

Abstract
Hypoxic damage to the prefrontal cortex (PFC) has been implicated in the frontal lobe dysfunction found in various neuropsychiatric disorders. The underlying subcortical mechanisms, however, have not been well explored. In this study, we induced a PFC-specific hypoxia-like damage by cobalt-wire implantation to demonstrate that the role of the mediodorsal thalamus (MD) is critical for the development of frontal lobe dysfunction, including frontal lobe-specific seizures and abnormal hyperactivity. Before the onset of these abnormalities, the cross talk between the MD and PFC nuclei at theta frequencies was enhanced. During the theta frequency interactions, burst spikes, known to depend on T-type Ca2+ channels, were increased in MD neurons. In vivo knockout or knockdown of the T-type Ca2+ channel gene (CaV3.1) in the MD substantially reduced the theta frequency MD–PFC cross talk, frontal lobe-specific seizures, and locomotor hyperactivity in this model. These results suggest a two-step model of prefrontal dysfunction in which the response to a hypoxic lesion in the PFC results in abnormal thalamocortical feedback driven by thalamic T-type Ca2+ channels, which, in turn, leads to the onset of neurological and behavioral abnormalities. This study provides valuable insights into preventing the development of neuropsychiatric disorders arising from irreversible PFC damage.
Introduction
Abnormal oscillations or excitability has been a key neurological sign for frontal lobe dysfunction in neurological and psychiatric disorders (McAllister and Price, 1987; Barry et al., 2003; Gucuyener et al., 2003; Wienbruch et al., 2003; Tan and Appleton, 2005; Beleza et al., 2009); for example, the 1–7 Hz oscillations seen in schizophrenia (Elbert et al., 1992), the enhanced theta rhythms in attention deficit hyperactivity disorder (ADHD) (Barry et al., 2003), and the frontal lobe-specific paroxysms in frontal lobe epilepsy (FLE), which lead to ADHD-like abnormalities in childhood (Hernandez et al., 2001; Gonzalez-Heydrich et al., 2007) and schizophrenia-like symptoms in adults (Adachi et al., 2000; Helmstaedter, 2001; Patrikelis et al., 2009).
Considering the critical role of thalamic neurons in controlling cortical excitability, synchrony, and resonance in various physiological and pathophysiological states of the brain (McCormick and Bal, 1994; Steriade and Contreras, 1995; Steriade, 2006; Ernst et al., 2009), the abnormal frontal lobe rhythms seen in patients with frontal lobe dysfunction could be attributed to the thalamus. This hypothesis has not been explored, probably due to an assumption that frontal lobe excitability can be explained by impairments or damage to the neurons in the frontal cortex (Luria et al., 1964; McAllister and Price, 1987; Stuss and Gow, 1992; Paradiso et al., 1999; Kellinghaus and Lüders, 2004; Max et al., 2005; Millichap, 2008).
Hypoxic damage in the frontal cortex, which is induced by various conditions including traumatic damages, tumors, and fetal hypoxia (Ishige et al., 1987; Choi, 1990), has been one of the most frequent causes underlying the development of frontal lobe dysfunctions (Damasio and Anderson, 1985; Owen et al., 1990, 1993; Shallice and Burgess, 1991; Stuss and Gow, 1992; Pigula et al., 1993; Höckel and Vaupel, 2001). Based on the fact that cobalt (Co) ions trigger signaling pathways which are activated by hypoxia (Piret et al., 2002), we establish a murine model of frontal lobe dysfunction by implanting a wire made of Co in the prefrontal cortex (PFC) and demonstrate how the role of the thalamus is involved in the development of frontal lobe dysfunction in the model.
Materials and Methods
Animals
Animal care and handling were performed according to the directives of the Animal Care and Use Committee of Korea Advanced Institute of Science and Technology. The CaV3.1−/− and wild-type littermate control mice were generated by mating heterozygotes from C57BL/6J backgrounds. Mice were maintained with free access to food and water under a 12 h light/dark cycle.
Mouse model of FLE generated using a wire form of cobalt
C57BL/6J male and female mice (10–20 weeks old) were prepared for Co-wire implantation. Under avertin (0.2%) anesthesia (tribromoethanol, 20 mg/ml, i.p.), we implanted a piece of Co-wire (500 μm diameter, Alfa Aesar) with the radial orientation (vertical to the cortical surface) into the right dorsolateral PFC (anteroposterior, 2.6 mm; lateral, 1.8 mm; ventral, 1.3 mm) using a stereotaxic device (David Kopf Instruments). An epidural electrode for EEG recordings was implanted in the right frontal cortex, where the cobalt was implanted, and another electrode was implanted in the left frontal and temporal cortexes (right frontal: anteroposterior, 2.8 mm; lateral, 0.8 mm; left frontal: anteroposterior, 2.8 mm; lateral, 0.8 mm; right temporal: anteroposterior, −2.4 mm; lateral, −2.4 mm; left temporal: anteroposterior; −2.4 mm, lateral, 2.4 mm). A grounding electrode was implanted in the occipital region of the skull. After 3 d of recovery, the EEG signal and video recording were acquired for 30 d, during which mice were free to explore an EEG chamber (a square-floor rectangular box). EEG signals were amplified (model 7H polygraph, Grass Technologies) and digitized at a sampling rate of 500 Hz (DIGIDATA 1320A, Molecular Devices). pClamp9.2 software (Molecular Devices) was used for data acquisition. To induce secondary generalization, low-dose homocysteine thiolactone (HT) (Fluka) was injected intraperitoneally (550 mg/kg), 7 d after Co-wire implantation (7.0 ± 0.8 d on average) (Walton and Treiman, 1988).
Magnetic resonance imaging
For magnetic resonance imaging (MRI), mice with Co-wire implantation (5 d after Co-wire implantation) were anesthetized and set into an MR compatible cradle. During MRI, the animals were anesthetized by breathing 2% isoflurane into oxygen-enriched air with a facemask. The rectal temperature was carefully monitored and maintained at 36 ± 1°C. The MRI acquisitions were performed on a 4.7 T horizontal MRI (Biospec, Bruker) with a shielded gradient coil 65 mm in diameter. The experiments were performed using a 38 mm internal diameter birdcage coil (Bruker). The measurements were performed using the parameters given below. T1-weighted images were obtained using a fast spin-echo sequence with the following parameters: pulse repetition time (TR) = 1500 ms; echo time (TE) = 10 ms; matrix size = 256 × 256; field of view (FOV) = 25 mm; slice thickness (ST) = 1 mm; pixel resolution = 100 ×100 μm; and acquisition time for one set = 6.7 min.
T2-weighted images were obtained using a fast spin-echo sequence with the following parameters: TR = 4200 ms; TE = 36 ms; matrix size = 256 × 256; FOV = 25 mm; ST = 1 mm; pixel resolution = 100 ×100 μm; and acquisition time for one set = 11.5 min.
Histology and immunoblotting
For the histological analysis of cell death, brain was placed in phosphate-buffered 4% formalin for 24 h and embedded in paraffin for sectioning on a microtome into 5 μm slices, which were stained with hematoxylin and eosin after deparaffination. The brain slices were visualized under a bright-field microscope (Olympus). For immunoblotting, brains were isolated on ice and cut into 1 mm slices using a brain matrix (RBMA-200C, WPI). The presumed damaged region in right prefrontal cortex, with regard to the brain atlas, were separately removed and sonicated in homogenization buffer containing protease inhibitor (Complete-Mini, Roche) and protein phosphatase inhibitor (Phostop, Roche). Proteins (30–50 μg) were loaded onto 10% polyacrylamide Tris-HCl gel and transferred to a nitrocellulose membrane (Protran, Whatman). The following antibodies used: β-actin (sc-47778, Santa Cruz Biotechnology), VEGFa (sc-507, Santa Cruz Biotechnology). Anti-mouse (Abcam) and anti-rabbit secondary antibodies (Cell Signaling Technology) were used.
Behavioral analysis
Open-field test.
Each mouse was gently placed at the center of the open-field test arena (a square-floor rectangular box made of white acrylic, 40 × 40 × 50 cm) in a dark room. The distance of spontaneous movement during a 1.5 h time period was monitored at 5 min intervals by digital video recording. The test was performed between 6:00 P.M. and 10:00 P.M. EthoVision (Noldus) was used to analyze the video data.
Fear-conditioning test.
Fear conditioning was conducted in conditioning chambers (acrylic test station, dim light, metal grid floor; San Diego Instruments) and a testing chamber (a white plastic, circular cylinder). A video camera was mounted on top of the chambers. The fear-conditioning procedure was conducted over 2 d. On day 1, each mouse was acclimated to the conditioning chamber (4 min, 40 s) and then given three pairs of a conditional stimulus (tone, 20 s, 5 kHz, 75 dB) that coterminated with an unconditional stimulus (foot shock, 2 s, 0.7 mA). The trial interval was 60 s. On the day of testing, the freezing responses to the conditional stimulus were measured in the testing chamber with test tones (180 s). To test contextual conditioning, the mice were placed in the conditioning chamber and were allowed to explore for 5 min. Behavioral freezing was manually counted by an observer blinded to the previous treatments.
Electrolytic lesions
C57BL/6J male and female mice (10–20 weeks old) were anesthetized with avertin (tribromoethanol, 20 mg/ml) and placed in a stereotaxic device (David Kopf Instruments). A small hole was made in the skull with a dental drill. Electrolytic lesions were made in the mediodorsal thalamus (MD) (anteroposterior, −1.46; lateral, −0.25; ventral, 3.3 mm) with monopolar electrodes (stainless steel insulated with Epoxylite, Am Systems). Electrolytic lesions were made by passing a DC current (10 mA; A365, World Precision Instruments) for 15 s. A piece of Co-wire (Alfa Aesar) was then implanted in the right PFC. The epidural electrodes were implanted as described for the cobalt model of PFC damage. After 4 d of recovery, the EEG signal was acquired for 30 d. After EEG recordings, animals were humanely killed by an overdose of avertin and fixed with 10% formalin. Brain slices (40 μm) were cut on a cryostat and stained with cresyl violet.
Pharmacological analyses
Bicuculline methobromide test.
C57BL/6J male and female mice (10–20 weeks old) were prepared for injection of bicuculline methobromide (BMB). Under avertin (0.2%) anesthesia (tribromoethanol, 20 mg/ml, i.p.), electrolytic lesions were made in the contralateral and ipsilateral sides of the MD thalamus. After 4 d of recovery, the EEG signal was acquired for 2 h, during which BMB (30 mg/kg) was injected intraperitoneally.
Antiepileptic drug test.
C57BL/6J male and female mice (10–20 weeks old) were prepared for injection of ethosuximide (Coulter et al., 1989). Basal EEG activity was recorded for 8 ± 1.1 d after Co-wire implantation. After intraperitoneal injection of ethosuximide (150 mg/kg), the EEG signal was acquired for 2 h. Zonisamide (60 mg/kg)and phenytoin (100 mg/kg) were injected to test the effect on frontal seizure spikes.
Multiunit recording
C57BL/6J male and female mice (10–20 weeks old) were anesthetized with avertin (tribromoethanol, 20 mg/ml) and placed in a stereotaxic device (David Kopf instruments). A piece of Co-wire (Alfa Aesar) was then implanted in the right PFC (anteroposterior, 2.6 mm; lateral, 1.8 mm; ventral, 1.3 mm). After 4.2 ± 0.2 d, and 9.2 ± 0.2 d after cobalt implantation, mice were anesthetized by intraperitoneal injection of urethane (under light anesthesia; 1.35 g/kg) and placed in a stereotaxic device (Thomas RECORDING). The temperature was maintained at 37°C using a homothermic blanket system (Harvard Apparatus). A single incision was made in the scalp, the skull was exposed, and a hole made in the skull above the region where the MD nuclei are located. Quartz-coated tetrodes (0.5–2 MΩ; Thomas RECORDING) were positioned in the MD regions (anteroposterior, −1.46 mm; lateral, −0.25 mm; ventral, 3.1–3.3 mm). Signals were amplified ∼95-fold and bandpass filtered at 300–5000 Hz (for the measurement of multiunit activity) or at 0.50–50 Hz [for the measurement of local field potential (LFP)] by an AC amplifier (PGMA, Thomas RECORDING) digitized at the 10 kHz sampling rate (DT3010, Neuralynx). A burst was defined by the following criteria in NeuroExplorer (version 4): maximum interval to start burst (i.e., the first interspike interval within a burst) must be ≤6 ms; maximum interval to end burst, 10 ms; minimum interval between bursts, 200 ms; minimum duration of burst, 4 ms; and minimum number of spikes within a burst, two. Low threshold spike bursts were defined by a preceding silent period >100 ms, high-frequency spikes of 200–400 Hz with more than two spikes, a shortening of the first interspike interval (1–4 ms), and a progressive prolongation of successive interspike intervals, as described previously (Guido et al., 1992; Kim et al., 2003).
In vivo transduction of lentivirus
A lentiviral vector expressing short hairpin RNA (shRNA) to target the CaV3.1 T-type calcium channel was constructed. A synthetic double-oligonucleotide (5′–CGGAATTCCGGGAAGATCGTAGATA GCAAAttcaagagaTTTGCTATCTACGATCTTCTTTTTGATATCTAGACA–3′) was inserted into the shLentisyn3.4G lentiviral vector. The shLentisyn3.4G lentiviral vector was designed to express shRNAs from the U6 promoter and to express enhanced green fluorescent protein from the synapsin promoter. The target sequences did not overlap with any mRNAs in the database of the National Center for Biotechnology Information, other than CaV3.1. A scrambled version of the CaV3.1 shRNA oligonucleotide (5′–CGGAATTCCGGGTAAGTGAA CTGACAAGAAttcaagaga TTCTTGTCAGTTCACTTACT TTTTGATATCTAGACA–3′) was also inserted into the shLentisyn3.4G vector and used as a control. This sequence did not have homology to any known mammalian genes. The recombinant lentiviral vectors were produced and concentrated commercially (Macrogen LentiVector Institute). We used lentivirus titers of ∼2 × 106 transduction units/ml. The solution containing the viruses carrying the CaV3.1 shRNA or the scrambled control were injected into the ipsilateral MD with Nanofil 33G blunt needles and a Nanofil syringe (World Precision Instruments) using a micro syringe pump (Eicom). Seven days after viral transduction, a Co-wire with epidural electrodes for EEG recordings were implanted as described above. EEG signals were acquired for 30 d with video monitoring.
Immunohistochemistry
After EEG recordings, mice were anesthetized with avertin (tribromoethanol, 20 mg/ml) and perfused transcardially with heparin solution (10 units/ml) followed by 4% formaldehyde dissolved in PBS. For CaV3.1 immunohistochemistry, cryostat sections were prepared. Brain slices (20 μm) were incubated with CaV3.1 primary antibody (Alomone Labs), followed by avidin-biotin-peroxidase complex. The brain slices were visualized under a ScanScope CS/GL scanner (ss5060, Aperio Technologies).
Patch-clamping
Seven to 10 d after infection with lentiviruses harboring shC or sh3.1, mice (postnatal day 19–21) were intracardially perfused with an ice-cold solution containing 125 mm choline-Cl, 3.0 mm KCl, 1.25 mm NaH2PO4, 25 mm NaHCO3, 1.0 mm Ca Cl2, 7.0 mm MgCl2, 10 mm dextrose, 1.3 mm ascorbate acid, and 3.0 mm pyruvate (bubbled with 95% O2/5% CO2). The brain was then rapidly removed, and slices (350 μm thick) were made with a vibratome (Leica Microsystems). After incubation for at least 45 min in 32°C, slices were kept at room temperature in a holding chamber until they were transferred to a submersion-type recording chamber held at room temperature. The solution used for slice incubation contained 130 mm NaCl, 3.0 mm KCl, 2.0 mm MgCl2, 2.0 mm CaCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 10 mm glucose. After rupturing the MD neurons, cells were maintained at −60 mV and the recording solution was substituted with Ca2+ current recording solution composed of 115 mm NaCl, 3.0 mm KCl, 10 mm sucrose, 10 mm glucose, 26 mm NaHCO3, 2 mm MgCl2, 2.5 mm CaCl2, 0.5 mm 4-aminopyridine, 5 mm CsCl, 10 mm tetraethylammonium-Cl (TEA-Cl), and 0.001 mm TTX, pH 7.4 (gassed with 95% O2/5% CO2). The intracellular pipette solution contained 78 mm Cs-gluconate, 20 mm HEPES, 10 mm BAPTA-Cs4 (cell-impermeant), 0.5 mm CaCl2, 1.0 mm MgCl2, 4 mm Mg-ATP, 0.3 mm GTP-Tris, 6 mm phosphocreatine (Di-Tris salt), 4.0 mm NaCl, and 20 mm TEA-Cl, pH 7.3 (titrated with CsOH). Standard voltage protocols for inducing T-currents were applied (Perez-Reyes, 2003). Only cells with low membrane capacitance (40–100 pF) and high membrane resistance (<400 MΩ) were included in the analysis.
Data analysis
Coherence analysis.
The signals >600 s were analyzed to measure the coherence coefficients between PFC and MD thalamus. Bicoherence was analyzed using the higher-order spectral analysis toolbox and Matlab R2008a (Chua et al., 2007).
Granger causality analysis.
Granger causality (GC) analysis was used to analyze the strength and direction of functional connectivity between the right frontal cortex (FR) and MD thalamus. The GC analysis uses the prediction error in autoregressive (AR) modeling of the signal. It can be determined whether signal y influences signal x by comparing the prediction error of x obtained from AR modeling with the prediction error of x obtained from joint AR modeling of x and y. That is, when y influences x, the prediction error is significantly decreased for the joint AR modeling. This is quantified by the following relative prediction improvement, called the Granger–Sargent statistic:
 |
Here, εx2 is the mean-squared prediction error of the univariate AR model of x, and εxy2 is the multivariate mean error of predicting x obtained from the joint AR model of x and y. When the εxy22 is much less than εx2, the value of Syx2 becomes large, and it can be assumed that signal y influences signal x. The AR model order was determined from Bayesian information criteria considering the tradeoff between the number of model parameters and the accuracy of prediction (Pereda et al., 2005). For the GC analysis, it is essential to determine the epoch in which the signal can be assumed to be stationary (Granger, 1969). We could verify this and obtained reliable AR models using 5 s segmentation of our data. Therefore, the signals within 600 s were segmented into 5 s epochs (Sitnikova et al., 2008).
Event detection.
For detection of spikes, Clampfit 9.2 (Molecular Devices) was used. Template search analysis was used to obtain the number and property of each spike.
Unit (pixel) analysis.
For measuring the level of knockdown after transfection of shRNA, a Positive Pixel Count Algorithm (Aperio) was used. All data were analyzed using the t test, ANOVA, and the SigmaStat 3.1 software package (SPSS), followed by analysis using the post hoc Holm–Sidak test.
Results
Hypoxia-like damages in the PFC lesions by Co-wire implantation
To induce frontal lobe-specific damage, we implanted a Co-wire into the right PFC of adult mice. After 5 d of implantation, a bloody scar reflecting angiogenesis was formed where Co-wire had been implanted, but this was not found in mice with a tungsten (W)-wire implantation (Fig. 1a). T2-weighted magnetic resonance imaging (MRI) showed that Co-wire implantation led to more profound damage in the PFC than W-wire (Fig.1a) (tungsten, n = 4; cobalt, n = 4). Such damage was limited to the right frontal cortex, while no damage signals were found in the left frontal cortex and temporal cortex (Fig. 1a; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The damaged area detected in MR images was further characterized by hematoxylin/eosin staining, and we found that Co-wire implantation resulted in clear signs of hypoxic damage as measured by the increase of ghost cells (Ito et al., 2006) and inflammatory angiogenesis (Sharp and Bernaudin, 2004) visualized by blood vessels filled with red blood cells (RBCs) (Fig. 1b,c; supplemental Fig. S2a, available at www.jneurosci.org as supplemental material) (nonlesion control, n = 3; tungsten, n = 3; cobalt, n = 4; p < 0.05).
Figure 1.

PFC lesions caused by Co-wire lead to increased RBCs, ghost cells, and VEGF expression 5 d after Co-wire implantation. a, The brains of the cobalt-implanted group showed bloody scarring, and MRI displayed that Co-wire implantation led to more profound damage in the PFC than tungsten wire. b, Representative images with hematoxylin/eosin staining of the cobalt- and tungsten-implanted groups. The cobalt group showed increased neuronal death (images 1 and 2 in the Co-wire group) with increased blood cells (an arrow shows RBCs in image 1 in the Co-wire group), and ghost cells (four arrows show the pink ghost cells in image 2 in the Co-wire group), while the tungsten-implanted group showed damaged neuronal cells in limited regions (image 1 in tungsten-implanted group) with morphologically normal neurons in the surrounding area (image 2 in tungsten-implanted group). Left, Ipsilateral PFC (100×); middle, magnified image 1 of ipsilateral PFC (1000×); right, magnified image 2 of ipsilateral PFC (1000×). c, RBCs and ghost cells were significantly increased in cobalt-implanted group. Morphologically normal neurons were largely decreased in Co-wire-implanted group, compared with nonlesion control and tungsten-implanted groups. Arbitrary units (AU) were calculated by dividing the number of neurons in right PFC by the number of neurons in left PFC (FR/FL) d, The level of VEGF (65 kDa) was significantly increased in the cobalt-implanted group. Ghosts, Ghost cells. FR, right prefrontal cortex; FL, left prefrontal cortex. *p < 0.05. Data represent means ± SEM.
To measure the Co-wire-induced damage at molecular level, we examined vascular endothelial growth factor (VEGF), a key angiogenic modulator in response to hypoxia (Sharp and Bernaudin, 2004) and found that VEGF proteins were significantly increased in the PFC 5 d after Co-wire implantation when compared with tungsten-wire-implanted groups (Fig. 1d; supplemental Fig. S2b, available at www.jneurosci.org as supplemental material) (nonlesion control, n = 2; tungsten, n = 4, cobalt, n = 3). These results suggest that Co-wire led to a hypoxia-like damage in the PFC.
Co-wire-induced damage in the PFC leads to abnormal hyperactivities with stereotypic circling
Next, we wondered whether the Co-wire-induced PFC damage caused behavioral abnormalities known to be reported in patients with PFC dysfunctions (Benson, 1991; Helmstaedter, 2001; Patrikelis et al., 2009). An open-field test revealed a progressive development of hyperactivity (Fig. 2a,c) (control, n = 6; cobalt ≤6 d, n = 5; cobalt ≤14 d, n = 5; cobalt ≥28 d, n = 9; two-way ANOVA, F(3,192) = 30.018, p < 0.001; two-tailed t test, p < 0.05) when compared with control mice. In addition, they showed a decreased exploration in center areas of an open-field box (Fig. 2b,d) (two-tailed t test, p < 0.05) due to their stereotypic circling behaviors along the wall of test box (Fig. 2b,e) (two-tailed t test, p < 0.05). The abnormal hyperactivity and stereotypic behaviors have been associated with attention problems in patients with PFC dysfunctions (McGuire and Sylvester, 1987; Gainetdinov et al., 2001; Kates et al., 2005; Gilby, 2008). Consistently, they also showed reduced learning in a fear-conditioning experiment (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) (two-tailed t test, context, p < 0.01; cue, p < 0.05), which requires careful attention to cue or contextual stimuli (Armony and Dolan, 2002; Han et al., 2003). These results suggest that the implantation of a Co-wire in the PFC leads to behavioral abnormalities similar to those found in ADHD (Davids et al., 2003) and FLE childhood (Hernandez et al., 2001; Gonzalez-Heydrich et al., 2007).
Figure 2.

Locomotor hyperactivity was induced after Co-wire was implanted. a, Cobalt-implanted mice exhibited higher locomotor activity in an open field compared with nonepileptic controls. b, The example of locomotor patterns of the cobalt group and control group during the first 5 min (5 min) and last 5 min (60 min) of a 60 min recording. c, Cobalt-implanted mice showed increased total distance moved during 60 min. d, Center exploration was decreased in cobalt-implanted mice during first 5 min of the open-field test. e, The cobalt group of mice showed stereotypic circling, repetitive movement in the same direction, while the control mice were exploratory. *p < 0.05, ***p < 0.001. Data represent means ± SEM.
Co-wire-induced damage in the PFC leads to frontal lobe epilepsy
Since frontal lobe dysfunctions are associated with abnormal frontal lobe rhythms (Elbert et al., 1992; Barry et al., 2003) we tried to examine cortical electroencephalogram (EEG) and LFPs of MD thalamus (supplemental Fig. S4a, available at www.jneurosci.org as supplemental material). Approximately 5 to 6 d after Co-wire implantation in the right PFC (5.8 ± 0.7 d on average), the right frontal lobe showed single spike activities (Fig. 3a). However, other wires, made with metals such as tungsten, copper, and aluminum, did not induce seizure spikes when implanted in the PFC during the whole recording period (supplemental Figs. S4c, Fig. S5a, available at www.jneurosci.org as supplemental material) (cobalt, n = 10; tungsten, n = 4; copper, n = 4; aluminum, n = 3). Nine to 10 d after (9.0 ± 1.2 d on average), the frontal seizure spikes were propagated to the left frontal lobe and to the MD thalamus (Fig. 3a). The frontal lobe-specific spikes were continuously examined during the whole recording period (Fig. 4a), although the peak frequency of single-spike activities became faster as time passed by (supplemental Fig. S5b, available at www.jneurosci.org as supplemental material). Between 11 and 30 d postimplantation, mice intermittently showed a secondary generalization of single spikes (onset, 11.2 ± 1.4 d on average) or ictal discharges with whole-body convulsions (onset, 18.1 ± 2.5 d on average) (Fig. 3a).
Figure 3.
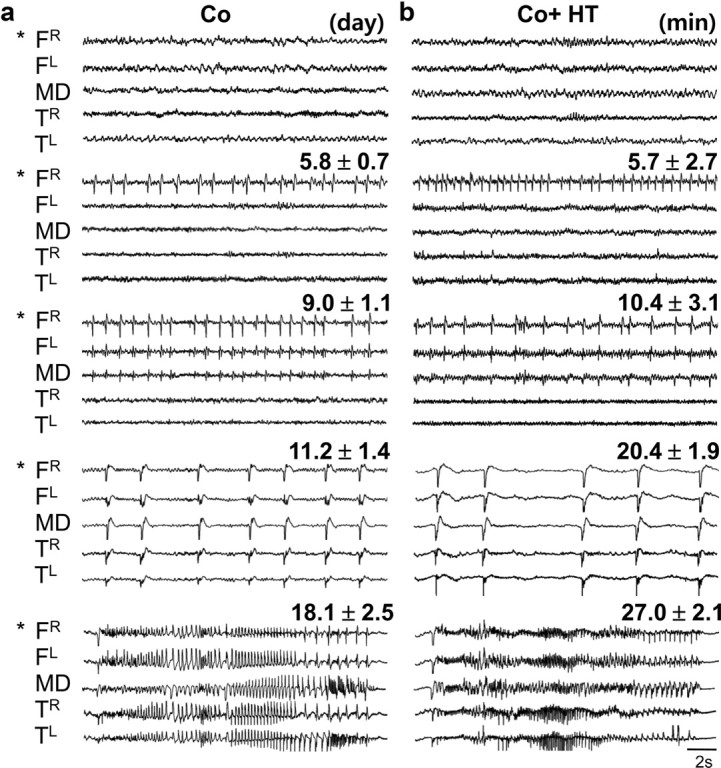
Frontal lobe-specific spikes were induced in a Co-wire model. a, Generation of frontal lobe-specific seizure spikes (n = 10). “Day” indicates the day of onset of seizure development. b, Seizure development was precisely reproduced within an hour by administration of HT at subconvulsive doses (550 mg/kg) (n = 12). “Minute” indicates onset of seizure development. FR, Right prefrontal cortex; FL, left prefrontal cortex; TL, left temporal cortex; TR, right temporal cortex. *Site of Co-wire implantation.
Figure 4.

The quantification of seizure development over time in each brain regions and the effect of FLE drugs on the frontal lobe seizure spikes. a, The number of spike events during seizure development in the cobalt-implanted group. b, The number of spike events after the systemic injection of HT. c, Representative EEG traces from 10 min before and 20 min after antiepileptic drugs were systemically injected. d, The Antiepileptic drugs alleviated frontal seizure spikes compared with the vehicle-injected group. Veh, Vehicle (0.85% normal saline); ZNS, zonisamide 60 mg/kg; PHT, phenytoin 100 mg/kg; Etho, ethosuximide, 150 mg/kg.
By administration of HT (Walton and Treiman, 1988) at a subconvulsive dose (550 mg/kg), the corticothalamic and corticocortical propagation of seizure spikes from the PFC with intermittent secondary generalizations was facilitated within an hour with a similar pattern (Figs. 3b, 4b) (n = 12). At that dose of HT, mice without Co-wire implantation did not show any seizure spikes (supplemental Fig. S6, available at www.jneurosci.org as supplemental material) (n = 5). Thus, the development of frontal lobe seizures in the Co-wire model seems to be activity- and neural pathway-dependent but not due to the time-dependent diffusion of Co ions from the PFC to the MD or other brain regions.
Since frontal lobe-specific seizure with secondary generalization is a well known symptom found in patients with FLE (Kellinghaus and Lüders, 2004), we tested whether the Co-wire-induced seizures could be inhibited by anti-FLE drugs including zonisamide and phenytoin. Systemic administration of FLE drugs robustly abolished the Co-wire-induced seizures (Fig. 4c,d). In addition, we found that ethosuximide, a drug for absence seizures known to inhibit thalamic burst firings (Coulter et al., 1989), also suppressed the Co-wire-induced seizure spikes (8 ± 1.1 d after Co-wire implantation) (Fig. 4d) (vehicle group, n = 4; zonisamide group, n = 3; phenytoin group, n = 3; ethosuximide group, n = 5; p < 0.05) (Frampton and Scott, 2005). These results suggest that the PFC damage by Co-wire leads to FLE in mice. In addition, the development of FLE in this model is temporally associated with the progressive increase of abnormal hyperactivities in this model (Fig. 2). Consistently, the abnormal hyperactivity has been reported in patients with FLE (Helmstaedter, 2001).
Lesions in the MD inhibit the onset of frontal lobe-specific spikes
The propagation of seizure spikes to the MD and the suppression of seizures by ethosuximide suggested a possibility that the thalamus is involved in the development of FLE. To address this issue, we examined the Co-wire-induced FLE in mice with a right lesion in the MD (Fig. 5a) and found that they were resistant to the generation of frontal spikes (Fig. 5b,c) (6 d after Co-wire implantation; contralateral, n = 5; ipsilateral, n = 5; two-tailed t test, p < 0.01) and secondary generalizations when compared with the left MD lesion. The right MD lesion, however, showed no effects on cortical seizures induced by BMB (30 mg/kg), a GABAA antagonist, which induces cortical seizures through thalamus-independent mechanisms (Steriade and Contreras, 1998) (Fig. 5b,c) (two-tailed t test, p > 0.05). These results suggest that the development of Co-wire-induced FLE demands the role of thalamic neurons. The hemisphere-specific interactions between the MD and PFC neurons through reciprocal connections (Kuroda et al., 1993) seem to be critical.
Figure 5.

Lesions in the ipsilateral MD thalamus abolish frontal lobe-specific spikes in the cobalt-implanted group. a, Coronal representation depicts the area of the electrolytic lesion. *Site of Co-wire implantation. Contr, Contralateral (left MD); ipsi, ipsilateral (right MD). b, Representative EEG traces of contralateral (left)/ipsilateral (right) lesions in the MD after Co-wire implantation, and contralateral(left)/ipsilateral (right) lesions in the MD group without Co-wire implantation after BMB injection. c, *Co: comparison of the effects of contralateral and ipsilateral MD lesions on spike generation, 6 d after Co-wire implantation. The number of events (Spike events) was calculated as the number of seizure spikes per hour. +Bic, Comparison of the effects of contralateral and ipsilateral MD lesions on spike generation induced by systemic injection of BMB. **p < 0.01. Data represent means ± SEM.
Coherence between the PFC and MD was increased before the onset of frontal-lobe-specific spikes
To probe the PFC–MD interactions, we tried to measure the activity coherence between the two nuclei during the development of frontal spikes. The bicoherence between the two regions reached a peak around theta frequencies before the onset of seizure spikes (4.2 ± 0.3 d on average) (Fig. 6a,b,d) (control, n = 3; cobalt, n = 5; two-tailed t test, p < 0.05). As frontal spikes were initiated and their interevent interval (IEI) was increased, the theta coherence between the two regions was decreased (IEI >2, 5.8 ± 0.2 d on average; IEI ≤1.5, 7.6 ± 0.5 d on average) (Fig. 6a,b,d) (control, n = 3; cobalt, n = 5; two-tailed t test, p < 0.01). To test the strength of the thalamocortical feedback in response to the corticothalamic drive before and after the onset of frontal spikes, Granger causality analysis was performed between MD and PFC. Consistent with coherence analysis, the MD–PFC interaction was increased before the onset of frontal spikes (4.2 ± 0.3 d after Co-wire implantation) (Fig. 6c; supplemental Table S1, available at www.jneurosci.org as supplemental material). These results suggest that the resonant cross talk between the MD and the PFC at theta frequencies preceded the onset of frontal lobe spikes.
Figure 6.
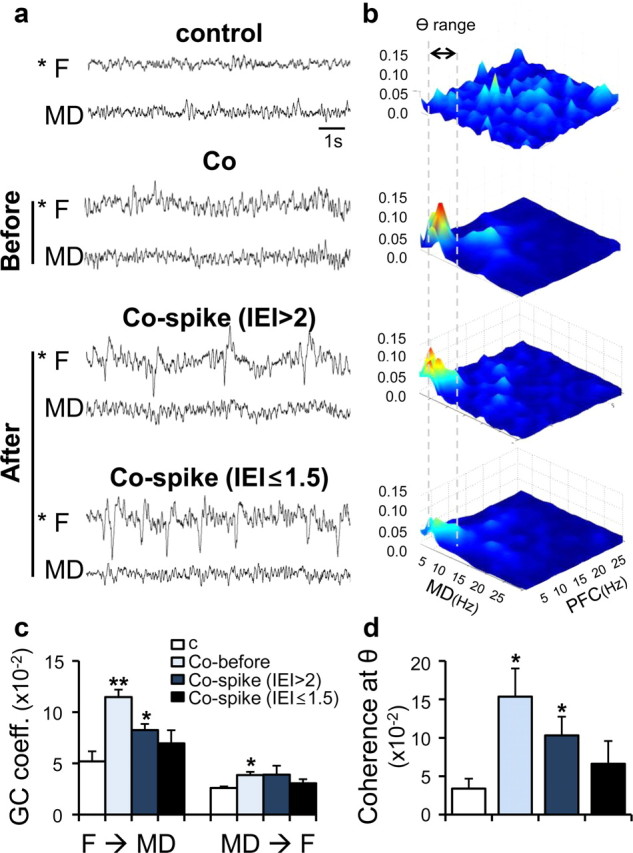
Enhanced thalamocortical resonance was observed before the generation of frontal lobe-specific spikes. a, Representative EEG traces after Co-wire implantation and nonepileptic control. Co-before, 4.2 ± 0.3 d after cobalt implanted; Co-spike (IEI >2), 5.8 ± 0.2 d after cobalt implanted; Co-spike (IEI ≤1.5), 7.6 ± 0.5 d after cobalt implanted. b, Averaged bicoherence analysis between the PFC and MD. Gray dotted lines show the range of theta frequency (4–7 Hz). c, Granger causality analysis between the PFC and MD during development of frontal lobe spikes. d, The comparision of the coherence value at theta range (4–7 Hz) during development of frontal lobe spikes. contr, Contralateral; ipsi, ipsilateral. *p < 0.05, **p < 0.01. Data represent means ± SEM.
Enhanced burst mode firing in the MD around the onset of FLE-like symptoms
Considering a previous study that thalamic burst firings are involved in the generation of thalamocortical interactions (Jeanmonod et al., 1996, 2001), we examined the firing mode of MD neurons in our model by in vivo extracellular recordings of the MD under partial urethane anesthesia (1.35 mg/kg). Before the onset of frontal lobe spikes (4.2 ± 0.2 d after Co-wire implantation), low-threshold burst spikes known to depend on the activation of T-type Ca2+ channels (Perez-Reyes et al., 1998; Llinás et al., 1999) significantly increased in the MD without changes in tonic spike activity (Fig. 7). In the period when the faster frontal spikes fully developed (9.0 ± 0.2 d after Co-wire implantation), burst spikes were decreased to the level of control group (Fig. 7b) [control, n = 14; before, n = 18; spike (IEI ≤1.5), n = 12; one-way ANOVA: burst, p < 0.05; tonic, p > 0.05]. These results suggest that burst firings of MD neurons support the PFC–MD cross talks at theta frequencies that are increased before the onset of frontal spikes (Fig. 6).
Figure 7.

Enhanced burst mode firing in the MD was observed around the onset of FLE-like symptoms. a, Representative local field potentials of the MD (5 s), raw spike traces, and raster plot of each mode after Co-wire implantation. b, Burst activity increased before the generation of frontal spikes. Before, 4.2 ± 0.2 d after cobalt implanted; Co-spike (IEI ≤1.5), 9.0 ± 0.2 d after cobalt implanted. *p < 0.05. Data represent means ± SEM.
FLE-like symptoms are abolished by either knockout or region-specific knockdown of the CaV3.1 gene in the MD
Considering that CaV3.1 is the major T-type Ca2+ channel subunit that supports burst firings in the thalamocortical relay neurons (Crunelli et al., 1989), we introduced lentiviruses harboring CaV3.1-specific shRNA into the MD; this infection significantly reduced CaV3.1 proteins and T-type Ca2+ currents in the MD neurons (Fig. 8a,b; supplemental Table S2, available at www.jneurosci.org as supplemental material) [lentivirus harboring shRNA-Control (shC), n = 5; lentivirus harboring shRNA Cav3.1 (sh3.1), n = 5; two-tailed ± test, p < 0.05]. Knockdown of CaV3.1 in the MD also significantly ameliorated neurological and behavioral abnormalities found in this model. The PFC power at theta range and MD–PFC cross talk was significantly decreased before spiking (4.6 ± 0.2 d) (Fig. 8c,d; supplemental Fig. S7, available at www.jneurosci.org as supplemental material) (shC, n = 5; sh3.1, n = 3; two-tailed t test, p < 0.01), compared with the control group. The number of frontal lobe-specific spikes on day 6 was decreased, compared with the control group (Fig. 8e,f) (shC, n = 5; sh3.1, n = 5; two-tailed t test, p < 0.05). MD-specific knockdown of the CaV3.1 gene also ameliorated the locomotor hyperactivity seen in the model (Fig. 8g,h) (shC, n = 6; sh3.1, n = 5; two-tailed t test, p < 0.01).
Figure 8.

Frontal lobe-specific oscillations and increased thalamocortical interaction at theta range with locomotor hyperactivity were abolished by a region-specific knockdown of the CaV3.1 gene in the MD. a, Lentivirus harboring CaV3.1-specific shRNA led to the reduction of T-type Ca2+ currents in MD neurons. Insets indicate infected neuron visualized with green fluorescence. b, Quantified T-current amplitude in both groups. *p < 0.05. c, Averaged bicoherence analysis between the PFC and MD in shC- and sh3.1-transfected group with Co-wire implantation (4.6 ± 0.2 d after cobalt implanted). Gray dotted lines show the range of theta frequency (4–7 Hz). d, The theta frequency of PFC was also decreased in the sh3.1 group, compared with shC group (4.6 ± 0.2 d after cobalt implanted). e, Representative EEG traces 6 d after Co-wire was implanted. f, The comparision of spike events between the shC/sh3.1 group,6 d after Co-wire implantation. g, The example of locomotor patterns of the sh3.1 group and control group during the first 5 min (5 min) and last 5 min (60 min) of a 60 min recording, 6 d after Co-wire implantation. h, The sh3.1 group showed decreased hyperactivity, compared with shC group (6 d after cobalt wire was implanted). *p < 0.05. Data represent means ± SEM.
In addition, the therapeutic effect of the MD-specific knockdown of the CaV3.1 gene on FLE-like symptoms was also observed after knockout of the gene (CaV3.1−/−), which is known to abolish burst spikes in the thalamus (Kim et al., 2001). CaV3.1−/− mice showed reduced MD–PFC interactions at theta frequencies (supplemental Fig. S8a, available at www.jneurosci.org as supplemental material) (CaV3.1+/+, n = 6; CaV3.1−/−, n = 5; two-tailed t test, p < 0.01), a lack of frontal lobe-specific spikes after the onset of spiking, and an absence of locomotor hyperactivity (6 d after Co-wire implantation) (supplemental Fig. S8b,c, available at www.jneurosci.org as supplemental material) (CaV3.1+/+, n = 5; CaV3.1−/−, n = 4; two-way ANOVA, p < 0.001).
Discussion
Since the first report on Phineas Gage, who suffered a severe frontal lobe injury in 1848 (Damasio et al., 1994; Macmillan, 2002), frontal lobe dysfunction characterized by a spectrum of cognitive impairments such as hyperactivity, inattention, impulsiveness, and, in severe cases, personality changes (Mateer and Williams, 1991; Owen et al., 1993; Rolls et al., 1994; Daffner et al., 2000) has been reported in patients with autism, schizophrenia, ADHD, and FLE (Levin, 1984; Benson, 1991; Gedye, 1991; Shue and Douglas, 1992; Niedermeyer, 1998; Kellinghaus and Lüders, 2004; Yoon et al., 2008). Here, we demonstrate that the implantation of Co-wire in the PFC in mice leads to neurological and behavioral abnormalities that have been reported in patients with FLE (Helmstaedter, 2001; Kellinghaus and Lüders, 2004).
FLE does not usually cause severe convulsions, despite frontal-lobe-specific spikes. Instead, patients with FLE show cognitive dysfunctions that have been reported in other neuropsychiatric disorders (Stores et al., 1991; Scheffer et al., 1995; Jobst and Williamson, 2005; Hermann et al., 2007). It is unclear how a local seizure is initiated in the frontal cortex and then leads to complex behavioral abnormalities. One of the obstacles to addressing this issue has been the lack of robust experimental models that allow researchers to induce frontal-lobe-specific spikes. In this study, we established a Co-wire model of PFC damage, which leads to neurological and behavioral symptoms that have been reported in patients with FLE (Figs. 2, 3), and found that the activation of thalamic T-type Ca2+ channels within the MD is critical for the onset of FLE (Figs. 7, 8).
PFC damage by Co-wire implantation mimics hypoxic conditions including increased neuronal death, ghost cells (Fig. 1b; supplemental Fig. S2a, available at www.jneurosci.org as supplemental material), and enhanced VEGF expression (Fig. 1d). In addition to VEGF, hypoxia induces many other molecules including Hif-1, Glut1, and lactate dehydrogenase A (Sharp and Bernaudin, 2004). It has been well known that hypoxic stress on brain slices increases a transient neuronal excitability as measured by EPSPs (Luhmann and Heinemann, 1992; Jensen et al., 1998). Those molecular and physiological changes in hypoxia-like states may lead to a local excitability of PFC neurons, which then stimulates MD neurons via corticothalamic pathways (Kuroda et al., 1993), although it remains to be studied how the Co-wire-induced damage triggers the local excitability of PFC neurons.
In this model, our results suggest that the activation of thalamic T-type Ca2+ channels within the MD is critical for the onset of FLE (Figs. 5–8). In contrast to our results, other studies show that the involvement of thalamic T-type Ca2+ channels in cortically induced seizures is not required, since the induction of cortical seizures by local injection of BMB is not associated with thalamic functions (Neckelmann et al., 1998; Steriade and Contreras, 1998; Destexhe et al., 1999). This difference can be explained, in part, by the capacity of BMB to directly increase the synchronous seizure spikes in cortical neurons (Gutnick et al., 1982). PFC damage caused by Co-wire, however, may not be enough to induce cortical synchrony, but may also require thalamic bursting activity modulated by T-type Ca2+ channels.
How can PFC damage cause activation of thalamic T-type Ca2+ channels? From an anatomical perspective, PFC neurons can stimulate two types of thalamic neurons [thalamocortical relay and GABAergic nucleus reticularis thalami (nRT) neurons] through reciprocal connections (Groenewegen, 1988; Huguenard and Prince, 1994; Zikopoulos and Barbas, 2006). The GABA neurotransmitter released into the MD by nRT neurons may be involved in de-inactivation mechanisms that allow T-type Ca2+ channels to be easily activated in response to excitatory inputs (Crunelli and Leresche, 1991; Cox et al., 1997). Consistent with this idea, both the corticothalamic and thalamocortical drives increased together (Fig. 6c) and at the same time as the low-threshold burst spikes of MD neurons were enhanced (Fig. 7), which reflects the activation of T-type Ca2+ channels (Huguenard, 1996; Kim et al., 2001; Crunelli and Leresche, 2002; Lee et al., 2004; Ernst et al., 2009). Since the thalamic burst spikes are known to be efficient in stimulating postsynaptic neurons (Lisman, 1997), the activation of T-type Ca2+ channels in the MD may specifically stimulate PFC neurons, thereby enhancing MD–PFC cross talk and leading to frontal lobe-specific seizures at the EEG level (Fig. 6).
In addition, thalamic burst spiking is thought to be a sensory gating mechanism, which can terminate the relay of sensory information to the cortex (McCormick and Bal, 1994). In this regard, it is plausible that the selective increases in burst spikes in the MD neurons cause disturbances in the processing of information in the MD–PFC pathways, leading to frontal lobe dysfunction. Therefore, the inhibition of thalamic T-type Ca2+ channels (Fig. 8) or drugs that weaken the thalamocortical interactions may be a novel option for preventing further cognitive dysfunction in patients with irreversible cortical damage. Consistent with this, ethosuximide, a T-type Ca2+ channel blocker used to treat absence seizures, efficiently abolished the onset of FLE in our model (Fig. 4d) (ethosuximide, n = 5; p < 0.01).
Finally, our results suggest a two-step model of PFC dysfunction in which the response to a lesion in the PFC results in abnormal thalamocortical feedback driven by thalamic T-type Ca2+ channels, which, in turn, leads to the onset of neurological and behavioral abnormalities. This model neatly explains why the progressive development of cortical dysfunction in patients with a lesion in the PFC takes time (Grattan and Eslinger, 1992; Anderson et al., 2000; McKinlay et al., 2002; Englander et al., 2003) and how the thalamus contributes to attention span when patients are awake, which is a controversial issue (Crick, 1984; Steriade et al., 1993; Sherman, 2001).
Footnotes
This work was supported by the National Honor Scientist Program of Korea (Grant 2010000002, to H.-S.S.), the Mid-Career Researcher Program of the National Research Foundation of Korea funded by the Korea government (Grant 20100000032, to D.K.), and by the 21st Century Frontier R&D Programs of the Korea Ministry of Education, Science and Technology (Grant 2010k000814, to D.K.). We thank H. S. Choi and S. B. Park for their useful comments and help with data analysis. We are grateful to S. Y. Bahk for her assistance with manuscript preparation.
References
- Adachi N, Onuma T, Nishiwaki S, Murauchi S, Akanuma N, Ishida S, Takei N. Inter-ictal and post-ictal psychoses in frontal lobe epilepsy: a retrospective comparison with psychoses in temporal lobe epilepsy. Seizure. 2000;9:328–335. doi: 10.1053/seiz.2000.0413. [DOI] [PubMed] [Google Scholar]
- Anderson SW, Damasio H, Tranel D, Damasio AR. Long-term sequelae of prefrontal cortex damage acquired in early childhood. Dev Neuropsychol. 2000;18:281–296. doi: 10.1207/S1532694202Anderson. [DOI] [PubMed] [Google Scholar]
- Armony JL, Dolan RJ. Modulation of spatial attention by fear-conditioned stimuli: an event-related fMRI study. Neuropsychologia. 2002;40:817–826. doi: 10.1016/s0028-3932(01)00178-6. [DOI] [PubMed] [Google Scholar]
- Barry RJ, Clarke AR, Johnstone SJ. A review of electrophysiology in attention-deficit/hyperactivity disorder: I. Qualitative and quantitative electroencephalography. Clin Neurophysiol. 2003;114:171–183. doi: 10.1016/s1388-2457(02)00362-0. [DOI] [PubMed] [Google Scholar]
- Beleza P, Bilgin O, Noachtar S. Interictal rhythmical midline theta differentiates frontal from temporal lobe epilepsies. Epilepsia. 2009;50:550–555. doi: 10.1111/j.1528-1167.2008.01780.x. [DOI] [PubMed] [Google Scholar]
- Benson DF. The role of frontal dysfunction in attention deficit hyperactivity disorder. J Child Neurol. 1991;6:S9–12. doi: 10.1177/0883073891006001s02. [DOI] [PubMed] [Google Scholar]
- Choi DW. Cerebral hypoxia: some new approaches and unanswered questions. J Neurosci. 1990;10:2493–2501. doi: 10.1523/JNEUROSCI.10-08-02493.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua CK, Chandran V, Acharya R, Lim CM. Higher order spectral (HOS) analysis of epileptic EEG signals. Conf Proc IEEE Eng Med Biol Soc. 2007;2007:6496–6499. doi: 10.1109/IEMBS.2007.4353847. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Huguenard JR, Prince DA. Characterization of ethosuximide reduction of low-threshold calcium current in thalamic neurons. Ann Neurol. 1989;25:582–593. doi: 10.1002/ana.410250610. [DOI] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA. Nucleus reticularis neurons mediate diverse inhibitory effects in thalamus. Proc Natl Acad Sci U S A. 1997;94:8854–8859. doi: 10.1073/pnas.94.16.8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Function of the thalamic reticular complex: the searchlight hypothesis. Proc Natl Acad Sci U S A. 1984;81:4586–4590. doi: 10.1073/pnas.81.14.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. A role for GABAB receptors in excitation and inhibition of thalamocortical cells. Trends Neurosci. 1991;14:16–21. doi: 10.1016/0166-2236(91)90178-w. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Leresche N. Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci. 2002;3:371–382. doi: 10.1038/nrn811. [DOI] [PubMed] [Google Scholar]
- Crunelli V, Lightowler S, Pollard CE. A T-type Ca2+ current underlies low-threshold Ca2+ potentials in cells of the cat and rat lateral geniculate nucleus. J Physiol. 1989;413:543–561. doi: 10.1113/jphysiol.1989.sp017668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffner KR, Mesulam MM, Holcomb PJ, Calvo V, Acar D, Chabrerie A, Kikinis R, Jolesz FA, Rentz DM, Scinto LF. Disruption of attention to novel events after frontal lobe injury in humans. J Neurol Neurosurg Psychiatry. 2000;68:18–24. doi: 10.1136/jnnp.68.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damasio A, Anderson S. The frontal lobes. Clin Neuropsychol. 1985:339–375. [Google Scholar]
- Damasio H, Grabowski T, Frank R, Galaburda AM, Damasio AR. The return of Phineas Gage: clues about the brain from the skull of a famous patient. Science. 1994;264:1102–1105. doi: 10.1126/science.8178168. [DOI] [PubMed] [Google Scholar]
- Davids E, Zhang K, Tarazi FI, Baldessarini RJ. Animal models of attention-deficit hyperactivity disorder. Brain Res Rev. 2003;42:1–21. doi: 10.1016/s0165-0173(02)00274-6. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M. Cortically-induced coherence of a thalamic-generated oscillation. Neuroscience. 1999;92:427–443. doi: 10.1016/s0306-4522(99)00024-x. [DOI] [PubMed] [Google Scholar]
- Elbert T, Lutzenberger W, Rockstroh B, Berg P, Cohen R. Physical aspects of the EEG in schizophrenics. Biol Psychiatry. 1992;32:595–606. doi: 10.1016/0006-3223(92)90072-8. [DOI] [PubMed] [Google Scholar]
- Englander J, Bushnik T, Duong TT, Cifu DX, Zafonte R, Wright J, Hughes R, Bergman W. Analyzing risk factors for late posttraumatic seizures: a prospective, multicenter investigation. Arch Phys Med Rehabil. 2003;84:365–373. doi: 10.1053/apmr.2003.50022. [DOI] [PubMed] [Google Scholar]
- Ernst WL, Zhang Y, Yoo JW, Ernst SJ, Noebels JL. Genetic enhancement of thalamocortical network activity by elevating α1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. J Neurosci. 2009;29:1615–1625. doi: 10.1523/JNEUROSCI.2081-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton JE, Scott LJ. Zonisamide: a review of its use in the management of partial seizures in epilepsy. CNS Drugs. 2005;19:347–367. doi: 10.2165/00023210-200519040-00010. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Mohn AR, Caron MG. Genetic animal models: focus on schizophrenia. Trends Neurosci. 2001;24:527–533. doi: 10.1016/s0166-2236(00)01886-5. [DOI] [PubMed] [Google Scholar]
- Gedye A. Frontal lobe seizures in autism. Med Hypotheses. 1991;34:174–182. doi: 10.1016/0306-9877(91)90189-6. [DOI] [PubMed] [Google Scholar]
- Gilby KL. A new rat model for vulnerability to epilepsy and autism spectrum disorders. Epilepsia. 2008;49(Suppl 8):108–110. doi: 10.1111/j.1528-1167.2008.01851.x. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Heydrich J, Dodds A, Whitney J, MacMillan C, Waber D, Faraone SV, Boyer K, Mrakotsky C, DeMaso D, Bourgeois B, Biederman J. Psychiatric disorders and behavioral characteristics of pediatric patients with both epilepsy and attention-deficit hyperactivity disorder. Epilepsy Behav. 2007;10:384–388. doi: 10.1016/j.yebeh.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger CWJ. Investigating causal relations by econometric models and cross-spectral methods. Econometrica. 1969;37:424–438. [Google Scholar]
- Grattan LM, Eslinger PJ. Long-term psychological consequences of childhood frontal lobe lesion in patient DT. Brain Cogn. 1992;20:185–195. doi: 10.1016/0278-2626(92)90068-w. [DOI] [PubMed] [Google Scholar]
- Groenewegen HJ. Organization of the afferent connections of the mediodorsal thalamic nucleus in the rat, related to the mediodorsal-prefrontal topography. Neuroscience. 1988;24:379–431. doi: 10.1016/0306-4522(88)90339-9. [DOI] [PubMed] [Google Scholar]
- Gucuyener K, Erdemoglu AK, Senol S, Serdaroglu A, Soysal S, Kockar AI. Use of methylphenidate for attention-deficit hyperactivity disorder in patients with epilepsy or electroencephalographic abnormalities. J Child Neurol. 2003;18:109–112. doi: 10.1177/08830738030180020601. [DOI] [PubMed] [Google Scholar]
- Guido W, Lu SM, Sherman SM. Relative contributions of burst and tonic responses to the receptive field properties of lateral geniculate neurons in the cat. J Neurophysiol. 1992;68:2199–2211. doi: 10.1152/jn.1992.68.6.2199. [DOI] [PubMed] [Google Scholar]
- Gutnick MJ, Connors BW, Prince DA. Mechanisms of neocortical epileptogenesis in vitro. J Neurophysiol. 1982;48:1321–1335. doi: 10.1152/jn.1982.48.6.1321. [DOI] [PubMed] [Google Scholar]
- Han CJ, O'Tuathaigh CM, van Trigt L, Quinn JJ, Fanselow MS, Mongeau R, Koch C, Anderson DJ. Trace but not delay fear conditioning requires attention and the anterior cingulate cortex. Proc Natl Acad Sci U S A. 2003;100:13087–13092. doi: 10.1073/pnas.2132313100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedter C. Behavioral aspects of frontal lobe epilepsy. Epilepsy Behav. 2001;2:384–395. doi: 10.1006/ebeh.2001.0259. [DOI] [PubMed] [Google Scholar]
- Hermann B, Jones J, Dabbs K, Allen CA, Sheth R, Fine J, McMillan A, Seidenberg M. The frequency, complications and aetiology of ADHD in new onset paediatric epilepsy. Brain. 2007;130:3135–3148. doi: 10.1093/brain/awm227. [DOI] [PubMed] [Google Scholar]
- Hernandez MT, Sauerwein HC, de Guise E, Lortie A, Jambaqué I, Dulac O, Lassonde M. Neuropsychology of frontal lobe epilepsy in children. Adv Behav Biol. 2001;50:103–111. [Google Scholar]
- Höckel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266–276. doi: 10.1093/jnci/93.4.266. [DOI] [PubMed] [Google Scholar]
- Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. Intrathalamic rhythmicity studied in vitro: nominal T-current modulation causes robust antioscillatory effects. J Neurosci. 1994;14:5485–5502. doi: 10.1523/JNEUROSCI.14-09-05485.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishige N, Pitts LH, Hashimoto T, Nishimura MC, Bartkowski HM. Effect of hypoxia on traumatic brain injury in rats. Part 1: changes in neurological function, electroencephalograms, and histopathology. Neurosurgery. 1987;20:848–853. doi: 10.1227/00006123-198706000-00005. [DOI] [PubMed] [Google Scholar]
- Ito U, Kuroiwa T, Nagasao J, Kawakami E, Oyanagi K. Temporal profiles of axon terminals, synapses and spines in the ischemic penumbra of the cerebral cortex: ultrastructure of neuronal remodeling. Stroke. 2006;37:2134–2139. doi: 10.1161/01.STR.0000231875.96714.b1. [DOI] [PubMed] [Google Scholar]
- Jeanmonod D, Magnin M, Morel A. Low-threshold calcium spike bursts in the human thalamus. Common physiopathology for sensory, motor and limbic positive symptoms. Brain. 1996;119:363–375. doi: 10.1093/brain/119.2.363. [DOI] [PubMed] [Google Scholar]
- Jeanmonod D, Magnin M, Morel A, Siegemund M, Cancro A, Lanz M, Llinás R, Ribary U, Kronberg E, Schulman J, Zonenshayn M. Thalamocortical dysrhythmia II. Clinical and surgical aspects. Thalamus Relat Syst. 2001;1:245–254. [Google Scholar]
- Jensen FE, Wang C, Stafstrom CE, Liu Z, Geary C, Stevens MC. Acute and chronic increases in excitability in rat hippocampal slices after perinatal hypoxia in vivo. J Neurophysiol. 1998;79:73–81. doi: 10.1152/jn.1998.79.1.73. [DOI] [PubMed] [Google Scholar]
- Jobst BC, Williamson PD. Frontal lobe seizures. Psychiatr Clin North Am. 2005;28:635–651. doi: 10.1016/j.psc.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Kates WR, Lanham DC, Singer HS. Frontal white matter reductions in healthy males with complex stereotypies. Pediatr Neurol. 2005;32:109–112. doi: 10.1016/j.pediatrneurol.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Kellinghaus C, Lüders HO. Frontal lobe epilepsy. Epileptic Disord. 2004;6:223–239. [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW, Shin HS. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking α1G T-type Ca2+ channels. Neuron. 2001;31:35–45. doi: 10.1016/s0896-6273(01)00343-9. [DOI] [PubMed] [Google Scholar]
- Kim D, Park D, Choi S, Lee S, Sun M, Kim C, Shin HS. Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science. 2003;302:117–119. doi: 10.1126/science.1088886. [DOI] [PubMed] [Google Scholar]
- Kuroda M, Murakami K, Oda S, Shinkai M, Kishi K. Direct synaptic connections between thalamocortical axon terminals from the mediodorsal thalamic nucleus (MD) and corticothalamic neurons the MD in the prefrontal cortex. Brain Res. 1993;612:339–344. doi: 10.1016/0006-8993(93)91683-j. [DOI] [PubMed] [Google Scholar]
- Lee J, Kim D, Shin HS. Lack of delta waves and sleep disturbances during nonrapid eye movement sleep in mice lacking α1G-subunit of T-type calcium channels. Proc Natl Acad Sci U S A. 2004;101:18195–18199. doi: 10.1073/pnas.0408089101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin S. Frontal lobe dysfunctions in schizophrenia–II. Impairments of psychological and brain functions. J Psychiatr Res. 1984;18:57–72. doi: 10.1016/0022-3956(84)90047-5. [DOI] [PubMed] [Google Scholar]
- Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- Llinás RR, Ribary U, Jeanmonod D, Kronberg E, Mitra PP. Thalamocortical dysrhythmia: a neurological and neuropsychiatric syndrome characterized by magnetoencephalography. Proc Natl Acad Sci U S A. 1999;96:15222–15227. doi: 10.1073/pnas.96.26.15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhmann HJ, Heinemann U. Hypoxia-induced functional alterations in adult rat neocortex. J Neurophysiol. 1992;67:798–811. doi: 10.1152/jn.1992.67.4.798. [DOI] [PubMed] [Google Scholar]
- Luria A, Pribram K, Homskaya E. An experimental analysis of the behavioral disturbance produced by a left frontal arachnoidal endothelioma (meningioma) Neuropsychologia. 1964;2:257–280. [Google Scholar]
- Macmillan M. MIT; 2002. An odd kind of fame: stories of Phineas Gage. [Google Scholar]
- Mateer C, Williams D. Effects of frontal lobe injury in childhood. Dev Neuropsychol. 1991;7:359–376. [Google Scholar]
- Max JE, Manes FF, Robertson BA, Mathews K, Fox PT, Lancaster J. Prefrontal and executive attention network lesions and the development of attention-deficit/hyperactivity symptomatology. J Am Acad Child Adolesc Psychiatry. 2005;44:443–450. doi: 10.1097/01.chi.0000156661.38576.0f. [DOI] [PubMed] [Google Scholar]
- McAllister TW, Price TR. Aspects of the behavior of psychiatric inpatients with frontal lobe damage: some implications for diagnosis and treatment. Compr Psychiatry. 1987;28:14–21. doi: 10.1016/0010-440x(87)90039-3. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Bal T. Sensory gating mechanisms of the thalamus. Curr Opin Neurobiol. 1994;4:550–556. doi: 10.1016/0959-4388(94)90056-6. [DOI] [PubMed] [Google Scholar]
- McGuire TL, Sylvester CE. Neuropsychiatry evaluation and treatment of children with head injury. J Learn Disabil. 1987;20:590–595. doi: 10.1177/002221948702001004. [DOI] [PubMed] [Google Scholar]
- McKinlay A, Dalrymple-Alford JC, Horwood LJ, Fergusson DM. Long term psychosocial outcomes after mild head injury in early childhood. J Neurol Neurosurg Psychiatry. 2002;73:281–288. doi: 10.1136/jnnp.73.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millichap JG. Etiologic classification of attention-deficit/hyperactivity disorder. Pediatrics. 2008;121:e358–e365. doi: 10.1542/peds.2007-1332. [DOI] [PubMed] [Google Scholar]
- Neckelmann D, Amzica F, Steriade M. Spike-wave complexes and fast components of cortically generated seizures. III. Synchronizing mechanisms. J Neurophysiol. 1998;80:1480–1494. doi: 10.1152/jn.1998.80.3.1480. [DOI] [PubMed] [Google Scholar]
- Niedermeyer E. Frontal lobe functions and dysfunctions. Clin Electroencephalogr. 1998;29:79–90. doi: 10.1177/155005949802900206. [DOI] [PubMed] [Google Scholar]
- Owen AM, Downes JJ, Sahakian BJ, Polkey CE, Robbins TW. Planning and spatial working memory following frontal lobe lesions in man. Neuropsychologia. 1990;28:1021–1034. doi: 10.1016/0028-3932(90)90137-d. [DOI] [PubMed] [Google Scholar]
- Owen AM, Roberts AC, Hodges JR, Summers BA, Polkey CE, Robbins TW. Contrasting mechanisms of impaired attentional set-shifting in patients with frontal lobe damage or Parkinson's disease. Brain. 1993;116:1159–1175. doi: 10.1093/brain/116.5.1159. [DOI] [PubMed] [Google Scholar]
- Paradiso S, Chemerinski E, Yazici KM, Tartaro A, Robinson RG. Frontal lobe syndrome reassessed: comparison of patients with lateral or medial frontal brain damage. J Neurol Neurosurg Psychiatry. 1999;67:664–667. doi: 10.1136/jnnp.67.5.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrikelis P, Angelakis E, Gatzonis S. Neurocognitive and behavioral functioning in frontal lobe epilepsy: a review. Epilepsy Behav. 2009;14:19–26. doi: 10.1016/j.yebeh.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Pereda E, Quiroga RQ, Bhattacharya J. Nonlinear multivariate analysis of neurophysiological signals. Prog Neurobiol. 2005;77:1–37. doi: 10.1016/j.pneurobio.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee JH. Molecular characterization of a neuronal low-voltage-activated T-type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- Pigula FA, Wald SL, Shackford SR, Vane DW. The effect of hypotension and hypoxia on children with severe head injuries. J Pediatr Surg. 1993;28:310–316. doi: 10.1016/0022-3468(93)90223-8. [DOI] [PubMed] [Google Scholar]
- Piret JP, Mottet D, Raes M, Michiels C. CoCl2, a chemical inducer of hypoxia-inducible factor-1, and hypoxia reduce apoptotic cell death in hepatoma cell line HepG2. Ann NY Acad Sci. 2002;973:443–447. doi: 10.1111/j.1749-6632.2002.tb04680.x. [DOI] [PubMed] [Google Scholar]
- Rolls ET, Hornak J, Wade D, McGrath J. Emotion-related learning in patients with social and emotional changes associated with frontal lobe damage. J Neurol Neurosurg Psychiatry. 1994;57:1518–1524. doi: 10.1136/jnnp.57.12.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer IE, Bhatia KP, Lopes-Cendes I, Fish DR, Marsden CD, Andermann E, Andermann F, Desbiens R, Keene D, Cendes F. Autosomal dominant nocturnal frontal lobe epilepsy: a distinctive clinical disorder. Brain. 1995;118:61–73. doi: 10.1093/brain/118.1.61. [DOI] [PubMed] [Google Scholar]
- Shallice T, Burgess PW. Deficits in strategy application following frontal lobe damage in man. Brain. 1991;114:727–741. doi: 10.1093/brain/114.2.727. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Bernaudin M. HIF1 and oxygen sensing in the brain. Nat Rev Neurosci. 2004;5:437–448. doi: 10.1038/nrn1408. [DOI] [PubMed] [Google Scholar]
- Sherman SM. Tonic and burst firing: dual modes of thalamocortical relay. Trends Neurosci. 2001;24:122–126. doi: 10.1016/s0166-2236(00)01714-8. [DOI] [PubMed] [Google Scholar]
- Shue KL, Douglas VI. Attention deficit hyperactivity disorder and the frontal lobe syndrome. Brain Cogn. 1992;20:104–124. doi: 10.1016/0278-2626(92)90064-s. [DOI] [PubMed] [Google Scholar]
- Sitnikova E, Dikanev T, Smirnov D, Bezruchko B, van Luijtelaar G. Granger causality: cortico-thalamic interdependencies during absence seizures in WAG/Rij rats. J Neurosci Methods. 2008;170:245–254. doi: 10.1016/j.jneumeth.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Steriade M. Grouping of brain rhythms in corticothalamic systems. Neuroscience. 2006;137:1087–1106. doi: 10.1016/j.neuroscience.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D. Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. J Neurosci. 1995;15:623–642. doi: 10.1523/JNEUROSCI.15-01-00623.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Contreras D. Spike-wave complexes and fast components of cortically generated seizures. I. Role of neocortex and thalamus. J Neurophysiol. 1998;80:1439–1455. doi: 10.1152/jn.1998.80.3.1439. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Stores G, Zaiwalla Z, Bergel N. Frontal lobe complex partial seizures in children: a form of epilepsy at particular risk of misdiagnosis. Dev Med Child Neurol. 1991;33:998–1009. doi: 10.1111/j.1469-8749.1991.tb14816.x. [DOI] [PubMed] [Google Scholar]
- Stuss DT, Gow CA. “Frontal dysfunction” after traumatic brain injury. Cogn Behav Neurol. 1992;5:272–282. [Google Scholar]
- Tan M, Appleton R. Attention deficit and hyperactivity disorder, methylphenidate, and epilepsy. Arch Dis Child. 2005;90:57–59. doi: 10.1136/adc.2003.048504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton NY, Treiman DM. Experimental secondarily generalized convulsive status epilepticus induced by d,l-homocysteine thiolactone. Epilepsy Res. 1988;2:79–86. doi: 10.1016/0920-1211(88)90023-x. [DOI] [PubMed] [Google Scholar]
- Wienbruch C, Moratti S, Elbert T, Vogel U, Fehr T, Kissler J, Schiller A, Rockstroh B. Source distribution of neuromagnetic slow wave activity in schizophrenic and depressive patients. Clin Neurophysiol. 2003;114:2052–2060. doi: 10.1016/s1388-2457(03)00210-4. [DOI] [PubMed] [Google Scholar]
- Yoon JH, Minzenberg MJ, Ursu S, Ryan Walter BS, Walters R, Wendelken C, Ragland JD, Carter CS. Association of dorsolateral prefrontal cortex dysfunction with disrupted coordinated brain activity in schizophrenia: relationship with impaired cognition, behavioral disorganization, and global function. Am J Psychiatry. 2008;165:1006–1014. doi: 10.1176/appi.ajp.2008.07060945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. Prefrontal projections to the thalamic reticular nucleus form a unique circuit for attentional mechanisms. J Neurosci. 2006;26:7348–7361. doi: 10.1523/JNEUROSCI.5511-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]