

Abstract
The analgesic properties of the opium poppy Papever somniferum were first mentioned by Hippocrates around 400 BC, and opioid analgesics remain the mainstay of pain management today. These drugs can cause the serious side-effect of respiratory depression that can be lethal with overdose, however the critical brain sites and neurochemical identity of the neurons mediating this depression are unknown. By locally manipulating neurotransmission in the adult rat, we identify the critical site of the medulla, the preBötzinger complex, that mediates opioid-induced respiratory depression in vivo. Here we show that opioids at the preBötzinger complex cause respiratory depression or fatal apnea, with anesthesia and deep-sleep being particularly vulnerable states for opioid-induced respiratory depression. Importantly, we establish that the preBötzinger complex is fully responsible for respiratory rate suppression following systemic administration of opioid analgesics. The site in the medulla most sensitive to opioids corresponds to a region expressing neurokinin-1 receptors, and we show in rhythmically active brainstem section in vitro that neurokinin-1 receptor-expressing preBötzinger complex neurons are selectively inhibited by opioids. In summary, neurokinin-1 receptor-expressing preBötzinger complex neurons constitute the critical site mediating opioid-induced respiratory rate depression, and the key therapeutic target for its prevention or reversal.
Introduction
The analgesic properties of the opium poppy Papaver somniferum has been recognized for centuries, and opioid analgesics remain the mainstay of pain management today (Gutstein, 2001). The most widely recognized and potentially fatal acute side-effect of opioid analgesics is respiratory depression (Gutstein, 2001; Pattinson, 2008), and this is of particular concern when opioid drugs are abused (Gutstein, 2001; Hall et al., 2008). Enhanced susceptibility to opioid-induced respiratory depression among a subpopulation of patients also limits the degree of analgesia that can be provided (Agrò et al., 2004; Desrosiers, 2006). Yet despite this serious problem, the key sites in the brain where opioid analgesics act to suppress breathing have not been identified (Gutstein, 2001; Pattinson, 2008).
Rhythmic breathing is essential to life in mammals, and this behavior is generated by a complex neuronal network in the brainstem. Embedded in this network is the preBötzinger complex (preBötC), a region of the medulla that plays an essential role in generating the basic respiratory rhythm in vitro (Smith et al., 1991; Gray et al., 1999) and in vivo (Gray et al., 2001; Tan et al., 2008). Destruction of neurokinin-1 receptor (NK1R)-expressing preBötC neurons leads to irregular breathing (Gray et al., 2001; McKay et al., 2005), with this and other evidence suggesting that preBötC neurons are essential for normal respiratory rhythm and have the capacity to even abolish breathing in the intact conscious organism (Tan et al., 2008). In neonatal rodents, NK1R and μ-opioid receptors are coexpressed in preBötC neurons, and application of opioids to the preBötC slows respiratory rate in vitro (Gray et al., 1999). Yet there are multiple sites in the intact brain, including neurons of the respiratory network other than the preBötC, that express μ-opioid receptors (Xia and Haddad, 1991; Gray et al., 1999; McCrimmon and Alheid, 2003), which when activated by systemic administration of opioids could directly or indirectly influence the respiratory network (Gray et al., 1999; Lalley, 2003; Zhang et al., 2007). Nevertheless, the severely depressed respiratory rate observed in the presence of opioid drugs indicates a key effect on the site(s) generating respiratory rhythm.
Given that opioids depress respiratory rate in vitro (Gray et al., 1999; Manzke et al., 2003), it has been suggested that the preBötC mediates opioid-induced respiratory depression. However, the critical role of the preBötC in mediating respiratory rate depression following systemic opioids has not been demonstrated. Here we propose that NK1R-expressing preBötC neurons constitute the critical site mediating opioid-induced respiratory rate depression. By manipulating neurotransmission in the medulla in vivo, we show that opioids at the preBötC cause slowing of respiratory rate, with anesthesia and deep-sleep being particularly vulnerable states for this depression. This effect is due to the action of opioids at a spatially restricted site corresponding to NK1R-expressing preBötC neurons. We then demonstrate that the preBötC fully mediates respiratory rate depression following a clinically relevant systemic administration of opioids. Furthermore, in rhythmically active brainstem sections containing the preBötC, stimulation of μ-opioid receptors preferentially inhibits NK1R-expressing preBötC neurons that includes a postsynaptic mechanism.
Materials and Methods
All procedures were performed in accordance with the recommendations of the Canadian Council on Animal Care, and were approved by the University of Toronto Animal Care Committee, and the University of Alberta Animal Welfare Committee.
In vivo modulation of the preBötC
Anesthetized preparations.
To determine the effect of μ-opioid receptor stimulation at the preBötC on respiratory activities, we used reverse-microdialysis to perfuse μ-opioid receptor agonists into the preBötC of anesthetized adult male Wistar rats (n = 21). The experimental procedures were adapted from a previous study (Steenland et al., 2008). Rats were anesthetized with isoflurane (2–2.5%) and tracheostomized. The rats spontaneously breathed a 50%-oxygen gas mixture (balance nitrogen), and the femoral artery was catheterized for blood pressure measurement. Core body temperature was monitored with a rectal probe and maintained between 36.5 and 38°C (TC-1000 Temperature Controller, CWE Inc.). Diaphragm and genioglossus muscle activities were recorded using modified needle electrodes (Grass Technologies) and electrical signals were amplified and filtered (BMA-400 Bioamplifier, CWE Inc.). The electrocardiogram was removed from the diaphragm signal using an electronic blanker (SB-1, CWE Inc.). The moving-time averages of the signals were also obtained (times constant = 100 ms, MA-821/RSP Moving Averager, CWE Inc.). Raw signals were recorded on a computer at a sampling rate of 2000 Hz, whereas the moving-time averaged signals were sampled at 200 Hz (Spike 2 software version 6, and Micro-1401, Cambridge Electronic Design). Then, the rat was placed in the prone position in a stereotaxic apparatus (model SAS-4100, ASI Instruments Inc.) with blunt ear bars. To ensure consistent positioning between animals, the flat skull position was achieved with an alignment tool (model 944, Kopf Instruments). Using a dorsal approach, a microdialysis probe (CMA11, Chromatography Sciences Company), continuously perfused at 3 μl/min with freshly made artificial CSF (aCSF), was inserted into the brainstem 2.0 mm dorsal to the preBötC, by placing the probe 12.2 mm posterior, 2 mm lateral and 8.5 mm ventral to bregma. The composition of the aCSF was (in mm): 125 NaCl, 3 KCl, 1 KH2PO4, 2 CaCl2, 1 MgSO4, 25 NaHCO3, and 30 glucose. The pH was adjusted at 7.4 by bubbling CO2 in the aCSF. The microdialysis probe was then progressively lowered into the brainstem while recording all physiological variables, and was left in place when genioglossus muscle activity decreased by ∼30% which typically occurred with the probe ∼ 10.5 mm ventral to bregma. With experience derived from extensive preliminary experiments and anatomical determinations, we note that this response is a good initial marker of the appropriate depth of the probe before subsequent confirmation by postmortem histology.
Baseline levels of the physiological variables were recorded for at least 60 min. Following this control period, the μ-opioid receptor agonists [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO, 5 μm, Tocris Bioscience) or fentanyl (fentanyl citrate, 100 μm, Sandoz Canada Inc.) were then added to the aCSF for perfusion of the preBötC. The responses to DAMGO (n = 9 rats) and fentanyl (n = 8 rats) were recorded for the next 30–60 min and were then followed by perfusion of the μ-opioid receptor antagonist naloxone (naloxone hydrochloride solution, 100 μm, Sandoz Canada Inc.) in aCSF for another 30 min. In a separate set of experiments, we also perfused a solution of 200 μm DAMGO into the preBötC to determine whether this dose would abolish respiratory activity (n = 3). In the event of cessation of breathing, the rat was artificially ventilated and naloxone (100 μm) was perfused into the preBötC until respiratory activity resumed. The rat was then killed by overdose with isoflurane and the brain was fixed with formalin, cut with cryostat at 50 μm, and stained with neutral red for subsequent histology. Breath-by-breath measurements of respiratory rate, inspiratory and expiratory durations, diaphragm and genioglossus muscle amplitudes were averaged over the last 5 min of microdialysis perfusion of the preBötC with aCSF, DAMGO or fentanyl, and naloxone.
Freely behaving preparations.
To determine the effects of μ-opioid receptor stimulation at the preBötC on respiratory activity in conscious adult rats across sleep–wake states, we perfused DAMGO bilaterally into both preBötC in freely behaving male adult Wistar rats (n = 3). One week before the experiments, sterile surgery was performed under isoflurane anesthesia to implant the rats with electroencephalogram (EEG) and postural (neck) muscle electrodes to identify sleep–wake states, and diaphragm and genioglossus electrodes for respiratory muscle recordings. Two insulated, multistranded stainless steel wires (AS631, Cooner Wire) were implanted bilaterally and directly into the genioglossus muscle and secured with sutures and tissue glue. To record diaphragm activity, two wires were sutured onto the costal diaphragm via an abdominal approach. Tests for the accurate placement of the genioglossus electrodes and their function throughout the experiments included electromyogram recordings during surgery and observing tongue movements in response to electrical stimulation of the electrodes (Morrison et al., 2003). The rat was placed in the prone position in the stereotaxic apparatus (model SAS-4100) with blunt ear bars, and three holes were drilled into the skull for the placement of the EEG electrodes. Two stainless steel screws (size 0–80 × 1/16, Plastics One Inc.) were placed ∼2 mm to the right and 2 mm anterior to bregma, and 2 mm to the left and 3 mm posterior to bregma for EEG activity; the third electrode was placed 3 mm to the left and 3 mm anterior to bregma. Insulated multistranded stainless steel wires were also sutured on the dorsal neck muscles to record the electromyogram. Finally, two microdialysis guide cannulas (AG-X, Eicom Corp.) were positioned 5.0 mm above the preBötC by placing them 12.2 mm posterior, 2 mm either side and 5.5 mm ventral to bregma with the guide cannulas secured in place with dental acrylic. Postsurgical care consisted of a subcutaneous injection of an anti-inflammatory drug (ketoprofen, 2 mg kg−1) and an analgesic (buprenorphine, 1 mg/kg). The rats recovered for 1 week before experiment.
On the day of the experiment, the rat was transiently anesthetized (isoflurane 2–2.5%) for the careful and accurate placement of the microdialysis probes (CMA-11). The microdialysis probes were perfused with aCSF at 3 μl/min, and were lowered slowly into the guides using the same approach as in the anesthetized preparation, i.e., to ensure consistent placements within and between preparations. These careful placements also used the change in genioglossus muscle activity as the marker of appropriate probe depth, and so also required an immobile animal as spontaneous changes in motor activity in the behaving animal would obscure the signal and preclude slow and progressive lowering of the probe. Following placements of the microdialysis probes the rat was connected to the recording apparatus which allowed electrophysiological signals to be recorded while the rat moved freely in a Plexiglas bowl filled with fresh bedding. The Plexiglas bowl was placed on a rotating turntable (Raturn, BASi) which automatically adjusts its position when the rat moves to avoid entanglements of the microdialysis tubing and recording cable. After a recovery period from anesthesia of at least 120 min (with the rat apparently behaving normally after ∼45 min), the EEG, neck and diaphragm muscle activities were recorded for 30 min during bilateral perfusion of aCSF into both preBötC. DAMGO (20 μm) was then added to the perfusing solution for 90 min followed by perfusion of naloxone (100 μm) into the preBötC for a further 90 min. Measurements started at ∼11.00 and ended at ∼14.30. Data were amplified, filtered, moving-time averaged and sampled as described in the anesthetized experiments. Respiratory rate, diaphragm and neck muscle amplitudes were averaged every 10 s time bin. EEG and neck muscle activities were also displayed every 10 s to identify the prevailing sleep/wake states according to standard criteria (Morrison et al., 2003). EEG frequencies in the following frequency bands: δ2 (0.5–2 Hz), δ1 (2–4 Hz), θ (4–7.5 Hz), α (7.5–13.5 Hz), β1 (13.5–20 Hz), β2 (20–30 Hz) were analyzed using Spike 2 software. In addition, ratio of high to low frequencies (i.e., the β2/δ1 ratio) was calculated for each 10 s bin as an index of relative EEG activation. Values were averaged for each sleep–wake state occurring over the last 30 min of aCSF, DAMGO, and naloxone perfusions.
Construction of correlation maps
We constructed correlation maps to relate the location of the intervention sites with the resultant effect on respiratory activity. The rationale for the construction of correlation maps is that for a locus of effect of opioids at any particular brainstem site, the latency for the drug to diffuse through the tissue and to progressively change respiratory activity will vary as a function of the distance of the probe from the effective site. We first determined the locations of the intervention (perfusion) sites by using anatomical markers and standard brain maps (Paxinos and Watson, 1998) (see supplemental Table 1, available at www.jneurosci.org as supplemental material for coordinates). To determine the anterior–posterior coordinates, we used the caudal end of the facial nucleus as the reference point which is located 11.6 mm posterior to bregma. The dorsal-ventral and medial-lateral coordinates were defined using the nucleus ambiguus and standard brain maps (Paxinos and Watson, 1998). For each animal, we then calculated the latencies to a 10% change in respiratory rate and genioglossus activity in response to fentanyl. For every possible sets of coordinates within a 3-dimensional grid (resolution 50 μm) spanning from 11.0 to 13.0 mm caudal to bregma, we then calculated the distances from the perfusion sites to the corresponding set of coordinates (supplemental Fig. 1, supplemental Table 1, available at www.jneurosci.org as supplemental material) and correlated these distances with the latencies for the effects on respiratory activities. Using Matlab 12 software (MathWorks), the correlation coefficients (0 < r2 < 1) were then calculated for every set of coordinates and plotted as color pixels (blue to red) in standard brain maps (Paxinos and Watson, 1998). To further identify the regions statistically most strongly associated with the latency for a change in respiratory rate or genioglossus activity, the locations of the “hot spot” regions were identified by replotting the correlation coefficients with p < 0.05 and r2 > 0.8.
Immunohistochemistry
We used NK1R immunohistochemistry to locate the preBötC from brainstem sections (Gray et al., 2001) (n = 3 male adult Wistar rats) using techniques previously described (Pagliardini et al., 2003). Following fixation in 4% paraformaldehyde and cryoprotection in 30% sucrose, the brains were frozen and cut in 50 μm sections. The antibodies used were rabbit anti-NK1R (1:1000, AB-N04, Advanced Targeting Systems) and donkey anti-rabbit immunoglobin G (1:100, Jackson ImmunoResearch Laboratories). Sections were stained with a chromogen diaminobenzidine solution and were mounted on slides, dried, and sealed with Cytoseal 280. Sections were digitized with a CCD camera (Infinity 1, Olympus) and microscope (BX-41, Olympus).
Bilateral perfusion of the preBötC and systemic administration of opioids
To determine whether the preBötC is the critical site where systemically administered opioids act to depress respiratory rate, we administered, in male adult rats, a single intravenous dose of fentanyl in the presence of bilateral microdialysis perfusion into the preBötC of aCSF (control, n = 4) or naloxone (n = 9). The microdialysis probes were inserted 12.2 mm posterior, 2 mm either side of, and 10.5 mm ventral to bregma as described previously. After baseline recording for 30 min with perfusion of aCSF into the preBötC, we then added naloxone (300 μm) to the perfusing solution for 45 min, or maintained aCSF perfusion as the control. We then injected fentanyl into the femoral vein at a dose of 1 μg/kg over 1 min, which produces a robust (∼30%) suppression of respiratory rate. As described previously (see section: Construction of correlation maps), we also correlated the distances from the perfusion sites to each preBötC with the capacity of naloxone at the preBötC to prevent the depression of respiratory rate, genioglossus muscle activity and blood pressure by the systemic intravenously administered opioids.
Medullary slice preparation and electrophysiology
Newborn male Sprague Dawley rats (postnatal days 1–4) were anesthetized by inhalation of isoflurane (2–3%), and the neuraxis was isolated in artificial CSF (aCSF-in vitro) (in mm): 128 NaCl, 3 KCl, 1.5 CaCl2, 1 MgSO4, 23.5 NaHCO3, 0.5 NaH2PO4, and 30 d-glucose equilibrated with 95% O2-5% CO2 at room temperature. Rhythmically active medullary slice preparations were produced as described previously (Smith et al., 1991). A single transverse slice containing the preBötC complex was cut (650–750 μm thick) and transferred to a chamber containing aCSF-in vitro. Tetramethylrhodamine-conjugated Substance P (0.5 μm, Alberta Peptide Institute) or carboxytetramethylrhodamine-conjugated Substance P (0.5 μm, Invitrogen) were used to label NK1R-expressing neurons for subsequent whole-cell patch-clamp recordings (Pagliardini et al., 2005). Preincubation of sections with the NK1R antagonist RP67580 (10 μm, Tocris Bioscience) before exposure to conjugated Substance P was performed to block receptor internalization. Recordings of population inspiratory activity from hypoglossal nerve roots were made via suction electrodes. Signals were amplified, filtered (0.3–3 kHz), rectified, integrated, and recorded on a computer using a Digidata 1322A analog-to-digital board and Axoscope software (Molecular Devices). NK1R-expressing and non-NK1R-expressing neurons in the preBötC were identified under epifluorescence using a CCD camera (ICD-47; Ikegami). Whole-cell patch recordings were then obtained from NK1R-expressing and nonexpressing neurons. Input resistance was measured by injecting small hyperpolarizing current pulses (0.5 s duration) and recording the resultant change in membrane potential. All drugs were made freshly from stock solution and bath applied. TTX (0.5–1 μm) and DAMGO (0.5 μm) were purchased from Sigma.
Statistical analysis
For the anesthetized experiments with local perfusion of DAMGO or fentanyl into the preBötC, we used a one-way repeated-measures ANOVA design with post hoc Bonferroni t tests for comparison with the aCSF controls. For the freely behaving experiments, we used a Kruskal–Wallis ANOVA on Ranks with Dunn's post hoc test for comparison with the aCSF controls. Correlations were performed using the Spearman's Rank test. For experiments with intravenous injection of fentanyl and aCSF versus naloxone at the preBötC, we used a two-way ANOVA with one repeated-measure (before and after intravenous fentanyl) and post hoc Bonferroni t tests for comparison with the aCSF controls. For the in vitro experiments, we used z tests for comparison of NK1R versus non-NK1R neurons, and Student's paired t tests for comparing the changes in input resistance after drug application. All values are reported as means ± SEM. Differences were considered significant if p < 0.05.
Results
Opioids at the preBötC depress breathing and cause persistent apnea
To determine whether the site in the medulla responsible for the generation of breathing in vivo (Gray et al., 2001; Tan et al., 2008) is sensitive to opioid analgesics, we applied μ-opioid receptor agonists into the preBötC in isoflurane-anesthetized adult rats. Continuous local unilateral application (Fig. 1A) of the synthetic μ-opioid receptor agonist DAMGO (5 μm) or fentanyl (100 μm) into the preBötC caused sustained slowing of respiratory rate, increased respiratory rate variability (ataxic breathing), and suppressed tongue (genioglossus) muscle, but not diaphragm muscle activation (Fig. 1B; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The significant reductions in respiratory rate with DAMGO (44.4 ± 2.0 to 25.6 ± 2.0 breaths · min−1, p < 0.0001, n = 9) were produced solely by increased expiratory duration (p < 0.0001, Fig. 1C), with no changes in inspiratory duration (p = 0.493). DAMGO at the preBötC also increased respiratory rate variability (coefficient of variation: 4.4 ± 0.6 to 38.6 ± 13.5%, p = 0.018), decreased genioglossus activity (0.58 ± 0.12 to 0.15 ± 0.12 arbitrary units, p = 0.022, n = 8) but did not affect the amplitude of diaphragm activation (0.54 ± 0.19 to 0.43 ± 0.15 arbitrary units, p = 0.86). The reduction in respiratory rate, increased respiratory rate variability, and prolonged expiratory duration were all reversed by the perfusion of μ-opioid receptor antagonist naloxone (100 μm) into the preBötC, i.e., the values with naloxone were returned to levels indistinguishable from aCSF controls (p = 0.089, p = 1.000, and p = 0.35, respectively, Fig. 1C). Such reversal of opioid-induced respiratory depression was also observed when naloxone was coapplied with DAMGO into the preBötC (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In some animals, naloxone at the preBötC increased genioglossus muscle amplitude to levels higher than aCSF controls, but the mean increase was not significant for the group (p = 0.075, aCSF versus naloxone). Heart rate and mean arterial blood pressure were unaffected by DAMGO at the preBötC (heart rate: 361 ± 38 to 350 ± 32, p = 0.070, n = 9, and mean arterial blood pressure: 76.2 ± 9.9 to 60.5 ± 8.8 mmHg, p = 0.075, n = 4).
Figure 1.

Opioid receptor mechanisms at the preBötC modulate breathing in vivo. A, Schema and histological section showing unilateral location of the tip of the microdialysis probe (arrow) in the medulla. B, Example showing that addition of 5 μm DAMGO to the aCSF perfusing the preBötC caused respiratory depression, effects that were reversed with naloxone. C, Group mean data (n = 9) for the effect of DAMGO (5 μm), and then the subsequent reversal of effects with naloxone (100 μm), on respiratory rate, duration of inspiration, duration of expiration, the amplitude of diaphragm (Dia) muscle activation, the amplitude of genioglossus (GG) muscle activation, and respiratory rate variability. Error bars are SEM. D, DAMGO (200 μm) caused complete respiratory arrest in the anesthetized adult rat. The animal was kept alive by artificial ventilation, and respiratory activity recovered after 30 min of perfusion with naloxone (100 μm). *p < 0.05, significant differences compared with aCSF controls. na, Nucleus ambiguus; io, inferior olive.
We then demonstrated that opioids at the preBötC at sufficient concentration (DAMGO, 200 μm) can cause complete cessation of breathing (Fig. 1D). Such respiratory arrest was identified as complete cessation of diaphragm and tongue (genioglossus) muscle activities (mean latency for complete suppression of genioglossus and diaphragm activities was 3.5 ± 1.3 and 3.8 ± 1.4 min respectively, n = 3). Cessation of breathing was reversed with artificial ventilation and perfusion of naloxone (100 μm) into the preBötC in 24.0 ± 3.1 min.
Opioids at the preBötC cause state-dependent respiratory depression
To determine the functional capacity of opioids to suppress breathing in conscious and freely behaving adult rats, we perfused opioids into the preBötC while recording respiratory muscle activity across sleep–wake states. Local bilateral application of DAMGO (20 μm) to both preBötCs caused respiratory rate slowing (Fig. 2A). In wakefulness, DAMGO decreased respiratory rate (mean change = 20.6 ± 2.6%, p = 0.01, n = 3, Figs. 2B, 3B). In non-rapid-eye-movement (non-REM) sleep, DAMGO also decreased significantly respiratory rate (mean change = 33.0 ± 2.6%, p < 0.001, n = 3, Figs. 2C, 3B), with these effects reversed by naloxone at the preBötC (Figs. 2A,C, 3B). The magnitude of this respiratory rate suppression with DAMGO was significantly correlated (p < 0.001, Fig. 3C) with the degree of cortical activation as quantified by EEG frequencies (ratio β2/δ1), with this suppression being most pronounced in state of deep non-REM sleep characterized by a low β2/δ1 ratio. DAMGO at the preBötC did not significantly change the percentage of time spent in wakefulness or non-REM sleep (both p > 0.899).
Figure 2.

In vivo modulation of breathing by the μ-opioid agonist DAMGO at the preBötC across sleep–wake states. A, Recordings of a whole experimental protocol with traces showing the prevailing sleep–wake states, electroencephalogram (EEG) activity, integrated neck muscle activity, mean respiratory rate (derived from successive 10 s time-bins), instantaneous (breath-by-breath) rate, and integrated diaphragm (Dia) muscle activity. The top line represents the time course for the continuous perfusion of the different drugs into the preBötC, with the dashed lines showing the 30 min periods used to calculate the mean data used in Figure 3. Note the persistent suppression of respiratory rate during perfusion of DAMGO, which is especially apparent during non-REM sleep, and the subsequent reversal of this rate depression by naloxone. B, C, DAMGO (20 μm) at the preBötC caused respiratory slowing in wakefulness (B) and non-REM sleep (C), with this effect reversed by naloxone.
Figure 3.
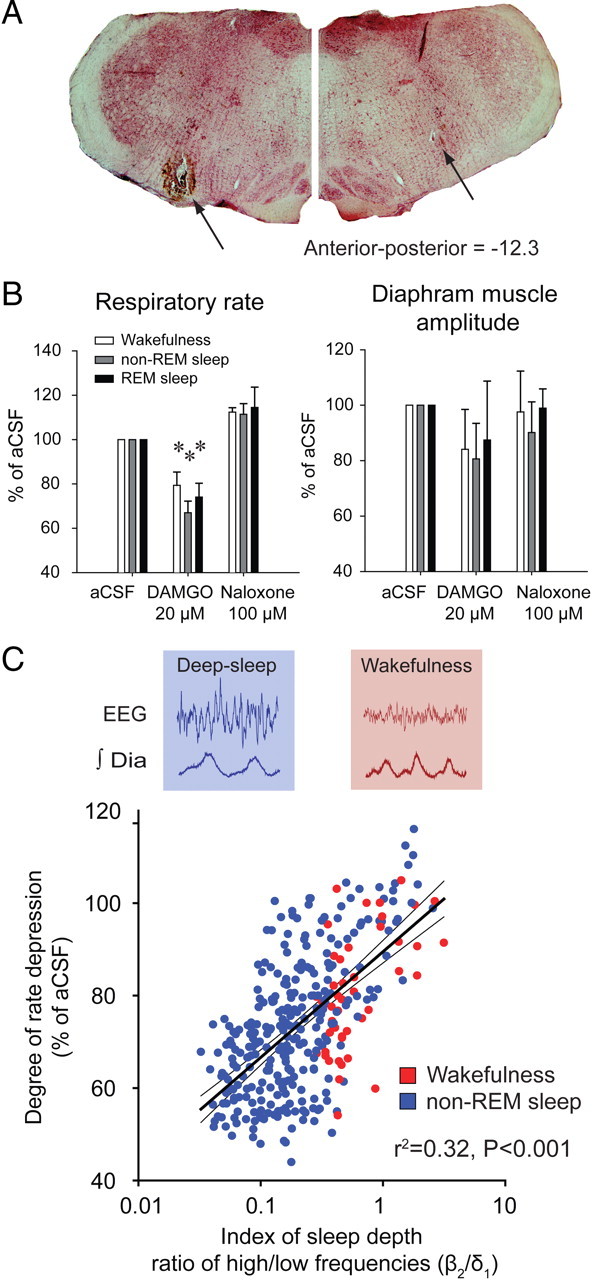
Bilateral microdialysis perfusion of the μ-opioid receptor agonist DAMGO into the preBötC of freely behaving adult rats. A, Bilateral sites of perfusion (tips of the microdialysis probes indicated by the arrows) identified from a section of medulla at 12.3 mm posterior to bregma. B, Group mean data for the effect of DAMGO (20 μm) at the preBötC on respiratory rate and the amplitude of diaphragm muscle activation across states in conscious freely behaving rats (n = 3). Error bars are SEM. The reversal of effects by bilateral application of the μ-opioid receptor antagonist naloxone (100 μm) is also shown. Note the significant depression of respiratory rate but not diaphragm muscle amplitude by DAMGO at the preBötC. C, Significant relationship between the degree of opioid-induced respiratory rate depression and the depth of sleep as indexed by the ratio of high (β2, 20–30 Hz) to low (δ1, 0.5–2 Hz) EEG frequencies. *p < 0.05, values significantly different from aCSF.
Anatomical hotspots for opioid-induced respiratory depression
To identify the opioid-sensitive “hotspot” regions for respiratory depression, we constructed correlation maps that relate the location of the intervention sites in the preBötC region with the resultant effect on respiratory activity (Fig. 4; supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The rationale for the construction of correlation maps is that for a locus of effect of opioids at any putative brainstem site, the latency for the μ-opioid receptor agonist fentanyl to diffuse through the tissue and to progressively change respiratory rate or genioglossus activity will vary as a function of the distance of the intervention site from the effective site(s) (supplemental Table 1, available at www.jneurosci.org as supplemental material). We first demonstrate that perfusion of opioids within the preBötC caused a faster decrease in respiratory rate whereas perfusion further from the preBötC induced a slower decrease in respiratory rate; there was a significant relationship between distance from preBötC to the intervention (microdialysis probe) site and the latency for a drug effect (r2 = 0.81, p = 0.001, n = 8, Fig. 4B). For all the possible sites within the volume of brainstem surrounding the preBötC (see Materials and Methods), construction of three dimensional anatomical maps reveal “hotspot” regions that are statistically highly correlated with the latencies for respiratory rate and genioglossus suppression (Fig. 4C). The anatomical region most strongly associated with respiratory rate slowing corresponds to the preBötC as identified by NK1R expression and in vitro electrophysiology (Smith et al., 1991; Gray et al., 2001), and this is further examined in vitro below. We also determine that the anatomical regions for opioid-induced respiratory slowing and suppression of genioglossus muscle activity originate from distinct regions of medulla, with the latter region located caudal, dorsal and medial to the preBötC (Fig. 4C,D; supplemental Fig. 1D–G, available at www.jneurosci.org as supplemental material).
Figure 4.

Identification of the brainstem sites affected by opioids. A, Site of microdialysis perfusion located in the preBötC region as identified by neurokinin-1 receptor (NK1R) expression (n = 3) and histology (n = 8). B, Correlations between distances from the preBötC (−12.3 mm posterior, 2.5 mm lateral, and 10.2 mm ventral to bregma) to each perfusion site, and the corresponding latencies to a 10% decrease in respiratory rate (shown in red) or genioglossus (GG) muscle amplitude (blue). Note that the correlation between the perfusion sites and the corresponding latencies for respiratory rate depression is high and statistically significant (red, r2 = 0.81, p = 0.002), but for genioglossus activity the correlation is weak and not significant (blue, r2 = 0.35, p = 0.164). See supplemental Table 1, available at www.jneurosci.org as supplemental material for additional information. C, Such correlations were performed for all regions on the histological sections using a 50 μm square grid, and the magnitude of these correlations are color coded. These correlation maps reveal “hot spots” for the region most strongly associated with opioid-induced respiratory rate suppression, with this region corresponding to the preBötC. A similar approach for a 10% suppression of GG activity shows that a distinct region located dorsomedial and caudal to the preBötC was most strongly associated with opioid-induced suppression of tongue muscle activity (supplemental Fig. 1F,G, available at www.jneurosci.org as supplemental material). D, Three-dimensional reconstruction showing only those regions with high correlations (r2 > 0.8), with the regions associated with suppression of respiratory rate (blue) and GG activity (green) being nonoverlapping. XIIn, Hypoglossal motor nucleus; io, inferior olive; na, nucleus ambiguus.
The preBötC mediates respiratory depression by systemically administered opioids
Here, we show that the fentanyl-induced respiratory slowing was prevented by local bilateral application of naloxone to the preBötC. Intravenous infusion of fentanyl at a clinically relevant dose (1 μg/kg over 1 min) produced a robust ∼30% reduction in respiratory rate (Fig. 5A,C) in the presence of bilateral perfusion of artificial CSF (control) at the preBötC (Fig. 6A), with this respiratory slowing fully prevented by addition of naloxone to the fluid locally perfusing the preBötC (Fig. 5B,D). With aCSF at the preBötC, intravenous application of fentanyl caused clear suppression of respiratory rate (mean decrease = 37.5 ± 5.1%, p = 0.007, n = 4), genioglossus muscle activity (mean decrease = 67.0 ± 18.6%, p = 0.037, n = 4) and blood pressure (mean decrease = 21.8 ± 4.6%, p = 0.018, n = 4, Fig. 6B). With naloxone (300 μm) at the preBötC, the suppression of respiratory rate following intravenous application of fentanyl (1 μg/kg over 1 min) was fully prevented (n = 4, p < 0.001, Fig. 6B), whereas the suppression of genioglossus muscle activity and blood pressure still occurred and were statistically unaffected by naloxone (p = 0.371 and p = 0.121). A higher concentration of naloxone was used in this set of experiments to minimize the chances of incomplete μ-opioid receptor blockade. Importantly, the data in Figures 5 and 6 also show that despite all the potential sites in the CNS and the periphery where the systemically administered fentanyl could potentially act to produce this respiratory slowing, bilateral local application of naloxone to only one critical site in the medulla prevented this respiratory rate depression. Moreover, the ability to fully prevent the respiratory slowing with local application of naloxone to the preBötC was significantly related to the proximity of the perfusion sites to the preBötC (r2 = 0.52, p = 0.028, n = 9, Fig. 6C; see supplemental Table 2, available at www.jneurosci.org as supplemental material for additional information), such that with successful placement of perfusion sites in both preBötC (as in Fig. 6A) the opioid-induced respiratory rate suppression was fully blocked. As expected, such significant relationships were not present for the genioglossus (r2 = 0.007, p = 0.83, n = 9, Fig. 6C) and blood pressure (r2 = 0.028, p = 0.66, n = 9) responses, which were unaffected by naloxone at the preBötC. Blocking μ-opioid receptors by bilateral perfusion of naloxone alone at the preBötC did not significantly change respiratory rate (p = 0.125, n = 4) or genioglossus muscle activity (p = 0.625, n = 4).
Figure 5.

The preBötC mediates opioid-induced respiratory rate suppression following systemic administration of fentanyl. A, With aCSF at the preBötC, intravenous application of fentanyl (1 μg/kg over 1 min) caused suppression of respiratory rate, genioglossus (GG) muscle activity and blood pressure. B, With naloxone at the preBötC, the suppression of respiratory rate following intravenous application of fentanyl (1 μg/kg over 1 min) was fully prevented, whereas the suppression of GG muscle activity and blood pressure still occurred. C, D, Tracings in a shorter time scale. Dia, diaphragm muscle amplitude.
Figure 6.
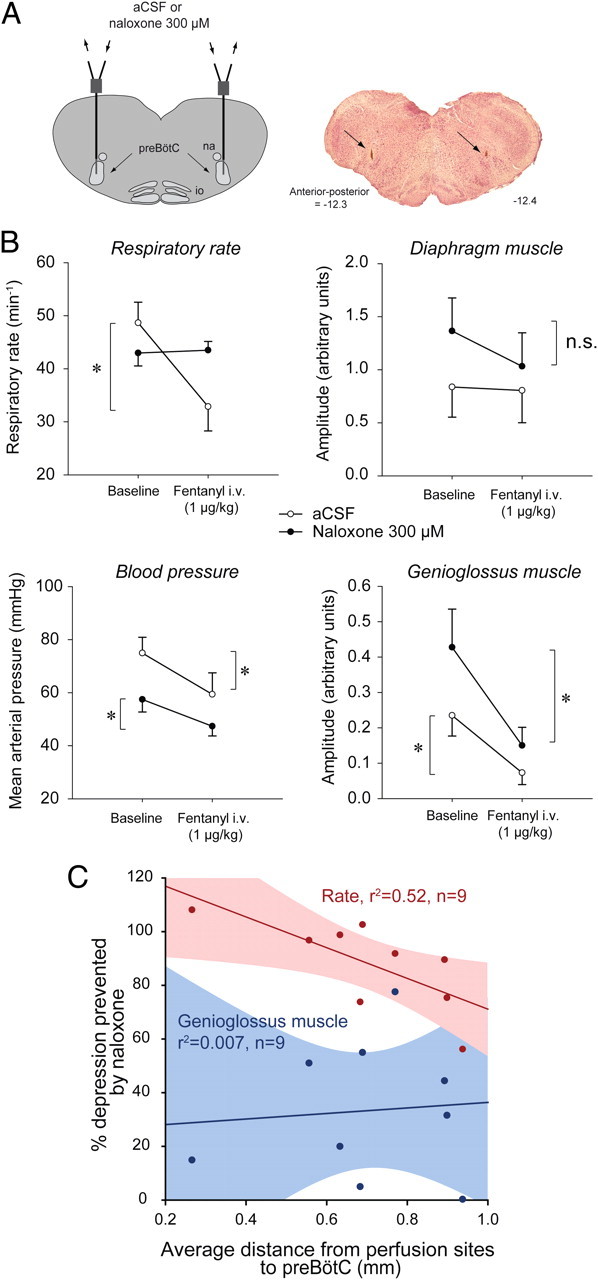
Prevention of opioid-induced respiratory depression by naloxone at the preBötC in anesthetized rats (n = 4). A, Schema and histological section showing location of probes (arrows) for bilateral application of aCSF or naloxone into both preBötCs. B, Full prevention of fentanyl-induced suppression of respiratory rate by naloxone (300 μm, filled circles compared with aCSF (control, open circles). Diaphragm muscle amplitude was slightly, but not significantly, reduced by intravenous administration of fentanyl. Naloxone at the preBötC failed to prevent the suppression of blood pressure and genioglossus muscle activity produced by intravenous fentanyl. Data are means ± SEM. C, Significant correlation (r2 = 0.52, p = 0.028) between degree of prevention of respiratory rate and average distance from perfusion sites to preBötCs (−12.5 mm posterior, 2.5 mm either side of the midline, and −10.2 mm ventral to bregma). These coordinates correspond to a region of high NK1R expression. Genioglossus muscle suppression by opioids was not correlated with the average distance from perfusion sites to preBötCs since naloxone did not prevent the suppression. supplemental Figure 2 (available at www.jneurosci.org as supplemental material) shows that respiratory rate depression following intravenous opioids was not fully prevented even when one of the two naloxone perfusion sites was at the preBötC, i.e., both are required and both mediate the suppression following systemic opioid administration. * indicates when fentanyl significantly decreases activity (p < 0.05, post hoc Bonferroni t tests).
NK1R-expressing preBötC neurons are opioid sensitive
NK1R-expressing preBötC neurons are essential to stable breathing and generating respiratory rhythm (Gray et al., 2001; McKay et al., 2005), and the opioid-sensitive site identified above corresponds to a region of the medulla expressing NK1R (Fig. 4C). Accordingly, we determined whether NK1R-expressing neurons are preferentially inhibited by DAMGO in vitro. We made targeted whole-cell recordings from preBötC neurons expressing NK1R identified following internalization of tetramethylrhodamine-conjugated Substance P, the natural ligand for NK1R, in medullary slice preparations expressing a spontaneous respiratory rhythm (Pagliardini et al., 2005) (Fig. 7A). In NK1R-expressing neurons, the discharge patterns in relation to the respiratory cycle were 67/141 (47.5%) inspiratory, 38/141 (27.0%) expiratory and 36/141 (25.5%) nonrespiratory. The distribution of neuronal discharge patterns among non-NK1R-expressing neurons in the region of the preBötC was 22/70 (31.4%) inspiratory, 14/70 (20%) expiratory and 34/70 (48.6%) nonrespiratory. We randomly sampled from NK1R-expressing and nonexpressing preBötC neurons, and found a marked difference in the electrophysiological responses to application of DAMGO in these two populations. Bath application of DAMGO (0.5 μm) caused the expected slowing of respiratory rate in this in vitro preparation (Greer et al., 1995; Gray et al., 1999) (Fig. 7B). In 9 of 10 NK1R-expressing preBötC inspiratory neurons, DAMGO caused membrane hyperpolarization (mean decrease in membrane potential = 4.6 ± 0.4 mV, n = 10, p = 0.001, Fig. 7B). In eight non-NK1R-expressing inspiratory preBötC neurons, six did not respond to DAMGO application and only two neurons were hyperpolarized by DAMGO, and overall membrane potential was not significantly affected (mean change = 0.8 ± 1.4 mV, n = 8, Fig. 7C). In the presence of tetrodotoxin (1 μm), to isolate preBötC neurons from any action potential driven synaptic transmission, DAMGO hyperpolarized 6 of 7 NK1R-expressing preBötC neurons (mean decrease in membrane potential = 4.1 ± 0.9 mV) and decreased their input resistance (271 ± 43 vs 223 ± 26 MΩ, p = 0.041, Fig. 7D).
Figure 7.
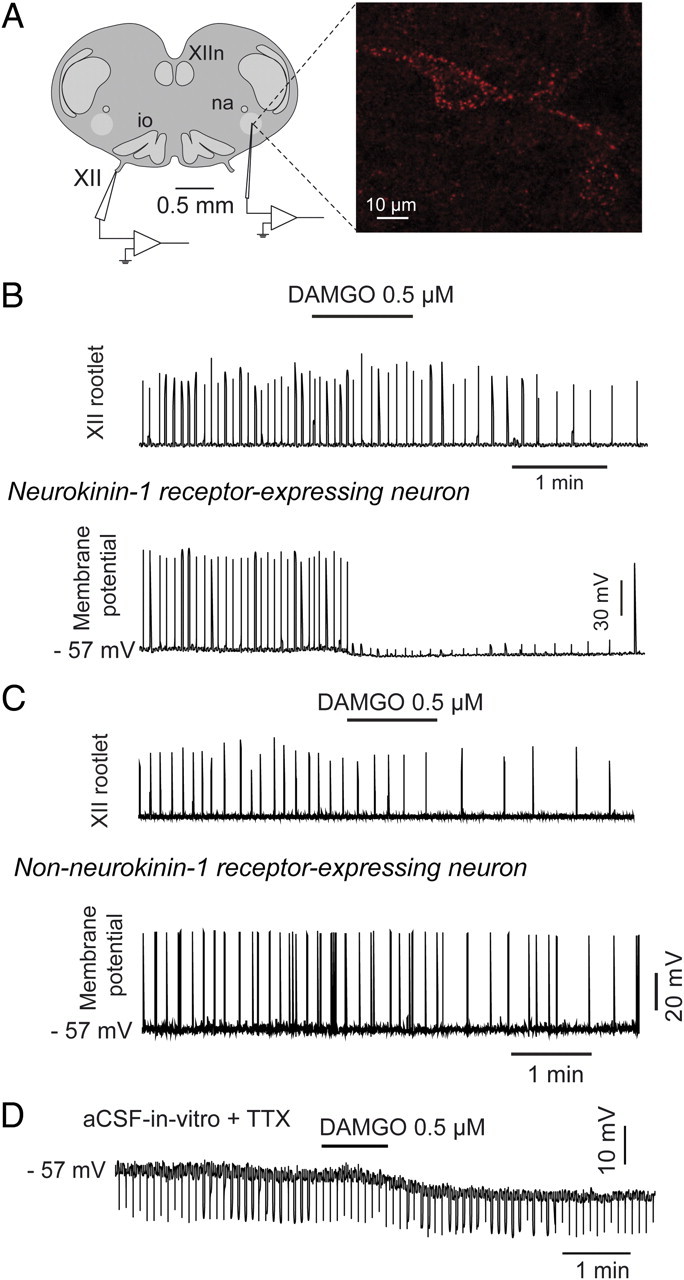
μ-Opioid receptor-induced inhibition of neurokinin-1 receptor-expressing (NK1R) inspiratory preBötC neurons in vitro. A, Whole-nerve recording from hypoglossal (XII) rootlet and whole-cell intracellular recording of NK1R-expressing preBötC neurons (red fluorescent labeling from internalization of tetramethylrhodamine-conjugated Substance P). B, Application of DAMGO (0.5 μm) to the bathing medium of the tissue slice decreased respiratory rate (as shown by decreased frequency of respiratory bursts in the XII nerve rootlet). Simultaneous current-clamp recording of a NK1R-expressing preBötC neuron showed membrane hyperpolarization, and clear suppression of inspiratory depolarization such that the threshold for action potential generation was not reached. C, Recordings of a non-NK1R-expressing preBötC neuron showing its lack of response to bath application of the μ-opioid receptor agonist DAMGO (0.5 μm). D, DAMGO-induced membrane hyperpolarization persisted in the presence of TTX (1 μm). na, Nucleus ambiguus; io, inferior olive, XIIn, hypoglossal motor nucleus; XII hypoglossal nerve rootlet.
Discussion
Rhythmic breathing is essential to life in mammals. Accordingly, identifying and defining the brain sites underlying the generation of breathing, and their responses to commonly used drugs such as opioid analgesics, is of fundamental importance to understand respiratory depression. Although the critical function of the preBötC in generating respiratory rhythm is well established in neonatal rodents in vitro (Smith et al., 1991; Gray et al., 1999), its role in modulating rhythm in the adult organism in vivo has been controversial (Richter and Spyer, 2001; Feldman and Del Negro, 2006). Here we first demonstrate that nondestructive reversible manipulation of the preBötC by opioids at sufficient concentration (tested under anesthesia) can arrest respiratory rhythm, abolish respiratory muscle activity and cause fatal apnea unless reversed by naloxone. This finding is in agreement with data showing that destruction or silencing of preBötC neurons induces ataxic breathing or outright central apneas in freely behaving adult rats (Gray et al., 2001; McKay et al., 2005; Tan et al., 2008). Importantly, we also determine that the preBötC participates in the modulation of normal rhythmic breathing in vivo, previously controversial, because opioids at the preBötC produce reversible slowing of respiratory rate i.e., not ataxia or apnea as occur with destruction or silencing of preBötC neurons (Gray et al., 2001; McKay et al., 2005; Tan et al., 2008).
The present study is the first to describe and validate methods for the manipulation of the preBötC region in a freely behaving mammal and so has widespread implications for studies investigating the role of this region in respiratory rhythmogenesis in vivo. Previous attempts to modulate respiratory activity with application of opioids to the preBötC in vivo have yielded contradictory results (Lonergan et al., 2003; Krause et al., 2009), because either the preBötC neurons were not identified by NK1R expression so making the locations of intervention sites difficult to identify, or the use of an acute microinjection of a single dose of opioids did not affect a sufficient number of preBötC neurons. Here we show that opioids were applied to the preBötC region expressing NK1R, these NK1R-expressing preBötC neurons are selectively inhibited by opioids in vitro, and this rate suppression effect is recapitulated in vivo with anatomical and functional specificities to the preBötC region expressing NK1R.
Opioid drugs are in widespread clinical use for the management of acute and chronic pain, especially after surgery when patients also spend a significant time asleep (Gutstein, 2001). Here we show that anesthesia and deep non-REM sleep are particularly vulnerable states for respiratory rate depression when opioids are present at the CNS sites responsible for the generation of breathing. This principle of state-dependent respiratory depression fits with the concept that wakefulness can provide a significant excitatory drive to the respiratory system which can compensate, at least in great part, for otherwise outright defects in the expression of respiratory activity (Phillipson and Bowes, 1986). For instance, cessation of breathing is pronounced during non-REM sleep in congenital central hypoventilation syndrome (Shea, 1997) and destruction of NK1R-expressing preBötC neurons causes a progressive deterioration of breathing, with this deterioration first appearing in sleep (McKay et al., 2005).
To overcome the spatial limitations inherent to the localized pharmacological manipulation of selected neuronal groups, we developed a novel functional and anatomical approach to identify the sites in the medulla most sensitive to the applied agents. Using the capacity of opioids to diffuse through the tissue and progressively induce specific physiological effect depending on physical proximity, we found a unique site in the medulla that was the most highly statistically associated with opioid-induced respiratory rate suppression, with this “hotspot” region overlapping with the NK1R expression that identifies the preBötC anatomically (Gray et al., 2001), and with NK1R-expressing neurons in this region being selectively inhibited by opioids in vitro (Fig. 7). The anatomical specificity for the suppression of respiratory rate by locally applied opioids is further supported by the absence of a change in the amplitude of respiratory-related diaphragm activity (Figs. 1, 2), indicating that μ-opioid responsive phrenic premotor neurons in the more caudal regions of the ventral respiratory column (Lalley, 2003; Stucke et al., 2008) are not appreciably affected by the interventions at the preBötC.
We also identify a separate distinct site of the medulla that was the most highly statistically associated with opioid-induced suppression of tongue muscle activity. Therefore, in addition to the inhibitory action of opioids at the hypoglossal motor pool (Hajiha et al., 2009), we now identify a region that is statistically highly associated with opioid-induced suppression of tongue muscle activity which corresponds to the site of hypoglossal premotor neurons identified in neonates in vitro (Smith et al., 2007) and the parahypoglossal nucleus in adults (Tan et al., 2010). This region is located caudal, dorsal and medial to the preBötC and is innervated by preBötC neurons (Koizumi et al., 2008; Tan et al., 2010) and projects to the hypoglossal motor nucleus (Koizumi et al., 2008). Identification of this opioid-responsive region is significant because upper airway obstruction is a serious clinical concern associated with administration of opioid drugs during sleep and anesthesia such that current recommendation is that opioid drugs should only be administered if the airway is under control (Kryger, 2000).
A major unanswered question in physiology and medicine has been to identify the key site(s) in the brain where opioid drugs act to suppress breathing and produce fatal apnea. Although there are multiple sites in the brain that express μ-opioid receptors which when activated could directly or indirectly influence the respiratory network (Xia and Haddad, 1991; Gray et al., 1999; Lalley, 2003; Manzke et al., 2003; McCrimmon and Alheid, 2003), here we demonstrate that the preBötC can fully mediate respiratory rate depression following systemic opioid administration because this rate depression was entirely prevented by naloxone at the preBötC only, with the efficacy of this prevention depending upon proximity of the intervention to the preBötC (Fig. 6C; supplemental Table 2, available at www.jneurosci.org as supplemental material). The specificity of the naloxone intervention selectively preventing rate suppression is further supported by the fact that the preBötC per se does not mediate opioid-induced suppression of tongue muscle activity or blood pressure since these suppressions persist despite naloxone at the preBötC; in agreement with the separate opioid-sensitive site identified above and shown in Figure 4. In two previous studies attempting to antagonize opioid-sensitive preBötC neurons (Stucke et al., 2008; Mustapic et al., 2010), it is not clear whether the interventions were targeted to the NK1R-expressing region and whether single micro-injections affected a sufficient number of neurons, so explaining the lack of prevention.
In the preBötC we also found inspiratory, expiratory, and nonrespiratory neurons (Johnson et al., 1994; Pagliardini et al., 2005) with a subpopulation expressing NK1R (Gray et al., 2001). Here, we show that approximately half of the NK1R-expressing preBötC neurons sampled were active during inspiration and that these neurons were preferentially inhibited by opioids. A recent report using in vitro preparations of rat medullary slice concluded that opioid-mediated suppression of preBötC activity is mediated solely by presynaptic mechanisms (Ballanyi et al., 2010). However, our data indicate significant postsynaptic hyperpolarization mainly among NK1R-expressing preBötC neurons. A postsynaptic, in addition to a previously described presynaptic (Haji et al., 2003), action is consistent with immunohistochemical detection of μ-opioid receptors on NK1R-expressing preBötC neurons (Gray et al., 1999; Manzke et al., 2003) and previous reports of DAMGO-induced membrane hyperpolarization of a nonidentified subset of preBötC neurons (Gray et al., 1999; Takeda et al., 2001; Lorier et al., 2008). With the additional in vitro data in this report, we identify the neurochemical phenotype of the opioid-sensitive preBötC neurons. In further support of this conclusion, in mice lacking the tachykinin 1 (tac1) gene encoding for the precursor protein of Substance P, the natural ligand for NK1R, the severity of morphine-induced respiratory depression is reduced compared with wild-type animals (Bilkei-Gorzo et al., 2010).
In summary, this report establishes that NK1R-expressing preBötC neurons constitute the critical site mediating opioid-induced respiratory rate depression in vivo. In principle, these findings are essential to develop new pharmacological approaches to prevent life-threatening respiratory depression, without reducing the beneficial analgesic properties of opioid drugs, by a strategy of targeted reactivation of NK1R-expressing preBötC neurons.
Footnotes
G.M. was supported by Postdoctoral Research Awards from the Canadian Lung Association, the Quebec Health Research Funds (FRSQ), and the Canadian Thoracic Society. R.L.H. holds the Canada Research Chair in Sleep and Respiratory Neurobiology. Research was funded by the Canadian Institutes of Health Research and the Ontario Thoracic Society—Canadian Lung Association. We thank G. D. Funk and A. G. Huxtable for their help with immunohistochemistry. We also thank B. A. Orser, P. W. Frankland, and S. W. Hughes for their careful reading and comments on the manuscript.
References
- Agrò F, Salvinelli F, Casale M, Gherardi S. Difficulty in airway management during sedation of patients affected by obstructive sleep apnea. Can J Anaesth. 2004;51:279. doi: 10.1007/BF03019116. [DOI] [PubMed] [Google Scholar]
- Ballanyi K, Panaitescu B, Ruangkittisakul A. Indirect opioid actions on inspiratory pre-Botzinger complex neurons in newborn rat brainstem slices. Adv Exp Med Biol. 2010;669:75–79. doi: 10.1007/978-1-4419-5692-7_16. [DOI] [PubMed] [Google Scholar]
- Bilkei-Gorzo A, Berner J, Zimmermann J, Wickström R, Racz I, Zimmer A. Increased morphine analgesia and reduced side effects in mice lacking the tac1 gene. Br J Pharmacol. 2010;160:1443–1452. doi: 10.1111/j.1476-5381.2010.00757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers G. When opioid analgesia kills. Perspect Infirm. 2006;4:6–9. [PubMed] [Google Scholar]
- Feldman JL, Del Negro CA. Looking for inspiration: new perspectives on respiratory rhythm. Nat Rev Neurosci. 2006;7:232–242. doi: 10.1038/nrn1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray PA, Rekling JC, Bocchiaro CM, Feldman JL. Modulation of respiratory frequency by peptidergic input to rhythmogenic neurons in the preBotzinger complex. Science. 1999;286:1566–1568. doi: 10.1126/science.286.5444.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray PA, Janczewski WA, Mellen N, McCrimmon DR, Feldman JL. Normal breathing requires preBotzinger complex neurokinin-1 receptor-expressing neurons. Nat Neurosci. 2001;4:927–930. doi: 10.1038/nn0901-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer JJ, Carter JE, al-Zubaidy Z. Opioid depression of respiration in neonatal rats. J Physiol. 1995;485:845–855. doi: 10.1113/jphysiol.1995.sp020774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutstein HB. Opioid analgesics. In: Hardman JG, Limbird LE, Gilman AG, editors. Goodman and Gilmans's the pharmacological basis for therapeutics. New-York: McGraw-Hill; 2001. pp. 569–618. [Google Scholar]
- Haji A, Okazaki M, Ohi Y, Yamazaki H, Takeda R. Biphasic effects of morphine on bulbar respiratory neuronal activities in decerebrate cats. Neuropharmacology. 2003;45:368–379. doi: 10.1016/s0028-3908(03)00154-0. [DOI] [PubMed] [Google Scholar]
- Hajiha M, DuBord MA, Liu H, Horner RL. Opioid receptor mechanisms at the hypoglossal motor pool and effects on tongue muscle activity in vivo. J Physiol. 2009;587:2677–2692. doi: 10.1113/jphysiol.2009.171678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AJ, Logan JE, Toblin RL, Kaplan JA, Kraner JC, Bixler D, Crosby AE, Paulozzi LJ. Patterns of abuse among unintentional pharmaceutical overdose fatalities. JAMA. 2008;300:2613–2620. doi: 10.1001/jama.2008.802. [DOI] [PubMed] [Google Scholar]
- Johnson SM, Smith JC, Funk GD, Feldman JL. Pacemaker behavior of respiratory neurons in medullary slices from neonatal rat. J Neurophysiol. 1994;72:2598–2608. doi: 10.1152/jn.1994.72.6.2598. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Wilson CG, Wong S, Yamanishi T, Koshiya N, Smith JC. Functional imaging, spatial reconstruction, and biophysical analysis of a respiratory motor circuit isolated in vitro. J Neurosci. 2008;28:2353–2365. doi: 10.1523/JNEUROSCI.3553-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause KL, Neumueller SE, Marshall BD, Kiner T, Bonis JM, Pan LG, Qian B, Forster HV. Micro-opioid receptor agonist injections into the presumed pre-Botzinger complex and the surrounding region of awake goats do not alter eupneic breathing. J Appl Physiol. 2009;107:1591–1599. doi: 10.1152/japplphysiol.90548.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryger MH. Management of obstructive sleep apnea-hypopnea syndrome: overview. In: Kryger MH, Roth T, Dement WC, editors. Principles and practice of sleep medicine. Philadelphia: Saunders; 2000. pp. 940–954. [Google Scholar]
- Lalley PM. Mu-opioid receptor agonist effects on medullary respiratory neurons in the cat: evidence for involvement in certain types of ventilatory disturbances. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1287–R1304. doi: 10.1152/ajpregu.00199.2003. [DOI] [PubMed] [Google Scholar]
- Lonergan T, Goodchild AK, Christie MJ, Pilowsky PM. Mu opioid receptors in rat ventral medulla: effects of endomorphin-1 on phrenic nerve activity. Respir Physiol Neurobiol. 2003;138:165–178. doi: 10.1016/s1569-9048(03)00173-3. [DOI] [PubMed] [Google Scholar]
- Lorier AR, Lipski J, Housley GD, Greer JJ, Funk GD. ATP sensitivity of preBotzinger complex neurones in neonatal rat in vitro: mechanism underlying a P2 receptor-mediated increase in inspiratory frequency. J Physiol. 2008;586:1429–1446. doi: 10.1113/jphysiol.2007.143024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzke T, Guenther U, Ponimaskin EG, Haller M, Dutschmann M, Schwarzacher S, Richter DW. 5-HT4(a) receptors avert opioid-induced breathing depression without loss of analgesia. Science. 2003;301:226–229. doi: 10.1126/science.1084674. [DOI] [PubMed] [Google Scholar]
- McCrimmon DR, Alheid GF. On the opiate trail of respiratory depression. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1274–R1275. doi: 10.1152/ajpregu.00428.2003. [DOI] [PubMed] [Google Scholar]
- McKay LC, Janczewski WA, Feldman JL. Sleep-disordered breathing after targeted ablation of preBotzinger complex neurons. Nat Neurosci. 2005;8:1142–1144. doi: 10.1038/nn1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JL, Sood S, Liu H, Park E, Nolan P, Horner RL. GABAA receptor antagonism at the hypoglossal motor nucleus increases genioglossus muscle activity in NREM but not REM sleep. J Physiol. 2003;548:569–583. doi: 10.1113/jphysiol.2002.033696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustapic S, Radocaj T, Sanchez A, Dogas Z, Stucke AG, Hopp FA, Stuth EA, Zuperku EJ. Clinically relevant infusion rates of mu-opioid agonist remifentanil cause bradypnea in decerebrate dogs but not via direct effects in the pre-Botzinger complex region. J Neurophysiol. 2010;103:409–418. doi: 10.1152/jn.00188.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliardini S, Ren J, Greer JJ. Ontogeny of the pre-Botzinger complex in perinatal rats. J Neurosci. 2003;23:9575–9584. doi: 10.1523/JNEUROSCI.23-29-09575.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliardini S, Adachi T, Ren J, Funk GD, Greer JJ. Fluorescent tagging of rhythmically active respiratory neurons within the pre-Botzinger complex of rat medullary slice preparations. J Neurosci. 2005;25:2591–2596. doi: 10.1523/JNEUROSCI.4930-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattinson KT. Opioids and the control of respiration. Br J Anaesth. 2008;100:747–758. doi: 10.1093/bja/aen094. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Phillipson EA, Bowes G. Control of breathing during sleep. In: Cherniak NS, Widdicombe JG, editors. Handbook of physiology, Section III, The respiratory system. vol. II. Bethesda, MD: American Physiological Society; 1986. pp. 649–989. [Google Scholar]
- Richter DW, Spyer KM. Studying rhythmogenesis of breathing: comparison of in vivo and in vitro models. Trends Neurosci. 2001;24:464–472. doi: 10.1016/s0166-2236(00)01867-1. [DOI] [PubMed] [Google Scholar]
- Shea SA. Life without ventilatory chemosensitivity. Respir Physiol. 1997;110:199–210. doi: 10.1016/s0034-5687(97)00084-4. [DOI] [PubMed] [Google Scholar]
- Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Pre-Botzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Abdala AP, Koizumi H, Rybak IA, Paton JF. Spatial and functional architecture of the mammalian brain stem respiratory network: a hierarchy of three oscillatory mechanisms. J Neurophysiol. 2007;98:3370–3387. doi: 10.1152/jn.00985.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenland HW, Liu H, Horner RL. Endogenous glutamatergic control of rhythmically active mammalian respiratory motoneurons in vivo. J Neurosci. 2008;28:6826–6835. doi: 10.1523/JNEUROSCI.1019-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucke AG, Zuperku EJ, Sanchez A, Tonkovic-Capin M, Tonkovic-Capin V, Mustapic S, Stuth EA. Opioid receptors on bulbospinal respiratory neurons are not activated during neuronal depression by clinically relevant opioid concentrations. J Neurophysiol. 2008;100:2878–2888. doi: 10.1152/jn.90620.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Eriksson LI, Yamamoto Y, Joensen H, Onimaru H, Lindahl SG. Opioid action on respiratory neuron activity of the isolated respiratory network in newborn rats. Anesthesiology. 2001;95:740–749. doi: 10.1097/00000542-200109000-00029. [DOI] [PubMed] [Google Scholar]
- Tan W, Janczewski WA, Yang P, Shao XM, Callaway EM, Feldman JL. Silencing preBotzinger complex somatostatin-expressing neurons induces persistent apnea in awake rat. Nat Neurosci. 2008;11:538–540. doi: 10.1038/nn.2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W, Pagliardini S, Yang P, Janczewski WA, Feldman JL. Projections of preBotzinger Complex neurons in adult rats. J Comp Neurol. 2010;518:1862–1878. doi: 10.1002/cne.22308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Haddad GG. Ontogeny and distribution of opioid receptors in the rat brainstem. Brain Res. 1991;549:181–193. doi: 10.1016/0006-8993(91)90457-7. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Xu F, Zhang C, Liang X. Activation of opioid mu receptors in caudal medullary raphe region inhibits the ventilatory response to hypercapnia in anesthetized rats. Anesthesiology. 2007;107:288–297. doi: 10.1097/01.anes.0000270760.46821.67. [DOI] [PubMed] [Google Scholar]