

Abstract
Neuroadaptation in the nucleus accumbens (NAc), a central component of the mesolimbic dopamine (DA) system, has been implicated in the development of cocaine-induced psychomotor sensitization and relapse to cocaine seeking. However, little is known about the cellular and synaptic mechanisms underlying such adaptation. Using a mouse model of behavioral sensitization, we show that animals withdrawn from repeated cocaine exposure have a selective deficit in the ability to elicit metabotropic glutamate receptor (mGluR)-dependent long-term depression (LTD) in the shell of the NAc in response to bath application of the group I mGluR agonist (S)-3,5-dihydroxyphenylglycine (DHPG). Experiments conducted in the presence of the selective mGluR1 antagonists 7-(hydroxyimino)cyclopropachromen-carboxylate ethyl ester and (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid, or the mGluR5 antagonist 2-methyl-6-(phenylethynyl)-pyridine, demonstrated that the impaired DHPG-LTD is likely attributable to a loss of mGluR5 function. Quantitative real-time reverse transcriptase-PCR and Western blot analysis revealed significant downregulation of mGluR5, but not mGluR1, mRNA and protein levels in the NAc shell. The inhibitory effect of repeated cocaine exposure on DHPG-LTD was selectively prevented when cocaine was coadministered with the selective D1-like DA receptor antagonist (R)-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine. Furthermore, the levels of brain-derived neurotrophic factor (BDNF) protein in the NAc shell increased progressively after cocaine withdrawal, and the impairment of DHPG-LTD in the NAc shell was not found in slices from BDNF-knock-out mice after cocaine withdrawal. These results suggest that withdrawal from repeated cocaine exposure may result in increased BDNF levels in the NAc shell, which leads to a selective downregulation of mGluR5 and thereby impairs the induction of mGluR-dependent LTD.
Introduction
Drug addiction is a pathological condition characterized by compulsive drug seeking and use, despite severe adverse consequences. Emerging evidence suggests that addiction to drugs of abuse usurps reward-related learning and memory processes, ultimately producing long-lasting neuroadaptation that may underlie relapse to drug seeking (Shaham and Hope, 2005; Thomas et al., 2008). One major brain reward circuitry that has been implicated in the development and expression of addictive behaviors is the mesolimbic dopamine (DA) pathway, which comprises the ventral tegmental area (VTA) and its projections to the nucleus accumbens (NAc). Although the neural mechanisms underlying drug addiction are primarily associated with long-lasting changes in DA transmission, recent evidence has implicated the synaptic plasticity of glutamatergic transmission in both the VTA and NAc as the cellular substrate of addictive drug-induced alterations (Hyman et al., 2006; Schmidt and Pierce, 2010). For example, a single cocaine injection has been reported to produce a rapid but transient potentiation of excitatory inputs onto VTA DA neurons (Ungless et al., 2001; Argilli et al., 2008). In the NAc, cocaine-induced synaptic plasticity also occurs, but on a slower and more long-lasting time course. In contrast to the robust effect of a single cocaine injection on VTA DA neurons, this treatment does not change synaptic strength in the NAc (Thomas et al., 2001; Fourgeaud et al., 2004). With repeated cocaine exposure, however, synaptic strength in medium spiny neurons (MSNs) of the NAc shell is depressed. During the course of cocaine withdrawal, cocaine-induced long-term depression (LTD)-like plasticity in the NAc transforms into a potentiation through an increase in cell surface expression of AMPA receptors (Boudreau et al., 2007).
In addition to triggering persistent changes in glutamatergic transmission in the VTA and NAc, it was recently shown that withdrawal from cocaine self-administration impairs the ability of subsequent stimuli to induce long-term potentiation (LTP) and LTD in the NAc, suggesting cocaine-induced metaplasticity (Moussawi et al., 2009). Furthermore, systemic administration of N-acetylcysteine effectively reversed cocaine-induced metaplasticity and restored the ability to induce LTP and LTD by indirectly stimulating metabotropic glutamate receptor (mGluR) 2/3 and mGluR5. To date, two main forms of LTD have been identified in the NAc, and these are distinguished on the basis of their induction requirements. The first form of LTD is induced by trains of stimuli at 5 Hz for 3 min, and this LTD induction depends on NMDA receptor (NMDAR) activation (Martin et al., 2006). The second form, known as mGluR-dependent LTD, is induced by the mGluR2/3 agonist (+)-2-aminobicyclo-[3.1.0]hexane-2,6-dicarboxylic acid (LY354740) (Kasanetz et al., 2010). There is evidence that repeated cocaine exposure impairs NMDAR-dependent LTD induction in the NAc (Thomas et al., 2001; Kasanetz et al., 2010), but it remains unknown whether and how withdrawal from repeated cocaine exposure affects the induction of mGluR-dependent LTD in the NAc. In this study, we demonstrate a novel form of mGluR-dependent LTD that is induced by pharmacological activation or synaptic stimulation of mGluR5 in MSNs in the NAc. We also provide the first evidence that cocaine withdrawal may selectively impair mGluR5-dependent LTD in the shell of the NAc.
Materials and Methods
Animals and treatment.
Male C57BL/6J mice, homozygous brain-derived neurotrophic factor (BDNF)-floxed (Bdnftm3Jae/J), and calmodulin-kinase II (CamK)-cAMP response element (Cre) transgenic mice were originally obtained from The Jackson Laboratory and bred within our animal facility. Bdnftm3Jae/J mice were crossed with CamK-Cre mice to generate BDNF conditional knock-out mice. Bdnftm3Jae/J mice were used as the wild-type control littermates for comparison with BDNF conditional knock-out mice. Mice were genotyped by a PCR-based method using genomic DNA isolated from tail samples. Mice (26–28 d old) were housed under a 12 h light/dark cycle (lights on at 6:00 A.M.) and had ad libitum access to food and water. After a 3 d acclimation period, mice were assigned randomly to two groups that received an intraperitoneal injection of saline (10 ml/kg) or cocaine HCl (15 mg/kg) once per day for 5 consecutive days, followed by a withdrawal period of 1–28 d before the experiments (WD1–28). In some experiments to assess the persistence of the effect of repeated cocaine exposure followed by WD14, mice received cocaine (15 mg/kg) injections, and locomotor activities were assessed with a behavioral tracking system (Ethovision). All comparisons between saline- and cocaine-treated groups were performed by experimenters blind to group assignment. Animal care was consistent with the guidelines set by the Laboratory Animal Center of National Cheng Kung University. All experiments were approved by the National Cheng Kung University Institutional Animal Care and Use Committee governing the participating laboratories.
Locomotor activity.
Following each intraperitoneal injection of cocaine (15 mg/kg) or saline, mice were immediately placed in the activity chamber (45 × 20 × 20 cm3), and horizontal locomotor activity was monitored with the video tracking system for 15 min under dim light, sound-attenuated conditions. Distance traveled was analyzed for estimates of locomotor response. For locomotor habituation to the activity chamber, mice were placed in the chamber 15 min/d for 4 consecutive days. Following habituation, all mice received 2 d of saline injections and then were divided into two groups that received five daily injections of cocaine or saline (from 10:00 A.M. to 12:00 P.M.). To assess the persistence of the effect of repeated cocaine administration following WD14, all groups received cocaine (15 mg/kg) injections, and locomotor activities were reassessed.
Slice preparation and electrophysiology.
Slice preparation and whole-cell patch-clamp recordings were conducted as described previously (Martin et al., 2006). In brief, mice were anesthetized with halothane and decapitated with a guillotine, and coronal slices (250 μm thick) containing the NAc were prepared using a vibrating microtome (VT1200S; Leica). The slices were placed in a holding chamber of artificial CSF (aCSF) oxygenated with 95% O2-5% CO2 and maintained at room temperature for at least 1 h before recording. The composition of the aCSF solution was as follows (in mm): NaCl 117, KCl 4.7, CaCl2 2.5, MgCl2 1.2, NaHCO3 25, NaH2PO4 1.2, and glucose 11, pH 7.3–7.4, and equilibrated with 95% O2-5% CO2. For recording, one slice was transferred to a recording chamber and fixed at the glass bottom of the chamber with a nylon grid on a platinum frame. The chamber consisted of a circular well of low volume (1–2 ml) and was perfused constantly at 32.0 ± 0.5°C at a rate of 2–3 ml/min. Whole-cell patch-clamp recordings were performed from visualized MSNs located in the NAc shell or core. The MSNs were voltage-clamped at −70 mV. Recordings were made using a patch-clamp amplifier (Axopatch 200B; Molecular Devices) under an infrared-differential interference contrast microscope. Electrical signals were low-pass filtered at 2 kHz and digitized at 10 kHz using a 12 bit analog-to-digital converter (Digidata 1320; Molecular Devices). An Intel Pentium-based computer with pCLAMP software (version 8.0; Molecular Devices) was used for on-line acquisition and off-line analysis of the data. For measurement of synaptically evoked EPSCs, a bipolar stainless steel stimulating electrode was placed 150–200 μm rostral to the recording electrode to stimulate excitatory afferents at 0.05 Hz, and the superfusate routinely contained picrotoxin (100 μm) to block inhibitory synaptic responses. The strength of synaptic transmission was quantified by measuring the amplitude of EPSCs. In some experiments, LTD was induced by 900 PP-LFS at 1 Hz, with a 50 ms interstimulus interval in the presence of d-2-amino-5-phosphonovalerate (d-APV; 50 μm). The electrode resistance was typically 3–6 MΩ. The composition of intracellular solution was as follows (in mm): K-gluconate 115, KCl 20, HEPES 10, MgCl2 2, EGTA 0.5, Na2ATP 3, Na3GTP 0.3, QX-314 5, and sucrose to bring the osmolarity to 290–300 mOsM and pH to 7.3. Series resistance and input resistance were monitored on-line throughout the whole-cell recording, with a 5 mV depolarizing step given after every afferent stimulus; data were discarded if access resistance changed by >20%.
Quantitative real-time RT-PCR.
Total RNA was isolated from NAc shell or core tissue samples using a TriReagent kit (Molecular Research Center) and treated with RNase-free DNase (RQ1; Promega) to remove potential contamination by genomic DNA. Total RNA (2 μg) from samples was reverse transcribed using a SuperScript cDNA synthesis kit (Invitrogen). Real-time reverse transcriptase (RT)-PCR was performed on a LightCycler instrument (Roche Diagnostics), using the FastStart DNA Master SYBR Green I kit (Roche Applied Science) according to the manufacturer's instructions. The primers used in this experiment were as follows: mGluR1, 5′-GGGCAGGGAATGCCAATT-3′ (forward) and 5′-GGCGCCTGTCTTCCACC-3′ (reverse); mGluR5, 5′-CTCCTGGCCCACTGACGA-3′ (forward) and 5′-GGTCACCCCAGCGAAGATAC-3′ (reverse); and 18S ribosomal RNA (rRNA), 5′-CAACTTTCGATGGTAGTCGC-3′ (forward) and 5′-CGCTATTGGAGCTGGAATTAC-3′ (reverse). Crude total RNA samples were amplified for 40 cycles. Cycling conditions were 50°C (2 min), 60°C (20 min) for RT step, followed by 95°C (5 min), and a three-step PCR with 60 cycles of 94°C (15 s), 59°C (1 min), and 72°C (1 min), as previously described (Kirschstein et al., 2007) with minor changes. After amplification, equal volumes of PCR products were subjected to electrophoresis on 1% (w/v) agarose gels and visualized with ethidium bromide. PCR amplifications were repeated in duplicate. A melting curve was created at the end of the PCR cycle to confirm that a single product had been amplified. Data were analyzed by LightCycler quantification software to determine the threshold cycle above background for each reaction. The relative transcript amount of the gene of interest, which was calculated using standard curves of serial RNA dilutions, was normalized to that of 18S rRNA.
Western blotting.
Microdissected NAc shell or core tissue samples were lysed in ice-cold Tris-HCl buffer solution (TBS), pH 7.4, containing a mixture of protein phosphatase and proteinase inhibitors (50 mm Tris-HCl, 100 mm NaCl, 15 mm sodium pyrophosphate, 50 mm sodium fluoride, 1 mm sodium orthovanadate, 5 mm EGTA, 5 mm EDTA, 1 mm phenylmethylsulfonyl fluoride, 1 μm microcystin-LR, 1 μm okadaic acid, 0.5% Triton X-100, 2 mm benzamidine, 60 μg/ml aprotinin, and 60 μg/ml leupeptin), to avoid dephosphorylation and degradation of proteins, and ground with a pellet pestle (Kontes Glassware). Samples were sonicated and spun down at 15,000 × g at 4°C for 10 min. The supernatant was then assayed for total protein concentration using Bio-Rad Bradford Protein Assay Kit. Each sample from tissue homogenate was separated in a 6.5% (for mGluR analysis) or 12% (for BDNF analysis) SDS-PAGE gel. Following the transfer on nitrocellulose membranes, blots were blocked in buffer solution containing 5% milk and 0.1% Tween-20 in PBS (124 mm NaCl, 4 mm KCl, 10 mm Na2HPO4, and 10 mm KH2PO4), pH 7.2, for 1 h and then blotted for 2 h at room temperature with the rabbit polyclonal antibodies that recognize mGluR1 (1:1000; BD Transduction Laboratories), mGluR5 (1:2000; Millipore Bioscience Research Reagents) or BDNF (1:500; Santa Cruz Biotechnology). It was then probed with HRP-conjugated secondary antibody for 1 h and developed using the ECL immunoblotting detection system (GE Healthcare), according to manufacturer's instructions. Immunoblots were analyzed by densitometry using BioLight Bio-profil PC software. Only film exposures in the linear range of the ECL reaction were used for quantification analysis. The expression of mGluR1 or mGluR5 was evaluated relative to that of β-actin. Background correction values were subtracted from each lane to minimize variability across membranes.
BDNF immunoassay.
BDNF levels were measured with a conventional two-site ELISA system according to the manufacturer's protocol (Promega). Briefly, the NAc shell tissue samples were weighed and homogenized in 20 volumes of ice-cold lysis TBS, and the homogenates were centrifuged for 20 min at 15,000 × g at 4°C. Tissue samples were acidified for 15 min and neutralized. BDNF concentrations were calculated from regression analysis of human recombinant BDNF standard curves run in each assay.
Drug treatment.
Cocaine HCl (15 mg/kg), (R)-(+)-chloro-8-hydroxy-3-methyl-1-phenyl-2, 3,4,5-tetrahydro-1H-3-benzazepine (SCH23390; 0.5 mg/kg), raclopride (0.5 mg/kg), and 8-[5-(2, 4-dimethoxy-5-(4-trifluoromethylphenylsulfonamido)phenyl-5-oxopentyl)-1,3,8-triazaspiro[4.5]decane-2,4-dione (RS102221; 2 mg/kg) were dissolved in 0.9% NaCl and administered intraperitoneally. Drug doses were selected on the basis of published studies (Essman et al., 1994; Filip et al., 2001; Wellman et al., 2002; Dong et al., 2004; Conductier et al., 2005; Berg et al., 2006; Huang et al., 2007). All drugs used for in vitro experiments were applied by manually switching the superfusate. Drugs were diluted from stock solutions just before application. (S)-3,5-dihydroxyphenylglycine (DHPG), 2-methyl-6-(phenylethynyl)-pyridine (MPEP), (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385), 7-(hydroxyimino)cyclopropachromen-carboxylate ethyl ester (CPCCOEt), and LY354740 were dissolved in dimethyl sulfoxide (DMSO) stock solutions, and stored at −20°C until the day of the experiment. Other drugs used in this study were dissolved in distilled water. The concentration of DMSO in the perfusion medium was 0.1%, which alone had no effect on basal synaptic transmission. DHPG, MPEP, LY367385, CPCCOEt, LY354740, pirenzepine, SCH23390, raclopride, RS102221, and d-APV were purchased from Tocris Cookson; cocaine HCl, human recombinant BDNF, and picrotoxin were obtained from Sigma.
Statistical analysis.
The data for each experiment were normalized relative to baseline and are presented as the mean ± SEM. The number of animals used is indicated by n. The significance of the difference between the groups was calculated by one-way ANOVA with Bonferroni's post hoc analyses or Student's t test where appropriate. p values of <0.05 were considered to represent significant differences.
Results
Cocaine withdrawal selectively blocks DHPG-LTD in the NAc shell
The group I mGluR agonist DHPG has been shown to effectively induce mGluR-dependent LTD at many central synapses (Lüscher and Huber, 2010). To determine the effect of withdrawal from repeated cocaine exposure on mGluR-dependent LTD induction in the NAc, we compared DHPG-LTD in NAc slices prepared from saline- and cocaine-treated mice. Mice were administered daily injections of saline (10 ml/kg) or cocaine (15 mg/kg) intraperitoneally for 5 d, followed by a WD14. The effectiveness of repeated cocaine exposure on mice was indicated by their heightened locomotor sensitivity to cocaine challenge (saline: 2026 ± 239 cm, n = 16; cocaine: 4968 ± 372 cm, n = 16; p < 0.01). Because a previous study has shown that a single re-exposure to cocaine during withdrawal period is a potent stimulus for NAc synaptic transmission (Kourrich et al., 2007), our electrophysiological recordings and biochemical assays were performed using mice that were left undisturbed (no cocaine challenge) before experiments. In slices from saline-treated mice, bath application of DHPG (100 μm) for 5 min reliably induced a robust LTD of EPSCs in MSNs in the shell (64.5 ± 5.6% of baseline, n = 8; p < 0.05) (Fig. 1A, representative experiment, C, averaged data) and core (68.4 ± 6.3% of baseline, n = 8; p < 0.05) of the NAc (Fig. 1B, representative experiment, D, averaged data). In contrast, the induction of DHPG-LTD in the NAc shell was impaired in slices from cocaine-treated mice (95.4 ± 3.2% of baseline, n = 12) (Fig. 1A, representative experiment, C, averaged data). However, no change in the extent of DHPG-LTD was observed in the NAc core in slices from cocaine-treated mice (76.5 ± 6.2% of baseline, n = 12) (Fig. 1B, representative experiment, D, averaged data). To determine whether withdrawal period is a critical parameter for the effect of cocaine, we measured the extent of DHPG-LTD in slices from mice at different WDs. We found the inhibitory effect of repeated cocaine exposure on DHPG-LTD in the NAc shell was evident on WD7, reached maximal levels on WD14, and remained evident on WD28 (F(5,35) = 8.97, p < 0.001) (Fig. 1E). This impairment was not found in the NAc core in slices from cocaine-treated mice examined on WD1–28 (F(5,24) = 0.38, p = 0.86) (Fig. 1F). These results suggest that repeated cocaine exposure selectively impairs DHPG-LTD induction in the shell of the NAc and that the effect only emerges during extended withdrawal.
Figure 1.

Cocaine withdrawal inhibits the induction of DHPG-LTD in MSNs in the NAc shell. A–D, Example and summary of normalized EPSC amplitude before and after brief application of DHPG (100 μm) for 5 min in the NAc shell (A, C) and core (B, D) MSNs from saline (SAL)- or cocaine (COC)-treated mice on WD14. E, F, Summary bar graph comparing the effect of withdrawal period on the induction of DHPG-LTD in the shell (E) and core (F) of the NAc. The magnitude of LTD was measured 30 min after washout of DHPG. Representative traces of EPSCs were taken at the time indicated by number. The total of animals examined is indicated by n. Horizontal bar denotes the period of delivery of DHPG. Dashed line indicates baseline level. *p < 0.05 compared with SAL control, unpaired Student's t test.
DHPG-LTD in the NAc is mediated by mGluR5
To investigate which subtype of group I mGluR underlies DHPG-LTD, we examined the effects of specific antagonists for mGluR1 and mGluR5 on this LTD induction. Application of the highly selective mGluR1 antagonists CPCCOEt (10 μm) or LY367385 (100 μm) had no effect on the induction of DHPG-LTD in slices from saline-treated mice in the NAc shell (CPCCOEt: 71.6 ± 6.3% of baseline, n = 5; LY367385: 72.5 ± 5.4% of baseline, n = 4; p > 0.05) (Fig. 2A,B,E). Likewise, blockade of mGluR1 did not affect the impairment of DHPG-LTD in slices from cocaine-treated mice (CPCCOEt: 96.5 ± 3.4% of baseline, n = 4; LY367385: 97.2 ± 3.8% of baseline, n = 4; p > 0.05). However, the induction of DHPG-LTD was completely blocked by the specific mGluR5 antagonist MPEP (10 μm) in slices from both saline-treated (96.3 ± 2.8% of baseline, n = 5) and cocaine-treated mice (97.6 ± 3.2% of baseline, n = 4) (Fig. 2C).
Figure 2.

Cocaine withdrawal inhibits mGluR5-dependent LTD induction in MSNs in the NAc shell. A, B, Summary of experiments showing the effects of mGluR1 antagonists, CPCCOEt (A) or LY367385 (B), on DHPG-LTD induction in the NAc shell in slices from saline (SAL)- or cocaine (COC)-treated mice on WD14. C, Summary of experiments showing the effect of mGluR5 antagonist MPEP on DHPG-LTD induction in the NAc shell in slices from SAL- or COC-treated mice. D, Summary of experiments showing the effect of M1 muscarinic acetylcholine antagonist pirenzepine on DHPG-LTD induction in the NAc shell in slices from SAL- or COC-treated mice. E, Summary of experiments showing the effects of different mGluR antagonists on DHPG-LTD induction for data in A–D. #p < 0.05 compared with SAL-control; *p < 0.05 compared with SAL-drug alone, unpaired Student's t test.
Recent studies have shown that the induction of DHPG-LTD in the hippocampal CA1 region relies on the activation of M1 muscarinic receptors (Kamsler et al., 2010). We next examined a possible effect of the selective M1 antagonist pirenzepine (75 nm) on DHGP-LTD. No change in the extent of DHPG-LTD was observed in slices from saline-treated mice (69.3 ± 5.2% of baseline, n = 4) (Fig. 2D). Likewise, blocking M1 muscarinic receptors did not affect the impairment of DHPG-LTD in slices from cocaine-treated mice. These results suggest that activation of mGluR5 is required for DHPG-LTD induction in the NAc.
The results thus far were obtained with exogenous application of DHPG, and it is unclear whether the release of glutamate within the NAc can effectively activate mGluR5 to elicit LTD. Because paired-pulse low-frequency stimulation (PP-LFS) in hippocampal slices can apparently elicit LTD via activation of mGluR5 (Kemp and Bashir, 1999), we used this protocol to test our hypothesis. As shown in Figure 3, A and B, delivery of PP-LFS (50 ms interstimulus interval) at 1 Hz for 15 min in the presence of d-APV (50 μm) produced a stable form of LTD (PP-LFS-LTD) in the NAc shell (61.5 ± 5.7% of baseline, n = 5; p < 0.05) and core (63.4 ± 6.9% of baseline, n = 4; p < 0.05) from saline-treated mice. Consistent with DHPG-LTD observed in cocaine-treated mice, the induction of PP-LFS-LTD was impaired in the NAc shell (91.2 ± 7.2% of baseline, n = 5) but not in the NAc core (73.6 ± 6.4% of baseline, n = 4). Moreover, PP-LFS-LTD was completely blocked by bath application of MPEP (10 μm), but not by LY367385 (100 μm), suggesting that PP-LFS-LTD was mainly mediated by activation of mGluR5 (Fig. 3C,D).
Figure 3.

Withdrawal from repeated cocaine exposure inhibits the induction of PP-LFS-LTD in the NAc shell MSNs. A, B, Summary of experiments showing the induction of PP-LFS-LTD (50 ms interstimulus interval, 1 Hz/15 min, in the presence of d-APV 50 μm) in the shell (A) and core (B) MSNs from saline (SAL)- or cocaine (COC)-treated mice on WD14. C, D, Summary of experiments showing the effects of mGluR1 antagonist LY367385 (C) and mGluR5 antagonist MPEP (D) on the induction of PP-LFS-LTD in the NAc shell in slices from SAL- or COC-treated mice. Representative traces of EPSCs were taken at the time indicated by number. Horizontal bar denotes the period of delivery of PP-LFS. Dashed line indicates baseline level.
A previous study has shown that a presynaptic mGluR2/3-dependent LTD was abolished in the NAc of morphine-withdrawal mice (Robbe et al., 2002). To test for changes in mGluR2/3 function in the NAc after cocaine withdrawal, we compared the magnitude of LTD induced by a 10 min application of the selective mGluR2/3 agonist LY354740 (200 nm) between groups. In contrast to what was observed with DHPG-LTD and PP-LFS-LTD, the shell and core MSNs from cocaine-treated mice showed similar levels of LTD as the saline-treated mice (Fig. 4). Together, these findings indicate that cocaine withdrawal selectively impairs mGluR5-mediated LTD in MSNs in the NAc shell.
Figure 4.
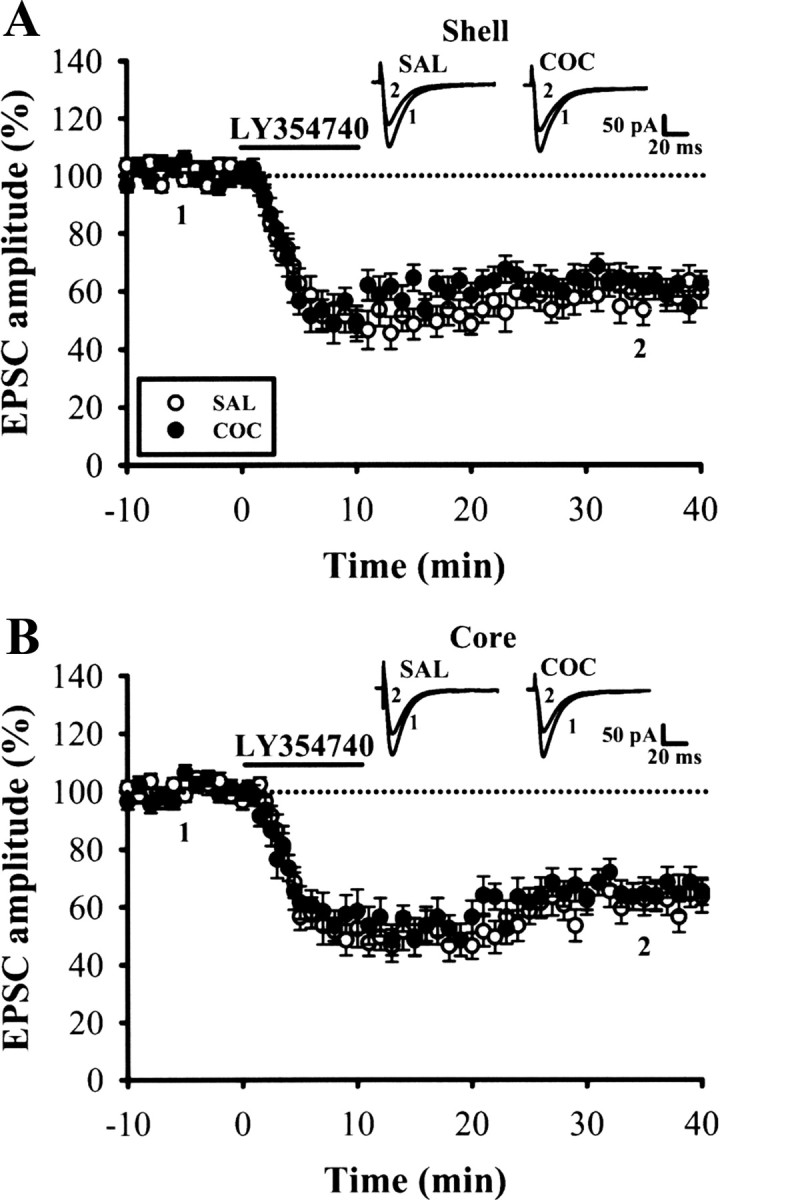
The induction of LY354740-LTD in the NAc shell and core MSNs after withdrawal from repeated cocaine exposure. A, B, Summary of normalized EPSC amplitude before and after a brief application of mGluR2/3 agonist LY354740 (200 nm) for 10 min in the NAc shell (A) and core (B) MSNs from saline (SAL)- or cocaine (COC)-treated mice on WD14. Representative traces of EPSCs were taken at the time indicated by number. Horizontal bar denotes the period of delivery of LY354740. Dashed line indicates baseline level.
Cocaine withdrawal impairs DHPG-LTD via the activation of D1-like receptors
We next investigated how repeated cocaine exposure alters DHPG-LTD. Considering that cocaine blocks the DA reuptake transporter and thereby increases local DA concentrations in brain areas receiving DA inputs (Hyman, 1996), it is therefore possible that activation of DA receptors in critical brain areas is required for the aforementioned cocaine-induced impairment of DHPG-LTD. To examine the role of DA receptors, we administrated specific D1- or D2-like receptor antagonists before the daily injection of cocaine. As shown in Figure 5, A and B, neither the D1 receptor antagonist SCH23390 (0.5 mg/kg) nor the D2 receptor antagonist raclopride (0.5 mg/kg) had a significant effect on DHPG-LTD when administered with saline (SCH23390, 69.8 ± 3.5% of baseline, n = 4; raclopride, 72.7 ± 4.7% of baseline, n = 4; p > 0.05). In the presence of SCH23390, the ability of cocaine to impair the induction of DHPG-LTD was blocked (i.e., DHPG-LTD was intact) in slices from cocaine-treated mice (SCH23390, 78.2 ± 5.3% of baseline, n = 6). In contrast, the ability of cocaine to impair DHPG-LTD was not diminished in the presence of raclopride (raclopride, 96.2 ± 3.5% of baseline, n = 5) (Fig. 5A,B,D).
Figure 5.

Withdrawal from repeated cocaine exposure impairs DHPG-LTD induction via the activation of D1-like dopaminergic receptors. A, Summary of experiments showing the induction of DHPG-LTD in the NAc shell in slices from mice receiving SCH23390 15 min before saline (SAL) or cocaine (COC) injections for 5 d and then examined on WD14. B, Summary of experiments showing the induction of DHPG-LTD in the NAc shell in slices from mice receiving raclopride before SAL or COC injections. C, Summary of experiments showing the induction of DHPG-LTD in the NAc shell in slices from mice receiving RS102221 before SAL or COC injections. D, Summary of experiments showing the effects of different antagonist pretreatment on the development of cocaine-induced impairment of DHPG-LTD for data in A–C. *p < 0.05 compared with SAL-drug alone, unpaired Student's t test.
Ramos et al. (2005) showed that administration of SCH23390 into the medial prefrontal cortex (mPFC) prevents the expression of 3,4-methylenedioxymethamphetamine-induced behavioral sensitization through the blockade of serotonin 5-HT2C, but not D1-like receptors. To exclude a role for the 5-HT2C receptor in the development of cocaine-induced impairment of DHPG-LTD, we administered the specific 5-HT2C receptor antagonist RS102221 (2 mg/kg) before the daily injection of cocaine. However, in contrast to SCH23390, RS102221 did not significantly affect the impairment of DHPG-LTD in the slice from cocaine-treated mice (95.5 ± 2.3% of baseline, n = 4) (Fig. 5C). RS102221 had no effect on DHPG-LTD when coadministered with saline (73.6 ± 5.3% of baseline, n = 4) (Fig. 5C,E). Together, these findings indicate that the activation of D1-like receptors is responsible for the inhibitory effect of repeated cocaine exposure on DHPG-LTD in the NAc shell.
Downregulation of mGluR5 after cocaine withdrawal
The preceding results point to an involvement of mGluR5 in the induction of DHPG-LTD. We reasoned that the loss of DHPG-LTD observed in cocaine-treated mice could be due to a downregulation of mGluR5 in the NAc. To answer this question, we determined the expression of mGluR5 mRNA and protein levels in the NAc of saline- and cocaine-treated mice on WD14. In accord with this hypothesis, we found that mGluR5 protein was decreased selectively in the shell, but not in the core, of the NAc in cocaine-treated mice. However, the levels of mGluR1 protein in the NAc shell and core were not altered by cocaine withdrawal (Fig. 6A). In agreement with the reduction in content of mGluR5 protein produced by cocaine withdrawal, the post hoc analysis revealed a significant decrease in mGluR5 mRNA content in the NAc shell (Fig. 6B). However, cocaine-treated mice showed normal levels of mGluR1 mRNA in the NAc shell and core (Fig. 6C). Furthermore, the effects of cocaine were prevented when cocaine was coadministered with SCH23390 (0.5 mg/kg), but not with raclopride (0.5 mg/kg) (Fig. 6D,E). Neither SCH23390 nor raclopride had any obvious effects on the expression of mGluR5 or mGluR1 mRNA and protein levels in the NAc (Fig. 6D–F). These results strongly suggest a functional correlation between the occurrence of DHPG-LTD and mGluR5 expression levels in the NAc.
Figure 6.

Cocaine withdrawal selectively decreases the expression of mGluR5 mRNA and protein levels in the NAc shell. A, Representative Western blot and summary bar graph depicting levels of mGluR1 and mGluR5 protein in the NAc core and shell from saline (SAL)- or cocaine (COC)-treated mice on WD14. B, C, Summary bar graph depicting levels of mGluR5 (B) and mGluR5 (C) mRNA in the NAc core and shell from SAL- or COC-treated mice on WD14. D, Representative Western blot and summary bar graph depicting levels of mGluR1 and mGluR5 protein in the NAc shell from mice receiving vehicle, SCH23390, or raclopride 15 min before SAL or COC injections. E, F, Summary bar graph depicting levels of mGluR5 (E) and mGluR5(F) mRNA in the NAc shell from mice receiving vehicle, SCH23390, or raclopride 15 min before SAL or COC injections. *p < 0.05 compared with SAL or SAL-drug alone, unpaired Student's t test.
BDNF-dependent synaptic adaptation in the NAc after cocaine withdrawal
Previous studies have shown that the infusion of BDNF into the rat NAc enhances cocaine-induced behavioral sensitization and cocaine self-administration (Horger et al., 1999), and that cocaine withdrawal increases BDNF protein levels in the NAc (Grimm et al., 2003; Lu et al., 2004). To examine whether the impaired DHPG-LTD in the NAc after cocaine withdrawal is caused by the elevation of BDNF, we examined DHPG-LTD induction in the NAc shell in slices from BDNF conditional knock-out mice that also received daily intraperitoneal injections of saline or cocaine for 5 d followed by WD14. To confirm that the ablation of BDNF expression occurs in BDNF conditional knock-out mice, we used Western blot analyses to examine BDNF protein levels of dissected brain regions from knock-out and wild-type littermate control mice. The BDNF conditional knock-out mice displayed an almost complete loss of BDNF protein expression in the hippocampus, amygdala, PFC, VTA, NAc, and striatum compared with wild-type control mice (Fig. 7A). In contrast, BDNF conditional knock-out mice showed normal BDNF expression in the cerebellum, confirming that the deletion of BDNF was restricted to the forebrain. Having confirmed an almost complete ablation of NAc BDNF in conditional knock-out mice, we then compared the induction of DHPG-LTD in slices from saline- and cocaine-treated BDNF conditional knock-out mice. We observed that the extent of DHPG-LTD in the NAc shell is similar in slices from saline-treated (72.5 ± 5.6% of baseline, n = 5) and cocaine-treated BDNF conditional knock-out mice (78.9 ± 4.2% of baseline, n = 6; p > 0.05) (Fig. 7B, representative experiment, C, averaged data). We also examined whether BDNF, by itself, is capable of impairing DHPG-LTD induction in the NAc shell in slices from naive mice. Application of BDNF (200 ng/ml) had no effect on the EPSC (96.5 ± 2.8% of baseline, n = 4) or DHPG-LTD (74.3 ± 3.9% of baseline, n = 5) (Fig. 7D), suggesting that acute application of BDNF is not sufficient to block the synaptic expression of DHPG-LTD in naive mice. The data obtained from the ELISA of the NAc shell tissues showed that BDNF protein levels increased significantly on WD14 and WD28, but were not detectable on WD1–7 (F(5,22) = 4.01, p < 0.01) (Fig. 7E). In addition, we found that the effect of cocaine on BDNF expression in the NAc was prevented when cocaine was coadministered with SCH23390, but not raclopride (Fig. 7F). Furthermore, cocaine-treated BDNF conditional knock-out mice showed normal mGluR5 mRNA and protein levels in the NAc shell and core when compared with saline-treated controls (Fig. 7G,H). No significant difference was detected in the expression of mGluR5 mRNA and protein in the NAc between wild-type control and BDNF conditional knock-out mice. These results suggest that the elevated BDNF may underlie the impairment of DHPG-LTD in the NAc after cocaine withdrawal.
Figure 7.

Time-dependent increase in BDNF levels after cocaine withdrawal is essential for cocaine-induced impairment of DHPG-LTD. A, Western blot of homogenates from various brain regions of wild-type (WT) and BDNF conditional knock-out (BDNF-KO) mice shows relative levels of BDNF expression in the hippocampus (Hip), amygdala (Amy), PFC, VTA, NAc, dorsal striatum (Str), and cerebellum (Cere). B, C, Example (B) and summary (C) of the induction of DHPG-LTD in the shell MSNs from saline (SAL)- or cocaine (COC)-treated BDNF-KO mice on WD14. D, Summary of experiments showing application of BDNF (200 ng/ml) had no effect on either the basal synaptic transmission or DHPG-LTD induction in the NAc shell in slices from naive mice. E, Summary bar graph comparing BDNF protein levels in the NAc shell tissues from mice that were injected daily with SAL or COC for 5 d and then examined on WD1–28. F, Summary bar graph depicting BDNF protein levels in the NAc shell tissue samples from mice receiving SCH23390 or raclopride 15 min before SAL or COC injections for 5 d and then examined on WD14. G, Representative Western blot and summary bar graph depicting levels of mGluR1 and mGluR5 protein in the NAc shell from SAL- or COC-treated WT and BDNF-KO mice on WD14. H, Summary bar graph depicting levels of mGluR5 and mGluR5 mRNA in the NAc shell from SAL- or COC-treated WT and BDNF-KO mice on WD14. *p < 0.05 compared with SAL or SAL-drug alone, unpaired Student's t test.
To examine further the relationship between BDNF and cocaine-induced behavioral sensitization, we measured the locomotor responses to acute and repeated cocaine administration in BDNF conditional knock-out mice. Previous studies have shown that BDNF-null mutants (Kernie et al., 2000) and conditional knock-out mice (Rios et al., 2001; Monteggia et al., 2007) exhibit locomotor hyperactivity. Consistent with these findings, we found that BDNF conditional knock-out mice exhibited significant increased locomotor activity compared with wild-type control littermates when first placed in the locomotor activity chamber, but showed equivalent habituation in locomotor activity after repeated exposures to chamber. By day 4, the BDNF conditional knock-out mice were indistinguishable from wild-type control mice (F(1,38) = 1.41, p = 0.24) (Fig. 8A). To minimize the influence of locomotor hyperactivity seen in the BDNF conditional knock-out mice, all mice received 4 d of habituation paradigm and 2 d of saline injections. Following habituation, the immediate locomotor response to five daily cocaine injections (15 mg/kg) was recorded. Fourteen days later, all mice were challenged with a single injection of 15 mg/kg cocaine. As shown in Figure 8B, wild-type (n = 12) and BDNF conditional knock-out mice (n = 12) showed equivalent locomotor responses to the daily injection of cocaine. However, BDNF conditional knock-out mice showed a significantly reduced increase in locomotor response to a challenge dose of cocaine administered 14 d following the last dose of the treatment regimen (F(1,22) = 13.1, p < 0.01). These findings demonstrate that BDNF is required for the expression of cocaine-induced behavioral sensitization.
Figure 8.
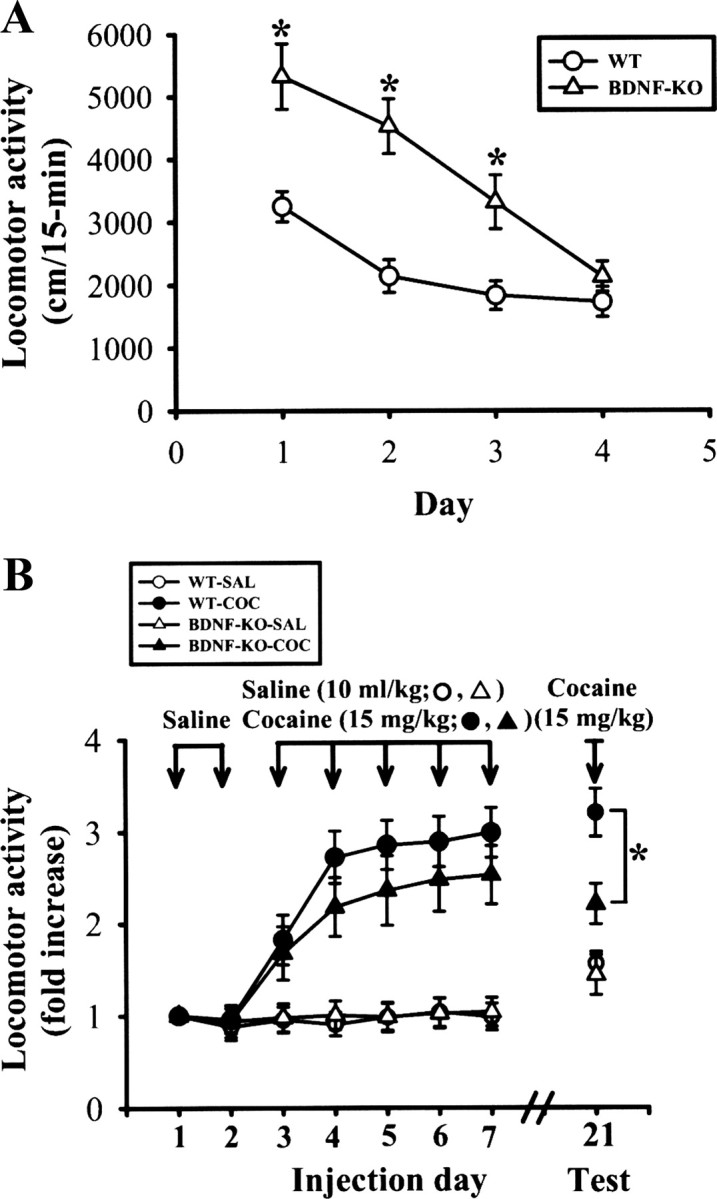
Behavioral sensitization induced by repeated cocaine administration. A, Locomotor activity was measured in the activity chamber for 15 min on 4 consecutive days. BDNF conditional knock-out (BDNF-KO) mice exhibited an increase in locomotor activity relative to wild-type (WT) control littermates on on days 1–3. No significant difference was observed between BDNF-KO and WT mice on day 4. B, Following each intraperitoneal injection of cocaine or saline, mice were immediately placed in the chamber and locomotor activity was monitored for 15 min. After 2 d of saline injections, BDNF-KO and WT mice were divided into two groups that received daily injections of saline or cocaine for 5 d. To examine the persistence of the effect of repeated cocaine administration, following 14 d without injections, all groups received cocaine (15 mg/kg) injections and locomotor activities were assessed. *p < 0.05 compared with WT, unpaired Student's t test.
Discussion
The present study shows that withdrawal from repeated cocaine exposure selectively impairs the induction of mGluR5-dependent LTD in MSNs in the NAc shell and that D1-like receptors play a major role in the process. The molecular processes mediating this change involves a progressive elevation in the level of BDNF protein after cocaine withdrawal, which leads to an ensuing subtype-specific decrease in mGluR5 transcript and protein expression, leading to a marked reduction in the ability of DHPG to induce LTD. To our knowledge, this is the first study to correlate the development of behavioral sensitization to cocaine with an alteration in mGluR5-dependent LTD of excitatory synaptic transmission in the NAc.
The NAc can be divided into two major anatomically and functionally distinct subregions, the core and the shell (Groenewegen et al., 1999). The NAc core is often considered to be a functional extension of the dorsal striatum and may be important for instrumental learning, including cue-induced reinstatement of drug seeking (Ito et al., 2000; Floresco et al., 2006). Alternatively, the NAc shell is thought to be a transitional region between the striatum and extended amygdala and is particularly relevant in mediating the primary reinforcing effects of drugs of abuse as well as behavioral sensitization (Pierce and Kumaresan, 2006). Previous studies indicate that cocaine preferentially increases extracellular DA level in the shell compared with the core of the NAc (Pontieri et al., 1995), and microinjections of D1-like receptor antagonists into the shell specifically reduce the reinstatement of cocaine seeking (Anderson et al., 2003). The present observation that cocaine withdrawal selectively impairs mGluR5-dependent LTD in the shell reinforces the idea that the shell of the NAc is particularly important for mediating the neuroadaptation that underlies cocaine addiction (Pierce and Kumaresan, 2006). Our results are in agreement with a previous study showing that behavioral sensitization to cocaine selectively causes a long-lasting depression of excitatory synaptic transmission in the NAc shell and occludes the further induction of NMDA receptor-dependent LTD (Thomas et al., 2001). There is evidence, however, that withdrawal from cocaine self-administration impairs the ability of PFC stimulation to produce LTP and LTD in the NAc core (Moussawi et al., 2009) and that extinction training after cocaine self-administration selectively abolishes the induction of LTD in the NAc core elicited by PFC stimulation (Knackstedt et al., 2010). Thus, our results should not be taken to indicate that cocaine-induced synaptic adaptations in other subregions are not required to mediate the addictive behaviors of cocaine. Considered together, the present and previous studies imply a possibility that passive cocaine injections and self-administration of cocaine protocols may hijack subregion-specific synaptic plasticity mechanisms in the NAc to promote the development and persistence of addictive behaviors.
The present data indicate that withdrawal from repeated exposure causes a subtype-specific downregulation of mGluR5 mRNA and protein in the NAc shell. Furthermore, decreased mGluR5 mRNA expression is unlikely to simply represent cell loss, as there is no difference in mRNA and protein levels measured for mGluR1. Thus, we believe that the transcriptional downregulation of mGluR5 is the most plausible explanation for the loss of mGluR5 expression in the NAc shell after withdrawal from repeated cocaine exposure. However, our findings are not consistent with a recent report showing that surface expression of mGluR5 was reduced only in the NAc core of cocaine-extinguished rats (Knackstedt et al., 2010). The reason for this discrepancy is unclear but could be attributable to the use of different animal models of addiction (cocaine self-administration versus passive cocaine injections) or animal species (rats versus mice), resulting in the stimulation of different cellular processes that may vary in their mode of action.
Cocaine blocks the reuptake of DA, norepinephrine, and serotonin. Despite the enormous research focus on DA, norepinephrine and serotonin have also been implicated in the processes that underlie cocaine-induced behavioral sensitization (Drouin et al., 2002). However, our results demonstrate that cocaine-induced impairment of mGluR5-dependent LTD in the NAc is dependent on D1-like receptor activation, since the coadministration of SCH23390, but not raclopride, with cocaine prevented the blockade of DHPG-LTD. Recently, it was shown that administration of SCH23390 into the mPFC prevents the expression of 3,4-methylenedioxymethamphetamine-induced behavioral sensitization through the blockade of 5-HT2C, but not D1-like, receptors (Ramos et al., 2005). Given that SCH23390 also exhibits a high affinity for 5-HT2C receptors (Millan et al., 2001), it might be argued that the antagonizing effect of SCH23390 on cocaine seen in the present study takes place, in part, through its action on 5-HT2C receptors. However, this possibility appears unlikely, because the selective 5-HT2C receptor antagonist RS102221 did not affect the cocaine-induced impairment of DHPG-LTD. It remains an open question as to where in the brain D1-like receptors are required to trigger the effect of cocaine on mGluR5-dependent LTD.
We found that time-dependent increase in BDNF levels in the NAc after cocaine withdrawal may mediate some of the molecular and cellular events that underlie the impairment of mGluR5-dependent LTD. The increase in BDNF levels occurred over a time course similar to that found for the impaired DHPG-LTD. We also noted that the same cocaine regimen had no effect on the induction of DHPG-LTD in the NAc in slices from BDNF conditional knock-out mice. More importantly, BDNF conditional knock-out mice exhibited a significantly reduced expression of cocaine-induced behavioral sensitization. These findings provide further support for the hypothesis that elevated BDNF expression after cocaine withdrawal may drive synaptic changes underlying drug craving and seeking behavior (McGinty et al., 2010). It remains unclear whether the elevation of BDNF results from an increase in the local synthesis of BDNF by NAc neurons or through anterograde transport to the NAc via afferent axons. In fact, BDNF mRNA is barely detectable in NAc neurons under normal conditions (Altar et al., 1997; Conner et al., 1997). It is therefore possible that the delayed emergence of BDNF elevation in the NAc after cocaine withdrawal is due to the timing required for BDNF transport from other brain regions, such as the VTA and PFC (Seroogy et al., 1994; Guillin et al., 2001), where high expression of BDNF has been found after cocaine withdrawal (Grimm et al., 2003; Pu et al., 2006). Given that our BDNF conditional knock-out mice showed an almost complete loss of BDNF in the VTA, PFC, and NAc, it is difficult to predict which brain region may be the major source of NAc BDNF contributing to the development of the persistent impairment of DHPG-LTD induction after cocaine withdrawal. Moreover, the impairment of LTD may reflect prior in vivo LTD induction or a disruption of induction mechanisms. Although we cannot exclude the possibility that withdrawal from repeated cocaine exposure may elicit a form of LTD that shares expression mechanisms with DHPG-LTD and reduces the capacity to induce further LTD, our results strongly suggest that the impairment in LTD is caused, at least in part, by disrupting the induction mechanisms. It is noteworthy that, although BDNF conditional knock-out mice exhibited normal DHPG-LTD in the NAc shell after cocaine withdrawal, the exogenous application of BDNF had no effect on DHPG-LTD induction in slices from naive mice. The most straightforward explanation for this observation is that the disruption of LTD induction mechanisms after cocaine withdrawal is not mediated by a rapid effect of BDNF, but rather represents a prolonged effect of BDNF.
Since BDNF conditional knock-out mice showed normal mGluR5 mRNA and protein levels after cocaine withdrawal, it is tempting to speculate that the activation of downstream signaling through BDNF may mediate a decrease in mGluR5 transcript. An open question that remains to be explored is how the BDNF signaling cascade suppresses the transcription of mGluR5 in the NAc. Because the genomic sequence spanning exon IB, corresponding to the mGluR5 5′-untranslated region, contains a negative regulatory sequence that represses gene transcription (Corti et al., 2003), it seems plausible that elevated levels of transcriptional silencers may contribute, at least in part, to the BDNF-induced suppression of mGluR5 expression. Further studies, however, are needed to test this possibility.
In conclusion, our data indicate that withdrawal from repeated cocaine exposure selectively impairs the induction of mGluR5-dependent LTD in the NAc. Our data also indicate that elevated BDNF expression after cocaine withdrawal may cause a transcriptional silencing of the mGluR5 gene, resulting in a decrease in mGluR5 expression, and thereby impair DHPG-LTD induction. What might be the functional consequences of a loss of NAc mGluR5-dependent LTD in the context of cocaine withdrawal? It has been shown that constitutive mGluR5 knock-out mice fail to self-administer cocaine and are insensitive to the locomotor stimulant effects of cocaine (Chiamulera et al., 2001). In addition, intra-NAc shell administration of mGluR5 antagonists attenuates cue-induced reinstatement of cocaine seeking (Kumaresan et al., 2009). Thus, the downregulation of mGluR5 function in the NAc after cocaine withdrawal more likely represents a compensatory response to behavioral sensitization. Moreover, considering the importance of mGluR-dependent LTD in goal-directed learning (Lüscher and Huber, 2010), failure to elicit this form of LTD in the NAc shell may reflect the difficulties experienced by addicts in acquiring new behaviors to compete with drug seeking.
Footnotes
This work was supported by research Grants NSC99-2321-B-006-012-MY3 (C.C.H.), NSC98-2321-B-006-001 (K.S.H.), and NSC98-2321-B-182A-004 (S.H.C.) from the National Science Council, Taiwan, Republic of China.
References
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Anderson SM, Bari AA, Pierce RC. Administration of the D1-like dopamine receptor antagonist SCH-23390 into the medial nucleus accumbens shell attenuates cocaine priming-induced reinstatement of drug-seeking behavior in rats. Psychopharmacology. 2003;168:132–138. doi: 10.1007/s00213-002-1298-5. [DOI] [PubMed] [Google Scholar]
- Argilli E, Sibley DR, Malenka RC, England PM, Bonci A. Mechanism and time course of cocaine-induced long-term potentiation in the ventral tegmental area. J Neurosci. 2008;28:9092–9100. doi: 10.1523/JNEUROSCI.1001-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KA, Navailles S, Sanchez TA, Silva YM, Wood MD, Spampinato U, Clarke WP. Differential effects of 5-methyl-1-[[2-[(2-methyl-3-pyridyl)oxyl]-5-pyridyl]carbamoyl]-6-trifluoromethylindone (SB 243213) on 5-hydroxytryptamine2C receptor-mediated responses. J Pharmacol Exp Ther. 2006;319:260–268. doi: 10.1124/jpet.106.104448. [DOI] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiamulera C, Epping-Jordan MP, Zocchi A, Marcon C, Cottiny C, Tacconi S, Corsi M, Orzi F, Conquet F. Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5-null mutant mice. Nat Neurosci. 2001;4:873–874. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- Conductier G, Crosson C, Hen R, Bockaert J, Compan V. 3,4-N-methlenedioxymethamphetamine-induced hypophagia is maintained in 5-HT1B receptor knockout mice, but suppressed by the 5-HT2C receptor antagonist RS102221. Neuropsychopharmacology. 2005;30:1056–1063. doi: 10.1038/sj.npp.1300662. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti C, Clarkson RW, Crepaldi L, Sala CF, Xuereb JH, Ferraguti F. Gene structure of the human metabotropic glutamate receptor 5 and functional analysis of its multiple promoters in neuroblastoma and astroglioma cells. J Biol Chem. 2003;278:33105–33119. doi: 10.1074/jbc.M212380200. [DOI] [PubMed] [Google Scholar]
- Dong Y, Saal D, Thomas M, Faust R, Bonci A, Robinson T, Malenka RC. Cocaine-induced potentiation of synaptic strength in dopamine neurons: behavioral correlates in GluRA(−/−) mice. Proc Natl Acad Sci U S A. 2004;101:14282–14287. doi: 10.1073/pnas.0401553101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouin C, Darracq L, Trovero F, Blanc G, Glowinski J, Cotecchia S, Tassin JP. α1b-Adrenergic receptors control locomotor and rewarding effects of psychostimulants and opiates. J Neurosci. 2002;22:2873–2884. doi: 10.1523/JNEUROSCI.22-07-02873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essman WD, Singh A, Lucki I. Serotonergic properties of cocaine: effects on a 5-HT2 receptor-mediated behavior and on extracellular concentrations of serotonin and dopamine. Pharmacol Biochem Behav. 1994;49:107–113. doi: 10.1016/0091-3057(94)90463-4. [DOI] [PubMed] [Google Scholar]
- Filip M, Nowak E, Papla I. On the role of serotonin2A/2C receptors in the sensitization to cocaine. J Physiol Pharmacol. 2001;52:471–481. [PubMed] [Google Scholar]
- Floresco SB, Ghods-Sharifi S, Vexelman C, Magyar O. Dissociable roles for the nucleus accumbens core and shell in regulating set shifting. J Neurosci. 2006;26:2449–2457. doi: 10.1523/JNEUROSCI.4431-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourgeaud L, Mato S, Bouchet D, Hémar A, Worley PF, Manzoni OJ. A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J Neurosci. 2004;24:6939–6945. doi: 10.1523/JNEUROSCI.0671-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JW, Lu L, Hayashi T, Hope BT, Su TP, Shaham Y. Time-dependent increases in brain-derived neurotrophic factor protein levels within the mesolimbic dopamine system after withdrawal from cocaine: implications for incubation of cocaine craving. J Neurosci. 2003;23:742–747. doi: 10.1523/JNEUROSCI.23-03-00742.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenewegen HJ, Wright CI, Beijer AV, Voorn P. Convergence and segregation of ventral striatal inputs and outputs. Ann N Y Acad Sci. 1999;877:49–63. doi: 10.1111/j.1749-6632.1999.tb09260.x. [DOI] [PubMed] [Google Scholar]
- Guillin O, Diaz J, Carroll P, Griffon N, Schwartz JC, Sokoloff P. BDNF controls dopamine D3 receptor expression and triggers behavioural sensitization. Nature. 2001;411:86–89. doi: 10.1038/35075076. [DOI] [PubMed] [Google Scholar]
- Horger BA, Iyasere CA, Berhow MT, Messer CJ, Nestler EJ, Taylor JR. Enhancement of locomotor activity and conditioned reward to cocaine by brain-derived neurotrophic factor. J Neurosci. 1999;19:4110–4122. doi: 10.1523/JNEUROSCI.19-10-04110.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Yang PC, Lin HJ, Hsu KS. Repeated cocaine administration impairs group II metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. J Neurosci. 2007;27:2958–2968. doi: 10.1523/JNEUROSCI.4247-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE. Addiction to cocaine and amphetamine. Neuron. 1996;16:901–904. doi: 10.1016/s0896-6273(00)80111-7. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Ito R, Dalley JW, Howes SR, Robbins TW, Everitt BJ. Dissociation in conditioned dopamine release in the nucleus accumbens core and shell in response to cocaine cues and during cocaine-seeking behavior in rats. J Neurosci. 2000;20:7489–7495. doi: 10.1523/JNEUROSCI.20-19-07489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamsler A, McHugh TJ, Gerber D, Huang SY, Tonegawa S. Presynaptic M1 muscarinic receptors are necessary for mGluR long-term depression in the hippocampus. Proc Natl Acad Sci U S A. 2010;107:1618–1623. doi: 10.1073/pnas.0912540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasanetz F, Deroche-Gamonet V, Berson N, Balado E, Lafourcade M, Manzoni O, Piazza PV. Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science. 2010;328:1709–1712. doi: 10.1126/science.1187801. [DOI] [PubMed] [Google Scholar]
- Kemp N, Bashir ZI. Induction of LTD in the adult hippocampus by the synaptic activation of AMPA/kainate and metabotropic glutamate receptors. Neuropharmacology. 1999;38:495–504. doi: 10.1016/s0028-3908(98)00222-6. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Liebl DJ, Parada LF. BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 2000;19:1290–1300. doi: 10.1093/emboj/19.6.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschstein T, Bauer M, Müller L, Rüschenschmidt C, Reitze M, Becker AJ, Schoch S, Beck H. Loss of metabotropic glutamate receptor-dependent long-term depression via downregulation of mGluR5 after status epilepticus. J Neurosci. 2007;27:7696–7704. doi: 10.1523/JNEUROSCI.4572-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Moussawi K, Lalumiere R, Schwendt M, Klugmann M, Kalivas PW. Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J Neurosci. 2010;30:7984–7992. doi: 10.1523/JNEUROSCI.1244-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Caine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaresan V, Yuan M, Yee J, Famous KR, Anderson SM, Schmidt HD, Pierce RC. Metabotropic glutamate receptor 5 (mGluR5) antagonists attenuate cocaine priming- and cue-induced reinstatement of cocaine seeking. Behav Brain Res. 2009;202:238–244. doi: 10.1016/j.bbr.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Dempsey J, Liu SY, Bossert JM, Shaham Y. A single infusion of brain-derived neurotrophic factor into the ventral tegmental area induces long-lasting potentiation of cocaine seeking after withdrawal. J Neurosci. 2004;24:1604–1611. doi: 10.1523/JNEUROSCI.5124-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group I mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9:868–869. doi: 10.1038/nn1713. [DOI] [PubMed] [Google Scholar]
- McGinty JF, Whitfield TW, Jr, Berglind WJ. Brain-derived neurotrophic factor and cocaine addiction. Brain Res. 2010;1314:183–193. doi: 10.1016/j.brainres.2009.08.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ, Newman-Tancredi A, Quentric Y, Cussac D. The “selective” dopamine D1 receptor antagonist, SCH23390, is a potent and high efficacy agonist at cloned human 5-HT2C receptors. Psychopharmacology. 2001;156:58–62. doi: 10.1007/s002130100742. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, Parada LF, Nestler EJ. Brain-derived neurotrophic factor conditional knockout show gender differences in depression-related behaviors. Biol Psychiatry. 2007;61:187–197. doi: 10.1016/j.biopsych.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, Kalivas PW. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–189. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Kumaresan V. The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse? Neurosci Biobehav Rev. 2006;30:215–238. doi: 10.1016/j.neubiorev.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Pontieri FE, Tanda G, Di Chiara G. Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the “shell” as compared with the “core” of the rat nucleus accumbens. Proc Natl Acad Sci U S A. 1995;92:12304–12308. doi: 10.1073/pnas.92.26.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L, Liu QS, Poo MM. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci. 2006;9:605–607. doi: 10.1038/nn1687. [DOI] [PubMed] [Google Scholar]
- Ramos M, Goñi-Allo B, Aguirre N. Administration of SCH 23390 into the medial prefrontal cortex blocks the expression of MDMA-induced behavioral sensitization in rats: an effect mediated by 5-HT2C receptor stimulation and not by D1 receptor blockade. Neuropsychopharmacology. 2005;30:2180–2191. doi: 10.1038/sj.npp.1300735. [DOI] [PubMed] [Google Scholar]
- Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, Lechan RM, Jaenisch R. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol. 2001;15:1748–1757. doi: 10.1210/mend.15.10.0706. [DOI] [PubMed] [Google Scholar]
- Robbe D, Bockaert J, Manzoni OJ. Metabotropic glutamate receptor 2/3-dependent long-term depression in the nucleus accumbens is blocked in morphine withdrawn mice. Eur J Neurosci. 2002;16:2231–2235. doi: 10.1046/j.1460-9568.2002.02273.x. [DOI] [PubMed] [Google Scholar]
- Schmidt HD, Pierce RC. Cocaine-induced neuroadaptations in glutamate transmission: potential therapeutic targets for craving and addiction. Ann N Y Acad Sci. 2010;1187:35–75. doi: 10.1111/j.1749-6632.2009.05144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seroogy KB, Lundgren KH, Tran TM, Guthrie KM, Isackson PJ, Gall CM. Dopaminergic neurons in rat ventral midbrain express brain-derived neurotrophic factor and neurotrophin-3 mRNAs. J Comp Neurol. 1994;342:321–334. doi: 10.1002/cne.903420302. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Hope BT. The role of neuroadaptations in relapse to drug seeking. Nat Neurosci. 2005;8:1437–1439. doi: 10.1038/nn1105-1437. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Kalivas PW, Shaham Y. Neuroplasticity in the mesolimbic dopamine system and cocaine addiction. Br J Pharmacol. 2008;154:327–342. doi: 10.1038/bjp.2008.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Wellman P, Ho D, Cepeda-Benito A, Bellinger L, Nation J. Cocaine-induced hypophagia and hyperlocomotion in rats are attenuated by prazosin. Eur J Pharmacol. 2002;455:117–126. doi: 10.1016/s0014-2999(02)02616-x. [DOI] [PubMed] [Google Scholar]