

Abstract
The modification of the neural cell adhesion molecule (NCAM) with polysialic acid (polySia) is tightly linked to neural development. Genetic ablation of the polySia-synthesizing enzymes ST8SiaII and ST8SiaIV generates polySia-negative but NCAM-positive (II−/−IV−/−) mice characterized by severe defects of major brain axon tracts, including internal capsule hypoplasia. Here, we demonstrate that misguidance of thalamocortical fibers and deficiencies of corticothalamic connections contribute to internal capsule defects in II−/−IV−/− mice. Thalamocortical fibers cross the primordium of the reticular thalamic nucleus (Rt) at embryonic day 14.5, before they fail to turn into the ventral telencephalon, thus deviating from their normal trajectory without passing through the internal capsule. At postnatal day 1, a reduction and massive disorganization of fibers traversing the Rt was observed, whereas terminal deoxynucleotidyl transferase dUTP nick end labeling and cleaved caspase-3 staining indicated abundant apoptotic cell death of Rt neurons at postnatal day 5. Furthermore, during postnatal development, the number of Rt neurons was drastically reduced in 4-week-old II−/−IV−/− mice, but not in the NCAM-deficient N−/− or II−/−IV−/−N−/− triple knock-out animals displaying no internal capsule defects. Thus, degeneration of the Rt in II−/−IV−/− mice may be a consequence of malformation of thalamocortical and corticothalamic fibers providing major excitatory input into the Rt. Indeed, apoptotic death of Rt neurons could be induced by lesioning corticothalamic fibers on whole-brain slice cultures. We therefore propose that anterograde transneuronal degeneration of the Rt in polysialylation-deficient, NCAM-positive mice is caused by defective afferent innervation attributable to thalamocortical pathfinding defects.
Introduction
Polysialic acid (polySia) is a posttranslational modification of the neural cell adhesion molecule (NCAM) and synthesized by the two polysialyltransferases ST8SiaII and ST8SiaIV (Rougon and Hobert, 2003; Rutishauser, 2008; Hildebrandt et al., 2010). As shown in mice with genetic ablation of both enzymes, polySia is essential for brain development (Weinhold et al., 2005; Angata et al., 2007). Among other abnormalities, these mice display hypoplasia of major brain axon tracts like anterior commissure, corpus callosum, and internal capsule (Weinhold et al., 2005; Hildebrandt et al., 2009). Axonal defects develop because of a gain of polySia-negative NCAM, as they are not observed in NCAM-negative mice and correlate precisely with the level of NCAM erroneously devoid of polySia during development (Hildebrandt et al., 2009). Thalamocortical and corticothalamic projections form a major part of the internal capsule and numerous studies deal with their formation (for examples, see Métin and Godement, 1996; Métin et al., 1997; Molnár and Cordery, 1999; Ma et al., 2002; Molnár et al., 2003; Andrews et al., 2006; López-Bendito et al., 2007; Wright et al., 2007; Demyanenko et al., 2010). As shown before, polySia is crucial for branch formation of thalamocortical axons within cortical layer 4 (Yamamoto et al., 2000). In contrast, the role of polySia or NCAM in early thalamocortical development has not yet been addressed. In the current study, we asked whether polySia deficiency causes internal capsule hypoplasia by affecting the development of connections between cortex and thalamus. We also analyzed a possible developmental relationship with the reticular thalamic nucleus (Rt), since all thalamocortical and corticothalamic projections pass through and give off collaterals within this structure (Mitrofanis and Guillery, 1993; Deng and Elberger, 2003).
The Rt forms a sheet of GABAergic neurons at the interface between thalamus and internal capsule (for review, see Guillery et al., 1998; Pinault, 2004). Its neurons originate from the ventral thalamus and migrate dorsally to envelop the dorsal thalamus. The Rt consists of GABAergic inhibitory neurons, which are characterized by the expression of the calcium-binding protein parvalbumin (PV) (Csillik et al., 2006) and receive major excitatory input from thalamocortical and corticothalamic axon collaterals (Harris, 1987). Unlike most thalamic nuclei, the Rt does not project to the cerebral cortex. Instead, its axons terminate within the Rt itself and, in a highly topographical manner, project to all thalamic nuclei (Pinault, 2004). Providing a powerful means for inhibiting thalamocortical activity, the Rt is involved in sensory processing (McAlonan and Brown, 2002; Nagaeva and Akhmadeev, 2006) and, as first hypothesized by Crick (1984), seems to mediate attentional modulation of thalamic neurons (McAlonan et al., 2008). Moreover, dysfunction of the Rt in gating thalamic transmission has been postulated to contribute to the emergence of hallucinations in schizophrenia (Behrendt, 2006; Byne et al., 2009).
Here, we first demonstrate that defects of thalamocortical and corticothalamic axons contribute to the hypoplasia of the internal capsule in polysialylation-deficient mice. Asking for possible consequences of the deficient projections, we found a substantial loss of neurons in the Rt, which occurred subsequent to the prominent reduction and defasciculation of thalamocortical and corticothalamic fibers. In addition, massive apoptosis of Rt neurons could be induced by deafferentation lesions in whole-brain slice cultures. We therefore propose that degeneration of the Rt occurs because of defective input by thalamocortical and corticothalamic axon collateral projections.
Materials and Methods
Mice.
C57BL/6J and transgenic mice were bred at the central animal facility at Hannover Medical School. All protocols for animal use were in compliance with German law and approved by the responsible animal welfare officer. St8siaII, St8siaIV, and NCAM1 single knock-out strains, which have been backcrossed with C57BL/6J mice for six generations, were crossbred to obtain double (St8siaII−/−St8siaIV−/−; II−/−IV−/−) or triple knock-out animals (St8siaII−/−St8siaIV−/−NCAM1−/−; II−/−IV−/−N−/−) (Weinhold et al., 2005). Genotyping was performed by PCR as described previously (Weinhold et al., 2005). For staging of embryos, the morning of the vaginal plug was considered as embryonic day 0.5 (E0.5). Mice of both sexes were used for analyses of juvenile stages up to postnatal day 20 (P20). Unless stated otherwise, males were used for analyses at P30.
Immunohistochemistry.
Mice were deeply anesthetized with 200 mg/kg ketamine, 8 mg/kg xylazine in 0.9% NaCl and transcardially perfused with 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer, pH 7.4, before brains were removed and postfixed overnight. Embryonic brains were directly fixed by immersion of the head in 4% PFA. Fifty micrometer serial coronal sections were cut on a vibrating microtome. Free-floating sections were permeabilized with 0.4% Triton X-100 for 15 min and blocked with 10% fetal calf serum for 1 h. The following monoclonal (mAb) or polyclonal (pAb) antibodies were used: polySia-specific mouse mAb 735 (IgG2a; 5 μg/ml) (Frosch et al., 1985), Islet-1-specific mouse mAb 4D5 (IgG2b; 1:500), neurofilament (NF)-specific mouse mAb 2H3 (IgG1; 1:500), TAG-1-specific mouse mAb 4D7 (IgM; 1:100; all three from Developmental Studies Hybridoma Bank), PV-specific mouse mAb (1:5000; IgG1; Swant), L1-specific rabbit pAb (1:1000; kind gift from F. Rathjen, Berlin, Germany), Pax-6-specific rabbit pAb (1:300; Covance), GABA-specific rabbit pAb (1:200; Sigma-Aldrich), and cleaved caspase-3-specific rabbit pAb (1:200; Cell Signaling Technology). Rabbit and mouse IgG-specific, IgM-specific, and subtype-specific Cy3- (Millipore Bioscience Research Reagents), Alexa 488-, Alexa 555-, Alexa 568-, and Alexa 647- (Invitrogen) conjugated antibodies were used at 1:500 for IgG-specific and 1:200 for subtype- and IgM-specific antibodies. Perineuronal nets were labeled with biotinylated Wisteria floribunda agglutinin (1:1000; Sigma-Aldrich). Biotinylation was detected with Cy3-conjugated streptavidin (1:1000; Rockland Immunochemicals). As first layer controls, sections were incubated in blocking solution lacking primary antibody. In double-stained immunofluorescence samples, cross-reactivity of secondary antibodies was controlled by omitting either of the two primary antibodies. Stained sections were washed with ddH2O and mounted on glass object slides (SuperFrostPlus; Menzel) using Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories).
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling.
DNA strand breaks were detected by terminal deoxynucleotidyl transferase-mediated digoxigenin-dUTP nick end labeling (TUNEL) as described by Herzog et al. (2007). After preincubation in 1× terminal deoxytransferase (TdT) buffer containing 0.2 m cacodylate, 25 mm Tris-HCl, 1 mm CoCl2, and 0.01% Triton X-100 (Fermentas), sections were labeled using 1× TdT buffer, 4 U of TdT, 1 μm DigdUTP, and 0.1 mm dTTP for 1 h at 37°C. The reaction was stopped by washing with 2× SSC (sodium citrate buffer) and PBS plus 0.4% Triton X-100. For Islet-1 double staining, DigdUTP-labeled sections were incubated with Islet-1-specific mouse mAb 4D5 overnight. Subsequently, sections were washed with PBS plus 0.4% Triton X-100, and digoxygenin was visualized using an anti-Dig-rhodamine antibody (1:100; Roche). The secondary antibody against Islet-1-specific mouse mAb was used as described for immunohistochemistry and incubated simultaneously with the anti-Dig-rhodamine antibody.
Axonal tracing from the thalamus with carbocyanine dye.
Coronal sections of 300 μm thickness of P30 control and II−/−IV−/− mice were cut on a vibrating microtome. Small crystals of the fluorescent carbocyanine dye 1,1′-didodecyl-3,3,3′,3′-tetramethylindocarbo-cyanine perchlorate (DiI) (Invitrogen) were placed unilaterally into the thalamus to label thalamocortical projections. The sections were covered with 0.1 m phosphate buffer, pH 7.4, and incubated for 10 d at 37°C.
Whole-brain slice cultures.
Slice cultures were obtained from 4-d-old C57BL/6J mice. Mice were decapitated; brains were transferred to ice-cold HBSS (Invitrogen) and cut into 300-μm-thick sections using a vibrating microtome. Slice cultures were prepared to retain thalamocortical and corticothalamic projections, as discussed by Agmon and Connors (1991). Slices were transferred to Millicell culture inserts (Millipore) in six-well culture plates. A scalpel (no. 15; Feather) was used to create a unilateral lesion of ∼1 mm between cortical plate and Rt. The contralateral side served as a control. In addition, slices with lesions that leave thalamus–cortex connectivity unaffected were used as sham-lesioned controls. Slices were incubated at 37°C in MEM (Invitrogen) with 25% heat-inactivated horse serum (Invitrogen) as originally described by Stoppini et al. (1991). Cultures were maintained for 22 h, since deafferentation of cortical sensory neurons resulted in a maximum of apoptotic cell death after this period (Capurso et al., 1997). After fixation, TUNEL staining combined with Islet-1 immunohistochemistry was performed as outlined above. A minimum of five slice cultures per condition were quantitatively evaluated. TUNEL-positive cells within the Rt (identified by Islet-1 staining and morphological criteria) were counted and plotted relative to the Rt area.
Microscopy, area measurements, cell counting, and statistics.
Microscopy, area measurements, and counting of Islet-1-, TUNEL-, and PV-positive cells was performed with a Zeiss Axiovert 200 M equipped with MosaiX and ApoTome module, AxioCam MRm digital camera, and AxioVision software (Carl Zeiss). Out of serial sections of brains, every third section was Islet-1, PV, or TUNEL stained and per animal four of these spaced-serial sections containing the Rt were evaluated. Optical sections of 5.1 μm thickness located ∼10 μm above the bottom (caudal level) of each of the 50-μm-thick sections were obtained by ApoTome technology using a 10× Plan Apochromat objective with 0.45 numerical aperture (Zeiss). Micrographs of entire sections were acquired using the MosaiX module of the AxioVision software. Thus, counting covered 100% of the sample area within each section, and therefore there was no need to make use of a counting frame, which is typically used in the optical dissector method. On each optical slice, the number of Islet-1-, PV-, or TUNEL-positive cells within the Rt was counted. As the aim of this study was not the determination of absolute cell numbers or densities, but a comparison between polySia-positive and polySia-deficient animals, there was no need to correct for the overcount produced by counting rather big objects in relatively thin optical sections [as discussed by, for example, Guillery (2002)]. Statistical evaluation was performed using Prism software (GraphPad). Differences between two groups (as for Islet-1- and TUNEL-positive cells) were analyzed using Student's t test (two-tailed). Values of p < 0.05 were considered significant. Multiple comparisons were evaluated using one-way ANOVA with Newman–Keuls multiple comparison post hoc test (two-tailed). Values of p < 0.05 were considered significant.
Results
Misrouting of thalamocortical and corticothalamic fibers in II−/−IV−/− mice
The connectivity between thalamus and cortex was investigated during embryonic and postnatal development. PolySia-negative, NCAM-positive II−/−IV−/− mice and polySia-negative, NCAM-negative II−/−IV−/−N−/− triple knock-out mice were compared with C57BL/6J wild-type or II+/−IV+/− controls with normal levels of polySia and NCAM (Weinhold et al., 2005). In mice, the first thalamocortical fibers start to grow out of the thalamus at E13 (Molnár and Blakemore, 1995). NF staining at E14.5 revealed abundant thalamocortical fibers crossing the presumptive internal capsule in control and II−/−IV−/−N−/− mice (Fig. 1A,B,D). In II−/−IV−/− mice, a highly fasciculated bundle of thalamofugal fibers turned ventrally, and therefore these axons failed to find their way toward the cortical plate (Fig. 1C). Axons within the internal capsule as well as cell surfaces in the thalamus and surrounding structures are polySia-positive (Fig. 1E), whereas polySia staining was absent in II−/−IV−/− and II−/−IV−/−N−/− mice (Fig. 1F,G). In controls, thalamocortical axons, labeled by the cell adhesion molecule L1 (Denaxa et al., 2001; Molnár et al., 2003) and axons from the cortical plate, labeled by TAG-1 (Del Río et al., 2004), met in the basal telencephalon, as described before (Fig. 1A,H) (Blakemore and Molnár, 1990; Molnár and Blakemore, 1991). The encounter of thalamocortical and corticofugal fibers did not take place in II−/−IV−/− mice at this developmental stage (Fig. 1I). In II−/−IV−/−N−/− mice, thalamocortical axons turned correctly toward the cortical plate, but at E14.5 these fibers had not yet extended toward the pallial–subpallial boundary (Fig. 1J). Corticothalamic axons approaching the basal telencephalon were labeled by TAG-1. Compared with the controls (Fig. 1H,H′), the number of these TAG-1-positive fibers was clearly reduced in II−/−IV−/−, but not in II−/−IV−/−N−/− mice (Fig. 1 I′,J′). At E14.5, corticothalamic and thalamocortical axons were investigated in seven control, four II−/−IV−/−, and three II−/−IV−/−N−/− mice, and defects were found with 100% penetrance in only the II−/−IV−/− animals. Double immunohistochemistry for Islet-1 and NF was performed to study the misrouted trajectories of thalamocortical fibers in II−/−IV−/− animals in relation to the permissive corridor of Islet-1-positive cells in the striatum that is essential for thalamocortical axon pathfinding (López-Bendito et al., 2006). The distribution of Islet-1-positive cells appeared mostly unaltered in II−/−IV−/− and II−/−IV−/−N−/− mice at E14.5, but thalamocortical fibers did not approach the Islet-1-positive corridor in the II−/−IV−/− animals (Fig. 1K–M). As mice deficient for the transcription factor Pax-6 show defects of thalamocortical axons (Kawano et al., 1999; Simpson et al., 2009), Pax-6 expression was analyzed. Comparable with Islet-1 immunohistochemistry, the Pax-6 staining pattern was unaltered in II−/−IV−/− and II−/−IV−/−N−/− mice (Fig. 1N–P).
Figure 1.
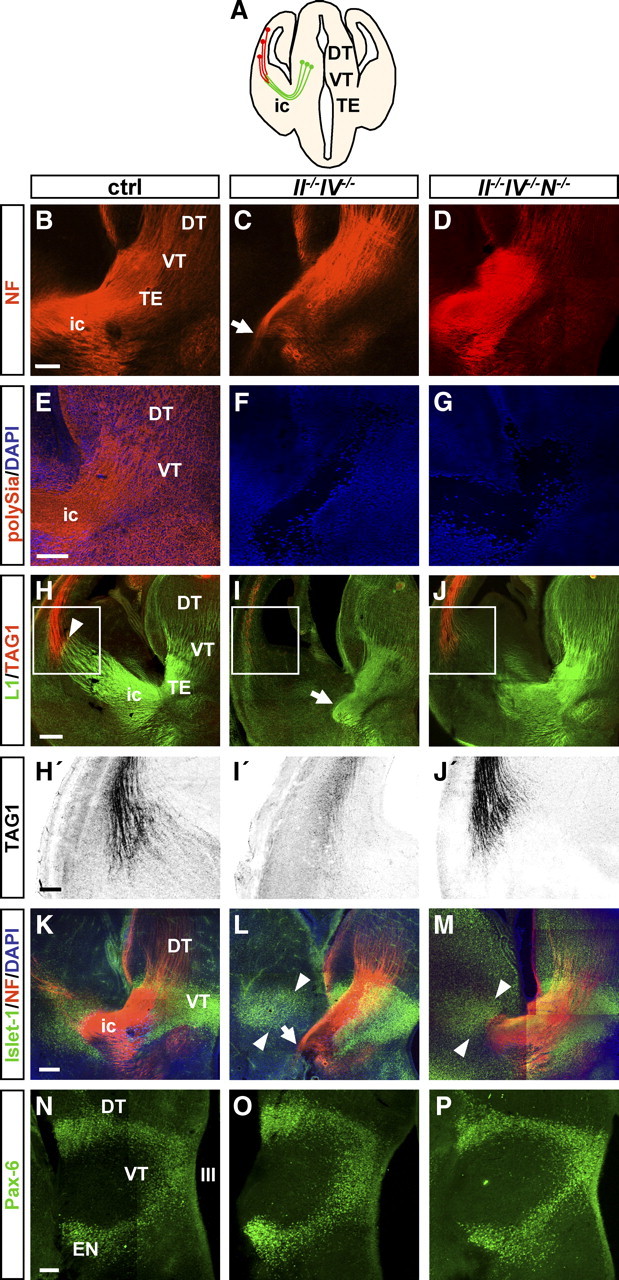
Malformation of reciprocal connectivity between cortex and thalamus in II−/−IV−/− mice at E14.5. A, Schematic drawing of a coronal section through the E14.5 mouse brain at the rostrocaudal level analyzed in B–P. B–P, MosaiX images of 5.1-μm-thick optical sections matched for rostrocaudal level showing NF, polySia, L1, TAG-1, Islet-1, and Pax-6 immunohistochemistry of wild-type controls (ctrl), II−/−IV−/−, and II−/−IV−/−N−/− mice at E14.5. B–D, NF labeling. Thalamocortical axons project through the internal capsule (ic) toward the cortical plate in control (ctrl) (B) and II−/−IV−/−N−/− mice (D), but do not cross the internal capsule in II−/−IV−/− mice (C, arrow). E–G, PolySia labeling. In controls, axons within the internal capsule as well as cells in the thalamus and surrounding structures are polySia positive (E). In II−/−IV−/− and II−/−IV−/−N−/− animals, polySia staining is completely absent (F, G). H–J, L1 and TAG-1 labeling of thalamocortical and corticothalamic axons. Thalamocortical axons meet corticothalamic axons in the basal telencephalon in control mice (H, arrowhead). In II−/−IV−/− mice, these axons do not meet. Instead, thalamocortical axons turn ventrally and intermingle (I, arrow). In II−/−IV−/−N−/− mice, thalamocortical axons turn toward the cortical plate at E14.5 but do not yet meet the corticothalamic axons (J). H′–J′, Higher magnification of TAG-1 staining corresponding to the sites boxed in H–J showing a clear reduction of corticothalamic axons exclusively in II−/−IV−/− mice. K–M, Double immunohistochemistry for Islet-1 and NF. In contrast to controls, NF-positive fibers in II−/−IV−/− mice turn ventrally (L, arrow) and fail to extend toward the Islet-1-positive corridor of the striatum (L, arrowheads). Similar to the control, the NF-positive fibers approach the Islet-1-positive corridor in II−/−IV−/−N−/− animals (M, arrowheads). N–P, Pax-6 immunohistochemistry. Staining pattern was indistinguishable between ctrl, II−/−IV−/−, and II−/−IV−/−N−/− mice. III, Third ventricle; DT, dorsal thalamus; EN, entopeduncular nucleus; TE, thalamic eminence; ic, internal capsule; NF, neurofilament; VT, ventral thalamus. Scale bars: B–G, N–P, H′–J′, 100 μm; H–J, K–M, 200 μm.
At P1, NF staining highlights the typical horseshoe shape of thalamocortical and corticothalamic fibers within the internal capsule in control and II−/−IV−/−N−/− brains (Fig. 2A,B,D). In II−/−IV−/− mice, fibers originating from the thalamus did not cross the internal capsule (Fig. 2C). In addition, corticofugal fibers were completely absent from the internal capsule of II−/−IV−/− mice at this rostrocaudal level. TAG-1 immunohistochemistry confirmed the absence of corticothalamic axons exclusively in II−/−IV−/−, but not II−/−IV−/−N−/− mice at P1 (Fig. 2E–G). At P1, the fiber tract defects were found in 7 of 7 II−/−IV−/− mice that were compared with 10 control and 3 II−/−IV−/−N−/− animals. To corroborate the persistence of this misrouting in II−/−IV−/− mice, anterograde tracing of thalamocortical and retrograde tracing of corticothalamic projections was performed on fixed brain sections of P30 mice by placing DiI into the dorsal thalamus (Fig. 2H,I). In controls, all projections turned dorsally toward the cortex (Fig. 2H), whereas the vast majority of labeled fibers in II−/−IV−/− mice did not cross the internal capsule and failed to turn dorsally (Fig. 2I). Together, these observations reveal severe guidance defects of thalamocortical and corticothalamic axons in specifically the polySia-negative, NCAM-positive II−/−IV−/− mice and demonstrate that the loss of these fibers contributes substantially to the hypoplasia of the internal capsule described previously in P1 and in 4- to 6-week-old II−/−IV−/− but not II−/−IV−/−N−/− mice (Weinhold et al., 2005; Hildebrandt et al., 2009).
Figure 2.
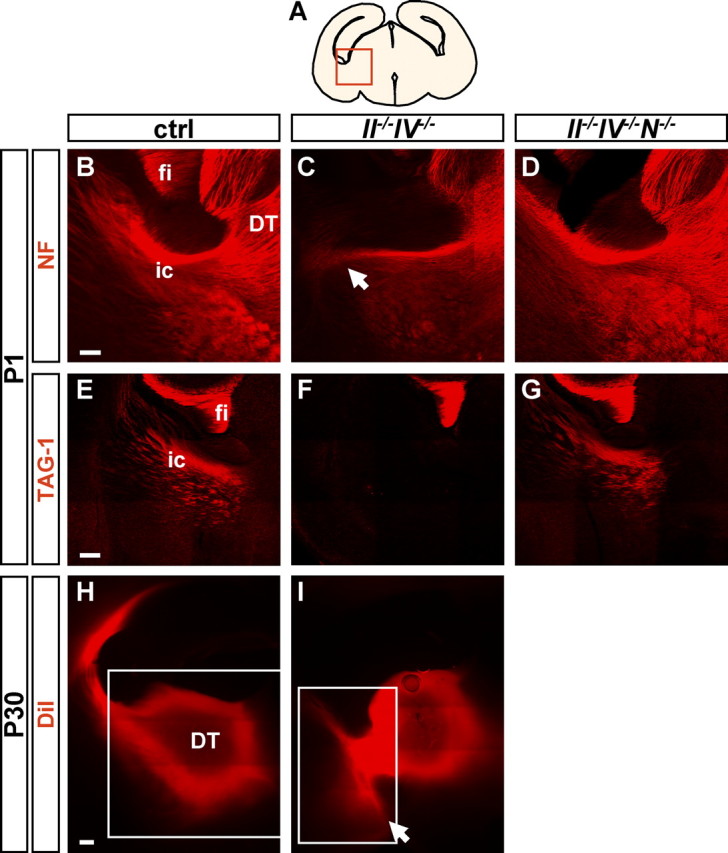
Aberrant trajectory of thalamocortical and corticothalamic fibers in II−/−IV−/− mice. A, Schematic drawing of a coronal section through the P1 mouse brain at the rostrocaudal level analyzed in B–G. B–D, NF labeling of thalamocortical and corticothalamic axons at P1. Fibers reciprocally connect cortex and thalamus in control mice (exemplarily shown for II+/−IV+/− with normal levels of polySia and NCAM (B) but do not cross the internal capsule in II−/−IV−/− mice (C, arrow). In II−/−IV−/− N−/− mice, NF staining was indistinguishable from controls (D). E–G, TAG-1 immunohistochemistry at P1 demarcates corticothalamic axons in control mice that arrive in the thalamus (E). In II−/−IV−/− mice, no TAG-1-positive corticothalamic axons are present (F). In II−/−IV−/−N−/− mice, TAG-1-labeled corticothalamic axons arrive in the thalamus (G). H, I, A small crystal of the lipophilic tracer DiI was placed into the thalamus in 300-μm-thick sections of P30, perfused animals. The white frames indicate adjustment of brightness to avoid overshadowing of labeled fibers. H, In control mice, DiI-labeled fibers turn dorsally toward the cortex. The brightness of the image in the boxed area was reduced compared with the rest of the picture. I, In II−/−IV−/− mice, some of these fibers turn ventrally instead (I, arrow). The brightness of the image in the boxed area was increased compared with the rest of the picture. DT, Dorsal thalamus; ic, internal capsule; fi, fimbria; NF, neurofilament. Scale bars: B–D, 100 μm; E–I, 200 μm.
Reduction and defasciculation of fibers traversing the ventral thalamus at P1
Comparable with the situation at E14.5, thalamocortical fibers traversed the Islet-1-positive area in the ventral thalamus and passed the diencephalic–telencephalic boundary in P1 II−/−IV−/− mice before they failed to turn (Fig. 3A,B). In control mice, NF-positive fibers cross this Islet-1-immunoreactive area as tightly fasciculated bundles (Fig. 3C). Together with the prominent reduction of traversing fibers, this fasciculated pattern was disrupted in II−/−IV−/− mice, and fibers were highly disorganized (Fig. 3D). As shown on mRNA and protein level, the transcription factor Islet-1 is expressed by neurons of the developing ventral thalamus and the adult Rt in mice and rats (Nakagawa and O'Leary, 2001; Wang and Liu, 2001). To substantiate the assumption that the Islet-1-immunoreactive cells of the ventral thalamus constitute the Rt, double immunolabeling of PV and Islet-1 was performed. Since Rt neurons express PV not before P10 (Mitrofanis, 1992) and because Islet-1 immunoreactivity was almost undetectable at P30 (data not shown), this was done in P20 wild-type mice (Fig. 4). Islet-1 was detected in all PV-positive cells of the Rt (Fig. 4A–D), and, as expected, higher magnification revealed Islet-1 immunoreactivity in the nucleus, whereas PV staining was located in the cytosol (Fig. 4B′–D′). Thus, Islet-1 staining in the ventral thalamus marks the developing Rt and the obvious reduction and defasciculation of traversing fibers point toward defective innervation of the Rt by thalamocortical and corticothalamic axon collaterals.
Figure 3.
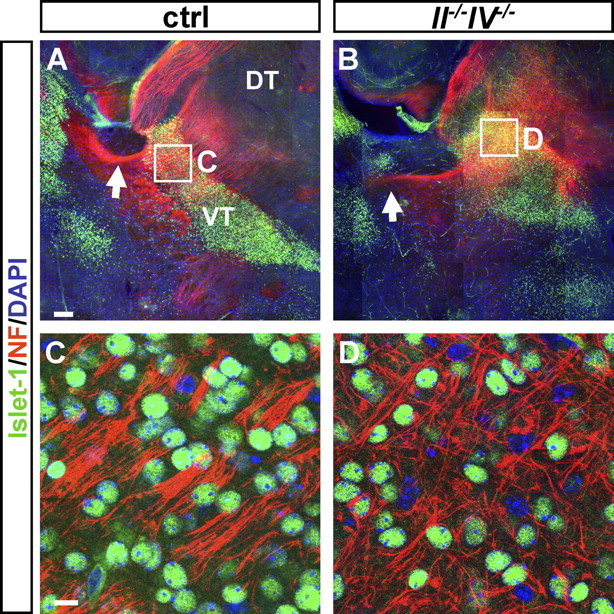
Defasciculation of thalamocortical fibers in II−/−IV−/− mice at P1. A, B, MosaiX images of 5.1-μm-thick optical sections showing double immunohistochemistry for Islet-1 and NF. Whereas NF-positive thalamocortical and corticothalamic fibers cross the Islet-1-positive VT and traverse the internal capsule in control mice (A, arrow), fibers deviate from their normal trajectory in II−/−IV−/− animals (B, arrow), and no corticofugal fibers approach the VT. C, D, Higher magnification of the area boxed in A and B. Fibers cross the Islet-1-positive ventral thalamus in tightly fasciculated bundles in control animals (C) but are disorganized in II−/− IV−/− mice (D). DT, Dorsal thalamus; NF, neurofilament; VT, ventral thalamus. Scale bars: A, B, 200 μm; C, D, 10 μm.
Figure 4.
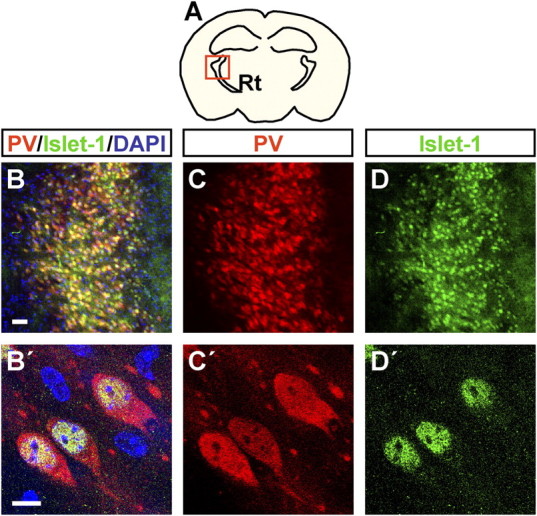
Islet-1 is coexpressed with PV in the Rt. A, Schematic drawing of a coronal section through the P20 mouse brain at the rostrocaudal level analyzed in B–D′. B–D′, Double immunohistochemistry for PV and Islet-1 at P20 in control mice. B–D, Low magnification of the Rt. All PV-positive cells coexpress Islet-1. B′–D′, The 5.1-μm-thick optical sections of P20 control mice. PV and Islet-1 are colocalized in the same cells; PV is expressed in the cytosol, and Islet-1 in the nucleus of Rt cells. Scale bars: B–D, 50 μm; B′–D′, 10 μm.
Postnatal degeneration of Rt neurons in II−/−IV−/− mice
Consistent with a general distortion and size reduction of the brain in II−/−IV−/− mice (Weinhold et al., 2005; Schiff et al., 2009), the overall shape of the Rt at P1 was more rounded than in controls (Fig. 5A,B). Despite this difference in shape, comparable levels of Islet-1 staining were consistently detected on II−/−IV−/− and control sections displaying the Rt at various rostrocaudal positions. Quantitative evaluation confirmed equal numbers of Islet-1-positive cells in the developing Rt (Fig. 5E). In addition to Islet-1, the Rt could be specifically labeled by PV immunofluorescence at P10 (Fig. 5C–D′). Between P10 and P30, the Rt formed a curved sheet of PV-positive cells around the dorsal thalamus of control brains (Fig. 5C,F). Compared with the controls, the PV- and Islet-1-labeled Rt at P10 and the PV-labeled Rt at P30 was considerably smaller in II−/−IV−/− mice (Fig. 5D,G). Quantitative evaluation of Islet-1-positive cells at P10 revealed a significant reduction in II−/−IV−/− mice (Fig. 5E), and the dramatic decrease of PV-positive Rt neurons was observed in seven of seven II−/−IV−/− animals at an age between P10 and P30. PV-containing neurons are surrounded by perineuronal nets that can be labeled with Wisteria floribunda agglutinin (WFA) (Härtig et al., 1992). Using this lectin, we found a dramatic loss of WFA-labeled perineuronal nets in the Rt of II−/−IV−/− mice (Fig. 5H,I). Together with the decrease of PV staining, this points toward a loss of Rt neurons. However, developmental maturation of perineuronal nets is activity dependent (Pizzorusso et al., 2002), and thus reduced WFA staining not necessarily indicates a loss of neurons. To further corroborate the loss of neurons as suggested by reduced Islet-1, PV, and WFA staining, GABA immunofluorescence was analyzed. GABA and PV staining colocalized in the Rt of control and II−/−IV−/− mice and was strongly diminished in the II−/−IV−/− situation (Fig. 5J–K′).
Figure 5.
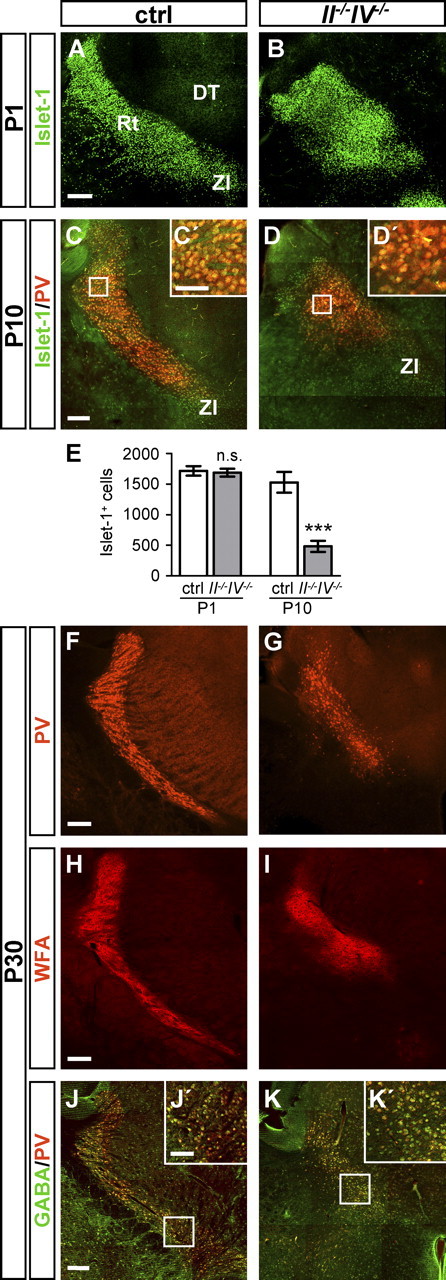
Time course of Rt disappearance. A, B, P1 brain sections of control (ctrl) and II−/−IV−/− mice at the level of the Rt, labeled by Islet-1. The Rt displays a more rounded shape in II−/−IV−/− brains compared with control animals. C, D, Double immunohistochemistry for Islet-1 and PV at the level of the Rt at P10. C′, D′, Higher magnification of the areas boxed in C and D. E, Quantification of Islet-1-positive cells within the Rt of ctrl and II−/−IV−/− mice at P1 and P10. Per hemisection, Islet-1-positive cells within the Rt of ctrl and II−/−IV−/− mice were counted (for details, see Materials and Methods). Per animal, four sections were evaluated and per group, mean values ± SEM of n = 3–4 animals are plotted (ctrl, wild-type and II+/−IV+/− controls with normal polySia levels). n.s., p > 0.1; ***p < 0.001 (t test). F, G, Brain sections of ctrl and II−/−IV−/− mice at the level of the Rt, labeled by PV at P30. The number of Rt cells is drastically reduced in II−/−IV−/− mice. H, I, Wisteria floribunda agglutinin staining of P30 mouse brain sections labels perineuronal nets around PV-positive neurons. J, K, Brain sections of ctrl and II−/−IV−/− mice at the level of the Rt, double labeled by PV and GABA at P30. J′, K′, Higher magnification of the areas boxed in J and K. DT, Dorsal thalamus. Scale bars: A–K, 200 μm; C′, D′, J′, K′, 100 μm.
These results suggest that cells of the Rt are generated in normal numbers but degenerate rapidly during postnatal development of II−/−IV−/− mice. To test this assumption, we investigated apoptotic cell death in the Rt using TUNEL and cleaved caspase-3 immunohistochemistry in combination with Islet-1 staining to identify the Rt. Analyses were performed at P5, as cell numbers in the Rt of II−/−IV−/− mice declined rapidly between P1 and P10 and stayed approximately constant from P10 onward (Fig. 5). In control animals, TUNEL staining indicated almost complete absence of apoptotic cells in the Rt (Fig. 6A). In clear contrast, II−/−IV−/− mice displayed abundant TUNEL-positive cells (Fig. 6B,B′). Quantitative evaluation confirmed significantly higher numbers of TUNEL-positive cells in the Rt (Fig. 6E). The apoptotic state of TUNEL-positive cells was supported by the pyknotic nuclei or DNA fragmentation observed in these cells by DAPI stain. Confirming this observation, cleaved caspase-3 immunostaining showed abundant positive dots in II−/−IV−/− mice that were not present in control mice (Fig. 6C,D,D′). Islet-1 labeling was absent in TUNEL- and cleaved caspase-3-positive cells. This is consistent with the observation that cell death can be accompanied by the loss of marker proteins. For example, a similar loss of PV was shown during cardiac arrest-induced apoptosis in the Rt (Kawai et al., 1995; Böttiger et al., 1998). In summary, the abundant cell death observed at P5, together with the dramatic loss of Islet-1- or PV-labeled cells between P1 and P30, strongly suggests that the Rt degenerates because of apoptotic cell death during early postnatal development of II−/−IV−/− mice.
Figure 6.
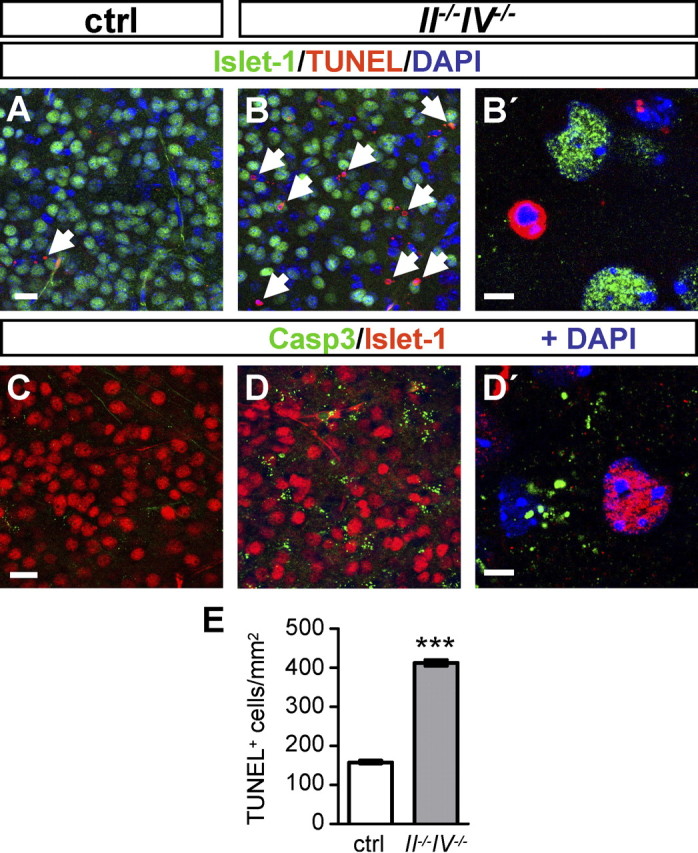
Apoptotic cell death in the Rt. A–B′, TUNEL staining combined with Islet-1 immunohistochemistry in control (ctrl) and II−/−IV−/− mice at P5. A, Very few TUNEL-positive cells were detected in the Rt of control mice at P5 (arrow). B, TUNEL-positive cells were abundant in the Rt of II−/−IV−/− mice (B, arrows; higher magnification in B′). C–D′, Double immunohistochemistry for cleaved caspase-3 and Islet-1 in ctrl and II−/−IV−/− mice at P5. Enhanced cleaved caspase-3 immunoreactivity in II−/−IV−/− (D, D′) compared with control (C) mice. E, Per hemisection, TUNEL-positive cells within the Rt were counted and expressed relative to the Rt area (in square millimeters) (for details, see Materials and Methods). Per animal, four sections were evaluated and per group, mean values ± SEM of n = 3 animals are plotted (ctrl, wild-type and II+/−IV+/− controls with normal polySia levels). ***p < 0.001, t test. Casp3, Cleaved caspase-3. Scale bars: A–D, 20 μm; B′, D′, 5 μm.
Dopaminergic innervation of the Rt
Beside the major glutamatergic projections from cortex and thalamus, the Rt receives dopaminergic input from collaterals of nigrostriatal fibers (Anaya-Martinez et al., 2006). Dopaminergic innervation of the Rt was investigated by double immunohistochemistry for PV and tyrosine hydroxylase (TH), a marker for dopaminergic cell bodies and fibers. TH-positive fibers were detected exclusively in the ventral, but not the dorsal part of the Rt in P30 control brains (Fig. 7A,C,E). Likewise, in II−/−IV−/− animals, dopaminergic fibers were absent from the area medial to the internal capsule, where the few PV-positive cells constituting the remaining Rt were located (Fig. 7B,D). More ventrally and medial to the globus pallidus, PV-positive cells were completely absent but dopaminergic fibers were present at a similar density as in control animals (Fig. 7F). These images suggest uncompromised dopaminergic projections in II−/−IV−/− mice. In addition, the pattern of TH-positive profiles also provides a landmark, indicating a complete loss of PV-positive cells in the ventral parts of the Rt, whereas the remaining PV-positive cells belong to its dorsal portion.
Figure 7.
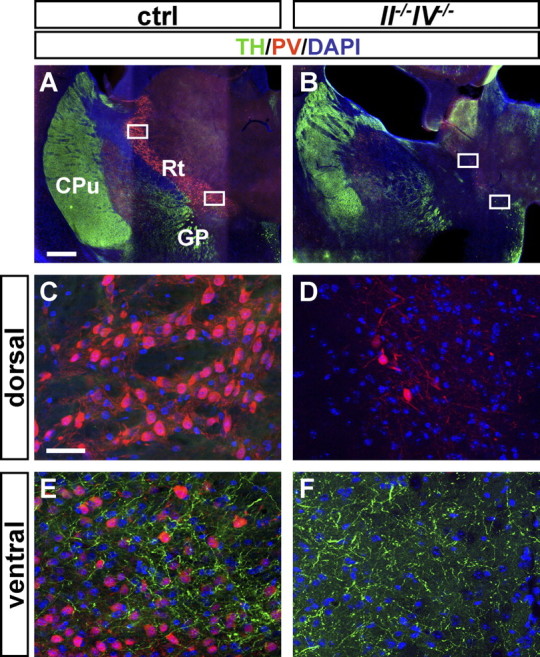
No lack of dopaminergic input in the Rt of II−/−IV−/− mice. A–F, PV and TH immunohistochemistry of P30 mouse brain sections. A, B, Overview of brain sections of control (ctrl) and II−/−IV−/− mice at the level of the reticular thalamic nucleus. PV+ cells are diminished in II−/−IV−/− brains. C, D, Higher magnification of the top boxed areas in A and B. In control and II−/−IV−/− mice, the dorsal part of the Rt lacks TH+ fibers. E, F, Higher magnification of the bottom boxed areas in A and B. In controls, the ventral part of the Rt is innervated by TH+ dopaminergic fibers (E). In II−/−IV−/− mice, TH+ fibers are present but PV+ cells are completely absent from this ventral area (F). CPu, Caudate–putamen; GP, globus pallidus. Scale bars: A, B, 500 μm; C–F, 50 μm.
Loss of Rt neurons occurs only in polySia-negative mice with internal capsule defects
Defective thalamocortical and corticothalamic projections (this study) and hypoplasia of the internal capsule (Weinhold et al., 2005; Hildebrandt et al., 2009) were found specifically in the polySia-negative, NCAM-positive II−/−IV−/−, but not in the polySia- and NCAM-deficient II−/−IV−/−N−/− triple knock-out mice. To correlate this distribution of the internal capsule defect with the loss of Rt neurons, we evaluated PV-positive Rt neurons in both polySia-negative animal models at P30 and also included the strains deficient for only NCAM or one of the polysialyltransferases (N−/−, II−/−, and IV−/−). In II−/−IV−/− animals, the numbers of PV-positive cells were clearly reduced when compared with control brains (Fig. 8). Consistent with the lack of internal capsule defects, no significant reduction of PV-positive cells was detected in II−/−IV−/−N−/− or N−/− mice. Moreover, no defects were observed in the single polysialyltransferase knock-out mice (Fig. 8).
Figure 8.

Loss of Rt neurons is restricted to polySia-negative, NCAM-positive mice at P30. Per hemisection, PV-positive cells of the Rt were counted and expressed relative to the total section area (in square millimeters) (for details, see Materials and Methods). Per animal, four sections were evaluated and per group, mean values ± SEM of n = 3 animals are plotted (ctrl, wild-type and II+/−IV+/− controls with normal polySia levels; all males, except for one female II−/−IV−/−, N−/−, and IV−/− specimen, respectively). One-way ANOVA indicated significant differences (p < 0.01) and Newman–Keuls post test was applied. *p < 0.05 versus all other groups. N−/−, NCAM deficiency.
Apoptotic cell death after corticothalamic lesion
The data presented so far indicate that degeneration of the Rt in II−/−IV−/− mice occurs subsequent to the malformation of afferent innervation by thalamocortical and corticothalamic axon collaterals. To test whether deafferentation is able to induce apoptotic cell death of Rt neurons, axons between Rt and cortex were lesioned by a unilateral incision into whole-brain slice cultures from P4 wild-type mice (Fig. 9A). Cultures were maintained for 22 h and TUNEL-positive cells within the Islet-1-positive Rt were counted (see Material and Methods for details). Islet-1 immunohistochemistry revealed a mostly intact pattern of Rt staining in the control hemisphere. In contrast, Islet-1 staining disappeared almost completely from the dorsal Rt of the lesioned side (Fig. 9B,D–F). On the same sections, TUNEL-positive cells were scarce in the unlesioned hemisphere, but became abundant within the Rt after the lesion (Fig. 9C–F). Quantification of TUNEL-positive cells revealed a significantly increased number of TUNEL-positive cells after lesion, if compared with the contralateral side as well as compared with control incisions leaving connections between Rt and cortex intact (Fig. 9G) (for details, see Materials and Methods). These results provide proof of principle that loss of afferent innervation causes apoptotic death of Rt neurons.
Figure 9.
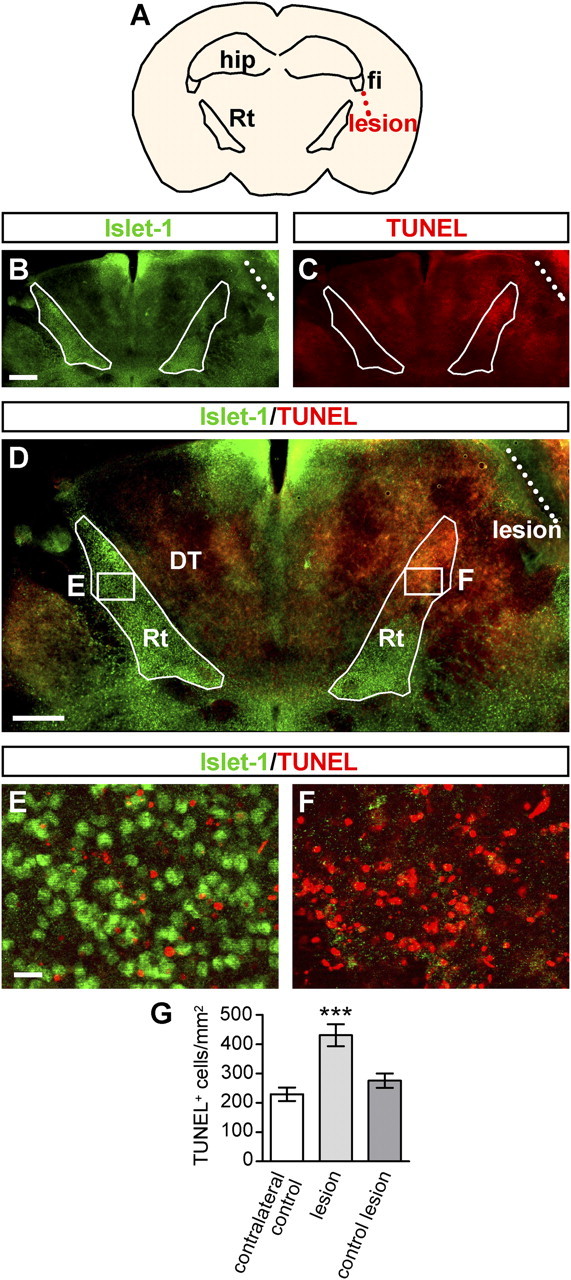
Apoptotic cell death in the Rt after deafferentation on P4 wild-type slice cultures. A, Schematic drawing illustrating the position of the lesion lateral to the Rt and ventral to the fimbria. B, Islet-1 immunohistochemistry 22 h after lesioning of axons between cortex and thalamus. Borders of the Rt and location of the lesion are indicated by white filled and dashed lines, respectively. C, TUNEL staining 22 h after the lesion. D, TUNEL staining combined with Islet-1 immunohistochemistry. E, Higher magnification of left boxed area in D (control hemisphere). TUNEL-positive cells are scarce here. F, Higher magnification of right boxed area in D (lesioned hemisphere), in which TUNEL staining is abundant. G, Per hemisection, TUNEL-positive cells of the Rt were counted and expressed relative to the Rt area (in square millimeters). Per group, mean values ± SEM of at least n = 5 slice cultures are plotted. One-way ANOVA indicated significant differences (p < 0.001), and Newman–Keuls post test was applied. ***p < 0.001 versus all other groups. fi, Fimbria; hip, hippocampus. Scale bars: B–D, 500 μm; E, F, 20 μm.
Discussion
In the present work, we demonstrate the postnatal disappearance of the Rt in polySia-deficient II−/−IV−/− mice because of increased apoptotic cell death. The degenerative process is preceded by pathfinding defects of thalamocortical axons, which in the embryonic and perinatal brain traverse the Rt correctly but then fail to cross the internal capsule. Thus, degeneration of the Rt may be a consequence of disturbed afferent pathways.
In mice, thalamocortical fibers start to grow toward the cortical plate as early as E13 (Molnár and Blakemore, 1995). They meet axons from the cortical plate in the basal telencephalon at E14 and intermingle in a process that has been described as the “handshake” (Blakemore and Molnár, 1990; Molnár and Blakemore, 1991). This process of selective fasciculation and contact guidance has been proposed to be required for the highly organized entry of thalamocortical fibers into the cortical plate as well as for corticothalamic fibers to be guided toward their targets (Molnár, 2000; Molnár et al., 2003). Other experimental data, however, suggest that the reciprocal connections between thalamus and cortex can develop independently of each other (Garel et al., 2002; Torii and Levitt, 2005). In II−/−IV−/− mice, the misguidance of thalamocortical fibers prevents the encounter of thalamocortical and corticofugal fibers and no entry of NF-positive corticothalamic projections into the internal capsule was observed at E14.5 and P1. Thus, the prominent reduction and defasciculation of axons traversing the Rt seems to be mainly caused by a lack of corticothalamic fibers. Although the exact contributions of thalamocortical and corticothalamic fiber deficiencies to the previously observed hypoplasia of the internal capsule remain open, the glutamatergic innervation of the Rt by collaterals of these axons is clearly disturbed in II−/−IV−/− mice.
A number of mouse models with thalamocortical projection defects have been described, but fiber densities and fasciculation within the Rt as well as potential consequences on Rt integrity have not been studied. Conditional ablation of Sonic hedgehog in the developing thalamus and hypothalamus completely prevents the emergence of the ventral thalamus, the largest part of which is the Rt, as well as the development of thalamocortical axons (Szabó et al., 2009a,b). In small eye rats and mice harboring a Pax6 mutation, thalamocortical axons turn ventrally directly after invading the ventral thalamus (Kawano et al., 1999; Simpson et al., 2009). In a number of other mouse models with defective thalamocortical projections, the developmental pathfinding errors are comparable with those observed in polySia-deficient mice. In Mash-1-, Robo1/2-, Slit1/2-, Emx2-, and Sema6A-null mutants, thalamocortical axons pass the Rt correctly but then take an aberrant route at the diencephalic–telencephalic junction (Tuttle et al., 1999; Bagri et al., 2002; López-Bendito et al., 2002, 2007; Plump et al., 2002; Andrews et al., 2006; Little et al., 2009). Perinatal lethality, however, prevents analysis of postnatal Rt development in Mash-1, Emx2, Robo1/2, and Slit1/2 mutants. In Sema6A-null mutants, thalamocortical connectivity recovers postnatally, leading to a grossly normal pattern of internal capsule projections (Little et al., 2009). In contrast, the pronounced deficits of the internal capsule in polySia-deficient mice are maintained until the age of 4–6 weeks, the latest time point amenable for analysis before most of the II−/−IV−/− mice die (Weinhold et al., 2005).
Consistent with the previously observed integrity of the internal capsule in mice lacking not only polySia but also its major protein carrier NCAM (Weinhold et al., 2005; Hildebrandt et al., 2009), trajectories of thalamocortical and corticothalamic fibers were normal in the II−/−IV−/−N−/− triple knock-out mice. In line with the proposed mechanism leading to axon tract defects (Hildebrandt et al., 2009), these findings indicate that thalamocortical misprojection and Rt degeneration are caused by a gain of non-polysialylated NCAM and not by a loss of polySia-mediated responses to guidance cues or other polySia-specific functions. Furthermore, altered functions of other polySia carriers, such as neuropilin-2 or SynCAM 1 (Curreli et al., 2007; Galuska et al., 2010), appear to be not involved. As polySia was found on thalamocortical axons and on surrounding structures, aberrant NCAM interactions caused by the loss of polySia may occur between axons as well as between an axon and its environment. Competing interactions, therefore, may either lead to hyperfasciculation, as observed for the aberrant thalamofugal projections or cause defasciculation, as seen for fibers crossing the Rt. It remains open, which of the homophilic or heterophilic interactions of NCAM (for review, see Nielsen et al., 2010) may be affected by the loss of polySia. Remarkably, however, one of the known binding partners of NCAM, L1, associates with neuropilin-1 to form a semaphorin 3A receptor implicated in axon guidance and, as shown recently, cooperates with its close homolog CHL1 in thalamocortical axon targeting (Castellani et al., 2000, 2002; Demyanenko et al., 2010). Thus, altered NCAM interactions with, for example, L1 may affect the response of the axons to guidance cues in the ventral telencephalon.
As for the defects of the internal capsule, reduced numbers of Rt neurons were only observed in polySia-negative, NCAM-positive II−/−IV−/−, but not in polySia- and NCAM-deficient N−/− and II−/−IV−/−N−/− mice. Since polySia is ubiquitously expressed in all thalamic nuclei during embryonic and perinatal development (Mazzetti et al., 2007; and our unpublished observations), the reasons for the conspicuous degeneration of Rt neurons are puzzling at first sight. Cell death may result from a pronounced vulnerability of the Rt as described for hypoxia-induced neuronal damage (Freund et al., 1990; Kawai et al., 1995; Böttiger et al., 1998). However, it appears unlikely that polySia-free NCAM interferes directly with the survival of Rt neurons. As demonstrated previously, a gain of polySia-free NCAM promotes survival of neuronal precursors. On the contrary, the combined deletion of NCAM and polySia reduces survival of these precursors (Gascon et al., 2007; Röckle et al., 2008). Islet-1-positive cells of the Rt are generated in the ventral thalamus and migrate tangentially to form the Rt. Similarly, cells of the Islet-1-positive permissive corridor migrate tangentially within the ventral telencephalon (López-Bendito et al., 2006). Although polySia is prominently involved in neuronal migration (for review, see Hildebrandt et al., 2007; Rutishauser, 2008), the initially normal localization of Islet-1-positive cells in the Rt of embryonic and perinatal II−/−IV−/− mice excludes the possibility that the loss of Rt neurons is caused by defective migration. Moreover, in contrast to the pathfinding defects, which together with Rt degeneration occur in II−/−IV−/− but not in NCAM-deficient animals, the features of II−/−IV−/− mice caused by disturbed migration are also seen N−/− and II−/−IV−/−N−/− animals (Weinhold et al., 2005).
Together, the data imply that the impact of polySia-deficient NCAM on Rt neurons is indirect and not caused by a migration deficit. Instead, we demonstrate pathfinding errors of thalamocortical axons in polySia-negative, NCAM-positive mice, which contribute to the hypoplasia of the internal capsule and precede apoptotic cell death in the Rt. As shown in rats, reticulothalamic projections are formed as early as E14 (Mitrofanis and Baker, 1993), which corresponds approximately to E12 in mice. In contrast, collaterals of thalamocortical and corticothalamic axons within the Rt are not detectable until birth (Mitrofanis and Guillery, 1993). Thus, degeneration of the Rt in II−/−IV−/− mice seems to start only after the time point when under normal conditions glutamatergic input is completed. Based on the coincidence of the two defects in specifically II−/−IV−/− mice, the close integration of Rt neurons into thalamocortical circuits, and the observed sequence of events, we propose that a lack of excitatory innervation causes apoptotic death of Rt neurons in polySia-negative, NCAM-positive mice. Although not as prominent as retrograde mechanisms after target deprivation (Buss et al., 2006), there are abundant examples for such a mechanism of anterograde transneuronal degeneration [as first defined by Cowan (1970)], which is now widely accepted (Oppenheim, 1991). As discussed by Capurso et al. (1997), anterograde degenerative effects have been observed in, for example, auditory and vestibular nuclei, the visual system, somatosensory cortex, and the olfactory system. Prominent examples of transneuronal anterograde degeneration after experimental deafferentation comprise apoptosis of cortical sensory neurons after deafferentation injury (Capurso et al., 1997) and degeneration of hippocampal granule cells after loss of input in the pilocarpine model of epilepsy (Marques-Mari et al., 2007). As shown before, PV-expressing neurons of the superior colliculus and the spinal cord are particularly vulnerable to afferent lesions (Barker and Dreher, 1998; Dekkers et al., 2002). In the current study, we provide direct evidence that also the PV-positive neurons of the Rt can be the target of transneuronal anterograde degeneration. Closely resembling the time course of cell death after deafferentation in vivo (Capurso et al., 1997), abundant apoptosis occurred within 24 h after lesioning corticothalamic axons in whole-brain slice cultures. The degeneration of the Rt in II−/−IV−/− mice, therefore, could indeed be caused by a loss of glutamatergic input because of malformation of thalamocortical and corticothalamic fibers.
Variations in the Ncam1 and St8siaII gene as well as dysregulation of NCAM protein have been linked to schizophrenia (Vawter, 2000; Arai et al., 2006; Atz et al., 2007; Sullivan et al., 2007; Tao et al., 2007). On the one hand, incomplete polysialylation of NCAM during brain development has been proposed as a mechanism for a predisposition to schizophrenia because it causes defects of cortical connectivity, including internal capsule defects, resembling those seen in schizophrenic patients (Hildebrandt et al., 2009). On the other hand, dysfunction of the Rt has been linked to schizophrenia (O'Donnell and Grace, 1998; Sharp et al., 2001; Krause et al., 2003; Behrendt and Young, 2004; Behrendt, 2006; Zikopoulos and Barbas, 2007). Since all connections between thalamus and cortex traverse the Rt, this nucleus is ideally positioned to play a role in feedback control of thalamocortical circuits and sensory gating. Concerning possible consequences of Rt dysfunction it would be worthwhile to analyze deficits of, for example, sensory gating in II−/−IV−/− mice. Unfortunately, the postnatal growth retardation and the cachectic state of even the juvenile II−/−IV−/− mice (Weinhold et al., 2005) prevent adequate testing of schizophrenia-like behavior. In conclusion, the current analysis of polysialylation-deficient mice suggests that Rt neurons are damaged in response to malformation of their glutamatergic input. This provides a potential neurodevelopmental mechanism of how sensory processing involving the Rt may be disturbed in association with internal capsule hypoplasia as observed in schizophrenia.
Footnotes
This work was funded by Deutsche Forschungsgemeinschaft and Deutsche Krebshilfe (H.H.). M.S. received a Georg-Christoph-Lichtenberg fellowship by the Zentrum für Systemische Neurowissenschaften Hannover. The monoclonal antibodies 2H3, 4D5, and 4D7 developed by Thomas M. Jessell, Jane Dodd, and Miyuki Yamamoto were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the Department of Biology of University of Iowa (Iowa City, IA). The polyclonal antibody against mouse L1 was a kind gift from Fritz Rathjen (Berlin, Germany). We thank U. Bernard, E. Kats, and D. Wittenberg for expert technical assistance.
References
- Agmon A, Connors BW. Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience. 1991;41:365–379. doi: 10.1016/0306-4522(91)90333-j. [DOI] [PubMed] [Google Scholar]
- Anaya-Martinez V, Martinez-Marcos A, Martinez-Fong D, Aceves J, Erlij D. Substantia nigra compacta neurons that innervate the reticular thalamic nucleus in the rat also project to striatum or globus pallidus: implications for abnormal motor behavior. Neuroscience. 2006;143:477–486. doi: 10.1016/j.neuroscience.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Andrews W, Liapi A, Plachez C, Camurri L, Zhang J, Mori S, Murakami F, Parnavelas JG, Sundaresan V, Richards LJ. Robo1 regulates the development of major axon tracts and interneuron migration in the forebrain. Development. 2006;133:2243–2252. doi: 10.1242/dev.02379. [DOI] [PubMed] [Google Scholar]
- Angata K, Huckaby V, Ranscht B, Terskikh A, Marth JD, Fukuda M. Polysialic acid-directed migration and differentiation of neural precursors is essential for mouse brain development. Mol Cell Biol. 2007;27:6659–6668. doi: 10.1128/MCB.00205-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai M, Yamada K, Toyota T, Obata N, Haga S, Yoshida Y, Nakamura K, Minabe Y, Ujike H, Sora I, Ikeda K, Mori N, Yoshikawa T, Itokawa M. Association between polymorphisms in the promoter region of the sialyltransferase 8B (SIAT8B) gene and schizophrenia. Biol Psychiatry. 2006;59:652–659. doi: 10.1016/j.biopsych.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Atz ME, Rollins B, Vawter MP. NCAM1 association study of bipolar disorder and schizophrenia: polymorphisms and alternatively spliced isoforms lead to similarities and differences. Psychiatr Genet. 2007;17:55–67. doi: 10.1097/YPG.0b013e328012d850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagri A, Marín O, Plump AS, Mak J, Pleasure SJ, Rubenstein JL, Tessier-Lavigne M. Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron. 2002;33:233–248. doi: 10.1016/s0896-6273(02)00561-5. [DOI] [PubMed] [Google Scholar]
- Barker DA, Dreher B. Spatiotemporal patterns of ontogenetic expression of parvalbumin in the superior colliculi of rats and rabbits. J Comp Neurol. 1998;393:210–230. doi: 10.1002/(sici)1096-9861(19980406)393:2<210::aid-cne6>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Behrendt RP. Dysregulation of thalamic sensory “transmission” in schizophrenia: neurochemical vulnerability to hallucinations. J Psychopharmacol. 2006;20:356–372. doi: 10.1177/0269881105057696. [DOI] [PubMed] [Google Scholar]
- Behrendt RP, Young C. Hallucinations in schizophrenia, sensory impairment, and brain disease: a unifying model. Behav Brain Sci. 2004;27:771–787. doi: 10.1017/s0140525x04000184. discussion 787–830. [DOI] [PubMed] [Google Scholar]
- Blakemore C, Molnár Z. Factors involved in the establishment of specific interconnections between thalamus and cerebral cortex. Cold Spring Harb Symp Quant Biol. 1990;55:491–504. doi: 10.1101/sqb.1990.055.01.048. [DOI] [PubMed] [Google Scholar]
- Böttiger BW, Schmitz B, Wiessner C, Vogel P, Hossmann KA. Neuronal stress response and neuronal cell damage after cardiocirculatory arrest in rats. J Cereb Blood Flow Metab. 1998;18:1077–1087. doi: 10.1097/00004647-199810000-00004. [DOI] [PubMed] [Google Scholar]
- Buss RR, Sun W, Oppenheim RW. Adaptive roles of programmed cell death during nervous system development. Annu Rev Neurosci. 2006;29:1–35. doi: 10.1146/annurev.neuro.29.051605.112800. [DOI] [PubMed] [Google Scholar]
- Byne W, Hazlett EA, Buchsbaum MS, Kemether E. The thalamus and schizophrenia: current status of research. Acta Neuropathol. 2009;117:347–368. doi: 10.1007/s00401-008-0404-0. [DOI] [PubMed] [Google Scholar]
- Capurso SA, Calhoun ME, Sukhov RR, Mouton PR, Price DL, Koliatsos VE. Deafferentation causes apoptosis in cortical sensory neurons in the adult rat. J Neurosci. 1997;17:7372–7384. doi: 10.1523/JNEUROSCI.17-19-07372.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellani V, Chédotal A, Schachner M, Faivre-Sarrailh C, Rougon G. Analysis of the L1-deficient mouse phenotype reveals cross-talk between Sema3A and L1 signaling pathways in axonal guidance. Neuron. 2000;27:237–249. doi: 10.1016/s0896-6273(00)00033-7. [DOI] [PubMed] [Google Scholar]
- Castellani V, De Angelis E, Kenwrick S, Rougon G. Cis and trans interactions of L1 with neuropilin-1 control axonal responses to semaphorin 3A. EMBO J. 2002;21:6348–6357. doi: 10.1093/emboj/cdf645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan WM. Anterograde and retrograde transneuronal degeneration in the central and peripheral nervous system. In: Nauta WJH, Ebbesson SOE, editors. Contemporary research methods in neuroanatomy. New York: Springer; 1970. pp. 217–251. [Google Scholar]
- Crick F. Function of the thalamic reticular complex: the searchlight hypothesis. Proc Natl Acad Sci U S A. 1984;81:4586–4590. doi: 10.1073/pnas.81.14.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csillik B, Mihaly A, Krisztin-Peva B, Fenyo R, Knyihar-Csillik E. Calcium-binding proteins in GABAergic calyciform synapses of the reticular nucleus. Neuroreport. 2006;17:575–578. doi: 10.1097/00001756-200604240-00004. [DOI] [PubMed] [Google Scholar]
- Curreli S, Arany Z, Gerardy-Schahn R, Mann D, Stamatos NM. Polysialylated neuropilin-2 is expressed on the surface of human dendritic cells and modulates dendritic cell-T lymphocyte interactions. J Biol Chem. 2007;282:30346–30356. doi: 10.1074/jbc.M702965200. [DOI] [PubMed] [Google Scholar]
- Dekkers J, Greensmith L, Navarrete R. Changes in the expression of parvalbumin immunoreactivity in the lumbar spinal cord of the rat following neonatal nerve injury. Dev Neurosci. 2002;24:283–293. doi: 10.1159/000066742. [DOI] [PubMed] [Google Scholar]
- Del Río JA, González-Billault C, Ureña JM, Jiménez EM, Barallobre MJ, Pascual M, Pujadas L, Simó S, La Torre A, Wandosell F, Avila J, Soriano E. MAP1B is required for Netrin 1 signaling in neuronal migration and axonal guidance. Curr Biol. 2004;14:840–850. doi: 10.1016/j.cub.2004.04.046. [DOI] [PubMed] [Google Scholar]
- Demyanenko GP, Siesser PF, Wright AG, Brennaman LH, Bartsch U, Schachner M, Maness PF. L1 and CHL1 cooperate in thalamocortical axon targeting. Cereb Cortex. 2010 doi: 10.1093/cercor/bhq115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denaxa M, Chan CH, Schachner M, Parnavelas JG, Karagogeos D. The adhesion molecule TAG-1 mediates the migration of cortical interneurons from the ganglionic eminence along the corticofugal fiber system. Development. 2001;128:4635–4644. doi: 10.1242/dev.128.22.4635. [DOI] [PubMed] [Google Scholar]
- Deng J, Elberger AJ. Corticothalamic and thalamocortical pathfinding in the mouse: dependence on intermediate targets and guidance axis. Anat Embryol (Berl) 2003;207:177–192. doi: 10.1007/s00429-003-0338-1. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsáki G, Leon A, Baimbridge KG, Somogyi P. Relationship of neuronal vulnerability and calcium binding protein immunoreactivity in ischemia. Exp Brain Res. 1990;83:55–66. doi: 10.1007/BF00232193. [DOI] [PubMed] [Google Scholar]
- Frosch M, Gorgen I, Boulnois GJ, Timmis KN, Bitter-Suermann D. NZB mouse system for production of monoclonal antibodies to weak bacterial antigens: isolation of an IgG antibody to the polysaccharide capsules of Escherichia coli K1 and group B meningococci. Proc Natl Acad Sci U S A. 1985;82:1194–1198. doi: 10.1073/pnas.82.4.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galuska SP, Rollenhagen M, Kaup M, Eggers K, Oltmann-Norden I, Schiff M, Hartmann M, Weinhold B, Hildebrandt H, Geyer R, Mühlenhoff M, Geyer H. Synaptic cell adhesion molecule SynCAM 1 is a target for polysialylation in postnatal mouse brain. Proc Natl Acad Sci U S A. 2010;107:10250–10255. doi: 10.1073/pnas.0912103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garel S, Yun K, Grosschedl R, Rubenstein JL. The early topography of thalamocortical projections is shifted in Ebf1 and Dlx1/2 mutant mice. Development. 2002;129:5621–5634. doi: 10.1242/dev.00166. [DOI] [PubMed] [Google Scholar]
- Gascon E, Vutskits L, Jenny B, Durbec P, Kiss JZ. PSA-NCAM in postnatally generated immature neurons of the olfactory bulb: a crucial role in regulating p75 expression and cell survival. Development. 2007;134:1181–1190. doi: 10.1242/dev.02808. [DOI] [PubMed] [Google Scholar]
- Guillery RW. On counting and counting errors. J Comp Neurol. 2002;447:1–7. doi: 10.1002/cne.10221. [DOI] [PubMed] [Google Scholar]
- Guillery RW, Feig SL, Lozsádi DA. Paying attention to the thalamic reticular nucleus. Trends Neurosci. 1998;21:28–32. doi: 10.1016/s0166-2236(97)01157-0. [DOI] [PubMed] [Google Scholar]
- Harris RM. Axon collaterals in the thalamic reticular nucleus from thalamocortical neurons of the rat ventrobasal thalamus. J Comp Neurol. 1987;258:397–406. doi: 10.1002/cne.902580308. [DOI] [PubMed] [Google Scholar]
- Härtig W, Brauer K, Brückner G. Wisteria floribunda agglutinin-labelled nets surround parvalbumin-containing neurons. Neuroreport. 1992;3:869–872. doi: 10.1097/00001756-199210000-00012. [DOI] [PubMed] [Google Scholar]
- Herzog KH, Schulz A, Buerkle C, Gromoll C, Braun JS. Radiation-induced apoptosis in retinal progenitor cells is p53-dependent with caspase-independent DNA fragmentation. Eur J Neurosci. 2007;25:1349–1356. doi: 10.1111/j.1460-9568.2007.05381.x. [DOI] [PubMed] [Google Scholar]
- Hildebrandt H, Mühlenhoff M, Weinhold B, Gerardy-Schahn R. Dissecting polysialic acid and NCAM functions in brain development. J Neurochem. 2007;103(Suppl 1):56–64. doi: 10.1111/j.1471-4159.2007.04716.x. [DOI] [PubMed] [Google Scholar]
- Hildebrandt H, Mühlenhoff M, Oltmann-Norden I, Röckle I, Burkhardt H, Weinhold B, Gerardy-Schahn R. Imbalance of neural cell adhesion molecule and polysialyltransferase alleles causes defective brain connectivity. Brain. 2009;132:2831–2838. doi: 10.1093/brain/awp117. [DOI] [PubMed] [Google Scholar]
- Hildebrandt H, Mühlenhoff M, Gerardy-Schahn R. Polysialylation of NCAM. Adv Exp Med Biol. 2010;663:95–109. doi: 10.1007/978-1-4419-1170-4_6. [DOI] [PubMed] [Google Scholar]
- Kawai K, Nowak TS, Jr, Klatzo I. Loss of parvalbumin immunoreactivity defines selectively vulnerable thalamic reticular nucleus neurons following cardiac arrest in the rat. Acta Neuropathol. 1995;89:262–269. doi: 10.1007/BF00309342. [DOI] [PubMed] [Google Scholar]
- Kawano H, Fukuda T, Kubo K, Horie M, Uyemura K, Takeuchi K, Osumi N, Eto K, Kawamura K. Pax-6 is required for thalamocortical pathway formation in fetal rats. J Comp Neurol. 1999;408:147–160. doi: 10.1002/(sici)1096-9861(19990531)408:2<147::aid-cne1>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Krause M, Hoffmann WE, Hajós M. Auditory sensory gating in hippocampus and reticular thalamic neurons in anesthetized rats. Biol Psychiatry. 2003;53:244–253. doi: 10.1016/s0006-3223(02)01463-4. [DOI] [PubMed] [Google Scholar]
- Little GE, López-Bendito G, Rünker AE, García N, Piñon MC, Chédotal A, Molnár Z, Mitchell KJ. Specificity and plasticity of thalamocortical connections in Sema6A mutant mice. PLoS Biol. 2009;7:e98. doi: 10.1371/journal.pbio.1000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Bendito G, Chan CH, Mallamaci A, Parnavelas J, Molnár Z. Role of Emx2 in the development of the reciprocal connectivity between cortex and thalamus. J Comp Neurol. 2002;451:153–169. doi: 10.1002/cne.10345. [DOI] [PubMed] [Google Scholar]
- López-Bendito G, Cautinat A, Sánchez JA, Bielle F, Flames N, Garratt AN, Talmage DA, Role LW, Charnay P, Marín O, Garel S. Tangential neuronal migration controls axon guidance: a role for neuregulin-1 in thalamocortical axon navigation. Cell. 2006;125:127–142. doi: 10.1016/j.cell.2006.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Bendito G, Flames N, Ma L, Fouquet C, Di Meglio T, Chedotal A, Tessier-Lavigne M, Marín O. Robo1 and Robo2 cooperate to control the guidance of major axonal tracts in the mammalian forebrain. J Neurosci. 2007;27:3395–3407. doi: 10.1523/JNEUROSCI.4605-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Harada T, Harada C, Romero M, Hebert JM, McConnell SK, Parada LF. Neurotrophin-3 is required for appropriate establishment of thalamocortical connections. Neuron. 2002;36:623–634. doi: 10.1016/s0896-6273(02)01021-8. [DOI] [PubMed] [Google Scholar]
- Marqués-Marí AI, Nacher J, Crespo C, Gutièrrez-Mecinas M, Martínez-Guijarro FJ, Blasco-Ibáñez JM. Loss of input from the mossy cells blocks maturation of newly generated granule cells. Hippocampus. 2007;17:510–524. doi: 10.1002/hipo.20290. [DOI] [PubMed] [Google Scholar]
- Mazzetti S, Ortino B, Inverardi F, Frassoni C, Amadeo A. PSA-NCAM in the developing and mature thalamus. Brain Res Bull. 2007;71:578–586. doi: 10.1016/j.brainresbull.2006.11.015. [DOI] [PubMed] [Google Scholar]
- McAlonan K, Brown VJ. The thalamic reticular nucleus: more than a sensory nucleus? Neuroscientist. 2002;8:302–305. doi: 10.1177/107385840200800405. [DOI] [PubMed] [Google Scholar]
- McAlonan K, Cavanaugh J, Wurtz RH. Guarding the gateway to cortex with attention in visual thalamus. Nature. 2008;456:391–394. doi: 10.1038/nature07382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métin C, Godement P. The ganglionic eminence may be an intermediate target for corticofugal and thalamocortical axons. J Neurosci. 1996;16:3219–3235. doi: 10.1523/JNEUROSCI.16-10-03219.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Métin C, Deléglise D, Serafini T, Kennedy TE, Tessier-Lavigne M. A role for netrin-1 in the guidance of cortical efferents. Development. 1997;124:5063–5074. doi: 10.1242/dev.124.24.5063. [DOI] [PubMed] [Google Scholar]
- Mitrofanis J. Patterns of antigenic expression in the thalamic reticular nucleus of developing rats. J Comp Neurol. 1992;320:161–181. doi: 10.1002/cne.903200203. [DOI] [PubMed] [Google Scholar]
- Mitrofanis J, Baker GE. Development of the thalamic reticular and perireticular nuclei in rats and their relationship to the course of growing corticofugal and corticopetal axons. J Comp Neurol. 1993;338:575–587. doi: 10.1002/cne.903380407. [DOI] [PubMed] [Google Scholar]
- Mitrofanis J, Guillery RW. New views of the thalamic reticular nucleus in the adult and the developing brain. Trends Neurosci. 1993;16:240–245. doi: 10.1016/0166-2236(93)90163-g. [DOI] [PubMed] [Google Scholar]
- Molnár Z. Conserved developmental algorithms during thalamocortical circuit formation in mammals and reptiles. Novartis Found Symp. 2000;228:148, 166. doi: 10.1002/0470846631.ch11. discussion 166–172. [DOI] [PubMed] [Google Scholar]
- Molnár Z, Blakemore C. Lack of regional specificity for connections formed between thalamus and cortex in coculture. Nature. 1991;351:475–477. doi: 10.1038/351475a0. [DOI] [PubMed] [Google Scholar]
- Molnár Z, Blakemore C. How do thalamic axons find their way to the cortex? Trends Neurosci. 1995;18:389–397. doi: 10.1016/0166-2236(95)93935-q. [DOI] [PubMed] [Google Scholar]
- Molnár Z, Cordery P. Connections between cells of the internal capsule, thalamus, and cerebral cortex in embryonic rat. J Comp Neurol. 1999;413:1–25. doi: 10.1002/(sici)1096-9861(19991011)413:1<1::aid-cne1>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Molnár Z, Higashi S, López-Bendito G. Choreography of early thalamocortical development. Cereb Cortex. 2003;13:661–669. doi: 10.1093/cercor/13.6.661. [DOI] [PubMed] [Google Scholar]
- Nagaeva DV, Akhmadeev AV. Structural organization, neurochemical characteristics, and connections of the reticular nucleus of the thalamus. Neurosci Behav Physiol. 2006;36:987–995. doi: 10.1007/s11055-006-0134-5. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, O'Leary DD. Combinatorial expression patterns of LIM-homeodomain and other regulatory genes parcellate developing thalamus. J Neurosci. 2001;21:2711–2725. doi: 10.1523/JNEUROSCI.21-08-02711.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J, Kulahin N, Walmod PS. Extracellular protein interactions mediated by the neural cell adhesion molecule, NCAM: heterophilic interactions between NCAM and cell adhesion molecules, extracellular matrix proteins, and viruses. Adv Exp Med Biol. 2010;663:23–53. doi: 10.1007/978-1-4419-1170-4_2. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA. Dysfunctions in multiple interrelated systems as the neurobiological bases of schizophrenic symptom clusters. Schizophr Bull. 1998;24:267–283. doi: 10.1093/oxfordjournals.schbul.a033325. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Pinault D. The thalamic reticular nucleus: structure, function and concept. Brain Res Brain Res Rev. 2004;46:1–31. doi: 10.1016/j.brainresrev.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science. 2002;298:1248–1251. doi: 10.1126/science.1072699. [DOI] [PubMed] [Google Scholar]
- Plump AS, Erskine L, Sabatier C, Brose K, Epstein CJ, Goodman CS, Mason CA, Tessier-Lavigne M. Slit1 and Slit2 cooperate to prevent premature midline crossing of retinal axons in the mouse visual system. Neuron. 2002;33:219–232. doi: 10.1016/s0896-6273(01)00586-4. [DOI] [PubMed] [Google Scholar]
- Röckle I, Seidenfaden R, Weinhold B, Mühlenhoff M, Gerardy-Schahn R, Hildebrandt H. Polysialic acid controls NCAM-induced differentiation of neuronal precursors into calretinin-positive olfactory bulb interneurons. Dev Neurobiol. 2008;68:1170–1184. doi: 10.1002/dneu.20649. [DOI] [PubMed] [Google Scholar]
- Rougon G, Hobert O. New insights into the diversity and function of neuronal immunoglobulin superfamily molecules. Annu Rev Neurosci. 2003;26:207–238. doi: 10.1146/annurev.neuro.26.041002.131014. [DOI] [PubMed] [Google Scholar]
- Rutishauser U. Polysialic acid in the plasticity of the developing and adult vertebrate nervous system. Nat Rev Neurosci. 2008;9:26–35. doi: 10.1038/nrn2285. [DOI] [PubMed] [Google Scholar]
- Schiff M, Weinhold B, Grothe C, Hildebrandt H. NCAM and polysialyltransferase profiles match dopaminergic marker gene expression but polysialic acid is dispensable for development of the midbrain dopamine system. J Neurochem. 2009;110:1661–1673. doi: 10.1111/j.1471-4159.2009.06267.x. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Tomitaka M, Bernaudin M, Tomitaka S. Psychosis: pathological activation of limbic thalamocortical circuits by psychomimetics and schizophrenia? Trends Neurosci. 2001;24:330–334. doi: 10.1016/s0166-2236(00)01817-8. [DOI] [PubMed] [Google Scholar]
- Simpson TI, Pratt T, Mason JO, Price DJ. Normal ventral telencephalic expression of Pax6 is required for normal development of thalamocortical axons in embryonic mice. Neural Dev. 2009;4:19. doi: 10.1186/1749-8104-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Keefe RS, Lange LA, Lange EM, Stroup TS, Lieberman J, Maness PF. NCAM1 and neurocognition in schizophrenia. Biol Psychiatry. 2007;61:902–910. doi: 10.1016/j.biopsych.2006.07.036. [DOI] [PubMed] [Google Scholar]
- Szabó NE, Zhao T, Zhou X, Alvarez-Bolado G. The role of Sonic hedgehog of neural origin in thalamic differentiation in the mouse. J Neurosci. 2009a;29:2453–2466. doi: 10.1523/JNEUROSCI.4524-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó NE, Zhao T, Cankaya M, Theil T, Zhou X, Alvarez-Bolado G. Role of neuroepithelial Sonic hedgehog in hypothalamic patterning. J Neurosci. 2009b;29:6989–7002. doi: 10.1523/JNEUROSCI.1089-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Li C, Zheng Y, Qin W, Zhang J, Li X, Xu Y, Shi YY, Feng G, He L. Positive association between SIAT8B and schizophrenia in the Chinese Han population. Schizophr Res. 2007;90:108–114. doi: 10.1016/j.schres.2006.09.029. [DOI] [PubMed] [Google Scholar]
- Torii M, Levitt P. Dissociation of corticothalamic and thalamocortical axon targeting by an EphA7-mediated mechanism. Neuron. 2005;48:563–575. doi: 10.1016/j.neuron.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Tuttle R, Nakagawa Y, Johnson JE, O'Leary DD. Defects in thalamocortical axon pathfinding correlate with altered cell domains in Mash-1-deficient mice. Development. 1999;126:1903–1916. doi: 10.1242/dev.126.9.1903. [DOI] [PubMed] [Google Scholar]
- Vawter MP. Dysregulation of the neural cell adhesion molecule and neuropsychiatric disorders. Eur J Pharmacol. 2000;405:385–395. doi: 10.1016/s0014-2999(00)00568-9. [DOI] [PubMed] [Google Scholar]
- Wang HF, Liu FC. Developmental restriction of the LIM homeodomain transcription factor Islet-1 expression to cholinergic neurons in the rat striatum. Neuroscience. 2001;103:999–1016. doi: 10.1016/s0306-4522(00)00590-x. [DOI] [PubMed] [Google Scholar]
- Weinhold B, Seidenfaden R, Röckle I, Mühlenhoff M, Schertzinger F, Conzelmann S, Marth JD, Gerardy-Schahn R, Hildebrandt H. Genetic ablation of polysialic acid causes severe neurodevelopmental defects rescued by deletion of the neural cell adhesion molecule. J Biol Chem. 2005;280:42971–42977. doi: 10.1074/jbc.M511097200. [DOI] [PubMed] [Google Scholar]
- Wright AG, Demyanenko GP, Powell A, Schachner M, Enriquez-Barreto L, Tran TS, Polleux F, Maness PF. Close homolog of L1 and neuropilin 1 mediate guidance of thalamocortical axons at the ventral telencephalon. J Neurosci. 2007;27:13667–13679. doi: 10.1523/JNEUROSCI.2888-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N, Inui K, Matsuyama Y, Harada A, Hanamura K, Murakami F, Ruthazer ES, Rutishauser U, Seki T. Inhibitory mechanism by polysialic acid for lamina-specific branch formation of thalamocortical axons. J Neurosci. 2000;20:9145–9151. doi: 10.1523/JNEUROSCI.20-24-09145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. Circuits for multisensory integration and attentional modulation through the prefrontal cortex and the thalamic reticular nucleus in primates. Rev Neurosci. 2007;18:417–438. doi: 10.1515/revneuro.2007.18.6.417. [DOI] [PMC free article] [PubMed] [Google Scholar]