

Abstract
Anxiety disorders, such as phobias and posttraumatic stress disorder, are among the most common mental disorders. Cognitive therapy helps in treating these disorders; however, many cases relapse or resist the therapy, which justifies the search for cognitive enhancers that might augment the efficacy of cognitive therapy. Studies suggest that enhancement of plasticity in certain brain regions such as the prefrontal cortex (PFC) and/or hippocampus might enhance the efficacy of cognitive therapy. We found that elevation of brain magnesium, by a novel magnesium compound [magnesium-l-threonate (MgT)], enhances synaptic plasticity in the hippocampus and learning and memory in rats. Here, we show that MgT treatment enhances retention of the extinction of fear memory, without enhancing, impairing, or erasing the original fear memory. We then explored the molecular basis of the effects of MgT treatment on fear memory and extinction. In intact animals, elevation of brain magnesium increased NMDA receptors (NMDARs) signaling, BDNF expression, density of presynaptic puncta, and synaptic plasticity in the PFC but, interestingly, not in the basolateral amygdala. In vitro, elevation of extracellular magnesium concentration increased synaptic NMDAR current and plasticity in the infralimbic PFC, but not in the lateral amygdala, suggesting a difference in their sensitivity to elevation of brain magnesium. The current study suggests that elevation of brain magnesium might be a novel approach for enhancing synaptic plasticity in a regional-specific manner leading to enhancing the efficacy of extinction without enhancing or impairing fear memory formation.
Introduction
Fear is essential for survival. However, excessive fear or the experience of strong fearful events can lead to anxiety disorders such as phobias and posttraumatic stress disorder (PTSD). In clinics, cognitive therapy can attenuate excessive fear; however, some cases relapse or resist the therapy (Foa et al., 2002).
In laboratory animals, the extinction of conditioned fear is the cognitive therapy-based experimental model for studying fear attenuation (Myers and Davis, 2007). Progress in understanding the brain circuits involved in fear extinction has been achieved by studying the circuits of Pavlovian fear conditioning in experimental animals (LeDoux, 2000; Myers and Davis, 2007). The amygdala is thought to contain circuits that form the conditioned fear memory and drive conditioned responses (LeDoux, 2000). After extinction, ventromedial prefrontal cortex is believed to retain the extinction memory in animals (Morgan and LeDoux, 1995; Milad and Quirk, 2002; for review, see Quirk and Mueller, 2008) and in humans (Phelps et al., 2004; Rauch et al., 2006). The hippocampus participates in the formation of conditioned fear by providing spatial (Sanders et al., 2003) and temporal (McEchron et al., 1998) information. It is also involved in the retention of extinction (Farinelli et al., 2006) and the modulation of extinction in a context-dependent manner (Hobin et al., 2006; Herry et al., 2008). Therefore, conditioned fear memories and responses are believed to be formed and generated by the amygdala, while fear expression is modulated adaptively by other brain regions such as the prefrontal cortex (PFC) and hippocampus.
Based on the above-mentioned fear memory and extinction circuitries, enhancement of activity/plasticity of the infralimbic prefrontal cortex (IL-PFC) and/or hippocampus could be an attractive strategy to augment the efficacy of extinction. Indeed, electrical stimulation in the IL-PFC (Milad and Quirk, 2002) and/or hippocampus (Farinelli et al., 2006) enhances the retention of extinction. Infusion of brain-derived neurotrophic factor (BDNF) into the IL-PFC or hippocampus attenuates fear responses even without extinction (Peters et al., 2010). However, long-term regional-specific enhancement of PFC and hippocampus functions without simultaneously enhancing/impairing amygdala functions has not been achieved.
We found that extracellular magnesium concentration ([Mg2+]o) is an important regulator of synaptic plasticity in vitro (Slutsky et al., 2004). Recently, a novel magnesium compound [magnesium-l-threonate (MgT)] that can elevate brain magnesium via chronic oral supplementation was developed (Slutsky et al., 2010). Using MgT as a tool, we began exploring the relationship between brain magnesium and cognition. We found that MgT treatment increases synaptic density and plasticity in the hippocampus and enhances learning abilities, working memory, and short- and long-term memory in both young and aged rats (Slutsky et al., 2010). Based on the enhancement of working memory, MgT treatment might also enhance synaptic plasticity in the PFC. If so, then MgT treatment may enhance the retention of extinction. Hence, we investigated the effects of an elevation of brain magnesium on fear memory and fear extinction processes and explored the molecular and cellular properties of brain regions implicated in these processes, namely the IL-PFC and basolateral amygdala.
Materials and Methods
Experimental animals
Male Sprague Dawley (4–8 month old) rats were obtained from Vital River Laboratory Animal Technology. All rats were individually housed with ad libitum access to food and water under a 12:12 h reversed light:dark cycle. Behavioral experiments were performed during the dark phase. All experiments involving animals were approved by Tsinghua University and University of Toronto Committees on Animal Care and Use.
MgT treatment
MgT (Magceutics) was administered via drinking water (604 mg/kg/d, 50 mg/kg/d elemental magnesium) as we described previously (Slutsky et al., 2010). The water intake (on daily basis) and body weight (every 3 d) were measured. The concentration of MgT in the drinking water was determined and adjusted based on these parameters to reach the target dose. Chronic MgT treatment at this dose did not influence water and food intake or the normal body weight gain, similar to our previous observations (Slutsky et al., 2010). Rats received rat-chow containing 0.15% elemental magnesium, which is broadly accepted as the basic nutritional magnesium concentration.
Fear conditioning
Apparatus.
Experiments were performed using a video-based fear conditioning system (Coulbourn Instruments). Rats were conditioned and tested in modular fear conditioning chambers (Rat Test Cage; Habitest) with metal stainless-steel rod floors connected to a shock generator. Each chamber was kept inside a sound-insulating cubicle. Stimulus presentation was automated using FreezeFrame2 software (Actimetrics). The conditioning chambers were modified to change the context from A to B. Context A (conditioning context) consisted of the original conditioning chamber with white light on the left side, background noise was ∼54 dB, and 20% ethanol odor. In context B (test context), the stainless-steel grid floor was removed and the chambers were covered with black acrylic platforms from inside. The white light was mounted on the right side of the chamber, background noise was reduced to ∼42 dB, and a 2% acetic acid odor was introduced.
Delay, trace fear conditioning, and testing.
Rats underwent either delay or trace fear conditioning (Fig. 1A). For both procedures, rats were placed in Context A and left to explore for 10 min (habituation to establish baseline) before tone onset (20 s, 5 kHz, 85 ± 3 dB). For delay conditioning experiments, the tone coterminated with foot shock (0.5 s, 0.7 mA). Training sessions consisted of one (Fig. 1C), three (Figs. 1B; 2A,C; 3) or seven (Fig. 2B) tone–shock trials. For trace conditioning experiments, tone and foot shock were separated by either 15 (Fig. 1D) or 30 s (Fig. 1E) trace intervals. During training sessions, rats received three tone–shock trials of trace fear conditioning. The intertrial interval [ITI; defined as the time between conditional stimulus (CS) onset and the next CS onset] was 180 s. Rats were removed from the conditioning chambers and returned to their home cages 180 s following the last tone.
Figure 1.

Effects of MgT on delay and trace fear conditioning. A, Top, Experimental design showing, after 4 weeks of MgT treatment, the fear conditioning (day 1) and an LTM test (day 2). Bottom, Illustration of the delay and trace fear conditioning protocols. B, Left, Freezing behavior of control and MgT-treated rats during baseline (BL) and three trials of delay fear conditioning. Right, Freezing behavior during LTM test before tone (Pre-CS) and after tone (Post-CS) of control and MgT-treated rats (n = 8). C, Freezing behavior during LTM test before and after tone of control and MgT-treated rats (n = 9). Test was conducted 24 h after a single trial of delay fear conditioning. D, Left, Freezing behavior of control and MgT-treated rats during baseline and three trials of trace fear conditioning (trace interval = 15 s). Right, Freezing behavior during LTM test before and after tone of control and MgT-treated rats (n = 10). E, Freezing behavior during LTM test before and after tone of control and MgT-treated rats (n = 9). The LTM test was conducted 24 h after trace fear conditioning with trace interval = 30 s. *p < 0.05. Data presented as mean ± SEM.
Figure 2.

Effects of MgT on extinction learning, retention, and retrieval. A, Top, Experimental design to test extinction (Ext.) learning and retention. Bottom left, Freezing behavior of MgT-treated and control rats during the first long-term memory test (LTM1) conducted 24 h after fear conditioning (Cond.; day 2). Middle, Freezing behavior of MgT-treated and control rats during extinction learning conducted 48 h after conditioning (day 3). Right, Freezing behavior of MgT-treated and control rats (n = 8) during a retention of extinction test (LTM2) conducted 3 d after extinction learning (day 6). B, Top, Experimental design to test the effects of MgT treatment on retention of extinction when treatment was given after fear conditioning for 4 weeks. Bottom, Freezing behavior of rats during conditioning, LTM1, extinction learning, and LTM2. In the LTM2 test, MgT-treated rats exhibited significantly lower freezing behavior than controls (n = 8). C, Top, Experimental design to test the effects of MgT on retrieval of the extinction memory. Bottom, Freezing behavior of untreated rats during three trials of fear conditioning (day 1) and 10 trials of extinction (day 2). Freezing behavior of rats assigned as control (white bar) and MgT-treated (lined bar) during retention of extinction test (LTM1) conducted 24 h after extinction learning (day 3). Right, Freezing behavior of MgT-treated (n = 13) and control (n = 14) rats during LTM2 conducted 4 weeks after the beginning of MgT treatment (day 32). *p < 0.05. Data presented as mean ± SEM.
Figure 3.

Effects of MgT on spontaneous recovery, renewal and reinstatement. A, Spontaneous recovery test. Freezing behavior of MgT-treated (n = 9) and control (n = 10) rats during fear conditioning (Cond.; day 1), extinction learning (Ext.; day 2; average of the first and the last two of 14 trials is presented), LTM test (day 3), and spontaneous recovery test (day 30). All experiments were performed in the same context, namely context A. B, Renewal test. Freezing behavior of MgT-treated (n = 17) and control (n = 18) rats during memory tests performed in the context where extinction learning (14 trials) was performed (context B) and in the context where fear conditioning was performed (context A). C, Reinstatement test. Freezing behavior of MgT-treated (n = 13) and control (n = 10) rats, during the last two trials of extinction performed on day 2 and during a reinstatement test performed 24 h (day 4) after exposure to five unsignaled foot-shocks (5-US) in the same context. *p < 0.05; **p < 0.01. Data presented as mean ± SEM.
During fear-memory-retention tone tests, rats were placed in the test context for 3 min to determine pre-CS freezing behavior. Test sessions began with the presentation of two tones (20 s each) at a CS–CS interval of 180 s. Rats were returned to their home cage 180 s after the second tone. Presented data are from the average of the two trials. The testing protocol applies to all testing phases except for renewal experiments, during which rats were tested using a single tone presentation followed by a 60 s interval in both contexts A and B. All tone tests were performed in context B except for certain experimental designs such as spontaneous recovery, renewal, and reinstatement (Fig. 3).
Extinction training
To ensure that extinction learning did not interfere with the original fear memory, as early extinction might permanently attenuate fear memory (Myers et al., 2006), we selected a long time interval between fear conditioning and extinction learning (48 h). Rats were placed in the extinction context for 3 min to determine pre-CS freezing behavior. Extinction training consisted of 10 presentations of tone without foot shock. The tone duration was 20 s and the ITI was 180 s. Rats were returned to their home cages 180 s after the last tone presentation. We also performed extinction training when MgT treatment was given after acquisition of fear conditioning (Fig. 2B) or after extinction training (Fig. 2C). In these experiments, the same extinction procedure was applied.
Spontaneous recovery, renewal, and reinstatement
All procedures were performed as described before (Monfils et al., 2009) with minor modifications. In a spontaneous recovery test 24 h after extinction, rats received a memory test to evaluate the retention of extinction. One month later, the same rats were tested again to evaluate their spontaneous recovery. In the renewal test, rats were conditioned in context A and then extinction was performed the following day in context B. Twenty-four hours later, rats were tested for the retention of extinction (using one CS presentation) in the extinction context (B). The next day, rats were tested again, but in context A. In the reinstatement test, following conditioning and 24 h after extinction, rats received five unsignaled foot-shocks in the same context and were removed immediately after the last foot-shock. The next day, rats were tested for the reinstatement of fear memory. Rats were fear conditioned (delay fear conditioning) using three conditioning trials, as described in Figure 1A. Extinction training also was same; however, we performed longer extinction training (14 presentations of tone without shock). The stronger extinction training was used to reduce the freezing behavior of control rats to a comparable level as MgT-treated rats during the first post extinction test.
Behavioral scoring
The total amount of CS-induced freezing as the percentage of total CS–CS interval [or CS–unconditional stimulus (US) trace interval for trace fear conditioning] was used as a measure of fear. In the post-fear-conditioning treatment experiments (Fig. 2B), the freezing behavior of all rats was more robust during the tone presentation. Thus, the freezing behavior in Figure 2B was assessed by measuring the percentage of time spent freezing during tone presentation. Freezing behavior was measured with an automated system with the aid of FreezeView2 software (Actimetrics). The software measures changes in pixel luminance intensity across successive video frames (taken at 1–5 Hz) and computes changes in motion as these parameters linearly correlated. A threshold is then applied to the data to yield a percentage freezing score. In our case, frames were taken at a rate of 3.12 Hz. We found significant correlation (p < 0.0001, r2 = 0.9710, Pearson test) between percentages of freezing from FreezeView2 and manual scoring, where immobility except for movement associated with breathing was manually scored as freezing (Blanchard and Blanchard, 1969).
Tissue dissection
To activate the NMDA receptors (NMDARs) signaling pathway specifically in the PFC without activating other brain regions (e.g., amygdala) involved in fear memory formation and extinction learning, which might indirectly influence the NMDAR signaling in the PFC, we chose the novel object recognition protocol. This memory test is a nonemotional task that is known to involve the NMDAR signaling pathway in the PFC (Akirav and Maroun, 2006).
We prepared two groups of rats (control and MgT-treated) and performed the novel object recognition test on them (24 h retention interval) with three objects, as we described previously (Slutsky et al., 2010). Following a memory test, rats were decapitated and the whole PFC (approximately first 3 mm, bregma 2.7) was dissected and frozen in the liquid nitrogen. To induce activation of NMDAR signaling in the basolateral amygdala, we exposed a new group of rats to the amygdala-dependent delay-fear-conditioning protocol (a single tone–shock pairing). To ensure that this stimulus activated amygdala, we tested the fear memory 24 h later and then rats were killed. Brains were frozen, mounted, and cut by a cryostat at −20°C (until approximately bregma −2.3), and then the BLA was delineated (∼1.5 mm depth) using a microscalpel, isolated, and immediately frozen in liquid nitrogen.
Western blot
Tissues were homogenized and equal amounts of proteins were resolved on polyacrylamide gel, then transferred to PVDF membranes (Millipore). Membranes were blocked and then probed with primary antibodies against NR2B (H-50, sc-9057), NR2A (C-17, sc-1468), NR1 (C-20, sc-1467) (all from Santa Cruz Biotechnology), α-calcium/calmodulin-dependent kinase (α-CaMKII), phospho-α-CaMKII, cAMP response element-binding protein (CREB), phospho-CREB, or β-actin (all from Cell Signaling Technology) overnight at 4°C. Membranes were then incubated with an HRP-conjugated secondary antibody (Jackson ImmunoResearch) at room temperature. Protein bands were detected by ECL detection reagent (Super Signal; Pierce) and captured on an autoradiography film (Kodak). Integrated optical density was determined using Image-Pro Plus software 6.0 (Media Cybernetics). Standard curves were constructed to establish that we operated within the linear range of the detection method. Codetection of β-actin on the same membrane served as a loading control. Experiments were performed by experimenters blind to the treated group.
ELISA assay of BDNF
ELISA assay of BDNF was performed using Chemikine BDNF ELISA kit (Millipore) with complete adherence to manufacturer's protocol. The total amount of BDNF was calculated as the percentage of tissue weight.
Fluorescent immunostaining and estimation of synaptophysin-positive puncta
Synaptophysin immunostaining and quantification of presynaptic puncta were performed as we described previously (Slutsky et al., 2010). Briefly, coronal brain sections were cut at 10 μm on a cryostat and anatomically matched cryosections were incubated with anti-synaptophysin primary antibody (Millipore) overnight at 4°C. Next, brain sections were rinsed with PBS and then incubated with Alexa 488-coupled rabbit anti-mouse IgG in PBS (Invitrogen) for 5 h at room temperature. Slides were coverslipped and given code numbers that were concealed until after image analyses.
Stained sections containing the medial prefrontal cortex or amygdala were imaged using the 60× water lens (NA = 1.2) at zoom ×3. Serial z-sectioning (0.9 μm) was performed and the best three z-sections (with highest number of puncta) were collected and merged into a single image. The strongest three images from each rat were taken and the density of synaptophysin-positive (Syn+) puncta in different regions was estimated using the Image-Pro Plus software 6.0. The mean puncta number per 1000 μm2 was used as an estimate of the density of presynaptic puncta.
Slice preparations and recordings
Slice preparation from MgT-treated and control rats.
Coronal brain slices (400 μm) from 3- to 4-months-old control and MgT-treated (for 4 weeks) rats, containing the medial prefrontal cortex or amygdala, were prepared. Slices were transferred to a submerged recovery chamber containing oxygenated (95% O2 and 5% CO2) artificial CSF (ACSF; 124 mm NaCl, 4.4 mm KCl, 2 mm CaCl2, 1 mm MgSO4, 25 mm NaHCO3, 1 mm NaH2PO4, and 10 mm glucose) at room temperature for at least 1 h.
Slice preparation for elevation of extracellular magnesium in vitro.
Coronal brain slices containing the medial prefrontal cortex or amygdala were prepared from untreated rats and were incubated for 5 h in ACSF containing either 0.8 or 1.2 mm extracellular magnesium concentration (referred to as 0.8-[Mg2+]o and 1.2-[Mg2+]o slices, respectively). Regardless of incubation conditions, [Mg2+]o in recording solution was set at 1.2 mm to exclude any potential effects on synaptic transmission/plasticity by acute elevation of [Mg2+]o.
Whole-cell recordings.
EPSCs were recorded from layer II/III neurons in the IL-PFC using an Axon 200B amplifier (Molecular Devices), and stimulations were delivered using a bipolar tungsten stimulating electrode placed in layer V of the IL-PFC (Zhao et al., 2005). In slices containing lateral amygdala, EPSCs were recorded in neurons in the dorsal part of lateral amygdala, while stimulations were delivered in the auditory thalamic inputs to lateral amygdala (Weisskopf and LeDoux, 1999).
For the NMDA receptor/AMPA receptor currents ratio (INMDA/AMPA) experiments in vitro, EPSCs were induced by repetitive stimulations at 0.03 Hz, and neurons were voltage clamped at −60 mV to record the EPSCAMPA, and then were clamped at +50 mV to record the EPSCNMDA (without liquid junction potential correction). The recording pipettes (3–5 MΩ) were filled with solution containing the following: 102 mm Cesium gluconate, 5 mm tetraethylammonium-chloride, 3.7 mm NaCl, 11 mm BAPTA, 0.2 mm EGTA, 20 mm HEPES, 2 mm MgATP, 0.3 mm NaGTP, and 5 mm QX-314 chloride (adjusted to pH 7.2 with CsOH). The solution contained high calcium buffer to minimize the calcium-dependent inactivation of the responses. The AMPA receptor-mediated currents were measured at the peak of the recorded currents and NMDA receptors-mediated currents were measured 50 ms after stimulation. The INMDA/AMPA ratio was calculated from the recorded data in individual neurons.
For long-term potentiation (LTP) experiments, EPSCs were induced by repetitive stimulations at 0.02 Hz and neurons were voltage clamped at −60 mV (without liquid junction potential correction). The recording pipettes (3–5 MΩ) were filled with solution containing the following: 120 mm K-gluconate, 5 mm NaCl, 1 mm MgCl2, 0.2 mm EGTA, 10 mm HEPES, 2 mm Mg-ATP, 0.1 mm Na3-GTP, and 10 mm phosphocreatine disodium (adjusted to pH 7.2 with KOH). After obtaining stable EPSCs for 10 min, LTP induction paradigm was used within 12 min after establishing the whole-cell configuration to prevent the wash-out effect on LTP induction. The protocol involved the following steps. First, the postsynaptic neurons were switched to current-clamp recording mode. Second, LTP was induced by three presynaptic stimuli (interstimulus interval = 33 ms) coupled with three postsynaptic action potentials (delivered 10 ms after the onset of each EPSC) and repeated 15 times (interburst interval = 5 s). Third, recording was switched back to voltage-clamp mode. Data were discarded if access resistance changed by >15% during an experiment. Picrotoxin (100 μm) was always applied to block the GABAA receptor-mediated response.
Statistical analysis
All experiments were statistically analyzed using GraphPad Prism software 5. Western-blot, LTP, and synaptophysin-positive puncta data were tested using two-tailed unpaired Student's t test. Memory retention tests were analyzed using two-tailed paired (within same group) or unpaired (between MgT and control) Student's t test. All training and extinction curves were analyzed using two-way ANOVA repeated measure, using trial as the within-subjects factor and drug (MgT vs control) as the groups' factor. ANOVAs were followed by Bonferroni's post hoc test. Difference was considered significant if p < 0.05. Data are presented as mean ± SEM.
Results
Effects of MgT on conditioned fear memory
To investigate the effects of MgT treatment on the fear memory process, we treated rats with MgT at a dose of 609 mg/kg for 4 weeks (∼50 mg/kg/d elemental magnesium; see Materials and Methods, above). The dose and time course were shown to be effective in elevating brain magnesium and enhancing hippocampus-dependent learning and memory (Slutsky et al., 2010).
After treatment, rats were fear conditioned using three trials in the conditioning chamber (Context A, day 1; Fig. 1A). Long-term fear memory (LTM) was evaluated on day 2 by measuring freezing behavior in response to tone presentations in a different chamber (context B; Fig. 1A).
First, we evaluated the acquisition and retention of delay fear conditioning, which is thought to be amygdala-dependent (LeDoux, 2000). The freezing behavior of MgT-treated and control rats increased equivalently during conditioning (Fig. 1B, left). Two-way ANOVA revealed significant effect of trials only (F(3,42) = 12.60, p < 0.0001). During the LTM test (day 2), both groups exhibited equivalent freezing behavior in response to tone presentations (two-tailed unpaired t test, t = 0.006, df = 14, p = 1.0; Fig. 1B, right). Therefore, MgT treatment appears to have no effects on the acquisition and retention of delay fear conditioning. To ensure that the lack of effect was not due to overtraining, we exposed another group of rats to a weaker conditioning protocol (single tone–shock pairing). Again, the freezing behavior in both groups was similar in the LTM test (t = 0.67, df = 16, p = 0.5; Fig. 1C).
Next, we evaluated the effects of MgT treatment on trace fear conditioning, which involves interplay between the hippocampus and amygdala (Solomon et al., 1986; McEchron et al., 1998). We conditioned rats with two different conditioning protocols (the trace interval between CS and US was either 15 or 30 s; Fig. 1A). The freezing behaviors of the MgT-treated and control rats were similar during conditioning at the 15 s trace interval (two-way ANOVA revealed a significant effect of trials only: F(3,54) = 96.10, p < 0.0001; Fig. 1D, left). During LTM test (24 h later), both groups exhibited equivalent freezing behavior in response to tone presentations (t = 1.10, df = 18, p = 0.3; Fig. 1D, right). When rats were conditioned at the longer trace interval (30 s), the freezing behavior of the control rats during the LTM test was significantly lower than that of the MgT-treated rats (t = 2.19, df = 16, p < 0.05; Fig. 1E). The improvement of trace fear memory at the longer trace interval might be due to the enhancement of the hippocampus-dependent memory capacity by the MgT treatment (Slutsky et al., 2010).
Effects of MgT on retention of extinction
Next, we tested the effects of MgT treatment on extinction learning and the retention of extinction, which is thought to involve the IL-PFC (Milad and Quirk, 2002; Quirk and Mueller, 2008). Twenty-four hours after delay fear conditioning (day 2; Fig. 2A, top), a long-term memory test was conducted to ensure that both groups had similar memories (t = 0.64, df = 14, p = 0.5; Fig. 2A, bottom left). During 10 trials of extinction (48 h after conditioning), the freezing of MgT-treated and control rats declined similarly under presentations of tone without foot-shock. Two-way ANOVA revealed significant effect of trials only (F(10,140) = 6.0, p < 0.0001; Fig. 2A, bottom middle), indicating that extinction learning was not facilitated by MgT treatment.
Interestingly, during a retention of extinction memory test (LTM, 72 h after extinction), the freezing of MgT-treated rats was significantly lower than controls (t = 2.29, df = 14, p < 0.05; Fig. 2A, bottom right), indicating a stronger retention of extinction 3 d after extinction learning.
In the above experiments, MgT was given before fear acquisition. We examined whether MgT could also enhance retention of extinction when given after the acquisition of fear memory (Fig. 2B, top). Another group of rats was conditioned with a stronger protocol (seven tone–shock pairings) and then randomly assigned to two groups; 48 h after conditioning, one group received MgT treatment. After a 4-week treatment, we compared long-term fear memories (LTM1; day 30) between control and MgT-treated rats and found similar freezing behavior (t = 0.07, df = 14, p = 0.9; Fig. 2B, bottom), suggesting that the MgT treatment did not interfere with long-term storage and later retrieval of previously acquired auditory fear memory. However, in the second long-term memory test, conducted 3 d after extinction (LTM2, day 34), MgT-treated rats exhibited significantly lower freezing behavior than controls (t = 2.81, df = 14, p < 0.05; Fig. 2B, bottom). Thus, MgT, when given after fear acquisition, still could enhance the retention of extinction.
Finally, we tested whether MgT treatment could enhance the retention of extinction when it was given after extinction learning. Another group of rats were conditioned using three tone–shock pairings. Twenty-four hours later, their fear memory was extinguished. The rats were assigned to two groups with similar levels of retention of extinction (Fig. 2C, bottom). Twenty-four hours later (48 h after extinction), one group received the MgT treatment and another group served as the control (Fig. 2C, bottom).
After a 4-week treatment, we evaluated the retention of extinction (LTM2, day 32) in the control and MgT-treated rats. Interestingly, MgT-treated and control rats exhibited similar, and high, freezing behavior in response to tone presentations (t = 0.55, df = 25, p = 0.6; Fig. 2C, bottom right), suggesting that MgT treatment could not enhance the retention of extinction when it was given after extinction learning. It is worth noting that these data excluded the possibility that the reduction in freezing behavior in MgT-treated rats following extinction learning (Fig. 2A,B) was due to general anxiolytic effect of the MgT treatment.
Effects of MgT on spontaneous recovery, renewal, and reinstatement
We examined whether the MgT treatment can prevent spontaneous recovery (Pavlov, 1927). Control and MgT-treated rats were conditioned as described above (delay fear conditioning; Fig. 1A). However, a stronger extinction protocol (14 presentations of tone without shock on day 2) was used to reduce the freezing behavior of control rats to levels comparable to MgT-treated rats on the first retention of extinction test. Both groups had low freezing behavior in the first retention of extinction test conducted 24 h after extinction; however, MgT-treated rats exhibited significantly less freezing than controls (LTM test, two-tailed unpaired t test, t = 2.29, df = 17, p = 0.04; Fig. 3A). During the second retention of extinction test conducted 27 d later (spontaneous recovery test, day 30), the freezing behavior of control rats significantly increased compared with the first test, suggesting the spontaneous recovery of extinguished fear memories (paired t test, t = 2.40, df = 9, p < 0.05, compared with the LTM test; Fig. 3A). In contrast, MgT-treated rats remained at a similar freezing level as in the first LTM test (paired t test, t = 0.70, df = 8, p = 0.5; Fig. 3A), indicating that MgT treatment prevented the spontaneous recovery of extinguished fear memories after passage of time.
For renewal (Bouton, 1993), control and MgT-treated rats were conditioned as described above. Twenty-four hours after extinction, a retention of extinction test was performed in context B (day 3; Fig. 3B). One day later, the same test was performed on the same rats, but in context A (renewal test, day 4; Fig. 3B). The freezing behavior of control rats in context B was low; however, it was significantly higher in context A (paired t test, t = 3.43, df = 17, p < 0.01, compared with test in context B; Fig. 3B). These data demonstrate a renewal of fear memory in control rats. In contrast, MgT-treated rats exhibited low freezing in both contexts A and B (paired t test, t = 0. 56, df = 16, p = 0.6; Fig. 3B), suggesting prevention of the renewal of extinguished fear memories by MgT treatment following contextual shift. Whether MgT can prevent renewal when given after fear conditioning or after extinction learning remains to be determined.
We considered the possibility that MgT treatment might lead to erasing fear memory following extinction learning. Thus, we tested whether extinguished fear memory can be reinstated (Westbrook et al., 2002) by reintroducing the harmful stimulus (foot-shock) in the same context. Twenty-four hours after extinction learning (day 3), we exposed the rats to five unsignaled foot-shocks. In the reinstatement test 24 h later (day 4), the control rats significantly increased their freezing in response to the tone compared with the average of the last two trials during extinction (paired t test, t = 3.22, df = 9, p < 0.05; Fig. 3C), indicating that the fear memory was reinstated. The freezing of MgT-treated rats also significantly increased following tone presentations (paired t test, t = 2.46, df = 12, p < 0.05; Fig. 3C). Thus, although MgT treatment prevented spontaneous recovery and renewal of extinguished fear memories, it did not erase the original fear memories, as the memories could be reinstated by recurring danger in the same context.
Effects of MgT on NMDAR signaling, presynaptic puncta density, and synaptic plasticity in the prefrontal cortex and amygdala
In our previous study, the increase in activation of NMDAR signaling, presynaptic puncta density, and synaptic plasticity in the hippocampus correlated with the improvement of memory functions in MgT-treated rats (Slutsky et al., 2010). To explore the molecular and cellular mechanisms that may underlie the observations on fear formation/modulation processes, we investigated the effects of MgT treatment on NMDAR signaling and presynaptic puncta and synaptic plasticity in the PFC and basolateral amygdala.
In the PFC, MgT treatment significantly increased expression of the NR2B subunit (by ∼42%, percentage above control, two-tailed unpaired t test, t = 2.64, df = 12, p < 0.05; Fig. 4A) without altering the expression of the NR2A (t = 0.65, df = 11, p = 0.5) or NR1 subunits (t = 0.91, df = 12, p = 0.4; Fig. 4A). Next, we checked the activation of downstream molecules of NMDAR signaling. To activate the NMDAR signaling in the PFC, rats were exposed to the novel object recognition test protocol (see Materials and Methods, above). MgT treatment did not change the protein expression of α-CaMKII and CREB (Fig. 4A, left). However, the ratios of p-CaMKII/CaMKII and p-CREB/CREB were significantly higher in MgT-treated rats than in controls (by ∼85%, t = 2.73, df = 10, p < 0.05; and by ∼52%, t = 2.64, df = 16, p < 0.05, respectively; Fig. 4A), suggesting higher activation of both proteins in the PFC of MgT-treated rats. BDNF expression was also increased in the PFC of MgT-treated rats (by ∼55%, t = 2.33, df = 18, p < 0.05; Fig. 4B).
Figure 4.

Effects of MgT on expression of NMDAR subunits, activation of NMDAR signaling, and BDNF expression in the prefrontal cortex and amygdala. A, Western blot analysis of expression of NR2B, NR2A, and NR1 subunits and activation of downstream molecules in the PFC. MgT treatment significantly increased NR2B expression only (n = 7) without increasing NR2A (n = 7) or NR1 (n = 7) subunits. The ratios of p-α-CaMKII/α-CaMKII (n = 6) and p-CREB/CREB (n = 9) were significantly higher in the prefrontal cortex of MgT-treated rats. Data are presented as the percentage of mean control and were statistically compared with the respective control (dashed line) for each experiment. β-actin was used as a loading control. B, Quantitative analysis of BDNF protein expression in the PFC using ELISA. BDNF level was significantly higher in MgT-treated rats in comparison with controls (n = 10). C, D, Same as A and B, but in the basolateral amygdala. MgT treatment did not alter NR2B (n = 7), NR2A (n = 6), or NR1 (n = 6) expression levels, p-CaMKII/CaMKII ratio (n = 7), p-CREB/CREB ratio (n = 7), or BDNF expression (n = 8) in the basolateral amygdala. *p < 0.05. Data presented as mean ± SEM.
Next, we examined NMDAR signaling in the basolateral amygdala. Interestingly, the MgT treatment did not alter NMDAR signaling based on the expression of NR2B (t = 0.54, df = 12, p = 0.6), NR2A (t = 0.61, df = 10, p = 0.56), or NR1 (t = 0.94, df = 10, p = 0.36). To activate the NMDAR signaling in the amygdala, we used the amygdala-dependent fear conditioning protocol (see Materials and Methods, above). The phosphorylation/expression of α-CaMKII (t = 0.03, df = 12, p = 1.0) and CREB (t = 1.00, df = 12, p = 0.34) were similar between MgT-treated and control rats (Fig. 4C). The MgT treatment also did not increase the BDNF expression (t = 0.12, df = 14, p = 0.9; Fig. 4D).
We also compared the densities of Syn+ puncta in MgT-treated and control rats. We focused our analysis on the lateral and basal amygdala, and on the prelimbic (PrL)-PFC and IL-PFC because those regions are implicated in the formation of fear memory, fear expression, and retention of extinction (Morgan and LeDoux, 1995; LeDoux, 2000; Quirk and Mueller, 2008; Sotres-Bayon and Quirk, 2010). The density of Syn+ puncta in MgT-treated rats was significantly higher than controls in the IL-PFC (by ∼30%, percentage above control, two-tailed unpaired t test, t = 5.58, df = 8, p < 0.001; Fig. 5A) and the PrL-PFC (by ∼45%, t = 6.52, df = 8, p < 0.001; Fig. 5B), whereas it did not change in the lateral amygdala (t = 0.40, df = 10, p = 0.7; Fig. 5C) or the basal amygdala (t = 1.37, df = 10, p = 0.2; Fig. 5D). Molecular results suggest that MgT treatment enhanced NR2B-containing NMDAR signaling of the PFC and hippocampus (Slutsky et al., 2010), but, surprisingly, not the basolateral amygdala functions.
Figure 5.

Effects of MgT on presynaptic boutons in the infralimbic and prelimbic prefrontal cortex and in basolateral amygdala. A, Left, Illustration of the medial prefrontal cortex showing the prelimbic and infralimbic regions (green). Middle, Syn+ puncta in the IL-PFC of control and MgT-treated rats. Right, Quantitative analysis of the density of Syn+ puncta in control and MgT-treated rats (n = 5). B, Quantitative analysis of the density of Syn+ puncta in the PrL-PFC of control and MgT-treated rats (n = 5). C, Left, Illustration of the lateral and basal amygdala (LA and BA, respectively; green). Middle, Syn+ puncta in the lateral amygdala of control and MgT-treated rats. Right, Quantitative analysis of the density of Syn+ puncta in the lateral amygdala of control and MgT-treated rats (n = 6). D, Quantitative analysis of the density of Syn+ puncta in the basal amygdala of control and MgT-treated rats (n = 6). The density was estimated as the number of immunostained puncta per 1000 μm2. ***p < 0.001. Data presented as mean ± SEM.
We next studied the impact of changing NMDAR signaling on synaptic plasticity in the IL-PFC and lateral amygdala of control and MgT-treated rats. After LTP induction, in the IL-PFC (Layer II/III), amplitude of EPSCs in slices from control rats increased modestly (by ∼19%, percentage of baseline; Fig. 6A). However, the increase in amplitude of EPSCs in slices from MgT-treated rats was significantly higher (∼59%, two-tailed unpaired t test, t = 3.47, df = 12, p < 0.01, compared with control; Fig. 6A). In contrast, after LTP induction in the dorsal part of the lateral amygdala, the amplitude of EPSCs did not differ between slices from control and MgT-treated rats (control: ∼36%; MgT: ∼43%, t = 0.30, df = 12, p = 0.8; Fig. 6B). Therefore, in line with the increase in NR2B expression, activation of NMDAR signaling, and BDNF expression in PFC, MgT treatment enhanced synaptic plasticity in the IL-PFC but not in the thalamolateral amygdala synapses.
Figure 6.
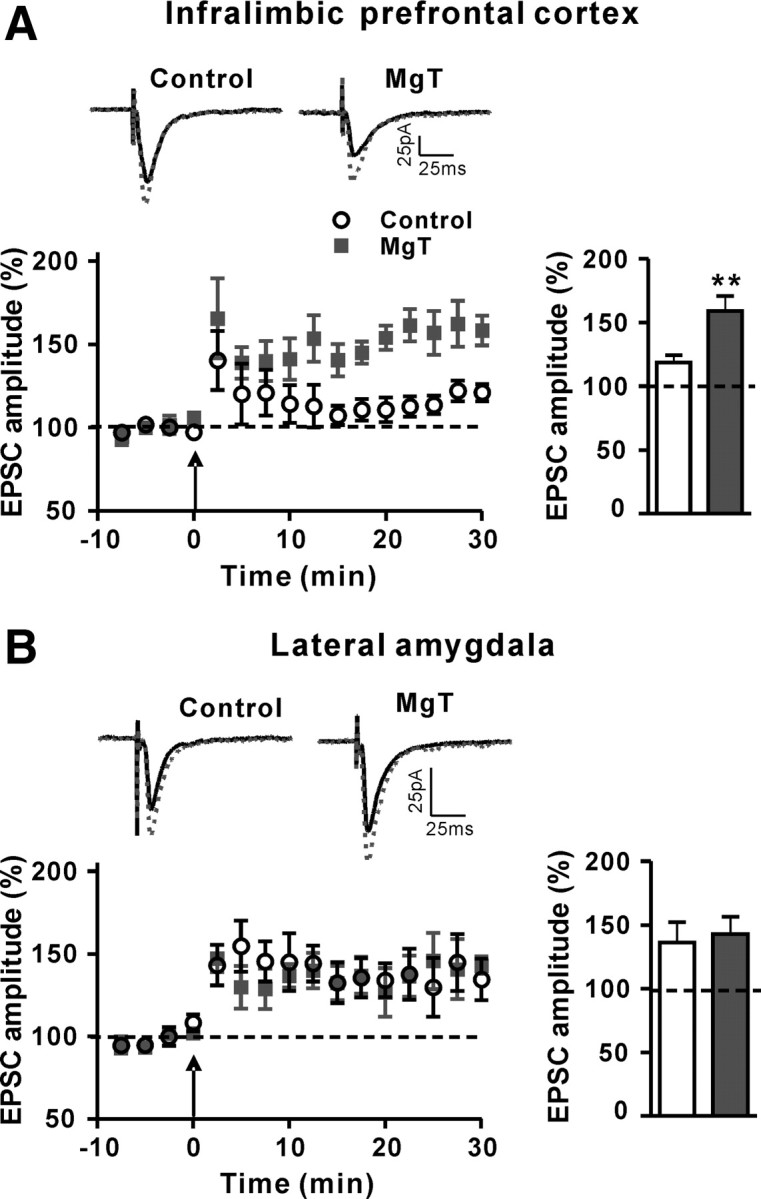
Effects of MgT treatment on synaptic plasticity in the infralimbic prefrontal cortex and lateral amygdala. A, Left, Long-term potentiation (as percentage of baseline) induced by the spike timing protocol (arrow) in pyramidal neurons in the infralimbic prefrontal cortex slices of control (n = 9) and MgT-treated (n = 5) rats. Insets, Representative traces of EPSCs are presented before (solid line) and after (dotted line) induction of long-term potentiation. Right, The magnitude of long-term potentiation (average over last 5 min). MgT treatment significantly increased the long-term potentiation in the infralimbic prefrontal cortex. B, Same as A, but in the lateral amygdala of control (n = 8) and MgT-treated (n = 6) rats. MgT treatment did not significantly change the long-term potentiation in the lateral amygdala. Dashed lines indicate the normalized basal synaptic responses. **p < 0.01. Data presented as mean ± SEM.
Effects of elevation of [Mg2+]o on synaptic NMDAR current and synaptic plasticity in the infralimbic prefrontal cortex and lateral amygdala in vitro
How can elevation of brain magnesium via MgT increase NR2B expression and enhance activation of NMDAR signaling leading to enhancement of synaptic plasticity in the IL-PFC and hippocampus (Slutsky et al., 2010) but not in the lateral amygdala? One possibility is that MgT treatment elevates the [Mg2+]o in the PFC and hippocampus but not in the lateral amygdala. Alternatively, [Mg2+]o may be homogenously elevated but the sensitivity of synapses to an elevation of [Mg2+]o may differ among these regions. To test this possibility, we investigated the effects of an elevation of [Mg2+]o on synaptic NMDAR current and synaptic plasticity in an acute slice containing the IL-PFC or lateral amygdala in vitro.
Our previous studies show that synaptic NMDAR current and plasticity in hippocampal acute slice preparation is enhanced 5 h following elevation of [Mg2+]o within a physiological range (0.8 to 1.2 mm) (Slutsky et al., 2010). Therefore, freshly cut IL-PFC or amygdala slices (from untreated rats) were incubated for 5 h in ACSF containing two different [Mg2+]o (either 0.8 mm or 1.2 mm; Fig. 7).
Figure 7.

Effects of elevation of [Mg2+]o in vitro on synaptic NMDAR current and synaptic plasticity in the infralimbic prefrontal cortex and lateral amygdala. A, Left, Representative traces of AMPA receptor EPSC (gray trace) and NMDAR EPSC (black traces) recorded at membrane potentials of −60 and +50 mV, respectively, in the infralimbic prefrontal cortex. Right, The ratio of amplitude of NMDAR EPSCs to amplitude of AMPA receptors EPSCs (INMDA/AMPA) calculated for each cell in the infralimbic prefrontal cortex slices incubated (5 h) under physiological extracellular magnesium concentration (0.8-[Mg2+]o, n = 7) and elevated [Mg2+]o (1.2-[Mg2+]o, n = 7). Elevation of [Mg2+]o in vitro significantly increased the INMDA/AMPA in the infralimbic prefrontal cortex. B, Left, Long-term potentiation (as percentage of baseline) induced by the spike timing protocol (arrow) in the infralimbic prefrontal cortex slices (0.8-[Mg2+]o slices, n = 6; 1.2-[Mg2+]o slices, n = 9). Insets, Representative traces of EPSC are presented before (solid line) and after (dotted line) induction of long-term potentiation. Right, The magnitude of long-term potentiation (average over last 5 min) of 0.8-[Mg2+]o and 1.2-[Mg2+]o slices. Elevation of [Mg2+]o in vitro significantly increased the long-term potentiation in the infralimbic prefrontal cortex. C, Same as A, but in the lateral amygdala (0.8-[Mg2+]o, n = 7; 1.2-[Mg2+]o, n = 8). D, Same as B, but in the lateral amygdala (0.8-[Mg2+]o, n = 6; 1.2-[Mg2+]o, n = 5). Elevation of [Mg2+]o in vitro did not significantly change the INMDA/AMPA ratio or long-term potentiation in the lateral amygdala. Dashed lines indicate the normalized basal synaptic responses. *p < 0.05. Data presented as mean ± SEM.
We first determined whether an elevation of [Mg2+]o changes NMDAR-mediated synaptic transmission in brain slice from the IL-PFC in vitro. The ratio of amplitude of NMDAR EPSCs to amplitude of AMPA receptors EPSCs (INMDA/AMPA) was calculated for each cell. The INMDA/AMPA in 0.8-[Mg2+]o slices was ∼0.50 (Fig. 7A), whereas the INMDA/AMPA in 1.2-[Mg2+]o slices was significantly higher (∼0.78, two-tailed unpaired t test, t = 2.20, df = 12, p < 0.05, compared with 0.8-[Mg2+]o slices; Fig. 7A). We next investigated the effects of elevating [Mg2+]o on LTP in the IL-PFC. The same protocol as above (Fig. 6) was applied to induce LTP. After LTP induction, the amplitude of EPSCs in 0.8-[Mg2+]o slices was increased modestly (∼10% from percentage of baseline; Fig. 7B). The increase in amplitude of EPSCs in the 1.2-[Mg2+]o slices, however, was significantly higher (∼58%, t = 2.85, df = 10, p < 0.05, compared with 0.8-[Mg2+]o slices; Fig. 7B). Therefore, elevation of [Mg2+]o in vitro significantly enhanced synaptic NMDAR current and synaptic plasticity in the IL-PFC, indicating that the IL-PFC synapses are sensitive to elevation of [Mg2+]o, similar to the hippocampal synapses (Slutsky et al., 2010).
In amygdala slices, the INMDA/AMPA in 0.8-[Mg2+]o slices was significantly higher than that in the IL-PFC (∼0.83, t = 2.45, df = 12, p < 0.05; Fig. 7A,C). In contrast to IL-PFC slices, elevation of [Mg2+]o did not increase the INMDA/AMPA in 1.2-[Mg2+]o slices (∼0.65, t = 1.22, df = 13, p = 0.24, compared with 0.8-[Mg2+]o slices; Fig. 7C). In line with these results, the increase in amplitude of EPSCs following LTP induction in lateral amygdala did not significantly differ between 0.8-[Mg2+]o and 1.2-[Mg2+]o slices (Fig. 7D). In fact, LTP appeared to be slightly, but not significantly, reduced by elevating [Mg2+]o (0.8-[Mg2+]o slices: ∼70%, 1.2-[Mg2+]o slices: ∼35%, t = 1.51, df = 9, p = 0.20). Therefore, the NMDAR-mediated synaptic transmission and synaptic plasticity in thalamolateral amygdala synapses might not be sensitive to an elevation of [Mg2+]o. These results are similar to the molecular and cellular results obtained after elevation of brain magnesium by MgT treatment in intact rats.
Discussion
A key finding from the current study was that elevation of brain magnesium enhanced the retention of extinction of fear memories without enhancing, impairing, or erasing original fear memory. This correlated with selective enhancement of NMDAR signaling, BDNF expression, and synaptic plasticity in the PFC, but not in the basolateral amygdala. This unique region-specific pattern of action might stem from a lack of sensitivity of NMDAR and its signaling, within the amygdala, to an elevation in the extracellular magnesium concentration in the brain.
Effects of MgT treatment on conditioned fear memories
In the current study, we found that MgT treatment did not affect the amygdala-dependent fear learning, fear memory consolidation (Fig. 1B,C), or its retrieval (Fig. 2B). The amygdala is widely implicated in the acquisition and retention of delay fear conditioning (Davis, 1992; LeDoux, 2000; Fanselow and Poulos, 2005; Maren, 2008). Synaptic modifications in the thalamolateral amygdala are considered to be the cellular basis of acquisition and retention of conditioned fear memory (LeDoux, 2000; Sigurdsson et al., 2007). Disruption of NMDAR function in the lateral amygdala impairs synaptic plasticity and the formation of fear memory (Bauer et al., 2002), whereas increasing NR2B-containing NMDARs in the forebrain (including the amygdala) enhances fear memory formation (Tang et al., 1999; Hawasli et al., 2007). Here, MgT treatment did not upregulate NMDAR signaling in the basolateral amygdala, which might explain the lack of enhancement of LTP, presynaptic puncta density, and the amygdala-dependent delay fear conditioning.
The hippocampus is involved in holding temporal (timing) information during trace conditioning (Solomon et al., 1986; McEchron et al., 1998). The disruption of NMDAR signaling in the hippocampus disrupts trace, but not delay, fear conditioning (Misane et al., 2005). Our previous studies indicate that MgT treatment enhances NMDAR signaling and synaptic plasticity in the hippocampus (Slutsky et al., 2010). Hence, it was expected that MgT treatment might improve the ability to associate the CS with US when the time interval between them was prolonged (Fig. 1E). Other studies show that other brain regions within the PFC, such as PrL-PFC (Gilmartin and Helmstetter, 2010) and/or anterior cingulate cortex (Han et al., 2003), also contribute to trace fear conditioning. Since MgT treatment enhanced NMDAR signaling in the PFC, it might have contributed to the improved ability to process temporal information in MgT-treated rats.
MgT treatment enhanced retention of extinction
In the current study, we found that elevating brain magnesium with MgT enhanced the efficacy of extinction, as indicated by the improved retention of extinction in MgT-treated rats, when the treatment was given before (Fig. 2A,B), but not after (Fig. 2C), extinction learning. The effects of extinction persisted up to 1 month after extinction learning (prevention of spontaneous recovery; Fig. 3A) and remained intact even after contextual shift (prevention of renewal; Fig. 3B). However, upon recurrence of the original harmful stimulus in the same context, MgT-treated rats exhibited a return of the original fear memory (reinstatement; Fig. 3C), indicating that MgT treatment did not erase the original fear memory.
Interestingly, MgT treatment enhanced the retention of extinction when it was given before, but not after, the extinction learning. We have shown that it takes ∼2 weeks to elevate brain magnesium levels and enhance memory by MgT (Slutsky et al., 2010). Memory consolidation typically occurs within less than 1 d following learning processes (Alberini, 2005). Hence, in Figure 2C experiments, the MgT treatment should have no effects on the consolidation processes of the extinction memory. Since MgT was given before the retention test (for 4 weeks), one would rather expect that the retrieval processes of the extinction memory would be influenced by elevation of brain magnesium. Based on the above logic, we speculate that the attenuation of fear responses by MgT treatment (following extinction learning) may not be due to the enhancement of the retrieval of the extinction memory; rather, it is more likely to be due to the enhancement of the consolidation of the extinction memory. However, we cannot exclude the possibility that MgT treatment might influence the fear memory decay processes and/or its vulnerability to extinction, despite the apparent lack of effects on the amygdala-dependent fear memory processes by the MgT treatment.
The fear extinction data suggest that MgT treatment might enhance functions of brain regions implicated in consolidating and retaining the extinction memory. Synaptic plasticity within the IL-PFC and/or hippocampus is believed to be crucial for extinction learning and retention of extinction (Myers and Davis, 2007; Quirk and Mueller, 2008). Increasing synaptic plasticity in the forebrain by increasing NR2B-containing NMDARs enhances extinction learning and the retention of extinction (Tang et al., 1999; Hawasli et al., 2007). Infusion of BDNF into the IL-PFC or hippocampus attenuates fear responses even without extinction (Peters et al., 2010). MgT treatment increased activation of NMDAR signaling, BDNF expression in the PFC, and presynaptic puncta and synaptic plasticity in the IL-PFC (Figs. 4–6) and hippocampus (Slutsky et al., 2010). Hence, we suggest that the effects of MgT treatment on retention of extinction are mediated, at least partially, by enhancement of the IL-PFC and/or hippocampus.
MgT treatment also increased the density of presynaptic puncta in the PrL-PFC. Recent studies show that an increase in activity within the PrL-PFC increases expression of conditioned fear, while inhibition of the PrL-PFC reduces fear expression (Corcoran and Quirk, 2007; Peters et al., 2009; Sierra-Mercado et al., 2011). However, we did not observe any enhancement in fear expression in MgT-treated rats. It has been suggested that the IL-PFC and PrL-PFC incorporate information from different brain structures, including the amygdala, to gate a proper expression of fear (Sotres-Bayon and Quirk, 2010). Therefore, the coenhancement of both IL-and PrL-PFC might result in the normal expression of conditioned fear in MgT-treated rats.
It is worth noting that our studies report on the effects of MgT treatment on three brain regions: the PFC, hippocampus, and amygdala. However, several studies suggest that these regions may not be the only regions controlling fear memory formation, fear expression, and/or retention of extinction (Sanders et al., 2003; Lebrón et al., 2004; Maren, 2008). Therefore, we cannot exclude the potential effects of MgT treatment on other brain regions that might also contribute to the behavioral readouts reported here.
Regional specificity of MgT
The most interesting finding of our studies is that an elevation of brain magnesium induced increases in the NR2B-containing NMDARs, activation of NMDAR signaling, BDNF expression, and synaptic plasticity in the PFC (present study) and hippocampus (Slutsky et al., 2010), but not in the amygdala (present study). The remaining question is: why are NMDARs and synaptic plasticity in the PFC and hippocampus sensitive to an elevation in [Mg2+]o, while in the lateral amygdala they are not?
We have shown that an increase in the blockage of NMDARs caused by elevating [Mg2+]o reduces the NMDAR current near resting membrane potentials, which triggers a compensatory upregulation of NR2B-containing NMDARs in the synapse, leading to enhancement of NMDAR current and synaptic plasticity (Slutsky et al., 2004, 2010). In this scenario, the sensitivity of NMDAR current to variations in [Mg2+]o is the key for the magnesium-induced effects. Interestingly, the synaptic NMDAR current in thalamic–amygdala synapses are strongly blocked by physiological [Mg2+]o such that elevation of [Mg2+]o has little influence on synaptic NMDAR current near resting membrane potentials (Weisskopf and LeDoux, 1999; Miwa et al., 2008). If the homeostatic regulation of synaptic NMDARs by [Mg2+]o is applicable to the lateral amygdala synapses, then one might predict that lateral amygdala synapses have higher synaptic NMDAR current and should have higher NR2B-containing NMDARs under physiological [Mg2+]o. In the current study, we did observe a higher INMDA/AMPA ratio in the lateral amygdala synapses than that in the IL-PFC synapses (by ∼66%; Fig. 7A,C) under physiological [Mg2+]o conditions (0.8 mm). Furthermore, the portion of NR2B-containing NMDAR current is ∼40% in the lateral amygdala, 20% in hippocampus (Miwa et al., 2008), and ∼15% in the PFC synapses (Zhao et al., 2005). Electron microscopy studies show that NMDARs at synapses in the lateral amygdala contain larger number of NR2B subunits than synapses in the hippocampal CA1 (Miwa et al., 2008). Since the degree of LTP is strongly correlated with the size of the synaptic NMDA current, the above data might also explain why the LTP magnitude in the lateral amygdala synapses is higher than that in the IL-PFC under control conditions (Figs. 6, 7B,D). Therefore, we speculate that the lack of upregulation of synaptic NMDA current under higher [Mg2+]o in vitro and NR2B subunit in MgT-treated animals might be due to higher sensitivity of synaptic NMDARs in the lateral amygdala to blockage by physiological [Mg2+]o. Hence, small elevations in [Mg2+]o may not be able to further increase synaptic NMDAR and LTP. These speculations might be applicable to the thalamolateral amygdala synapses, which are involved in the formation of conditioned fear memory (LeDoux, 2000). It might not apply to NMDARs in the corticolateral amygdala synapses, which have higher sensitivity to changes in [Mg2+]o (Weisskopf and LeDoux, 1999). Furthermore, the expression level of NR2B subunit is high in the dorsal part of lateral amygdala but was not homogenous throughout the basolateral amygdala (Rodrigues et al., 2000). The exact molecular mechanisms underlying the different sensitivity of NMDARs to changes in [Mg2+]o in amygdala remain to be determined. Identification of such mechanisms might open new avenues to differentially manipulate circuitries critical for fear memory and extinction processes, which might be helpful in developing new treatments for anxiety disorders such as phobias and PTSD.
Finally, emerging evidence shows that dietary factors might play a role in fear extinction and anxiety-like behavior. For example, dietary zinc restriction rescues the impairment in fear extinction in a mouse model of impaired fear extinction (Whittle et al., 2010). Dietary magnesium restriction induces an increase in anxiety-like behavior in mice (Singewald et al., 2004). Our studies suggest that long-term elevation of brain magnesium might enhance NMDARs functioning selectively in brain regions involved in what was previously described as “top-down control over amygdala” (Rauch et al., 2006) and might help to enhance the efficacy of fear extinction.
Footnotes
This work was supported by grants from Tsinghua-Yue-Yuen Medical Sciences Fund (to N.A.), China Scientific Foundation (Grant for Post Doc 023205002, to N.A.), the National Natural Science Foundation of China (Grant for Youth Scientists 30901796/H3101, to N.A; and 30630026, to G.L.), National Basic Research Program of China (Grant 2006CB3031, to G.L.), and The National High Technology Research and Development Program of China (2007AA02Z443, to G.L.). M.Z. was supported by grants from the EJLB-CIHR Michael Smith Chair in Neurosciences and Mental Health, Canada Research Chair, and CIHR operating grants (CIHR66975 and CIHR84256). X.Y.L. and T.C. were supported by postdoctoral fellowships from Fragile X Research Foundation of Canada. We thank Jack Feldman and David Glanzman for comments on the manuscript.
Guosong Liu declares that he is a cofounder of Magceutics, a company whose goal is to develop drugs to treat age-dependent memory decline and Alzheimer's disease. He also reports his United States patent application on Magnesium-l-Threonate. All other authors report no financial interests or potential conflicts of interest related to the current study.
References
- Akirav I, Maroun M. Ventromedial prefrontal cortex is obligatory for consolidation and reconsolidation of object recognition memory. Cereb Cortex. 2006;16:1759–1765. doi: 10.1093/cercor/bhj114. [DOI] [PubMed] [Google Scholar]
- Alberini CM. Mechanisms of memory stabilization: are consolidation and reconsolidation similar or distinct processes? Trends Neurosci. 2005;28:51–56. doi: 10.1016/j.tins.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE. NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci. 2002;22:5239–5249. doi: 10.1523/JNEUROSCI.22-12-05239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard RJ, Blanchard DC. Crouching as an index of fear. J Comp Physiol Psychol. 1969;67:370–375. doi: 10.1037/h0026779. [DOI] [PubMed] [Google Scholar]
- Bouton ME. Context, time, and memory retrieval in the interference paradigms of Pavlovian learning. Psychol Bull. 1993;114:80–99. doi: 10.1037/0033-2909.114.1.80. [DOI] [PubMed] [Google Scholar]
- Corcoran KA, Quirk GJ. Activity in prelimbic cortex is necessary for the expression of learned, but not innate, fears. J Neurosci. 2007;27:840–844. doi: 10.1523/JNEUROSCI.5327-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. The role of the amygdala in fear and anxiety. Annu Rev Neurosci. 1992;15:353–375. doi: 10.1146/annurev.ne.15.030192.002033. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Poulos AM. The neuroscience of mammalian associative learning. Annu Rev Psychol. 2005;56:207–234. doi: 10.1146/annurev.psych.56.091103.070213. [DOI] [PubMed] [Google Scholar]
- Farinelli M, Deschaux O, Hugues S, Thevenet A, Garcia R. Hippocampal train stimulation modulates recall of fear extinction independently of prefrontal cortex synaptic plasticity and lesions. Learn Mem. 2006;13:329–334. doi: 10.1101/lm.204806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foa EB, Franklin ME, Moser J. Context in the clinic: how well do cognitive-behavioral therapies and medications work in combination? Biol Psychiatry. 2002;52:987–997. doi: 10.1016/s0006-3223(02)01552-4. [DOI] [PubMed] [Google Scholar]
- Gilmartin MR, Helmstetter FJ. Trace and contextual fear conditioning require neural activity and NMDA receptor-dependent transmission in the medial prefrontal cortex. Learn Mem. 2010;17:289–296. doi: 10.1101/lm.1597410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han CJ, O'Tuathaigh CM, van Trigt L, Quinn JJ, Fanselow MS, Mongeau R, Koch C, Anderson DJ. Trace but not delay fear conditioning requires attention and the anterior cingulate cortex. Proc Natl Acad Sci U S A. 2003;100:13087–13092. doi: 10.1073/pnas.2132313100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawasli AH, Benavides DR, Nguyen C, Kansy JW, Hayashi K, Chambon P, Greengard P, Powell CM, Cooper DC, Bibb JA. Cyclin-dependent kinase 5 governs learning and synaptic plasticity via control of NMDAR degradation. Nat Neurosci. 2007;10:880–886. doi: 10.1038/nn1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herry C, Ciocchi S, Senn V, Demmou L, Müller C, Lüthi A. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- Hobin JA, Ji J, Maren S. Ventral hippocampal muscimol disrupts context-specific fear memory retrieval after extinction in rats. Hippocampus. 2006;16:174–182. doi: 10.1002/hipo.20144. [DOI] [PubMed] [Google Scholar]
- Lebrón K, Milad MR, Quirk GJ. Delayed recall of fear extinction in rats with lesions of ventral medial prefrontal cortex. Learn Mem. 2004;11:544–548. doi: 10.1101/lm.78604. [DOI] [PubMed] [Google Scholar]
- LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- Maren S. Pavlovian fear conditioning as a behavioral assay for hippocampus and amygdala function: cautions and caveats. Eur J Neurosci. 2008;28:1661–1666. doi: 10.1111/j.1460-9568.2008.06485.x. [DOI] [PubMed] [Google Scholar]
- McEchron MD, Bouwmeester H, Tseng W, Weiss C, Disterhoft JF. Hippocampectomy disrupts auditory trace fear conditioning and contextual fear conditioning in the rat. Hippocampus. 1998;8:638–646. doi: 10.1002/(SICI)1098-1063(1998)8:6<638::AID-HIPO6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature. 2002;420:70–74. doi: 10.1038/nature01138. [DOI] [PubMed] [Google Scholar]
- Misane I, Tovote P, Meyer M, Spiess J, Ogren SO, Stiedl O. Time-dependent involvement of the dorsal hippocampus in trace fear conditioning in mice. Hippocampus. 2005;15:418–426. doi: 10.1002/hipo.20067. [DOI] [PubMed] [Google Scholar]
- Miwa H, Fukaya M, Watabe AM, Watanabe M, Manabe T. Functional contributions of synaptically localized NR2B subunits of the NMDA receptor to synaptic transmission and long-term potentiation in the adult mouse CNS. J Physiol. 2008;586:2539–2550. doi: 10.1113/jphysiol.2007.147652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monfils MH, Cowansage KK, Klann E, LeDoux JE. Extinction-reconsolidation boundaries: key to persistent attenuation of fear memories. Science. 2009;324:951–955. doi: 10.1126/science.1167975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MA, LeDoux JE. Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behav Neurosci. 1995;109:681–688. doi: 10.1037//0735-7044.109.4.681. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2007;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- Myers KM, Ressler KJ, Davis M. Different mechanisms of fear extinction dependent on length of time since fear acquisition. Learn Mem. 2006;13:216–223. doi: 10.1101/lm.119806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov I. Conditioned reflexes: an investigation of the physiological activity of the cerebral cortex. London: Oxford UP; 1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Kalivas PW, Quirk GJ. Extinction circuits for fear and addiction overlap in prefrontal cortex. Learn Mem. 2009;16:279–288. doi: 10.1101/lm.1041309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–1290. doi: 10.1126/science.1186909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps EA, Delgado MR, Nearing KI, LeDoux JE. Extinction learning in humans: role of the amygdala and vmPFC. Neuron. 2004;43:897–905. doi: 10.1016/j.neuron.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch SL, Shin LM, Phelps EA. Neurocircuitry models of posttraumatic stress disorder and extinction: human neuroimaging research—past, present, and future. Biol Psychiatry. 2006;60:376–382. doi: 10.1016/j.biopsych.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Rodrigues SM, LeDoux JE, Morrison JH. Differential expression of the N-methyl-D-aspartate receptor NR2B subunit in the lateral nucleus divisions of the rat amygdala. Soc Neurosci Abstract. 2000;26:465–411. [Google Scholar]
- Sanders MJ, Wiltgen BJ, Fanselow MS. The place of the hippocampus in fear conditioning. Eur J Pharmacol. 2003;463:217–223. doi: 10.1016/s0014-2999(03)01283-4. [DOI] [PubMed] [Google Scholar]
- Sierra-Mercado D, Padilla-Coreano N, Quirk GJ. Dissociable roles of prelimbic and infralimbic cortices, ventral hippocampus, and basolateral amygdala in the expression and extinction of conditioned fear. Neuropsychopharmacology. 2011;36:529–538. doi: 10.1038/npp.2010.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson T, Doyère V, Cain CK, LeDoux JE. Long-term potentiation in the amygdala: a cellular mechanism of fear learning and memory. Neuropharmacology. 2007;52:215–227. doi: 10.1016/j.neuropharm.2006.06.022. [DOI] [PubMed] [Google Scholar]
- Singewald N, Sinner C, Hetzenauer A, Sartori SB, Murck H. Magnesium-deficient diet alters depression- and anxiety-related behavior in mice: influence of desipramine and Hypericum perforatum extract. Neuropharmacology. 2004;47:1189–1197. doi: 10.1016/j.neuropharm.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Slutsky I, Sadeghpour S, Li B, Liu G. Enhancement of synaptic plasticity through chronically reduced Ca2+ flux during uncorrelated activity. Neuron. 2004;44:835–849. doi: 10.1016/j.neuron.2004.11.013. [DOI] [PubMed] [Google Scholar]
- Slutsky I, Abumaria N, Wu L, Huang C, Zhang L, Li B, Zhao X, Govindarajan A, Zhao MG, Zhuo M, Tonegawa S, Liu G. Enhancement of learning and memory by elevating brain magnesium. Neuron. 2010;65:165–177. doi: 10.1016/j.neuron.2009.12.026. [DOI] [PubMed] [Google Scholar]
- Solomon PR, Vander Schaaf ER, Thompson RF, Weisz DJ. Hippocampus and trace conditioning of the rabbit's classically conditioned nictitating membrane response. Behav Neurosci. 1986;100:729–744. doi: 10.1037//0735-7044.100.5.729. [DOI] [PubMed] [Google Scholar]
- Sotres-Bayon F, Quirk GJ. Prefrontal control of fear: more than just extinction. Curr Opin Neurobiol. 2010;20:231–235. doi: 10.1016/j.conb.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, LeDoux JE. Distinct populations of NMDA receptors at subcortical and cortical inputs to principal cells of the lateral amygdala. J Neurophysiol. 1999;81:930–934. doi: 10.1152/jn.1999.81.2.930. [DOI] [PubMed] [Google Scholar]
- Westbrook RF, Iordanova M, McNally G, Richardson R, Harris JA. Reinstatement of fear to an extinguished conditioned stimulus: two roles for context. J Exp Psychol Anim Behav Process. 2002;28:97–110. [PubMed] [Google Scholar]
- Whittle N, Hauschild M, Lubec G, Holmes A, Singewald N. Rescue of impaired fear extinction and normalization of corticoamygdala circuit dysfunction in a genetic mouse model by dietary zinc restriction. J Neurosci. 2010;30:13586–13596. doi: 10.1523/JNEUROSCI.0849-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47:859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]