

Abstract
Activation of the extracellular signal-regulated kinase (ERK) signaling pathway in the striatum is crucial for molecular adaptations and long-term behavioral alterations induced by cocaine. In response to cocaine, ERK controls the phosphorylation levels of both mitogen and stress-activated protein kinase 1 (MSK-1), a nuclear kinase involved in histone H3 (Ser10) and cAMP response element binding protein phosphorylation, and Elk-1, a transcription factor involved in serum response element (SRE)-driven gene regulations. We recently characterized the phenotype of msk-1 knock-out mice in response to cocaine. Herein, we wanted to address the role of Elk-1 phosphorylation in cocaine-induced molecular, morphological, and behavioral responses. We used a cell-penetrating peptide, named TAT–DEF–Elk-1 (TDE), which corresponds to the DEF docking domain of Elk-1 toward ERK and inhibits Elk-1 phosphorylation induced by ERKs without modifying ERK or MSK-1 in vitro. The peptide was injected in vivo before cocaine administration in mice. Immunocytochemical, molecular, morphological, and behavioral studies were performed. The TDE inhibited Elk-1 and H3 (Ser10) phosphorylation induced by cocaine, sparing ERK and MSK-1 activation. Consequently, TDE altered cocaine-induced regulation of genes bearing SRE site(s) in their promoters, including c-fos, zif268, ΔFosB, and arc/arg3.1 (activity-regulated cytoskeleton-associated protein). In a chronic cocaine administration paradigm, TDE reversed cocaine-induced increase in dendritic spine density. Finally, the TDE delayed the establishment of cocaine-induced psychomotor sensitization and conditioned-place preference. We conclude that Elk-1 phosphorylation downstream from ERK is a key molecular event involved in long-term neuronal and behavioral adaptations to cocaine.
Introduction
By increasing striatal levels of dopamine (DA), virtually all drugs of abuse induce activation of the mitogen-activated kinase (MAP) kinase/extracellular signal-regulated kinase (ERK) signaling pathway within striatal neurons (Valjent et al., 2004; Girault et al., 2007). The pharmacological blockade of ERKs, using selective inhibitors of MEK, the kinase upstream from ERKs, prevents cocaine-induced expression of the immediate early genes (IEGs) c-Fos and Zif268, along with long-term behavioral alterations, including conditioned-place preference (CPP) (Valjent et al., 2000; Miller and Marshall, 2005), psychomotor sensitization (Pierce et al., 1999; Valjent et al., 2006a), and craving after late withdrawal (Lu et al., 2005). These results suggest that ERKs are critically involved in the control of neuronal plasticity that underlies long-term behavioral adaptations to cocaine (Lu et al., 2006). Nevertheless, the precise molecular events that drive, downstream from ERKs, such a vast range of behavioral processes remain essentially unknown.
In response to cocaine, activated ERK control phosphorylation levels of nuclear substrates involved in gene transcription and chromatin remodeling. These include the mitogen and stress-activated protein kinase 1 (MSK-1), a nuclear kinase involved in histone H3 (Ser10) and cAMP response element binding protein (CREB) phosphorylation (Brami-Cherrier et al., 2005; for review, see Brami-Cherrier et al., 2009), and the transcription factor Elk-1 (Valjent et al., 2000), a member of the ternary complex family (TCF) that controls serum response element (SRE)-driven gene regulations. We previously showed that knock-out mice for msk-1 had altered expression levels of the IEG c-Fos, but not Zif268, in response to cocaine. Furthermore, cocaine-induced locomotor sensitization, but not CPP, was decreased in these mice (Brami-Cherrier et al., 2005). These data suggested a possible involvement of Elk-1, downstream from ERKs, in cocaine-induced molecular and behavioral responses.
Constitutive knock-out mice for elk-1 do not present any sharp phenotype within the brain, probably because compensatory mechanisms mediated by other members of the TCF family are likely to occur (Cesari et al., 2004). Global inhibitors of ERKs act on a broad spectrum of substrates, including MSK-1. As such, these compounds cannot provide conclusive responses about the specific role of cocaine-induced Elk-1 phosphorylation in molecular and behavioral responses to cocaine. We thus developed a strategy aimed at interfering with Elk-1 phosphorylation downstream from ERKs without blocking ERK activity toward other substrates. We designed a cell-penetrating peptide, named TDE peptide (TDE), which interferes with the DEF docking domain of Elk-1 toward ERKs (Lavaur et al., 2007), and analyzed molecular, morphological, and behavioral responses to cocaine in mice.
Materials and Methods
Animals and drugs.
Mice C57BL/6 were purchased from Janvier. Ten-week-old males were maintained in a 12 h light/dark cycle in stable conditions of temperature (22°C) and humidity (60%). Three days before the experiments, mice were briefly handled each day. Animal care was conducted in accordance with the standard ethical guidelines (European Communities Guidelines on the Care and Use of Laboratory Animals: 86/608/EEC).
Cocaine hydrochloride (Sigma-Aldrich) dissolved in a 0.9% NaCl (w/v) aqueous solution (saline) was administrated at the indicated dose. The TDE peptide (GRKKRRQRRRPPSPAKLSFQFPSSGSAQVHI), its scrambled version (SCR) (GRKKRRQRRRPPQSKPSGSQHPIFSLAFVAS), and the TAT–DEJL–Elk-1 peptide (GRKKRRQRRRPPKGRKPRDLELPLSPSLL) were synthesized at the facility of the Institut Fédératif de Biologie Intégrative (IFR83, Université Pierre et Marie Curie, Paris, France). They were dissolved in saline solution and injected at the indicated dose 1 h before cocaine administration. In a set of experiments, kinetics of TDE administration were performed from 30 min to 8 hours before cocaine. NMDA receptor (MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate]; 0.3 mg/kg) and dopaminergic D1 receptor (SCH23390 [R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride]; 0.1 mg/kg) antagonists or an MEK inhibitor [SL327 (α-[amino[(4-aminophenyl)thio]methylene]-2-(trifluoromethyl)benzeneacetonitrile); 50 mg/kg] were administered 30 min before a saline or cocaine injection.
Tissue preparation and immunohistochemistry.
At the indicated times after drug treatments, mice were anesthetized rapidly with pentobarbital (50 mg/kg, i.p.; Sanofi-Aventis) and perfused transcardially with a fixative solution containing 4% paraformaldehyde (PFA) (w/v) in 0.1 m Na2HPO4/ Na2HPO4 buffer, pH 7.5 (4°C), delivered with a peristaltic pump at 20 ml/min for 5 min. Brains were postfixed overnight in the same solution and stored at 4°C. Sections (30 μm thick) were cut with a vibratome (Leica) and kept at −20°C in solution containing 30% ethylene glycol (v/v), 30% glycerol (v/v), and 0.1 m phosphate buffer. Sections were then processed as follows. On day 1, free-floating sections were rinsed three times for 10 min in Tris-buffered saline (TBS; 25 mm Tris-Cl and 150 mm NaCl, pH 7.5), followed by a permeabilization step 15 min in 0.2% Triton X-100 in TBS. For the detection of all phosphorylated proteins, 50 mm NaF was added in buffers and incubation solutions, as described previously (Sgambato et al., 1998). For phosphorylated MSK-1 (P-MSK-1) the sections were treated for 1 h with a blocking buffer [3% bovine serum albumin (BSA; w/v)]. After three rinses in TBS, the sections were incubated overnight at 4°C with the primary antibodies. On day 2, sections were rinsed three times for 10 min in TBS and incubated for 90 min with a goat anti-rabbit Cy3-coupled secondary antibody (1:500; GE Healthcare). For double-labeling experiments, goat anti-rabbit Alexa Fluor 546 (1:400; Invitrogen) and goat anti-mouse Alexa Fluor 488 antibody (1:400; Invitrogen) were used. Sections were rinsed three times for 10 min in TBS and three times for 10 min in Tris buffer (0.25 m Tris) before mounting in Vectashield (Vector Laboratories).
Cultured striatal neurons and transfection assays.
Striata were dissected out from 14-d-old Swiss mouse embryos (Janvier) and prepared as described previously (Brami-Cherrier et al., 2002). Cells were suspended in Neurobasal medium supplemented with B27 (Invitrogen; 500 nm l-glutamine, 60 g/ml penicillin G, and 25 μm β-mercaptoethanol (Sigma-Aldrich) and plated into 24-well (1.8 × 105 cells per well) Nunc (Dutscher) multiwell plates coated with 50 μg/ml poly-d-lysine (Sigma-Adrich). For all experiments, treatments were performed between DIV 7 and DIV 9, when the majority of cells were of neuronal phenotype with no detectable glial elements. For pharmacological treatments, drugs (see below, Chemical and reagents) were added directly to the medium, and the cells were put back at 37°C in a humidified atmosphere of 95% air and 5% CO2 for the appropriate time.
The cDNA constructs encoding HA-tagged Elk-1 either in its wild-type (WT) or mutated version in the DEF domain (DEF) or DEJL (docking site for ERKs and JNK, LXL) regions were provided by Dr. Pete Shaw (Zhang et al., 2008). Transient transfections of HA-tagged Elk-1 plasmids were performed on primary striatal neurons using Lipofectamine 2000 (Invitrogen) according to the instructions of the manufacturer. Cells (600/mm2) were transfected with 1 μg of DNA for 3.5 h. Neurons were then rinsed with fresh culture medium and put back to 37°C for 24 h before pharmacological treatments.
Immunocytochemistry.
After the appropriate time of glutamate treatment, cells were fixed with PBS containing 4% PFA (added directly to the medium to a final concentration of 2%) for 40 min at room temperature (RT) and then incubated with methanol/acetone solution (50:50) for 10 min at 4°C. After three rinses in PBS, cells were treated with blocking buffer (normal goat serum 10% in PBS) for 2 h at RT (except for P-ERK and P-H3 staining in which the blocking buffer contained 10% fetal calf serum, 1% BSA in PBS). Antibodies were incubated overnight at 4°C diluted in PBS or PBS containing 1% BSA, 0.05% Tween 20 for P-ERK and P-H3 labeling. Neurons were then incubated for 2 h at RT with anti-mouse Alexa Fluor 488 (1:400), anti-rabbit Alexa Fluor 488 (1:400), anti-mouse Alexa Fluor 568 (1:400), or anti-rabbit Alexa Fluor 546 (1:400; Invitrogen). After rinses, cells were counterstained with Hoescht (1:20,000; Invitrogen) and mounted under coverslips using Vectashield (Vector Laboratories).
Primary antibodies.
Polyclonal antibodies raised against diphospho-Thr-202/Tyr-204–ERK1/2 (1:400; Cell Signaling Technology), phospho-Thr-581–MSK-1 (1:750; Cell Signaling Technology), phospho-Ser10–H3 (1:500; Millipore), or a mouse monoclonal antibody raised against P-Ser383–Elk-1 (1:200; Santa Cruz Biotechnology) were used. Expression of IEGs was analyzed using rabbit polyclonal antibodies raised against c-Fos (1:1000) and Zif268 (1:800) from Santa Cruz Biotechnology.
Expression of HA-tagged Elk-1 constructs in transfected neurons was visualized by using a monoclonal anti-HA antibody (1:250; Roche Diagnostics) or a polyclonal anti-HA antibody (1:250; Zymed, Invitrogen).
Image analysis.
For single-staining experiments, immunoreactive cells were quantified for each marker in the dorsomedial striatum (DM) and the nucleus accumbens (NAcc) shell as indicated by black rectangles on mouse brain coronal sections at various bregma coordinates (Paxinos and Franklin, 2001): along the rostrocaudal axis (see Fig. 1E), in a total of 24 different images (421 × 336 μm) per animal (i.e., three adjacent images per structure, bilaterally, on two adjacent slices). For double-staining experiments, confocal images were acquired bilaterally using sequential laser scanning (SP5; Leica) with a 40× oil-immersion objective. Quantifications were performed using image analyzer software (Image-Pro Plus; Media Cybernetics), taking into account the cells with nuclear immunofluorescence above the background.
Figure 1.

Patterning Elk-1 phosphorylation in response to cocaine in the striatum. A–D, Cocaine activates Elk-1 and MSK-1 in the same subset of striatal neurons. P-Elk-1 (A, C, green) was detected along with P-ERKs (A, red) or P-MSK-1 (C, red) by immunohistochemical detection on the same DM striatal section, 10 min after an acute treatment with saline (top rows) or cocaine (20 mg/kg, i.p.; bottom rows). The nuclei were labeled by Hoechst staining (blue). Images are single confocal sections. Scale bar, 25 μm. Note the strong colocalization of P-Elk-1 and P-ERK1/2 (A, merge) as well as P-Elk-1 and P-MSK-1 (C, merge) immunofluorescence occurring in the same subset of striatal cells (Merge + Hoescht). B–D, Quantifications of P-ERK1/2 and P-Elk-1 (B) or P-ERK1/2 and P-MSK-1 (D) immunoreactive cells. E, Schematic representation of striatal regions analyzed for quantifications in the DM and the NACC shell. Cocaine-induced P-Elk-1 immunoreactivity in the DM (F) or NAcc shell (G) is abolished by pharmacological inhibition of NMDA (MK801; 0.1 mg/kg) or DA D1 (SCH23390; 0.1 mg/kg) receptors or MEK inhibition (SL327; 50 mg/kg). The inhibitors were administered intraperitoneally 30 min before cocaine. B–D, Data (means ± SEM; n = 4 mice per group) were analyzed using unpaired t test for each marker. **p < 0.01, cocaine group versus saline group. F–G, Data (means ± SEM; n = 3–6 mice per group) were analyzed using one-way ANOVA (between subjects) for the DM (F(4,14) = 34.38, p < 0.001) and NAcc shell (F(4,14) = 18.81, p < 0.001), followed by post hoc comparisons (Newman–Keuls test). ***p < 0.001, cocaine group versus saline group; °°°p < 0.001, cocaine group versus cocaine groups pretreated with antagonists or MEK inhibitor.
Striatal tissue collection and RNA isolation.
After treatments, mice were killed by decapitation, and heads were snapped frozen in liquid nitrogen. Striata were punched on dry ice and stored in individual tubes at −80°C until RNA isolation. Samples were homogenized in 500 μl of Qiazol reagent (Qiagen) using pestle. RNA isolation was performed with RNeasy Lipid Tissue Midi kit (Qiagen) according to the instructions of the manufacturer. RNA concentration and quality were measured on nanovue spectrophotometer (GE Healthcare). Reverse transcription was performed with Maxima First Strand cDNA synthesis kit for RT-qPCR (Fermentas) at 37°C for 30 min. Quantitative real-time PCR reactions were performed using LightCycler 480 SYBR Green I Master according to the protocol of the manufacturer and run on LightCycler 480 device (Roche Diagnostics). For each reaction, ∼ 20 ng of cDNA from an individual animal were used. The entire qPCR experiments were performed twice, on independent RNA samples (n = 6). To minimize the contribution of contaminating genomic DNA, primers were designed to span exon junctions. Amplification efficiency for each assay was determined by running a standard dilution curve. Expression of hypoxanthine guanine phosphoribosyltransferase 1 (Hprt1) transcript was used to normalize the cDNA amounts. The cycle threshold values were calculated automatically by LightCycler 480 SW 1.5 software with default parameters. All primers were designed using the NCBI primer tool ΔFosB (forward, TGCAGCTAAGTGCAGGAACCGT; reverse, GAGGACTTGAACTTCACTCGGCCA), srf (forward, GCAAACTGCAGCCCATGATCACCA; reverse, GCTGGCTTCAGTGTGTCCTTGGTT), β-actin (forward, GGGAAATCGTGCGTGACATCAAAGAG; reverse, GCCACAGGATTCCATACCCAAGAAGG), homer1a (forward, CAAACACTGTTTATGGACTG; reverse, TGCTGAATTGAATGTGTACC), arc (forward, AATGCAGCTGAAGCAGCAGACCTG; reverse, TCTCAGCAGCCTTGAGACCTGGTGT), and hprt1 (forward, TTGCTCGAGATGTCATGAAGGA; reverse, AGCAGGTCAGCAAAGAACTTATAG).
Diolistic labeling and dendritic spine analysis.
The tissue preparation consisted of a slightly modified version of the protocol used for immunohistochemistry. Briefly, animals were transcardially perfused with 30 ml of 1.5% PFA, pH 7.5 (4°C). Brains were postfixed for 1 h in the same solution and stored at 4°C. Sections (150 μm thick) were cut with a vibratome and kept at −20°C in a cryoprotector solution. Cartridge preparation and diolistic labeling were performed as described previously (Gan et al., 2000) with slight modifications. Three milligrams of DiI (Invitrogen) were dissolved in 300 μl of methylene chloride and added to 50 mg of tungsten beads (1.3 μm diameter; Bio-Rad), which were spread over a glass plate. The methylene chloride was left to evaporate. Beads were collected, resuspended in water (3.5 ml), and sonicated to prevent aggregates. Tefzel tube (Bio-Rad) was first filled with 10 mg/ml polyvinylpyrrolidone (Sigma-Aldrich) for 5 min before introduction of bead suspension (3 ml). The last 0.5 ml of the suspension containing bead aggregates was discarded. After 5 min of rotation, the solution was carefully removed from the tubing using a syringe, leaving the beads in the tubing. The rotating tubing was dried with a nitrogen flow for 10 min. Cartridges (13 mm length) obtained from the coated tubing were stored at 4°C and protected from light. Brain slices were transferred to glass surface, and the beads were delivered using the gene-gun device (Bio-Rad) through a 3 μm pore-sized filter (Millipore), using helium gas pressure (120–150 psi). Slices were kept in buffer at RT for at least 2 h before being mounted in Mowiol.
Images were collected in the NAcc shell as described previously. Image stacks were acquired using a confocal laser-scanning microscope (SP5; Leica), equipped with a 561 nm laser line for excitation. Images were collected in the range 570–620 nm through a 1.4 NA objective (oil immersion; Leica) with pinhole aperture set to 1 Airy unit, pixel size set to 60 nm, and z-step of 200 nm. To minimize noise, each image was acquired twice and averaged. Laser intensity and photomultiplier tube gain was set so that all of the images occupy the full dynamic range of the detector while minimizing noise level. Image stacks were deconvolved with Huygens 3.5 software (Scientific Volume Imaging) using an experimental point-spread function obtained from images of fluorescent beads (PS-Speck 170 nm; Invitrogen). Sections of dendrite (50–100 μm in length) from medium-sized spiny neurons were imaged 50–100 μm from the soma. Dendritic spines were analyzed with Neuronstudio software (Rodriguez et al., 2008). After automated detection, manual check was performed to avoid false positives. Head-spine diameter corresponds to ray-burst diameter (i.e., minimal diameter of the ellipse described by the head spine) calculated in the x–y axis.
Locomotor activity and sensitization.
Locomotor activity was measured by using rectangular activity boxes (20 × 15 × 25 cm) (Immetronic) located in a soundproof testing room with low luminosity. Locomotor activity was counted as travels detected by interruption of photocell beams located across the long axis, 15 mm (horizontal activity) and 30 mm (vertical activity) above the floor. Each box was connected to an electronic interface for data collection. In spontaneous exploration experiments, mice were injected 1 h before be exposed for the first time to the activity box; locomotor activity was recorded in 5 min intervals for 60 min. In cocaine sensitization experiments, mice were placed in the activity box for 60 min during 3 consecutive days for habituation before the actual experiments were performed. Mice were then repeatedly treated with saline or cocaine (10 mg/kg, i.p., every day) for 5 consecutive days. This repeated exposure was followed by 7 d of withdrawal and, on day 12, by a challenge injection of saline or cocaine (10 mg/kg). When indicated, mice were pretreated with either TDE or SCR at the dose of 8 mg/kg 60 min before saline or cocaine administration. Locomotor sensitization was evaluated as a recording of locomotor activity 30 min before and 30 min after the saline or cocaine injection.
Conditioned place preference.
The CPP was evaluated in a Plexiglas Y-shaped apparatus (Immetronic) located in a soundproof testing room with low luminosity. Two of the three chambers were used for the conditioning and distinguished by different patterns on floors and walls, separated by a central neutral area. The device is connected to an electronic interface for data collection. Entries and time spent in each chamber were measured, as well as the locomotor activity of the mice during the experiments. The CPP protocol included three different phases. (1) In the preconditioning (habituation) phase, mice were placed in the middle of the neutral central area and allowed to explore both chambers. The time spent in each chamber was measured during 18 min (1080 s). (2) In the conditioning phase, the treatments were counterbalanced between chambers to use an unbiased procedure. Mice were treated for 2 d (one-pairing protocol) or 6 d (three-pairing paradigm) consecutively with alternate injections of cocaine or saline. When indicated, mice were pretreated with either TDE or SCR at the dose of 8 mg/kg 60 min before saline or cocaine administration. After injections, mice were confined to a given chamber for a period of 20 min. In the case of three pairings, cocaine was administrated on days 1, 3, and 5 and saline on days 2, 4, and 6. Control mice received saline every day. (3) The postconditioning (test) phase was conducted exactly as the preconditioning phase with free access to both chambers. The time spent in each chamber was measured during 18 min (1080 s). Data were expressed as the difference between cocaine-paired chamber minus vehicle-paired chamber during preconditioning and postconditioning, as well as the time spent in each chamber during preconditioning and postconditioning.
Statistics.
Colabeling experiments were analyzed with unpaired Student's t test for each marker. Immunohistochemistry and real-time quantitative PCR experiments were analyzed using one-way ANOVA between subjects followed by the post hoc Newman–Keuls comparisons test, except for the time course of TDE effect, in which one-way ANOVA was followed by the post hoc Dunnett's multiple comparisons test. Immunocytochemistry, spine density, locomotor sensitization, and conditioned place preference experiments were analyzed with a mixed-factor two-way ANOVA followed by the post hoc Bonferroni's test. Spine morphology was analyzed using Kruskal–Wallis test followed by the post hoc Dunn's test. In all cases, significance was set at p < 0.05.
Results
Cocaine drives Elk-1 activation along with ERKs and MSK-1 in the same subset of striatal cells in vivo
We first analyzed whether cocaine-induced Elk-1 and MSK-1 phosphorylation occurred in the same subset of striatal cells. Colabeling experiments showed that cocaine induces an increase of the phosphorylated (i.e., activated) form of Elk-1 (P-Elk-1) along with phosphorylated ERKs (noted as P-ERK1/2 because ERK1 and ERK2 cannot be discriminated in our immunocytochemical conditions) in the same subset of striatal neurons (Fig. 1A,B). Similarly, the activation of Elk-1 and MSK-1 induced by cocaine occurred in the same subset of striatal neurons (Fig. 1C,D). Because ERK activation by cocaine depends on the concomitant stimulation of D1R and NMDARs (Valjent et al., 2000; Pascoli et al., 2011), we analyzed the contribution of these receptors in cocaine-induced striatal activation of Elk-1. For each treatment, P-Elk-1 immunoreactive neurons were analyzed in representative areas from the DM and NAcc shell parts of the striatum on three independent coronal sections (Fig. 1E). Cocaine-induced phosphorylation of Elk-1 was totally prevented by the selective D1R and NMDAR antagonists SCH23390 and MK801, respectively, administered before cocaine (Fig. 1F,G). The MEK inhibitor SL327, which totally prevents cocaine-induced ERK phosphorylation (Valjent et al., 2000), also prevents induction of P-Elk-1 immunoreactive neurons, thus confirming that Elk-1 phosphorylation induced by cocaine in the striatum is totally dependent on ERKs (Fig. 1F,G). Altogether, these data indicate that Elk-1 and MSK-1 are activated downstream from ERKs, at the same time in the same subset of medium spiny neurons (MSNs).
The TDE peptide interferes with Elk-1, but not ERK or MSK-1, phosphorylation induced by cocaine in vivo
We previously designed and validated, in primary striatal neurons in culture, a cell-penetrating peptide that mimics the DEF docking domain of Elk-1 toward ERKs (Lavaur et al., 1007). The DEF docking domain is necessary and sufficient to drive Elk-1 phosphorylation in response to ERK activation. The TAT–DEF–Elk-1 peptide (TDE) blocks this phosphorylation event without interfering with the global level of ERK activity or MSK-1 phosphorylation. We thus sought to use this TDE peptide in vivo and analyze its role in cocaine-induced responses. We first validated its effectiveness and performed dose–response curves in mice pretreated with increasing doses of TDE 60 min before cocaine administration. Ten minutes after cocaine injection, mice were killed and processed for immunohistochemical detection of P-ERK1/2, P-MSK-1, P-Elk-1, and P-H3 using phospho-specific antibodies on striatal sections (Fig. 2A–H). The TDE pretreatment induced a dose-dependent decrease in cocaine-induced P-Elk-1 and P-H3 immunoreactive cells, with a maximal effect at 8 mg/kg TDE for both markers (Fig. 2G,H). In contrast, the TDE failed to affect P-ERK and P-MSK-1 immunolabeling. The time course of TDE effectiveness was then determined on cocaine-induced P-Elk-1. Mice were pretreated with 8 mg/kg TDE for the indicated times before cocaine and then killed 10 min after cocaine administration. Mice pretreated with TDE from 0.5 to 2 h before cocaine showed a significant decrease in P-Elk-1 immunoreactivity (Fig. 2I,J). A maximal effect was found when TDE was administered 60 min before cocaine. In all the following experiments, TDE was thus intraperitoneally administered at 8 mg/kg 60 min before cocaine injection. The TDE effects were specific on its amino acid sequence because a scrambled version of the peptide, which corresponds to the same amino acid sequence distributed randomly, did not show any significant difference with the vehicle pretreated cocaine group (data not shown).
Figure 2.

Dose–response curves and kinetics of TDE effects on cocaine-induced phosphorylation of ERK, MSK-1, Elk-1, and H3 (Ser10). A–D, Immunohistochemical detection of cocaine-induced P-ERK1/2 (A), P-MSK-1 (B), P-Elk-1 (C), and P-H3 (Ser10) (D) was performed on striatal slices (illustrated here is the DM) 10 min after an acute treatment with saline or cocaine (20 mg/kg, i.p.) in mice. When indicated, the mice were pretreated with the TDE peptide (8 mg/kg), 60 min before cocaine treatment. Note the increased immunoreactivity for all markers in the cocaine group compared with saline control group. Note also the decreased P-Elk-1 and P-H3 (Ser10) immunoreactivities in the TDE-treated cocaine group when compared with cocaine alone. Scale bars, 50 μm. E–H, Quantifications of P-ERK1/2 (E), P-MSK-1 (F), P-Elk-1 (G), and P-H3 (Ser10) (H) immunoreactive cells in the DM, 10 min after an acute treatment with saline, cocaine alone (20 mg/kg), or cocaine and TDE (0–12 mg/kg) 60 min before cocaine. I, Illustration of the TDE effects on cocaine-induced phosphorylation of Elk-1. TDE (8 mg/kg) administered from 0.5 to 8 h before cocaine (20 mg/kg); mice were killed 10 min after cocaine administration. J, Quantification of P-Elk-1 immunoreactive cells in the DM in mice pretreated with TDE at indicated times (0–8 h) before a cocaine treatment occurring 10 min before being killed. Note the limited time window of TDE inhibitory effect between 30 and 120 min before cocaine administration. E–H, Data (means ± SEM; n = 8–16 mice per group) were analyzed using one-way ANOVA (between subjects) for P-ERK1/2 (F(5,62) = 22.29, p < 0.001), P-MSK-1 (F(5,62) = 31.76, p < 0.001), P-Elk-1 (F(5,62) = 17.05, p < 0.001), and P-H3 (Ser10) (F(5,62) = 6.665, p < 0.001), followed by post hoc comparisons (Newman–Keuls test). *p < 0.05, **p < 0.01, ***p < 0.001, cocaine groups versus saline group; °p < 0.05, °°p < 0.01, °°°p < 0.001, TDE pretreated cocaine groups versus saline pretreated cocaine group. J, Data (means ± SEM; n = 7–20 mice per group) were analyzed using one-way ANOVA (between subjects) (F(6,65) = 2.254, p < 0.05), followed by post hoc comparisons (Dunnett's multiple test). *p < 0.05, 0.5–8 h TDE pretreated groups versus immediately TDE pretreated group.
Molecular evidence that the DEF docking domain of Elk-1 is involved in H3 (Ser10) phosphorylation
We showed previously that MSK-1 is the major striatal kinase of H3 (Ser10) in response to an acute cocaine administration (Brami-Cherrier et al., 2005; for review, see Brami-Cherrier et al., 2009). Nevertheless, the present results indicate that H3 phosphorylation is significantly reduced by TDE (Fig. 2G,H), without any alteration of MSK-1 phosphorylation (Fig. 2F). These data suggest that the DEF domain of Elk-1 is necessary for histone H3 phosphorylation. Besides the DEF domain, Elk-1 contains a DEJL domain that is both responsible for the docking of Elk-1 to the MAP kinases/JNK and p38 (Sharrocks et al., 2000) and necessary for the recruitment of MSK-1 at the vicinity of histone H3 (Zhang et al., 2008). We thus reasoned that the inhibitory effect of TDE on H3 phosphorylation might be reproduced by a DEJL mutation on Elk-1. This point was addressed by transfecting cultured striatal neurons with cDNA constructs encoding HA-tagged Elk-1 in its WT or mutated versions on DEF or DEJL (Fig. 3A). Twenty-four hours after transfection, neurons were incubated in the absence (control) or presence of glutamate for 20 min. Transfected cells were detected with anti-HA antibody (Fig. 3B–D, top rows), and levels of ERK, MSK-1, Elk-1, and H3 activation were analyzed in transfected cells with phospho-specific antibodies (yellow) (Fig. 3B–D, middle rows). Glutamate-induced phosphorylation of Elk-1 (Fig. 3, compare B, C, left columns) was amplified in neurons overexpressing WT Elk-1 (Fig. 3C, arrowheads). We thus established a threshold to measure P-Elk-1 immunoreactivity that corresponded to a signal equal or above the signal obtained in WT Elk-1-transfected neurons. The DEF mutant of Elk-1 produced a significant reduction of P-Elk-1 immunoreactivity in transfected neurons (Fig. 3, compare B, C, see also G). Using thresholds for the measurements of P-ERK and P-MSK-1, we failed to observe any alteration of glutamate-induced phosphorylation of ERK and MSK-1 in striatal neurons transfected with the DEF mutant of Elk-1 (Fig. 3E,F). With regard to P-H3, the DEF mutant reduced glutamate-induced immunoreactivity (Fig. 3H). These in vitro data thus reproduce our in vivo results with the TDE peptide. In contrast, the DEJL mutant did not affect P-Elk-1 but significantly reduced P-H3 immunoreactivity. In all experimental conditions, blockade of glutamate-induced activation of ERK1/2 with the MEK inhibitor U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene] abrogated the phosphorylation of MSK-1, Elk-1, and H3, thus confirming the critical involvement of ERKs in the activation of these proteins. These in vitro data confirm the crucial implication of the DEF docking site for optimal activation of Elk-1. They also show that the mutation of either the DEF or DEJL docking domain affects glutamate-induced phosphorylation of H3, despite a preservation of MSK-1 activation (see Fig. 8 for schematic explanation).
Figure 3.

Mutations of Elk-1 DEF or DEJL domain alter glutamate-induced H3 (Ser10) phosphorylation but preserve MSK-1 activation in vitro. A, Schematic representation of the Elk-1 mutants encoded by the plasmids used for transfections. Striatal neurons were transfected at DIV6 with cDNAs encoding HA-tagged versions of WT Elk-1 (Elk-1 Wild-Type), Elk-1 carrying a deletion of the DEJL domain (Elk-1 DEJL deletion), or a mutation of the DEF domain (Elk-1 DEF mutation). Twenty-four hours later, neurons were incubated in the absence (Control) or presence of glutamate (Glutamate; 10 μm) for 20 min, with or without U0126 (10 μm) applied 30 min before and during glutamate (Glutamate + U0126) application. B–D, Representative pictures of striatal neurons transfected with the WT or HA-tagged DEF Elk-1 mutants treated or not with glutamate in the absence or presence of U0126. Transfected cells were detected with anti-HA antibody (red) or P-Elk-1 with a phospho-specific antibody (green). White arrows indicate transfected striatal neurons that are P-Elk-1 positive. E–H, Quantifications of P-ERK1/2 (E), P-MSK-1 (F), P-Elk-1 (G), and P-H3 (Ser10) (H) immunoreactive cells, in HA-transfected cells. Note the strong decrease in P-Elk-1 and P-H3 (Ser10) signals in cells transfected with the DEF mutant, whereas the DEJL mutant only affects P-H3 (Ser10). Note also the total inhibition of all markers in the case of a pretreatment with U0126. E–H, Data (means ± SEM; n = 3–4 independent experiments per group) were analyzed using two-way ANOVA: for P-ERK1/2, effect of treatment, F(2,18) = 95.28, p < 0.001; effect of mutation, F(2,18) = 0.059, NS; for P-MSK-1: effect of treatment, F(2,18) = 53.71, p < 0.001; effect of mutation, F(2,18) = 0.39, NS; for P-Elk-1: effect of treatment, F(2,27) = 120.4, p < 0.001; effect of mutation, F(2,27) = 29.54, p < 0.001; for P-H3 (Ser10): effect of treatment, F(2,18) = 19.05, p < 0.001; effect of mutation, F(2,18) = 7.36, p < 0.01, followed by post hoc comparisons (Bonferroni's test). **p < 0.01, ***p < 0.001, control (Ctrl) versus glutamate (Glu); +p < 0.05, +++p < 0.001, glutamate versus glutamate + U0126; °p < 0.05, °°°p < 0.001, wild-type Elk-1 versus Elk-1 construct (DEF mutation or DEJL deletion).
Figure 8.

Schematic representation of molecular events driven by Elk-1 phosphorylation in response to cocaine. A, During cocaine administration, combined activation of D1R and NMDAR drives ERK activation via calcium entry (Pascoli et al., 2011). This leads to the phosphorylation and nuclear translocation of Elk-1 along with ERKs. Within the nucleus, Elk-1 binds to the SRE sites of IEGs (including c-fos, zif268, and arc/arg3.1) and recruits ERK1/2 and MSK-1 at the proximity of the nucleosome. In turn, MSK-1 can phosphorylate histone H3 on Ser10 residue, allowing nucleosome repositioning and transcription. These molecular events are critically involved in MSN spine plasticity and long-term behavioral alterations in response to cocaine. B, In the presence of the TDE peptide, Elk-1 phosphorylation and nuclear translocation is impeded. Cocaine-induced ERK and MSK-1 phosphorylation persist, but MSK-1 is not recruited to the nucleosome. SRE-driven gene regulation is inhibited, along with spine plasticity and long-term behavioral responses.
These observations prompted us to design a penetrating peptide corresponding to the DEJL domain of Elk-1 along with its flanking amino acid sequences. The DEJL peptide was administered in the same experimental schedule as for the TDE (i.e., 8 mg/kg, 60 min before cocaine; Fig. 4A–H). In all cases, the DEJL peptide failed to alter cocaine-induced Elk-1, ERK, and MSK-1 phosphorylation in the DM striatum (Fig. 4A–C) and the NAcc shell (Fig. 4E–G). Despite a tendency to alter H3 (Ser10) phosphorylation levels, the DEJL peptide failed to reduce them significantly in the DM (Fig. 4D) and the NAcc shell (Fig. 4H). Because the DEJL peptide showed a marginal effect on cocaine-induced Elk-1 and H3 (ser10) phosphorylation, the following experiments were focused on the TDE peptide that significantly alters both P-Elk-1 and P-H3 (Ser10).
Figure 4.

Comparative effects of TDE and TAT–DEJL–Elk-1 peptide on molecular responses to cocaine. Quantification of P-ERK1/2 (A, E), P-MSK-1 (B, F), P-Elk-1 (C, G), and P-H3 (Ser10) (D, H) immunoreactive cells in the DM (A–D) or in the NAcc shell (E–H), 10 min after an acute treatment with saline or cocaine (20 mg/kg). When indicated, the mice were pretreated with the TDE or the TAT–DEJL–Elk-1 (TAT–DEJL) (8 mg/kg, i.p.) 60 min before cocaine treatment. Note the increased immunoreactivity for all of the markers in the cocaine groups compared with saline control groups. Note also the decreased P-Elk-1 and P-H3 (Ser10) immunoreactivities in the cocaine group pretreated with TDE when compared with cocaine alone and the absence of effect of TAT–DEJL–Elk-1 on P-Elk-1 immunoreactivity. A–H, Data (means ± SEM; n = 3–7 mice per group) were analyzed using one-way ANOVA (between subjects) in the DM for P-ERK1/2 (F(3,14) = 91.30, p < 0.001), P-MSK-1 (F(3,14) = 103.3, p < 0.001), P-Elk-1 (F(3,14) = 25.63, p < 0.001), and P-H3 (Ser10) (F(3,14) = 15.99, p < 0.001) and in the NAcc shell for P-ERK1/2 (F(3,14) = 5.99, p < 0.01), P-MSK-1 (F(3,14) = 11.46, p < 0.001), P-Elk-1 (F(3,14) = 4.52, p < 0.05), and P-H3 (Ser10) (F(3,14) = 41.20, p < 0.001), followed by post hoc comparisons (Newman–Keuls test). *p < 0.05, **p < 0.01, ***p < 0.001, cocaine groups versus saline group; °p < 0.05, TDE pretreated cocaine group versus saline pretreated cocaine group.
ERK-dependent Elk-1 phosphorylation controls SRE-driven gene regulation induced by cocaine
Elk-1 acts as a transcriptional activator by forming a ternary complex with the serum response factor (SRF) on the SRE at the promoter of several IEGs (Treisman, 1995). We analyzed how inhibition of Elk-1 phosphorylation could affect cocaine-induced expression levels of c-Fos and Zif268, two IEGs bearing SRE site(s) in their promoter. The number of c-Fos (Fig. 5A–C) and Zif268 (Fig. 5D–F) immunoreactive cells was quantified in the DM striatum and the NAcc shell 1 hour after cocaine administration in mice pretreated with the SCR or TDE peptide 1 hour before cocaine. For both c-Fos (Fig. 5A–C) and Zif268 (Fig. 5D–F), TDE partially (approximately one-third), but significantly, reduced the increased number of immuno-positive cells induced by cocaine when compared with SCR pretreated animals.
Figure 5.

Inhibition of Elk-1 phosphorylation alters SRE-driven gene regulation by cocaine in vivo. Immunohistochemical detections of c-Fos (A–C) and Zif268 (D–F) were performed in the DM and the NAcc shell 60 min after an acute treatment with saline or cocaine (10 mg/kg) in mice pretreated with TDE or SCR (8 mg/kg) 60 min before cocaine treatment. Note the increased immunoreactivity of c-Fos (A) and Zif268 (D) in mice pretreated with SCR before cocaine administration. Note also the decreased immunoreactivity in TDE pretreated cocaine groups. Scale bars, 50 μm. G, Real-time quantitative PCR was performed to evaluate levels of ΔfosB, arc/arg3.1, homer1a, srf, and actin mRNA 45 min after an acute cocaine injection in mice pretreated for 1 h with either TDE or SCR. B, C, E, F, Data (means ± SEM; n = 8 mice per group) were analyzed using one-way ANOVA (between subjects) in the DM for c-Fos (F(3,28) = 16.54, p < 0.001) and Zif268 (F(5,21) = 16.80, p < 0.001) and the NAcc shell for c-Fos (F(3,28) = 14.51, p < 0.001) and Zif268 (F(3,28) = 10.86, p < 0.001), followed by post hoc comparisons (Newman–Keuls test). *p < 0.05, **p < 0.01, ***p < 0.001, cocaine groups versus saline group; °p < 0.05, °°p < 0.01, TDE pretreated cocaine group versus SCR pretreated cocaine group. G, Data (means ± SEM; n = 5–6 mice per group) were analyzed using one-way ANOVA (between subjects) for ΔfosB (F(3,19) = 34.87, p < 0.001), arc/arg3.1 (F(3,19) = 16.64, p < 0.001), homer1a (F(3,19) = 5.349, p < 0.01), srf (F(3,19) = 0.581, NS), and actin (F(3,19) = 0.163, NS), followed by post hoc comparisons (Newman–Keuls test). *p < 0.05, **p < 0.01, ***p < 0.001, cocaine groups versus saline group; °p < 0.05, °°p < 0.01, TDE pretreated cocaine group versus SCR pretreated cocaine group.
Quantitative PCR were performed to analyze the implication of Elk-1 phosphorylation on mRNA levels of other IEGs. The pretreatment with TDE significantly decreased cocaine-induced ΔfosB and arc/arg3.1 (activity-regulated cytoskeleton-associated protein) mRNA. In contrast, TDE had no effect on homer1a expression, despite its rapid induction by cocaine (Fig. 5G). Because our previous studies in vitro indicated that chronic blockade of Elk-1 activation inhibits SRF and Actin protein levels during spontaneous differentiation of striatal neurons (Lavaur et al., 2007), we analyzed expression levels of these genes during acute cocaine administration. We did not detect any variation of srf and actin mRNA levels 45 min after acute cocaine, and the pretreatment with TDE did not affect the basal levels of mRNA encoding for both proteins (Fig. 5G, two right panels).
ERK-dependent Elk-1 phosphorylation regulates spine density and morphology of NAcc shell MSNs
Using the TDE or a dominant-negative version of Elk-1, we showed previously that Elk-1 activation controls dendritic elongation and cytoskeleton dynamics in primary striatal cultured cells (Lavaur et al., 2007). We thus asked whether Elk-1 phosphorylation could affect morphological remodeling induced by chronic cocaine treatments. For this purpose, we chronically pretreated different groups of mice with TDE or SCR before a saline or cocaine treatment. This procedure was repeated five times over a 3 d period. Chronic cocaine induced a significant increase in dendritic spine density that was totally and selectively blocked by TDE pretreatment (Fig. 6A,B). As a result of this diolistic staining, we also evaluated the impact of the treatments on head-spine diameters of NAcc shell MSNs (Fig. 6C). The chronic cocaine treatment did not modify head-spine diameters compared with the chronic saline group. Interestingly, chronic Elk-1 inhibition induced a decrease in head-spine diameters of NAcc shell MSNs in both saline- and cocaine-treated groups (Fig. 6C). These results confirm and extend the crucial contribution of Elk-1 in cytoskeleton dynamics under basal conditions, as well as in response to chronic cocaine-induced morphological rearrangements.
Figure 6.

Inhibition of Elk-1 phosphorylation inhibits cocaine-induced dendritic plasticity. A three-dimensional morphological analysis of DiI-labeled medium spiny neurons was performed in the NAcc shell of mice chronically treated after a chronic treatment with saline or cocaine (10 mg/kg; 5 injections over 3 d) in mice pretreated with TDE or SCR (8 mg/kg) 60 min before cocaine treatment. A, Representative pictures of DiI-stained dendritic sections in each experimental group. B, Quantifications of the number of dendritic spines detected per 10 μm of dendritic sections. Note that pretreatment with TDE totally abrogates cocaine-induced increase in dendritic spine density. C, Quantifications of head-spine diameters for each experimental group. Black dots represent the mean head-spine diameters per animal (n = 7 animals per group), and gray dots represent the median head-spine diameters per neuron (n = 10–14 neurons per animal). The black bar represents the mean head-spine diameters per group. Note the decreased head-spine diameters in chronically TDE pretreated groups independently of cocaine. B, Data (means ± SEM; n = 7 mice per group) were analyzed using two-way ANOVA: effect of pretreatment, F(1,24) = 5.443, p < 0.05; effect of treatment, F(1,24) = 5.342, p < 0.05, followed by post hoc comparisons (Bonferroni's test). **p < 0.01, cocaine groups versus saline group; °°p < 0.01, TDE pretreated cocaine group versus SCR pretreated cocaine group. C, Data (n = 72–79 neurons) were analyzed using Kruskal–Wallis test (p < 0.0001), followed by post hoc comparisons (Dunn's tests). **p < 0.01, ***p < 0.001; NS, not significant.
ERK-dependent Elk-1 phosphorylation regulates cocaine-induced behavioral adaptations
Consistent with the role of Elk-1 phosphorylation in gene transcription, the TDE had no effect on spontaneous horizontal or vertical locomotor activity (Fig. 7A). When administrated before the first exposure to a novel environment, the TDE did not modify the locomotor habituation measured 24 h after a re-exposition to the same environment (Fig. 7B).
Figure 7.
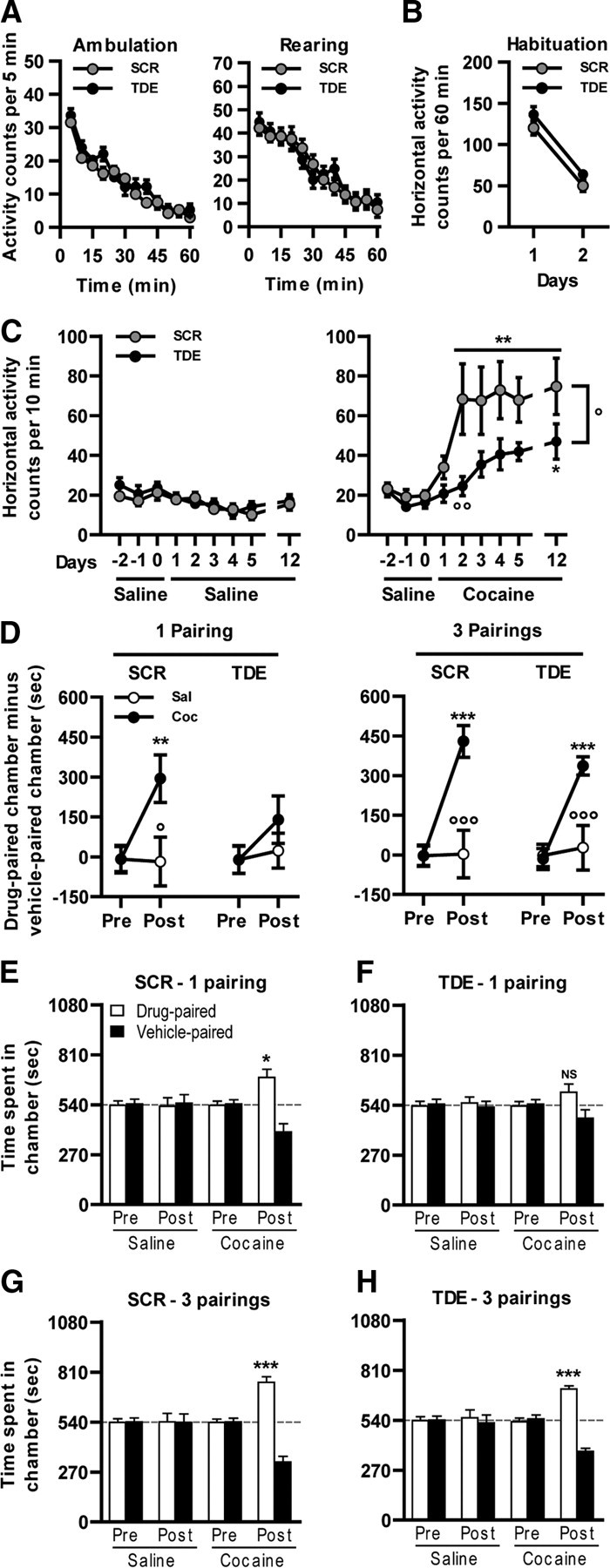
Inhibition of Elk-1 phosphorylation and cocaine-induced behavioral adaptations. Spontaneous locomotor activity: TDE compared with SCR injected at the dose of 8 mg/kg 60 min before the exposure to a novel environment (actimeter) does not alter either the spontaneous horizontal (ambulation) or vertical activity (rearing). B, Locomotor habituation. TDE compared with SCR injected at the dose of 8 mg/kg 60 min before the exposure to the same environment (actimeter) for 2 consecutive days does not alter the locomotor habituation. C, After 3 habituation days (days −2 to 0), mice were pretreated daily for 5 consecutive days, with either TDE or SCR at the dose of 8 mg/kg 60 min before saline (left) or cocaine (10 mg/kg; right) injection, and locomotor activity was measured. This procedure was also repeated on day 12 after a 7 d withdrawal. D, E, CPP induced by cocaine (n = 8 mice per group) was performed from mice pretreated during the conditioning phase of the protocol with TDE or its scramble version 1 h before cocaine administration in a one-pairing (D, left; E, F) or three-pairing (D, right; G, H) CPP paradigm. C, Data (means ± SEM; n = 14–15 mice per group) were analyzed using mixed-factor two-way ANOVA (repeated measure over time): saline: effect of time, F(8,224) = 7.53, p < 0.001; effect of pretreatment, F(1,224) = 0.35, NS; 10 mg/kg cocaine: effect of time, F(8,216) = 10.69, p < 0.001; effect of pretreatment, F(1,216) = 6.55, p < 0.05, followed by post hoc comparisons (Bonferroni's test). *p < 0.05, **p < 0.01, present day versus day 1; °p < 0.05, °°p < 0.01, TDE pretreated cocaine group versus SCR pretreated cocaine group. D, Data (means ± SEM; n = 8 mice per group) were analyzed using mixed-factor two-way ANOVA (repeated measure over conditioning session): one-pairing protocol SCR pretreatment: effect of conditioning, F(1,14) = 7.37, p < 0.05; effect of treatment, F(1,14) = 3.12, NS; one-pairing protocol TDE pretreatment: effect of conditioning, F(1,14) = 2.51, NS; effect of treatment, F(1,14) = 0.62, NS; three-pairing protocol SCR pretreatment: effect of conditioning, F(1,14) = 32.68, p < 0.001; effect of treatment, F(1,14) = 7.73, p < 0.05; three-pairing protocol TDE pretreatment: effect of conditioning, F(1,14) = 21.0, p < 0.001; effect of treatment, F(1,14) = 5.35, p < 0.05, followed by post hoc comparisons (Bonferroni's test). **p < 0.01, ***p < 0.001, posttest versus pretest; °p < 0.05, °°°p < 0.001, saline versus cocaine. E–H, Data (means ± SEM; n = 8 mice per group) were analyzed using paired t test for all of the groups before (Pre) and after (Post) conditioning. *p < 0.05, **p < 0.01, ***p < 0.001, drug-paired chamber versus vehicle-paired chamber. Dashed lines represent equal exploration of both chambers.
A single injection of psychostimulants induces hyperlocomotion in rodents, which may progressively and persistently increase after repeated exposure to a constant dose of the drug. This process, termed psychomotor sensitization, reflects drug-induced long-term behavioral alterations and involves transcriptional regulations. After a 3 d habituation period (days −2 to 0), the slight acute hyperlocomotion induced by a single cocaine administration (day 1) was not affected by the TDE pretreatment (Fig. 7C, right). In contrast, repeated exposure to cocaine induced a robust locomotor sensitization in the SCR pretreated group that was decreased in TDE pretreated mice (days 2–5). This sensitization remained observable even when the animals were challenged after a 7 d withdrawal (day 12), with a significant reduction in TDE pretreated mice (Fig. 7C, right). Of note, TDE did not alter basal locomotor activity of mice chronically treated with a saline solution (Fig. 7C, left). Altogether, these results demonstrate that Elk-1 phosphorylation is involved in locomotor sensitization induced by cocaine.
The ERK pathway is also involved in the establishment of CPP induced by cocaine (Valjent et al., 2000). Cocaine-induced CPP is a pavlovian associative learning based on the explicit association between the rewarding properties of the drug with environmental clues. Before conditioning, all the groups spent the same amount of time in each chamber (Fig. 7E–H, Pre). Cocaine was administered in a one-pairing (Fig. 7D, left, E,F) or three-pairing (Fig. 7D, right, G,H) protocol. Whereas SCR pretreated groups developed a clear preference for the drug-paired chamber, whatever the protocol (Fig. 7D–G), TDE pretreatment impeded the establishment of cocaine-induced CPP in the one-pairing (Fig. 7D, left, 7F) but not three-pairing (Fig. 7D, right, H) paradigm.
Discussion
In the present work, we demonstrate that the TDE, which interferes with cocaine-induced phosphorylation of Elk-1, but not MSK-1 or ERK, modifies molecular, morphological, and behavioral responses to cocaine (Fig. 8). In cultured striatal neurons, the TDE is a selective inhibitor of ERK-dependent Elk-1 phosphorylation induced by glutamate in vitro (Lavaur et al., 2007). One important constraint of our in vivo study was to determine whether ERK-dependent phosphorylation of Elk-1 and MSK-1 occurred in the same striatal population in response to cocaine. We demonstrated previously that the activation of Elk-1 downstream from ERKs occurs rapidly and transiently in the dorsal and ventral parts of the striatum (Valjent et al., 2000). Similarly, MSK-1 activation is transient and strictly dependent on ERK activation in response to cocaine (Brami-Cherrier et al., 2005). Furthermore, it occurs in D1R-expressing MSNs (Bertran-Gonzalez et al., 2008). Herein, we demonstrate that cocaine-induced Elk-1 phosphorylation is also strictly dependent on D1R and NMDAR converging onto ERKs. Finally, the activation of Elk-1, ERK, and MSK-1 occurs at the same time in the same MSNs. We thus conclude that cocaine, via a concomitant activation of D1R and NMDAR, activates two signaling modules downstream from ERKs in the same subpopulation of MSNs: the ERK/Elk-1 on one hand and the ERK/MSK-1 module on the other hand.
Systemic administration of TDE dose dependently altered cocaine-induced phosphorylation of Elk-1 in MSNs with a ceiling effect at the dose of 8 mg/kg. Although partial, this inhibition is specific because ERK1/2 and MSK-1 phosphorylations are spared. Time course experiments show that the optimal time point to inhibit Elk-1 phosphorylation is achieved when TDE is administered from 30 to 120 min before cocaine. This temporal window of effects is likely attributable to the short life of the synthetic peptide in vivo. The TDE also highlights the role of Elk-1 phosphorylation on chromatin remodeling via inhibition of histone H3 (Ser10) phosphorylation. Such inhibition was reproduced in vitro using a DEF-mutated version of Elk-1. These results were not expected a priori, because MSK-1 remained activated in the presence of TDE. MSK-1 is the major kinase for histone H3 (Ser10), and knock-out mice for msk-1 show a total inhibition of cocaine-induced H3 (Ser10) phosphorylation (Brami-Cherrier et al., 2005). A logical explanation to explain the inhibitory effect of TDE on cocaine-induced H3 phosphorylation is the recent demonstration that Elk-1 is able to recruit the ERK/MSK-1 module at the vicinity of the nucleosome located near the SRE site of c-fos and egr-1 (Zhang et al., 2008; Drobic et al., 2010). This recruitment involves a direct protein/protein interaction between activated ERKs and the DEJL domain of Elk-1. In our hands, H3 (Ser10) phosphorylation by glutamate was also reduced by mutation of the DEJL domain in vitro but only slightly reduced by the TDE peptide in vivo. Additional experiments are needed to characterize the sequential events that govern H3 (Ser10) phosphorylation in response to cocaine in neuronal cells.
Global inhibition of ERKs with MEK inhibitors leads to a downregulation of cocaine-induced expression of c-Fos and Zif268 (Valjent et al., 2000). In contrast, msk-1 knock-out mice showed inhibition of cocaine-induced c-Fos but not Zif268 induction (Brami-Cherrier et al., 2005). These data underlined the complexity of molecular events driven by ERKs and highlight the possible role of Elk-1 downstream from ERKs. We show here that Elk-1 phosphorylation is involved in both cocaine-induced c-Fos and Zif268 expression but also in the regulation of other IEGs bearing SRE regulatory elements in their promoter. Thus, TDE inhibited cocaine-induced arc/arg3.1 mRNA expression. arc/arg3.1 encodes a protein involved in dendritic spine morphology and AMPA receptor trafficking (Bramham et al., 2008). Transcription of arc/arg3.1 is known to be regulated by the ERK pathway (Waltereit et al., 2001), and mutation of the SRE site in its promoter strongly impairs long-term depression in Purkinje cells, thus placing this IEG at the interface between ERK signaling and long-term plasticity (Smith-Hicks et al., 2010). We propose that Elk-1 phosphorylation is an intermediate event between ERK activation and SRE-driven arc/arg3.1 induction. Finally, we found a downregulation of cocaine-induced expression of ΔFosB when Elk-1 phosphorylation was inhibited. Although ΔFosB does not contain an SRE site in its promoter, this effect might be an indirect one attributable to reduced levels of c-Fos-containing AP1 complexes (Zhang et al., 2002). As expected, cocaine-induced homer1a mRNAs, which are regulated by a PKA/CREB pathway, were not modified by TDE (Zhang et al., 2007). Finally, we determine that srf and actin, which bear SRE sites in their promoter, are not regulated by an acute cocaine administration. Our demonstration of an involvement of Elk-1 activation in cocaine-induced SRE-driven gene regulations is strengthened by a recent whole-genome microarray profiling of drugs of abuse showing that one important cluster of upregulated genes is under the control of SRF/SRE regulatory sites (Piechota et al., 2010). Induction of this group of genes was strongly impaired by SL327 pretreatment, thus showing the involvement of ERK signaling in the instatement of a large group of genes in response to cocaine.
Over the past decade, our group and others have accumulated experiments showing that Elk-1 is involved in structural plasticity. We originally described that overexpression of a non- phosphorylable version of Elk-1 impedes neuronal differentiation of PC12 cells (Vanhoutte et al., 2001). In contrast, PC12 cells overexpressing a constitutively nuclear mutant of Elk-1 show a facilitation of neuronal differentiation (Vanhoutte et al., 2001; Salinas et al., 2004). More recently, using the TDE peptide or a dominant-negative version of Elk-1 in primary striatal cultured cells, we found an inhibition of spontaneous dendritic outgrowth as well as cytoskeleton dynamics (Lavaur et al., 2007). As described previously (Shen et al., 2008), we found a global increase of spine density in NAcc shell MSNs after a chronic treatment with cocaine. Interestingly, a pretreatment with TDE totally blocked this structural plasticity, as demonstrated recently using a global inhibitor of the ERK pathway (Ren et al., 2010). TDE administration before each cocaine or saline injection also decreased NAcc shell MSNs head-spines diameter (Fig 6). These observations indicate that Elk-1 may also be engaged in the structural organization of dendritic spines. Together with Elk-1, SRF forms the ternary complex at SRE sites and is a prime transcriptional regulator of neurite outgrowth, axon guidance, and synaptic targeting (Knöll et al., 2006; Knöll and Nordheim, 2009). Nonetheless, a recent work reported that selective knockdown of srf in the NAcc did not alter cocaine-induced molecular and behavioral adaptations (Vialou et al., 2010).
Accumulating evidences have highlighted the key role of ERK signaling in cocaine-induced behavioral adaptations. We show here that ERK-mediated phosphorylation of Elk-1 is critically involved in these behavioral responses. Similar to global ERK inhibitors, the TDE strongly decreased the development of locomotor sensitization (Valjent et al., 2000). The TDE was also tested on the establishment of CPP. At the end of the conditioning, mice pretreated with TDE did not express any preference, nor aversion, using a mild (one pairing) or strong (three pairings) conditioning protocol. These data show that inhibition of Elk-1 activation per se did not exert either appetitive or aversive properties. Using a mild conditioning protocol (one pairing), but not a sustained one (three pairings), the TDE blocked cocaine-induced CPP. We showed previously that msk-1 knock-out mice displayed an enhanced CPP induced by a sustained protocol of cocaine administration (Brami-Cherrier et al., 2005). Cocaine-induced expression of c-Fos, which is dispensable for cocaine-induced CPP (Zhang et al., 2006), was inhibited in both TDE-treated and msk-1 knock-out mice. Contrasting with msk-1 knock-out mice, which displayed normal cocaine-induced Zif268 expression (Brami-Cherrier et al., 2005), the TDE partly inhibited cocaine-induced expression of this IEG. Zif268 is crucially implicated in cocaine-driven CPP (Valjent et al., 2006b). Interestingly, the authors reported a high correlation between Zif268 expression dosage and cocaine-induced CPP. Using a three-pairing protocol, the authors showed that cocaine-induced CPP was normal in zif268 heterozygous and totally abrogated in zif268 knock-out mice (Valjent et al., 2006b). Accordingly, when using a stronger protocol (three pairings), we showed that TDE pretreated mice developed a normal cocaine-induced CPP. Thus, by inducing a partial reduction of Elk-1 phosphorylation at the SRE site, the TDE reproduces a zif268 heterozygous phenotype in response to cocaine.
Altogether, the present work demonstrates that Elk-1 phosphorylation by ERKs is critically involved in chromatin remodeling and gene regulations driven by cocaine in vivo. The set of genes governed by the ERK/Elk-1 module can in turn control important events involved in striatal neuron plasticity and long-term behavior responses, such as locomotor sensitization and CPP in a mild paradigm.
Footnotes
This work has been supported by the Centre National de la Recherche Scientifique, l'Agence Nationale pour la Recherche Grant ANR-08-BLAN. A.B. has been supported by the Edmond Rothschild Chemical Dependency Institute Beth Israel Medical center and Fondation pour la Recherche Médicale. We also thank Christophe Piesse and Samuel Bornens from the Institut Federatif de Recherche de Biologie Integrative (IFR83, Paris) for their assistance in peptide synthesis and quantitative PCR. We are grateful to Dr. P. Shaw for the generous gift of the plasmids encoding for Elk-1 DEF and DEJL mutants. The TAT–DEF–Elk-1 peptide has been patented: EP 05 290 363 and WO 2006/087242.
The authors declare no competing financial interests.
References
- Bertran-Gonzalez J, Bosch C, Maroteaux M, Matamales M, Hervé D, Valjent E, Girault JA. Opposing patterns of signaling activation in dopamine D1 and D2 receptor-expressing striatal neurons in response to cocaine and haloperidol. J Neurosci. 2008;28:5671–5685. doi: 10.1523/JNEUROSCI.1039-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramham CR, Worley PF, Moore MJ, Guzowski JF. The immediate early gene arc/arg3.1: regulation, mechanisms, and function. J Neurosci. 2008;28:11760–11767. doi: 10.1523/JNEUROSCI.3864-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Garcia M, Pagès C, Hipskind RA, Caboche J. Dopamine induces a PI3-kinase-independent activation of Akt in striatal neurons: a new route to cAMP response element-binding protein phosphorylation. J Neurosci. 2002;22:8911–8921. doi: 10.1523/JNEUROSCI.22-20-08911.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Valjent E, Hervé D, Darragh J, Corvol JC, Pages C, Arthur SJ, Girault JA, Caboche J. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–11454. doi: 10.1523/JNEUROSCI.1711-05.2005. [Erratum (2006) 26:table of contents; Simon, Arthur J corrected to Arthur, Simon J] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Roze E, Girault JA, Betuing S, Caboche J. Role of the ERK/MSK1 signalling pathway in chromatin remodelling and brain responses to drugs of abuse. J Neurochem. 2009;108:1323–1335. doi: 10.1111/j.1471-4159.2009.05879.x. [DOI] [PubMed] [Google Scholar]
- Cesari F, Brecht S, Vintersten K, Vuong LG, Hofmann M, Klingel K, Schnorr JJ, Arsenian S, Schild H, Herdegen T, Wiebel FF, Nordheim A. Mice deficient for the ets transcription factor elk-1 show normal immune responses and mildly impaired neuronal gene activation. Mol Cell Biol. 2004;24:294–305. doi: 10.1128/MCB.24.1.294-305.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobic B, Pérez-Cadahía B, Yu J, Kung SK, Davie JR. Promoter chromatin remodeling of immediate-early genes is mediated through H3 phosphorylation at either serine 28 or 10 by the MSK1 multi-protein complex. Nucleic Acids Res. 2010;38:3196–3208. doi: 10.1093/nar/gkq030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW. Multicolor “DiOlistic” labeling of the nervous system using lipophilic dye combinations. Neuron. 2000;27:219–225. doi: 10.1016/s0896-6273(00)00031-3. [DOI] [PubMed] [Google Scholar]
- Girault JA, Valjent E, Caboche J, Hervé D. ERK2: a logical AND gate critical for drug-induced plasticity? Curr Opin Pharmacol. 2007;7:77–85. doi: 10.1016/j.coph.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Heck N, Betuing S, Vanhoutte P, Caboche J. A deconvolution method to improve automated 3D-analysis of dendritic spines: application to mouse model of Huntington's disease. Brain Struct Funct. 2011 doi: 10.1007/s00429-011-0340-y. [DOI] [PubMed] [Google Scholar]
- Knöll B, Nordheim A. Functional versatility of transcription factors in the nervous system: the SRF paradigm. Trends Neurosci. 2009;32:432–442. doi: 10.1016/j.tins.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Knöll B, Kretz O, Fiedler C, Alberti S, Schütz G, Frotscher M, Nordheim A. Serum response factor controls neuronal circuit assembly in the hippocampus. Nat Neurosci. 2006;9:195–204. doi: 10.1038/nn1627. [DOI] [PubMed] [Google Scholar]
- Lavaur J, Bernard F, Trifilieff P, Pascoli V, Kappes V, Pagès C, Vanhoutte P, Caboche J. A TAT-DEF-Elk-1 peptide regulates the cytonuclear trafficking of Elk-1 and controls cytoskeleton dynamics. J Neurosci. 2007;27:14448–14458. doi: 10.1523/JNEUROSCI.2279-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Hope BT, Dempsey J, Liu SY, Bossert JM, Shaham Y. Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat Neurosci. 2005;8:212–219. doi: 10.1038/nn1383. [DOI] [PubMed] [Google Scholar]
- Lu L, Koya E, Zhai H, Hope BT, Shaham Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006;29:695–703. doi: 10.1016/j.tins.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Miller CA, Marshall JF. Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron. 2005;47:873–884. doi: 10.1016/j.neuron.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Besnard A, Hervé D, Pagès C, Heck N, Girault JA, Caboche J, Vanhoutte P. Cyclic adenosine monophosphate-independent tyrosine phosphorylation of NR2B mediates cocaine-induced extracellular signal-regulated kinase activation. Biol Psychiatry. 2011;69:218–227. doi: 10.1016/j.biopsych.2010.08.031. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Piechota M, Korostynski M, Solecki W, Gieryk A, Slezak M, Bilecki W, Ziolkowska B, Kostrzewa E, Cymerman I, Swiech L, Jaworski J, Przewlocki R. The dissection of transcriptional modules regulated by various drugs of abuse in the mouse striatum. Genome Biol. 2010;11:R48. doi: 10.1186/gb-2010-11-5-r48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce RC, Pierce-Bancroft AF, Prasad BM. Neurotrophin-3 contributes to the initiation of behavioral sensitization to cocaine by activating the Ras/mitogen-activated protein kinase signal transduction cascade. J Neurosci. 1999;19:8685–8695. doi: 10.1523/JNEUROSCI.19-19-08685.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Sun WL, Jiao H, Zhang D, Kong H, Wang X, Xu M. Dopamine D1 and N-methyl-d-aspartate receptors and extracellular signal-regulated kinase mediate neuronal morphological changes induced by repeated cocaine administration. Neuroscience. 2010;168:48–60. doi: 10.1016/j.neuroscience.2010.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL. Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One. 2008;3:e1997. doi: 10.1371/journal.pone.0001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas S, Briançon-Marjollet A, Bossis G, Lopez MA, Piechaczyk M, Jariel-Encontre I, Debant A, Hipskind RA. SUMOylation regulates nucleo-cytoplasmic shuttling of Elk-1. J Cell Biol. 2004;165:767–773. doi: 10.1083/jcb.200310136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgambato V, Vanhoutte P, Pagès C, Rogard M, Hipskind R, Besson MJ, Caboche J. In vivo expression and regulation of Elk-1, a target of the extracellular-regulated kinase signaling pathway, in the adult rat brain. J Neurosci. 1998;18:214–226. doi: 10.1523/JNEUROSCI.18-01-00214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrocks AD, Yang SH, Galanis A. Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem Sci. 2000;25:448–453. doi: 10.1016/s0968-0004(00)01627-3. [DOI] [PubMed] [Google Scholar]
- Shen H, Sesack SR, Toda S, Kalivas PW. Automated quantification of dendritic spine density and spine head diameter in medium spiny neurons of the nucleus accumbens. Brain Struct Funct. 2008;213:149–157. doi: 10.1007/s00429-008-0184-2. [DOI] [PubMed] [Google Scholar]
- Smith-Hicks C, Xiao B, Deng R, Ji Y, Zhao X, Shepherd JD, Posern G, Kuhl D, Huganir RL, Ginty DD, Worley PF, Linden DJ. SRF binding to SRE 6.9 in the Arc promoter is essential for LTD in cultured Purkinje cells. Nat Neurosci. 2010;13:1082–1089. doi: 10.1038/nn.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman R. Journey to the surface of the cell: Fos regulation and the SRE. EMBO J. 1995;14:4905–4913. doi: 10.1002/j.1460-2075.1995.tb00173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Pages C, Besson MJ, Maldonado R, Caboche J. Involvement of the extracellular signal-regulated kinase cascade for cocaine-rewarding properties. J Neurosci. 2000;20:8701–8709. doi: 10.1523/JNEUROSCI.20-23-08701.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Pagès C, Hervé D, Girault JA, Caboche J. Addictive and non-addictive drugs induce distinct and specific patterns of ERK activation in mouse brain. Eur J Neurosci. 2004;19:1826–1836. doi: 10.1111/j.1460-9568.2004.03278.x. [DOI] [PubMed] [Google Scholar]
- Valjent E, Corvol JC, Trzaskos JM, Girault JA, Hervé D. Role of the ERK pathway in psychostimulant-induced locomotor sensitization. BMC Neurosci. 2006a;7:20. doi: 10.1186/1471-2202-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valjent E, Aubier B, Corbillé AG, Brami-Cherrier K, Caboche J, Topilko P, Girault JA, Hervé D. Plasticity-associated gene Krox24/Zif268 is required for long-lasting behavioral effects of cocaine. J Neurosci. 2006b;26:4956–4960. doi: 10.1523/JNEUROSCI.4601-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte P, Nissen JL, Brugg B, Gaspera BD, Besson MJ, Hipskind RA, Caboche J. Opposing roles of Elk-1 and its brain-specific isoform, short Elk-1, in nerve growth factor-induced PC12 differentiation. J Biol Chem. 2001;276:5189–5196. doi: 10.1074/jbc.M006678200. [DOI] [PubMed] [Google Scholar]
- Vialou V, Maze I, Renthal W, LaPlant QC, Watts EL, Mouzon E, Ghose S, Tamminga CA, Nestler EJ. Serum response factor promotes resilience to chronic social stress through the induction of ΔFosB. J Neurosci. 2010;30:14585–14592. doi: 10.1523/JNEUROSCI.2496-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltereit R, Dammermann B, Wulff P, Scafidi J, Staubli U, Kauselmann G, Bundman M, Kuhl D. Arg3.1/Arc mRNA induction by Ca2+ and cAMP requires protein kinase A and mitogen-activated protein kinase/extracellular regulated kinase activation. J Neurosci. 2001;21:5484–5493. doi: 10.1523/JNEUROSCI.21-15-05484.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang GC, Mao LM, Liu XY, Parelkar NK, Arora A, Yang L, Hains M, Fibuch EE, Wang JQ. In vivo regulation of Homer1a expression in the striatum by cocaine. Mol Pharmacol. 2007;71:1148–1158. doi: 10.1124/mol.106.028399. [DOI] [PubMed] [Google Scholar]
- Zhang HM, Li L, Papadopoulou N, Hodgson G, Evans E, Galbraith M, Dear M, Vougier S, Saxton J, Shaw PE. Mitogen-induced recruitment of ERK and MSK to SRE promoter complexes by ternary complex factor Elk-1. Nucleic Acids Res. 2008;36:2594–2607. doi: 10.1093/nar/gkn099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang D, McQuade JS, Behbehani M, Tsien JZ, Xu M. c-fos regulates neuronal excitability and survival. Nat Genet. 2002;30:416–420. doi: 10.1038/ng859. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang L, Jiao H, Zhang Q, Zhang D, Lou D, Katz JL, Xu M. c-Fos facilitates the acquisition and extinction of cocaine-induced persistent changes. J Neurosci. 2006;26:13287–13296. doi: 10.1523/JNEUROSCI.3795-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]