytes. We found that loss of SPARC caused a significant increase in surface β3-integrin (Fig. 6A,B) but not β1-integrin (Fig. 6C) (27.405 arbitrary units for WT vs 27.794 arbitrary units for SPARC-deficient cultures; two-tailed t test, p = 0.845) and that the changes in surface β3-integrin coincided with significant elevations in GluR2 subunits at synapses (Fig. 4I,J). Importantly, addition of SPARC to SPARC-deficient cultures reversed the abnormal accumulation of β3-integrin complexes (Fig. 6B).
Figure 6.

SPARC regulates β3-integrin complexes to control surface AMPARs. A, Representative images of surface β3-integrin expression showing increased intensity of surface β3-integrin puncta in SPARC-deficient cultures compared to WT cultures. Recombinant SPARC application (0.5 μg/ml, 48 h) restores surface β3-integrin levels in SPARC-deficient cultures. The boxed region is magnified below each image. B, Quantification of surface β3-integrin expression [ANOVA with post hoc Holm–Sidak test, *p = 0.000158 (n = 4)]. C, Surface β1-integrin levels are similar in WT and SPARC-deficient cultures [27.405 arbitrary units for WT versus 27.794 arbitrary units for SPARC-deficient cultures; 2-tailed t test, p = 0.845 (n = 3)]. D, HEK 293T cells expressing β3-integrin have reduced attachment to vitronectin in the presence of SPARC [ANOVA with post hoc Holm–Sidak test, *p = 0.0036, (n = 4)]. E, Diagram illustrating the domain structure of SPARC and location of Peptide 2.3. Peptide 2.3 reduces surface β3-integrin (F), GluR2 (G), and GluR1 (H) levels in SPARC-deficient cultures to a similar extent as the full-length protein [ANOVA with post hoc Holm–Sidak test, *p = 0.0081 for β3-integrin (n = 5), *p = 0.0127 for GluR2 (n = 5), and *p = 0.00511 for GluR1 (n = 4) vs control]. A peptide containing a scrambled sequence (scPep2.3) had no effect. I, Coapplication of the disintegrin Echistatin (300 nm) prevents SPARC rescue (0.5 μg/ml, 48 h) of surface GluR1 levels in SPARC-deficient cultures (ANOVA with post hoc Holm–Sidak test, *p = 0.003, SPARC-treated vs SPARC-treated and Echistatin condition). J, The β3-integrin function-blocking antibody Clone 2C9.G2 (10 μg/ml, 48 h) blocks SPARC-mediated rescue of surface GluR1 in SPARC-deficient cultures (ANOVA with post hoc Holm–Sidak test, *p = 0.006). An isotype-matched control antibody had no effect. Error bars indicate SEM. Scale bars, 20 μm.
To more directly assess the effect of SPARC on β3-integrin function, we used cell attachment assays of HEK 293T cells on vitronectin, a substrate for β3-integrin complexes. Endogenous β3-integrins are not expressed in HEK 293T cells and these cells have a low level of attachment to vitronectin (Simon et al., 1997). However, attachment can be significantly increased by transfection of β3-integrin (Simon et al., 1997). We found that application of SPARC reduced the ability of β3-integrin-overexpressing cells to attach to vitronectin (Fig. 6D). These results demonstrate that SPARC interacts with β3-integrin complexes and regulates their surface stability and function.
To further characterize the interaction between SPARC and β-integrins and its impact on AMPARs, we used a peptide comprising a small portion of the follistatin-like domain of SPARC that has been shown to interact with β-integrins on non-neuronal cells (peptide 2.3) (Fig. 6E) (Nie et al., 2008; Weaver et al., 2008) and can mimic the angiogenic activity of SPARC (Lane et al., 1994). Surprisingly, peptide 2.3 reduced the overaccumulation of surface β3-integrins and GluR2 on neurons grown with SPARC KO astrocytes (Fig. 6F,G). Coapplication of SPARC and peptide 2.3 failed to further reduce the levels of β3-integrin and GluR2, suggesting that these molecules operate on the same pathway. Peptide 2.3 also completely rescued surface GluR1 levels in the KO condition, indicating that engagement of β-integrins with peptide 2.3 does not preferentially reduce GluR2 over GluR1 (Fig. 6H). A peptide containing a scrambled sequence of peptide 2.3 was ineffective in rescuing the alterations. These findings further implicate SPARC in controlling surface AMPARs through β-integrins at synapses.
To test the requirement of β3-integrin function in SPARC regulation of surface AMPARs, we used two approaches to inhibit β3-integrin. We first used the peptide echistatin, an RGD-containing snake venom disintegrin that binds β3-integrins with high affinity and competitively blocks the ability of β3-integrin to interact with substrates (Gan et al., 1988; Dennis et al., 1990; Kumar et al., 1997). Second, we applied a characterized β3-integrin function-blocking antibody (Piali et al., 1995; Ashkar et al., 2000; Brandsma et al., 2006). Both echistatin and the β3-integrin function-blocking antibody prevented the ability of SPARC to restore surface GluR1 to control levels (Fig. 6I,J), indicating that β3-integrin function is needed for SPARC's ability to regulate surface GluR1. These findings, in addition to data showing SPARC's ability to regulate cell surface β3-integrin levels (Fig. 6A) and β3-integrin-mediated cell attachment (Fig. 6D), and the ability of Peptide 2.3 to rescue the effects of loss of SPARC (Fig. 6F–H), strongly implicate SPARC-β3-integrin interactions in regulating surface AMPAR levels.
Discussion
Synapse development is an intricate process involving a range of mechanisms. Clearly among these mechanisms is the requirement of ligands and receptors on neurons that facilitate target selection, synaptic induction, and maintenance (Waites et al., 2005). Neural activity and experience also shape the properties of developing synaptic connections (Zhu et al., 2000; Liao et al., 2001; Holtmaat and Svoboda, 2009); however, the molecular mechanisms underlying these processes remain unclear. In this study, we reveal that neurons require astrocytes to fine-tune the process of synaptic maturation. Specifically, we show that astrocytes produce the protein SPARC in response to changes in neural activity levels. SPARC, in turn, provides a necessary signal for tuning central synapses. SPARC acts as a “molecular brake” to prevent the overaccumulation of GluR1 and GluR2 at maturing synapses by regulating neuronal β-integrins. This regulation is essential for preserving a permissive working range for neurons to show synaptic plasticity. These findings establish a novel cellular and molecular pathway for controlling the properties of developing synapses in the brain.
The ability of glial-derived factors to promote synapse formation was discovered over a decade ago (Barres, 2008). However, the molecular identity of the factors that accomplish this and how they operate during synaptic development still remains poorly understood. In fact, thrombospondins are the only identified proteins secreted from glia that regulate synaptic development (Christopherson et al., 2005; Eroglu et al., 2009). Loss of thrombospondins 1 and 2 in mice results in decreased synaptic density without affecting dendritic growth. However, retinal neuron synapses induced by thrombospondin remain electrophysiologically “silent” and have little AMPAR content or function. Thus other glial-derived factors are likely mobilized to control the developmental progression of synapses.
Our work now shows that astrocytes produce SPARC to determine the physiological effectiveness of glutamate synapses. SPARC performs this function without impacting the initial steps of synapse formation as has been found for other proteins (Christopherson et al., 2005). Interestingly, SPARC is regulated by levels of neural activity and glutamate and is highly enriched during a brief time window in the developing hippocampus. We propose that SPARC constrains synaptic glutamate receptor levels during intense synapse formation and reorganization and when activity is especially important in refining connections. Hence, the ability of astrocytes to respond to glutamatergic transmission by increasing SPARC may regulate the time window for activity-dependent changes at synapses. The idea that astrocyte-derived signals govern the timing of developmental plasticity in regions of the brain was suggested by Müller and Best (1989). They found that injection of immature astrocytes into the adult visual cortex of cats reopened a window of ocular dominance plasticity. Whether SPARC is involved in this process is the subject of future investigation.
Our findings add to the growing body of evidence that glia secrete factors that are critical for synaptic plasticity (Volterra and Meldolesi, 2005). For example, glial-derived TNFα regulates homeostatic synaptic scaling (Stellwagen and Malenka, 2006) and glial-derived d-serine controls the threshold for plasticity in the hypothalamus and hippocampus (Yang et al., 2003; Panatier et al., 2006; Henneberger et al., 2010). Constitutive release of ATP from astrocytes and its subsequent degradation to adenosine also serves to globally suppress synaptic function to widen the range for neurons to undergo synaptic changes (Pascual et al., 2005). Our findings are unique in that SPARC functions at an early time point to confine the strength of maturing synapses within a range that permits them to respond to plasticity-inducing stimuli. In essence, synapses in the SPARC KO are “out of range” to undergo TTX-induced synaptic scaling and LTP during development.
SPARC may mediate its effects on synapses through multiple pathways (Brekken and Sage, 2001). More recently, SPARC was shown to bind to integrin subunits and modulate cell-substrate adhesion (Said et al., 2007; Nie et al., 2008; Nie and Sage, 2009). Our findings demonstrate that SPARC is an endogenously expressed regulator of β3-integrins at maturing synapses. Exogenous application of SPARC or a SPARC peptide known to bind β-integrins completely restored normal surface β3-integrin and AMPAR levels on WT neurons grown with SPARC KO astrocytes. Furthermore, inhibiting β3-integrin function with echistatin or a β3-integrin function-blocking antibody, blocked the ability of SPARC to regulate surface AMPARs. Consistent with this, Cingolani et al. (2008) recently showed that β3-integrins regulate AMPAR stability. Interestingly, neither echistatin nor the β3-integrin function-blocking antibody treatment alone had a significant effect on surface GluR1 levels (Fig. 6I,J). This suggests that merely inhibiting β3-integrin interactions with extracellular substrates alone during synaptic maturation is not sufficient to regulate AMPARs and requires additional conformational or signaling events induced by SPARC (Askari et al., 2009).
Accumulating evidence shows that SPARC is upregulated following injury or challenge to the CNS. Indeed, SPARC is increased in astrocytes following lesions of the perforant path (Liu et al., 2005) and upon epileptogenesis (Ozbas-Gerçeker et al., 2006). SPARC levels are also elevated following morphine administration to the amygdala, and exogenous administration of SPARC can increase the sensitivity of neurons to morphine (Ikemoto et al., 2000). SPARC produced by olfactory ensheathing cells also facilitates Schwann cell differentiation and promotes the repair of the injured spinal cord (Au et al., 2007). Thus SPARC likely has multiple roles in the CNS. During development, it plays an essential role in controlling levels of AMPARs at maturing synapses. In the adult, it may be upregulated in the brain and spinal cord to modulate neuronal health following injury or disease. Future studies will determine the role of SPARC in regulating glutamate receptors in these contexts.
Footnotes
This work was supported by the Canadian Institutes of Health Research, Canada Research Chairs Program, Canadian Foundation for Innovation, and the EJLB Foundation (K.K.M.) and the Natural Sciences and Engineering Research Council of Canada (T.P.W). E.V.J. was partially supported through a postdoctoral fellowship through the Research Institute of the McGill University Health Centre. Y.B. was supported by fellowships #PBLAB-118155 and #PA00A3-121419/1 from the Swiss National Science Foundation. We thank Dr. Roger Tsien for the mCherry construct, Dr. Paul Holland for the β3-integrin construct, and Drs. Donald van Meyel and David Stellwagen and members of the Murai laboratory for helpful discussions.
References
- Arnold SA, Brekken RA. SPARC: a matricellular regulator of tumorigenesis. J Cell Commun Signal. 2009;3:255–273. doi: 10.1007/s12079-009-0072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, Zawaideh S, Rittling SR, Denhardt DT, Glimcher MJ, Cantor H. Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science. 2000;287:860–864. doi: 10.1126/science.287.5454.860. [DOI] [PubMed] [Google Scholar]
- Askari JA, Buckley PA, Mould AP, Humphries MJ. Linking integrin conformation to function. J Cell Sci. 2009;122:165–170. doi: 10.1242/jcs.018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au E, Richter MW, Vincent AJ, Tetzlaff W, Aebersold R, Sage EH, Roskams AJ. SPARC from olfactory ensheathing cells stimulates Schwann cells to promote neurite outgrowth and enhances spinal cord repair. J Neurosci. 2007;27:7208–7221. doi: 10.1523/JNEUROSCI.0509-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Brandsma D, Ulfman L, Reijneveld JC, Bracke M, Taphoorn MJ, Zwaginga JJ, Gebbink MF, de Boer H, Koenderman L, Voest EE. Constitutive integrin activation on tumor cells contributes to progression of leptomeningeal metastases. Neuro Oncol. 2006;8:127–136. doi: 10.1215/15228517-2005-013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biol. 2001;19:816–827. doi: 10.1016/s0945-053x(00)00133-5. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Thalhammer A, Yu LM, Catalano M, Ramos T, Colicos MA, Goda Y. Activity-dependent regulation of synaptic AMPA receptor composition and abundance by beta3 integrins. Neuron. 2008;58:749–762. doi: 10.1016/j.neuron.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline H, Haas K. The regulation of dendritic arbor development and plasticity by glutamatergic synaptic input: a review of the synaptotrophic hypothesis. J Physiol. 2008;586:1509–1517. doi: 10.1113/jphysiol.2007.150029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis MS, Henzel WJ, Pitti RM, Lipari MT, Napier MA, Deisher TA, Bunting S, Lazarus RA. Platelet glycoprotein IIb-IIIa protein antagonists from snake venoms: evidence for a family of platelet-aggregation inhibitors. Proc Natl Acad Sci U S A. 1990;87:2471–2475. doi: 10.1073/pnas.87.7.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Simoni A, Griesinger CB, Edwards FA. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J Physiol. 2003;550:135–147. doi: 10.1113/jphysiol.2003.039099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia. 2006;54:676–690. doi: 10.1002/glia.20396. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Framson PE, Sage EH. SPARC and tumor growth: where the seed meets the soil? J Cell Biochem. 2004;92:679–690. doi: 10.1002/jcb.20091. [DOI] [PubMed] [Google Scholar]
- Gan ZR, Gould RJ, Jacobs JW, Friedman PA, Polokoff MA. Echistatin. A potent platelet aggregation inhibitor from the venom of the viper, Echis carinatus. J Biol Chem. 1988;263:19827–19832. [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of D-serine from astrocytes. Nature. 2010;463:232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Ikemoto M, Takita M, Imamura T, Inoue K. Increased sensitivity to the stimulant effects of morphine conferred by anti-adhesive glycoprotein SPARC in amygdala. Nat Med. 2000;6:910–915. doi: 10.1038/78675. [DOI] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner D, Morgan JL, Parker ED, Lewis RM, Wong RO. Neurotransmission selectively regulates synapse formation in parallel circuits in vivo. Nature. 2009;460:1016–1020. doi: 10.1038/nature08236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Lessmann V, Medina I, Gaiarsa JL. Activity-dependent dendritic release of BDNF and biological consequences. Mol Neurobiol. 2009;39:37–49. doi: 10.1007/s12035-009-8050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar CC, Nie H, Rogers CP, Malkowski M, Maxwell E, Catino JJ, Armstrong L. Biochemical characterization of the binding of echistatin to integrin alphavbeta3 receptor. J Pharmacol Exp Ther. 1997;283:843–853. [PubMed] [Google Scholar]
- Lane TF, Sage EH. Functional mapping of SPARC: peptides from two distinct Ca+(+)-binding sites modulate cell shape. J Cell Biol. 1990;111:3065–3076. doi: 10.1083/jcb.111.6.3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane TF, Iruela-Arispe ML, Johnson RS, Sage EH. SPARC is a source of copper-binding peptides that stimulate angiogenesis. J Cell Biol. 1994;125:929–943. doi: 10.1083/jcb.125.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature. 2000;405:955–959. doi: 10.1038/35016089. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Liao D, Zhang X, O'Brien R, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat Neurosci. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- Liao D, Scannevin RH, Huganir R. Activation of silent synapses by rapid activity-dependent synaptic recruitment of AMPA receptors. J Neurosci. 2001;21:6008–6017. doi: 10.1523/JNEUROSCI.21-16-06008.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linhoff MW, Laurén J, Cassidy RM, Dobie FA, Takahashi H, Nygaard HB, Airaksinen MS, Strittmatter SM, Craig AM. An unbiased expression screen for synaptogenic proteins identifies the LRRTM protein family as synaptic organizers. Neuron. 2009;61:734–749. doi: 10.1016/j.neuron.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ying G, Wang W, Dong J, Wang Y, Ni Z, Zhou C. Entorhinal deafferentation induces upregulation of SPARC in the mouse hippocampus. Brain Res Mol Brain Res. 2005;141:58–65. doi: 10.1016/j.molbrainres.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Mammen AL, Huganir RL, O'Brien RJ. Redistribution and stabilization of cell surface glutamate receptors during synapse formation. J Neurosci. 1997;17:7351–7358. doi: 10.1523/JNEUROSCI.17-19-07351.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch DH, Nägler K, Schumacher S, Göritz C, Müller EC, Otto A, Pfrieger FW. CNS synaptogenesis promoted by glia-derived cholesterol. Science. 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- Mendis DB, Malaval L, Brown IR. SPARC, an extracellular matrix glycoprotein containing the follistatin module, is expressed by astrocytes in synaptic enriched regions of the adult brain. Brain Res. 1995;676:69–79. doi: 10.1016/0006-8993(95)00101-u. [DOI] [PubMed] [Google Scholar]
- Müller CM, Best J. Ocular dominance plasticity in adult cat visual cortex after transplantation of cultured astrocytes. Nature. 1989;342:427–430. doi: 10.1038/342427a0. [DOI] [PubMed] [Google Scholar]
- Nägler K, Mauch DH, Pfrieger FW. Glia-derived signals induce synapse formation in neurones of the rat central nervous system. J Physiol. 2001;533:665–679. doi: 10.1111/j.1469-7793.2001.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie J, Sage EH. SPARC inhibits adipogenesis by its enhancement of beta-catenin signaling. J Biol Chem. 2009;284:1279–1290. doi: 10.1074/jbc.M808285200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie J, Chang B, Traktuev DO, Sun J, March K, Chan L, Sage EH, Pasqualini R, Arap W, Kolonin MG. IFATS collection: combinatorial peptides identify alpha5beta1 integrin as a receptor for the matricellular protein SPARC on adipose stromal cells. Stem Cells. 2008;26:2735–2745. doi: 10.1634/stemcells.2008-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL. Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron. 1998;21:1067–1078. doi: 10.1016/s0896-6273(00)80624-8. [DOI] [PubMed] [Google Scholar]
- Ozbas-Gerçeker F, Redeker S, Boer K, Ozgüç M, Saygi S, Dalkara T, Soylemezoglu F, Akalan N, Baayen JC, Gorter JA, Aronica E. Serial analysis of gene expression in the hippocampus of patients with mesial temporal lobe epilepsy. Neuroscience. 2006;138:457–474. doi: 10.1016/j.neuroscience.2005.11.043. [DOI] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- Piali L, Hammel P, Uherek C, Bachmann F, Gisler RH, Dunon D, Imhof BA. CD31/PECAM-1 is a ligand for alpha v beta 3 integrin involved in adhesion of leukocytes to endothelium. J Cell Biol. 1995;130:451–460. doi: 10.1083/jcb.130.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said N, Najwer I, Motamed K. Secreted protein acidic and rich in cysteine (SPARC) inhibits integrin-mediated adhesion and growth factor-dependent survival signaling in ovarian cancer. Am J Pathol. 2007;170:1054–1063. doi: 10.2353/ajpath.2007.060903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon KO, Nutt EM, Abraham DG, Rodan GA, Duong LT. The alphavbeta3 integrin regulates alpha5beta1-mediated cell migration toward fibronectin. J Biol Chem. 1997;272:29380–29389. doi: 10.1074/jbc.272.46.29380. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC. Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-α. J Neurosci. 2005;25:3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Sapperstein SK, Christopherson KS, Barres BA. Control of synapse number by glia. Science. 2001;291:657–661. doi: 10.1126/science.291.5504.657. [DOI] [PubMed] [Google Scholar]
- Vincent AJ, Lau PW, Roskams AJ. SPARC is expressed by macroglia and microglia in the developing and mature nervous system. Dev Dyn. 2008;237:1449–1462. doi: 10.1002/dvdy.21495. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Waites CL, Craig AM, Garner CC. Mechanisms of vertebrate synaptogenesis. Annu Rev Neurosci. 2005;28:251–274. doi: 10.1146/annurev.neuro.27.070203.144336. [DOI] [PubMed] [Google Scholar]
- Weaver MS, Workman G, Sage EH. The copper binding domain of SPARC mediates cell survival in vitro via interaction with integrin beta1 and activation of integrin-linked kinase. J Biol Chem. 2008;283:22826–22837. doi: 10.1074/jbc.M706563200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, Poo M, Duan S. Contribution of astrocytes to hippocampal long-term potentiation through release of D-serine. Proc Natl Acad Sci U S A. 2003;100:15194–15199. doi: 10.1073/pnas.2431073100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Martinez SJ, Haber M, Jones EV, Bouvier D, Doucet G, Corera AT, Fon EA, Zisch AH, Murai KK. EphA4 signaling regulates phospholipase Cγ1 activation, cofilin membrane association, and dendritic spine morphology. J Neurosci. 2007;27:5127–5138. doi: 10.1523/JNEUROSCI.1170-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Esteban JA, Hayashi Y, Malinow R. Postnatal synaptic potentiation: delivery of GluR4-containing AMPA receptors by spontaneous activity. Nat Neurosci. 2000;3:1098–1106. doi: 10.1038/80614. [DOI] [PubMed] [Google Scholar]