

Abstract
This report describes the behavioral and electrophysiological analysis of regulatable transgenic mice expressing mutant repeat domains of human Tau (TauRD). Mice were generated to express TauRD in two forms, differing in their propensity for β-structure and thus in their tendency for aggregation (“pro-aggregant” or “anti-aggregant”) (Mocanu et al., 2008). Only pro-aggregant mice show pronounced changes typical for Tau pathology in Alzheimer's disease (aggregation, missorting, hyperphosphorylation, synaptic and neuronal loss), indicating that the β-propensity and hence the ability to aggregate is a key factor in the disease. We now tested the mice with regard to neuromotor parameters, behavior, learning and memory, and synaptic plasticity and correlated this with histological and biochemical parameters in different stages of switching TauRD on or off. The mice are normal in neuromotor tests. However, pro-aggregant TauRD mice are strongly impaired in memory and show pronounced loss of long-term potentiation (LTP), suggesting that Tau aggregation specifically perturbs these brain functions. Remarkably, when the expression of human pro-aggregant TauRD is switched on for ∼10 months and off for ∼4 months, memory and LTP recover, whereas aggregates decrease moderately and change their composition from mixed human plus mouse Tau to mouse Tau only. Neuronal loss persists, but synapses are partially rescued. This argues that continuous presence of amyloidogenic pro-aggregant TauRD constitutes the main toxic insult for memory and LTP, rather than the aggregates as such.
Introduction
Alzheimer's disease (AD) is characterized by aggregation of Aβ, a derivative of amyloid precursor protein (APP), and Tau, a microtubule-associated protein. Several transgenic mouse models have been generated to study these aggregates (Ballatore et al., 2007; for review, see Götz and Ittner, 2008; Paulson et al., 2008; Small and Duff, 2008; Morrissette et al., 2009). Mouse models developing Aβ aggregates are based on mutations in APP or secretases but do not cause Tau aggregation. Mouse models with Tau aggregates were more difficult to obtain. Overexpression of Tau or Tau kinases did not induce aggregation because Tau is highly soluble (Götz et al., 1995). Aggregation was obtained only with Tau mutations causing frontotemporal dementias (for review, see Lee et al., 2001; McGowan et al., 2006). Promoters were used to express Tau in areas relevant for AD [e.g., calcium/calmodulin-dependent protein kinase II (CaMKIIα) promoter for forebrain] (Mayford et al., 1996), but avoiding expression in the spinal cord (because of motor side effects caused by axonal traffic inhibition) (Jaworski et al., 2010). This opened the way to study Tau pathology.
The mouse models used here were derived from the aggregation properties of Tau. (1) The mutation ΔK280 [a frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP17) mutation] (van Swieten et al., 2007) promotes aggregation because of enhanced propensity for β-structure (“pro-aggregant” Tau) (Barghorn et al., 2000). The protein was expressed either as full-length Tau (Eckermann et al., 2007) or as the four-repeat domain (TauRD) harboring the aggregation-prone motifs (Mocanu et al., 2008). Whereas full-length Tau protein binds tightly to microtubules, the repeat domain alone binds only weakly, with or without mutations (Goode and Feinstein, 1994; Gustke et al., 1994). Therefore, one can distinguish the effects of Tau expression from those of Tau-microtubule interactions. (2) Analogous Tau variants were expressed with additional proline mutations (ΔK280/PP) that prevent aggregation (“anti-aggregant” Tau) (Khlistunova et al., 2006). This allows us to discriminate effects of Tau expression from those of aggregation. (3) Expression was regulated via the tetracycline-responsive promoter tetracycline-responsive transactivator (tTA; tet-off system) (Gossen and Bujard, 2002), which allows switching expression on or off and observing the buildup and reversal of pathology.
The data revealed that Tau pathology developed only when TauRD constructs have a high propensity for β-structure, self-aggregation, and coaggregation with endogenous mouse Tau. Pro-aggregant TauRD mice showed AD-like features (Tau missorting, hyperphosphorylation, aggregation, synaptic and neuronal loss), whereas anti-aggregant TauRD mice did not (Mocanu et al., 2008). However, the biochemical and histological characterization of the TauRD mice left the question unanswered whether changes in Tau correlated with behavior, cognition, and long-term potentiation (LTP). Here we addressed these issues and extended the time span of the switch-off period to observe recovery. We find that pro-aggregant TauRD mice (but not anti-aggregant mice) develop cognitive deficits in telencephalon-dependent tasks, accompanied by loss of hippocampal LTP but without motor dysfunction. Notably, memory and LTP recover when pro-aggregant TauRD is switched off. In contrast, neuronal loss persists, and Tau aggregates remain (although now consisting only of mouse Tau), whereas synapses recover. This suggests that pro-aggregant TauRD with high β-propensity represents the toxic principle.
Materials and Methods
Generation of TauRD transgenic mice
Transgenic mice expressing the four-repeat domain of human Tau (TauRD, residues 244–372) were generated as described previously (Mocanu et al., 2008). The pro-aggregant form carried the mutation ΔK280, and the anti-aggregant form carried the mutations ΔK280/I277P/I308P. A DNA plasmid containing the Tet-operon-responsive element (tetO), bidirectional cytomegalovirus promoter (Ptet-bi), TauRD, and luciferase sequences (5.75kB) for detection of transgene expression by a luciferase reporter assay was constructed. For creating an inducible Tet-OFF system, the TauRD+luciferase transgenic mouse lines were crossed with CaMKIIα-tTA transgenic mice (Mayford et al., 1996). All mouse strains had an identical genetic background (C57BL/6) and were heterozygous. Nontransgenic littermates were used as controls. During gestation and in the first 3 weeks after birth, the mice were raised in the presence of doxycycline hydrochloride (50 μg/ml), dissolved in 5% sucrose supplied in the drinking water to suppress the exogenous TauRD during early development. For switch-off experiments, the pro-aggregant and anti-aggregant TauRD mice received 200 μg/ml doxycycline hydrochloride in the drinking water for ∼4 months. All animals were housed and tested according to standards of the German Animal Welfare Act.
To evaluate the expression level of TauRD, we used a luciferase reporter assay, based on primary cultures of fibroblasts derived from ear biopsies. The fibroblasts were grown to 90% confluency and were transiently transfected with 0.4 μg of tTA2-M2/pUHrT62–1 plasmid encoding the tetracycline transcriptional activators rtTA (tet-on) (for review, see Gossen and Bujard, 2002). Protein expression was induced by 1 μg/ml doxycycline, and the cell lysates were analyzed 24 h after gene expression. Luciferase activity was measured using the Lumat LB 9507 system (Berthold Technologies). Only mice that showed relative luminescence units (RLU) >100,000 were used for the experiments.
Western blotting
To estimate total Tau levels, fresh brain tissue or tissue snap-frozen in liquid N2 and stored at −80°C was homogenized in 8 vol of lysis buffer [50 mm Tris-HCl, pH 7.4, 10% glycerol, 1% NP-40, 5 mm DTT, 1 mm EGTA, 20 mm NaF, 1 mm Na3VO4, 150 mm NaCl, protease inhibitors (Complete Mini; Roche), 5 mm 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 100 U/ml benzonase, 5 μm okadaic acid]. Proteins were resolved by SDS-PAGE (17% polyacrylamide gels) and transferred to polyvinylidene difluoride membranes. The membranes were incubated in 5% nonfat milk in TBS-Tween for 1 h at room temperature. The blots were incubated at 4°C overnight with a primary antibody, followed by incubation 1 h at room temperature with a secondary antibody. Three micrograms of total protein were loaded per line for detection with pan-tau antibody K9JA (DAKO-0024, 1:20,000; Dako) or antibodies against synaptic proteins: synaptophysin (1:20,000; Sigma), PSD95 (1:1000; Dianova), drebrin (1:1000; Stressgen), and NMDAR1 (1:500; Millipore). Blots were analyzed by densitometry (LAS 3000, AIDA software; Raytest) and normalized by the concentration of actin (1:20,000; Sigma).
Extraction of sarcosyl-insoluble Tau
A sarcosyl-insoluble fraction of Tau was isolated from brain tissue as described previously (Greenberg and Davies, 1990). Briefly, the brain tissue was homogenized in 3 vol of cold buffer H (10 mm Tris-HCl, 1 mm EGTA, 0.8 m NaCl, 10% sucrose, pH 7.4) and centrifuged at 27,000 × g for 20 min at 4°C. The supernatant was collected, and the resulting pellet was homogenized in buffer H and centrifuged at 27,000 × g for 20 min at 4°C. Both supernatants were combined, adjusted to 1% (wt/vol) N-lauroylsarcosine, and incubated at 37°C with shaking for 60–90 min. After centrifugation at 150,000 × g for 35 min at 20°C, the pellet was resuspended in 50 mm Tris-HCl, pH 7.4, using 0.5 μl of buffer for each milligram of initial weight of the brain tissue. Western blotting was used to analyze the supernatant and the insoluble pellet. The ratio between the volumes of the supernatant and the sarcosyl-insoluble pellet loaded in 17% SDS gels was always 1:2 (the amount of protein in the insoluble fraction was doubled compared with the soluble fraction). For quantification of Tau levels in each fraction, the Western blots were probed with pan-tau antibody K9JA and analyzed by densitometry.
Histology
Samples for immunohistochemistry were prepared by paraffin embedding. Mice were anesthetized with 2-bromo-2-chloro-1,1,1-trifluoro ethane and killed by decapitation. The brains were removed and weighed. Briefly, one of the hemispheres of a mouse brain was placed in 4% formalin for 24 h and dehydrated by sequential incubation in a concentration series of ethanol and chloroform. The final embedding was done in a mixture of paraffin and paraplast. Five-micrometer coronal sections were obtained from each paraffin block. Endogenous peroxidase was quenched by treating the paraffin sections with 0.6% H2O2, and nonspecific binding of antibodies was eliminated by applying 10% horse serum for 30 min at room temperature. The primary antibody prepared in 1% horse serum was applied for 1 h at room temperature, followed by a secondary antibody for 30 min incubation at room temperature. All of the antibody dilutions and the washing steps were performed in 10 mm Tris and 154 mm NaCl, pH 7.5. For the secondary antibody and avidin-biotinylated peroxidase system, we used the Vectastain Universal Elite ABC kit (Vector Laboratories). Antigen retrieval was performed with citrate buffer for an efficient epitope exposure to the antibodies. The following antibodies were used: 12E8 (1:500) for phosphorylated S262/S356 Tau (gift from Dr. P. Seubert, Elan Pharma, South San Francisco, CA), MC-1 (1:5) and PHF-1 (1:50; both gifts from Dr. P. Davies, Albert Einstein College, Bronx, NY), NeuN (1:1000, neuronal marker; Millipore Bioscience Research Reagents), GFAP (1:1000, glial marker; Sigma), and AT8 (1:50) and AT270 (1:2000; both from Pierce). Rabbit polyclonal peptide antibodies were generated against pT231 (1:500, SA 5007), pS46 (1:50, SA 5010), pS202 (1:60; Biosource); and pS404 (1:100), pS199 (1:100), and pS214 (1:500; all from Biosource). Gallyas silver staining was performed on 5 μm paraffin sections stained with silver as described previously (Braak and Braak, 1991).
Electron microscopy and synapse count
Electron microscopic analysis of synapses was performed as described previously (Kretz et al., 2004). Briefly, 400 μm hippocampal slices were fixed in 1% glutaraldehyde and 1% paraformaldehyde in 0.1 m phosphate buffer, postfixed in 1% OsO4, dehydrated in ethanol, and embedded in Epon 820. Blocks were trimmed to contain only the stratum pyramidale and stratum radiatum of the CA1 region. Ultrathin sections were cut on a Reichert-Jung OmU3 ultramicrotome. The sections were stained with uranyl acetate, followed by lead citrate. The spine synapse density was calculated using stereological methods as described previously (Prange-Kiel et al., 2004).
Animals for behavioral and memory tests
For behavioral and cognitive tests, 15 pro-aggregant and 15 anti-aggregant mice were used, as well as 15 nontransgenic littermates that served as age-matched controls (equal numbers of both genders). All animals were kept in standard animal cages under conventional laboratory conditions (12 h light/dark cycle, 22°C, ad libitum access to food and water). Animals had ∼10 months of TauRD gene expression when they started in the behavioral experiments, which were performed during the light phase of the activity cycle. After behavioral testing, a small number of mice was used for electrophysiological studies (approximately four in each group). For switch-off experiments, half of the remaining mice in each of the three groups received 200 μg/ml doxycycline hydrochloride in the drinking water for ∼4 months, and the other half of mice received no doxycycline and continued to express the TauRD transgene for ∼4 months. All behavioral tests were repeated after switching expression off, using the same experimental protocols as used for the first series, unless stated otherwise. All protocols were reviewed and approved by the animal experiments committee of K.U. Leuven, Belgium, and were performed in accordance with the European Community Council Directive (86/609/EEC) and the German Animal Welfare Act.
Neuromotor tests
Grip strength was tested using a T-shaped bar connected to a digital dynamometer (Ugo Basile). Mice were placed on the apparatus so that they grabbed the bar spontaneously and gently pulled backward until they released the bar. Maximal strength (in millinewtons) was recorded 10 times per animal. Motor coordination and equilibrium were tested on an accelerating rotarod (MED Associates). Mice were placed on the rod with their body axis perpendicular to the rotation axis. Their head was directed against the direction of the rotation such that the animal had to progress forward to maintain its balance. Mice were first trained on a constant speed (4 rpm, 2 min) before starting with four test trials (intertrial interval, 10 min). During these trials, the mice had to balance on the rotating rod that accelerated from 4 to 40 rpm within 5 min. The latency period until falling from the rotating rod was recorded up to a maximum of 5 min. To measure cage activity, mice were put individually in 27 × 21 cm2 transparent cages that were placed between three photo beam sensors. Beam crossings (representing ambulatory cage activity) were registered for each 30 min interval during a 23 h recording period, using an interfaced personal computer counter.
Learning and memory tasks
Spatial memory abilities were examined in the standard hidden-platform acquisition and retention version of the Morris water maze. A 150 cm circular pool was filled with water opacified with nontoxic white paint and kept at 26°C as described previously (D'Hooge et al., 2005). A 15-cm-round platform was hidden 1 cm beneath the surface of the water at a fixed position. Four positions around the edge of the tank were arbitrarily designated 1–4, thus dividing the tank into four quadrants: target, adjacent 1, opposite, and adjacent 2. Each mouse was given four swimming trials per day (10 min intertrial interval) for 5 consecutive days. The start position was pseudo-randomized across trials. Mice that failed to find the submerged platform within 2 min were guided to the platform, where they remained for 15 s before returning to their cages. The time required to locate the hidden escape platform (escape latency), the distance traveled (path length), and swimming speed (velocity) were determined. Two days after the acquisition phase ended, a probe trial was conducted, during which the platform was removed and the search pattern of the mice was recorded for 100 s. After transgene switching off, and thus almost 5 months after the completion of the initial water maze training, reversal learning was assessed in half of the mice. Animals were tested again following the protocol of the previous acquisition phase, except that the escape platform was now placed in another quadrant (former adjacent to the target quadrant). Again, each mouse had to perform four trials per day in the reversal phase. At the end of this reversal training, animals were tested in a probe trial as described before. During acquisition and probe trials, the Ethovision video tracking system was used to record and analyze behavior (Noldus Information Technology).
Single-trial passive avoidance learning was examined in a step-through box with a small illuminated compartment and a larger dark compartment with a grid floor. The grid floor was connected to a constant current shocker (MED Associates). Animals were dark adapted for 30 min and placed in the small illuminated compartment. After 5 s, the sliding door to the dark compartment was opened, and the entry latency was recorded. When the mouse entered the dark compartment with four paws on the grid, the door was closed, and a slight foot shock (0.3 mA, 1 s) was delivered. Retention was tested 24 h later according to the same procedure, except that the animals did not receive a shock. The entry latency was recorded with a cutoff of 300 s.
Electrophysiology
CA1 recordings.
Mice were killed by cervical dislocation, and the hippocampus was rapidly dissected out into cold (4°C) artificial CSF (ACSF) saturated with carbogen (95% O2/5%CO2). ACSF consisted of (in mm) 124 NaCl, 4.9 KCl, 25.6 NaHCO3, 1.2 KH2PO4, 2.0 CaCl2, 2.0 MgCl2, and 10.0 d-glucose and was adjusted to pH 7.4. Transverse slices (400 μm thick) were prepared from the dorsal area of the right hippocampus with a tissue chopper and placed in a submerged-type chamber, where they were kept at 32°C and continuously perfused with ACSF at a flow rate of 2.2 ml/min. After a 90 min incubation, one slice was arbitrarily selected, and a tungsten electrode was placed in the CA1 stratum radiatum. For recording of field EPSPs (fEPSPs), a glass electrode (filled with ACSF, 3–7 MΩ) was placed in the stratum radiatum. Recordings were passed through a 3 kHz low-pass and a 10 kHz high-pass filter and sampled at 20 kHz with an 1800 amplifier (A-M Systems) and a Micro 1401 data acquisition unit (Cambridge Electronic Design). For stimulation, a 2100 Isolated Pulse Stimulator (A-M Systems) was used; for data analysis, a custom-made software was used. The time course of the fEPSP was measured as the descending slope function for all sets of experiments. About 1 h after electrode placement, input/output curves were established, and the stimulation strength was adjusted to elicit an fEPSP slope of 35% of the maximum, which was kept constant throughout the experiment. Paired-pulse facilitation (PPF) was investigated by applying two pulses in rapid succession (interpulse intervals of 10, 20, 50, 100, 200, and 500 ms, respectively) at 120 s intervals. During baseline recording, three single stimuli (0.1 ms pulse width, 10 s interval) were measured every 5 min and averaged. For LTP induction, a single theta burst stimulation (10 bursts of four stimuli at 100 Hz, applied every 200 ms; pulse width, 0.2 ms) was given. Such a weak stimulation protocol results in a decremental type of LTP that is more sensitive to any disturbance than a saturated LTP induced by repeated tetanizations (Wilsch et al., 1998; Balschun et al., 1999). Experiments of control and transgenic mice were interleaved with each other.
CA3 recordings.
Using standard procedures, transverse hippocampal slices (350 μm thick) were prepared from the brains of adult mice. All procedures were performed according to the guidelines and with the approval of the Animal Care Committee at the University of Kiel (Kiel, Germany). The slices were initially maintained in a high-sucrose solution containing (in mm) 75 sucrose, 87 NaCl, 3 KCl, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, and 10 d-glucose, constantly gassed with 95% O2/5% CO2. The solution was ice cold for cutting and warmed to 30°C for 20 min immediately afterward. The slices were then kept in ACSF containing (in mm) 125 NaCl, 3 KCl, 1 CaCl2, 3 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, and 10 d-glucose at room temperature (21–24°C) for at least 2 h before being transferred to the recording chamber. In the storage and recording chambers, slices were kept submerged, and ACSF was constantly gassed with 95% O2/5% CO2. Recordings were performed in modified ACSF containing (in mm) 125 NaCl, 3 KCl, 4 CaCl2, 4 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, and 10 d-glucose, pH 7.4.
For fEPSP recording, individual slices were transferred to a recording chamber mounted on the stage of an upright microscope (Axioskop; Zeiss). The bathing solution at room temperature was constantly exchanged by means of a gravity-driven superfusion system (flow rate, 2–3 ml/min) during experiments. Dodt infrared gradient contrast (Dodt et al., 1999) in conjunction with a contrast-enhanced CCD camera (Hamamatsu) was used to identify subregions in the hippocampal CA3 region. Recording pipettes were filled with modified ACSF, in which NaHCO3 was replaced with HEPES to avoid a pH change. Constant current pulses (pulse width, 0.1 ms) were delivered to a bipolar tungsten electrode located either in the granule cell layer/hilus to evoke mossy fiber fEPSPs in the CA3 stratum lucidum, or in the CA3 stratum radiatum to evoke associational/commissural (A/C) fEPSPs in the stratum radiatum, respectively. fEPSPs were filtered at 1 kHz and sampled at 10 kHz using a Multiclamp 700B amplifier in conjunction with a Digidata 1440A interface and pClamp 10 software (all from Molecular Devices).
Before and after tetanic stimulation, experimental pathways were stimulated at 0.1 Hz. The stimulus strength was adjusted to obtain 30–40% of the maximum EPSP amplitude under control conditions. LTP of CA3 mossy fiber fEPSPs was induced by tetanic stimulation at 25 Hz for 5 s. LTP of CA3 A/C fEPSPs was induced by three theta burst stimuli with an interval of 10 s. Each theta burst stimulus consists of 15 bursts of four pulses at 100 Hz, delivered at an interburst interval of 200 ms. Amplitudes of fEPSPs were normalized to 100% before tetanus and pooled across animals of the same genotype. Because long-term synaptic plasticity of A/C synapses in CA3 pyramidal cells is NMDA receptor dependent, the NMDA receptor antagonist d-2-amino-5-phosphonovaleric acid (50 μm; Tocris) was present in all experiments on mossy fiber LTP to prevent contamination from A/C fibers. Furthermore, the group II metabotropic glutamate receptor agonist (2S,1′R,2′R,3′R)-2(2,3-dicarboxycyclopropyl)glycine (DCG IV) (2.5 μm; Tocris) was applied at the end of each experiment to verify that the signal was generated by mossy fiber synapses. Data were included only when the responses were reduced by >90% with DCG IV. The amplitude of mossy fiber responses was measured after subtraction of the response remaining in the presence of DCG IV.
Statistics
Data are presented as mean ± SEM. Differences between mean values were determined using ANOVA procedures with Fisher's least significant difference corrections for post hoc comparisons. For interindividual comparisons of LTP, repeated-measures ANOVA (RM-ANOVA) was applied. For comparisons of paired-pulse ratios, t tests were used where differences of p < 0.05 were considered significant.
Results
Generation of inducible transgenic mice
The basic principles of generating the transgenic mice was to express the four-repeat domain of Tau (construct K18, 130 residues) in two forms (Mocanu et al., 2008): The pro-aggregant form contains the mutation ΔK280 (one of the FTDP17 mutations), which promotes β-structure and thus enhances aggregation, whereas the anti-aggregant form contains additional proline mutations that prevent β-structure and aggregation (Fig. 1A) (von Bergen et al., 2001). Although the constructs differ in a region important for microtubule binding (around residue 280) (Goode et al., 2000), they all have a similar low affinity for microtubules since they lack the domains flanking the repeats (Gustke et al., 1994). The expression was under control of the CaMKIIα promoter, which restricts the protein to the forebrain and thus avoids movement impairment that occurs with pan-neuronal promoters expressing Tau in the spinal cord (for review, see Götz and Ittner, 2008). The double-transgenic mice were regulatable under the control of a tTA so that the TauRD protein was expressed without doxycycline and suppressed with doxycycline in the drinking water (tet-off system) (Gossen and Bujard, 2002).
Figure 1.

Characterization of TauRD expression in mice. A, Bar diagrams illustrating the domains of full-length Tau (2N4R, 441 residues) and the repeat domain construct K18. Black segments indicate the hexapeptide motifs responsible for aggregation via β-structure. The pro-aggregant FTDP17 mutation ΔK280 and the anti-aggregant proline mutations (I277P, I308P) are highlighted. B, Expression level of TauRD/ΔK280 versus luciferase expression (RLU). Three mice with 40,000; 80,000; and 120,000 RLU are compared. The endogenous mouse Tau (Mr ∼ 55 kDa) is roughly comparable (top), as well as the actin control (bottom). The band of TauRD (Mr ∼ 14 kDa, middle) increases with the luciferase level. Tau bands are revealed by blotting with antibody K9JA. pro-aggr., Pro-aggregant. C, Quantitation of B (n = 4). White bars indicate that the protein ratio hTau:mTau deduced from blots varies almost linearly with luciferase readings. Orange bars show corresponding molar ratios calculated on the basis of molecular masses, ranging up to ∼3.4 at an RLU value of 120,000. D, Inverse relationship between loss of CA3–mossy fiber LTP and luciferase expression (RLU). This effect occurs only with pro-aggregant TauRD. The diagram depicts normalized mossy fiber fEPSPs before and after tetanic stimulation. In wild-type (WT) hippocampi, LTP attained 130 ± 4% (filled circles; n = 4), whereas transgenic hippocampi displaying 80,000–120,000 RLU failed to exhibit significant LTP (open circles; 105 ± 6%, n = 4). Representative fEPSP recordings from wild-type and transgenic hippocampi taken at the equivalent time points of the diagram are illustrated above. The inset depicts the inverse relationship between loss of LTP and RLU (40,000, n = 3; 80,000, n = 2; 120,000, n = 2). Note that the size of LTP at 40,000 RLU (138 ± 5%, n = 3) was not significantly different from the control.
One of the challenges in the study of Tau-transgenic mice is to ascertain that the expression level remains in a certain concentration range so that results from different animals can be compared and pooled. In initial studies, we found that the expression of TauRD could vary considerably. We therefore decided to preselect animals by culturing primary fibroblasts from ear biopsies from each double-transgenic animal. The fibroblasts were transfected with the transactivator rtTA (tet-on system) and induced by doxycycline, resulting in the expression of both TauRD and luciferase (both were cloned on the same plasmid but not as a fusion protein). The luciferase level could then be determined in cell culture by chemiluminescence. The RLU value was proportional to the expression level of TauRD, both in the fibroblast culture and subsequently in the mouse brain (Fig. 1B,C; supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). This tight link was also confirmed by the level of mossy fiber LTP in the CA3 region (for details, see below), which decreased proportionally to the RLU values and to the expression of pro-aggregant TauRD (Fig. 1C,D) (see below). Thus, to maintain an expression level sufficient for pathology, only animals with an RLU value of ∼100,000 or more were included in the study. The ratio of expressed TauRD to endogenous Tau protein was typically 0.7–1.2. Since TauRD comprises only a quarter of full-length Tau, this implies molar ratios of expressed versus endogenous Tau around 3:1 to 4:1. This elevated level explains the increased rate of aggregation of pro-aggregant TauRD.
Histopathological features during TauRD expression and after switch-off
In our previous study (Mocanu et al., 2008), we had determined the consequences of switching the expression of pro-aggregant TauRD on and then off again, as seen by the biochemical changes in Tau and the histopathology of the brain. Switching TauRD on for 5–10 months caused conformational changes, missorting into the somatodendritic compartment, hyperphosphorylation, aggregation, synaptic decay, and neuronal loss. These changes occurred only with pro-aggregant TauRD, showing that the ability to aggregate is important for toxicity. Surprisingly, the aggregates contained not only the exogenous pro-aggregant TauRD but also normal mouse Tau. In other words, the different forms of Tau were able to coaggregate in the cells, consistent with the similarity of the domains responsible for aggregation (Kampers et al., 1999). The endogenous mouse Tau became hyperphosphorylated and missorted, indicating that the exogenous human toxic TauRD was able to “poison” endogenous full-length mouse Tau. When TauRD was switched off for 1.5 months, the aggregation level decreased moderately. A surprising feature was that the remaining aggregates were composed exclusively of mouse Tau. This argued that Tau aggregates were in a dynamic equilibrium with their subunits and that nuclei of aggregation retained their nucleating power for extended periods of time. Given these observations, we wanted to clarify several issues: (1) How does the induction of TauRD or its repression affect the mice in terms of motoric performance, behavior, memory, or synaptic plasticity (LTP)? (2) Do these features depend on Tau aggregation? (3) How reversible are the changes when Tau is switched off for a long period of time? To answer these questions, we generated cohorts of pro-aggregant and anti-aggregant mice where expression of TauRD was switched on for ∼10 months and switched off for ∼4 months. At these time points (∼10 and ∼14 months), the mice were tested for behavior, memory, LTP, and histopathology.
At 10 months of TauRD expression, the pro-aggregant mice show histopathological features of AD, including pronounced Gallyas silver-stained Tau aggregates (Fig. 2A1,A3; supplemental Fig. S1A, available at www.jneurosci.org as supplemental material) and neuronal loss determined by NeuN staining (Fig. 2B2, arrows) in different regions of the hippocampus and cortex. In addition, the stainings with antibodies against phosphoepitopes of AD-Tau indicate a missorting of Tau into the somatodendritic compartment of cortical and hippocampal neurons (Fig. 2C2,E2, 12E8 and pT231, arrows and arrowheads) and a distinctive PHF1 immunoreactivity of the mossy fibers in the CA3 region of pro-aggregant mutant mice (Fig. 2D2, arrow, stratum lucidum). Note that the epitopes of the antibodies pT231 and PHF1 are only present in mouse Tau (Fig. 2G) and thus provide further evidence that mouse Tau participates in pathological pathways (see also Mocanu et al., 2008). Finally, GFAP staining against activated astrocytes indicates inflammatory processes inside the dentate gyrus (DG) of pro-aggregant mice (Fig. 2F2). Age-matched wild-type mice show neither Gallyas silver-positive neurofibrillary tangles (NFTs) (Fig. 2A2) nor neuronal loss (Fig. 2B1), no hyperphosphorylation of Tau (Fig. 2C1,D1,E1), and no astrogliosis (Fig. 2F1).
Figure 2.
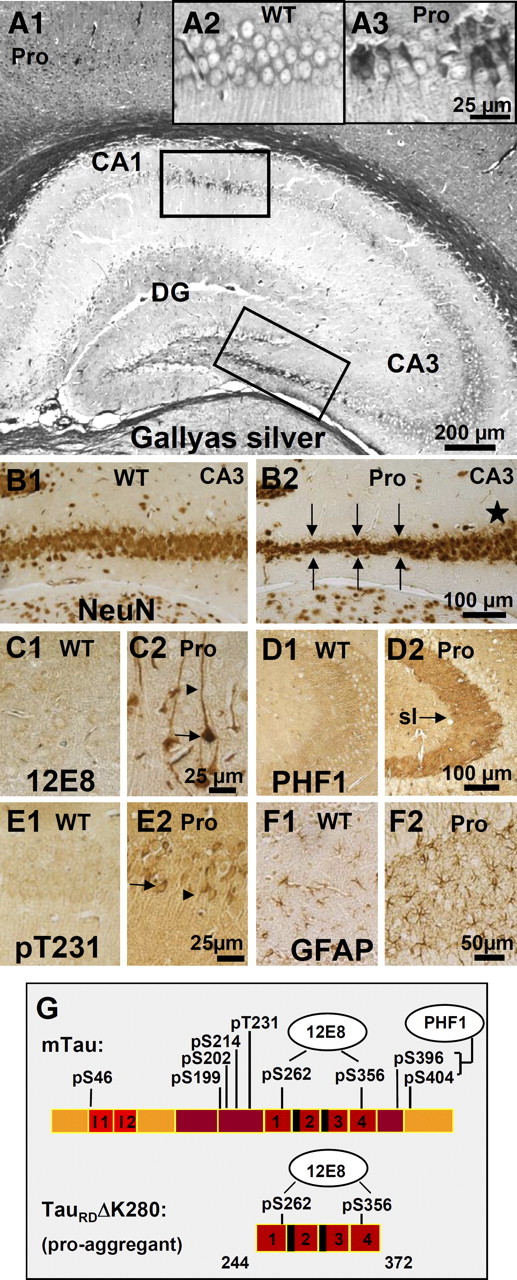
Histology of a pro-aggregant TauRD mouse at 10 months of expression. A, Gallyas silver staining of the hippocampus. Note prominent Tau aggregation in CA1, CA3, and DG regions of the pro-aggregant mutant mouse (A1). Insets show the boxed CA1 region with higher magnification for a Gallyas-negative wild-type (WT) animal (A2) compared with a pro-aggregant mutant mouse (A3). B, NeuN staining of neurons in the CA3 region (lower boxed area in A1) of a WT (B1) and a pro-aggregant mutant (B2) mouse. Note the loss of neurons in the pro-aggregant animal (B2, arrows) compared with the normal number of neurons in WT (B1) and the pro-aggregant mutant mouse within another part of the CA3 region (B2, star). C, Staining of somatosensoric cortical neurons by antibody 12E8 (against phosphorylated KXGS motifs inside the Tau repeat domain) for WT (C1) and pro-aggregant mutant (C2) mice. The phosphorylation (12E8 epitope) and missorting of Tau into the apical dendrites (arrowheads) and soma (arrows) appears only in the pro-aggregant mutant animal (C2). D, Pronounced PHF1 immunoreactivity (pS396, pS404) of the mossy fibers in the CA3 region of a pro-aggregant mutant mouse (D2, arrow; sl, stratum lucidum) compared with negatively stained mossy fibers of a WT mouse (D1). E, Stained CA1 neurons by an antibody against phosphorylated T231-Tau and the identification of missorted Tau into the soma (arrow) and apical dendrites (arrowhead) in the pro-aggregant mutant mouse (E2) compared with negative pyramidal neurons of the WT animal (E1). F, Staining by antibody against GFAP in the DG, revealing inflammatory processes by activated astrocytes for the pro-aggregant mutant mouse (F2) but not for the WT animal (F1). G, Bar diagram illustrating positions of phosphoepitopes inside and outside the Tau repeat domain. Staining with antibodies pS46, pS199, pS202, pS214, pT231, pS404, PHF1, and MC1 is positive, and staining with AT8 and AT270 is negative (data only partly shown) (see also Mocanu et al.,2008). Pro, Pro-aggregant mutant mouse; WT, Wild-type mouse. Scale bars: A1, 200 μm; B1, B2, D1, D2, 100 μm; F1, F2, 50 μm; A2, A3, C1, C2, E1, E2, 25 μm.
In contrast, mice expressing anti-aggregant TauRD showed only a low degree of Tau missorting and much less phosphorylation (Mocanu et al., 2008), but no aggregates (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material; see Fig. 4B), no neuronal loss (supplemental Fig. S2F, available at www.jneurosci.org as supplemental material), and only minor synaptic decay (Mocanu et al., 2008). These results imply that the Tau pathology is progressive, but only when the expressed foreign Tau is competent to aggregate (supplemental Fig. S1, available at www.jneurosci.org as supplemental material; see Fig. 8) (see also Mocanu et al., 2008). The data also argue that hyperphosphorylation of human and mouse Tau depends on an aggregation-prone Tau species, not on the expression as such, and does not require a high affinity of Tau with microtubules, since the repeat domain does not bind appreciably in cellular conditions (Preuss et al., 1997).
Figure 4.

Analysis of soluble and aggregated Tau. Pro-aggregant or anti-aggregant TauRD were expressed continuously for 14 months (ON mice) or for 10 months plus 4 months off (OFF mice). The brain tissue of the mice was subjected to sarcosyl fractionation, and Tau was analyzed by blotting with antibody K9JA and compared with wild-type mice with or without doxycycline treatment. A, Sarcosyl-soluble fraction. B, Insoluble fraction. In both cases, lanes 1 and 2 show control mice without or with doxycycline treatment, lanes 3 and 4 represent mice switched on for 14 months, lanes 5 and 6 show mice switched on for 10 months and off for 4 months, and lanes 7 and 8 represent anti-aggregant mutant mice without and with doxycycline treatment. A, Lanes 3 and 4, At 14 months switch-on, the soluble fraction contains pro-aggregant TauRD, full-length mouse Tau, and some fragments. Lanes 5 and 6, After 10 months switch-on plus 4 months switch-off, the exogenous, pro-aggregant TauRD has disappeared, and only mouse Tau remains, partly fragmented. Note an approximately similar TauRD expression in anti-aggregant ON mice (lane 7) and no TauRD expression neither in wild-type (lanes 1 and 2) nor in anti-aggregant OFF (lane 8) mice. B, Lanes 3 and 4, At 14 months switch-on, the insoluble fraction of pro-aggregant ON mice contains both human TauRD and mouse Tau. In the pro-aggregant ON mice, the insoluble fraction contains ∼40% exogenous TauRD and ∼60% endogenous mouse Tau. After 10 months switch-on plus 4 months switch-off, the insoluble fraction contains no exogenous TauRD, and only mouse Tau remains (lanes 5 and 6). Note there is no detection of Tau aggregates in wild-type (lanes 1 and 2) and anti-aggregant ON and OFF (lanes 7 and 8) mice. Pro-aggr., Pro-aggregant; Anti-aggr., anti-aggregant; WT, wild type; DOX, doxycycline; −, without doxycycline; +, with doxycycline.
Figure 8.

Cessation of pro-aggregant TauRD expression reverses the decrease of PTP and LTP at the mossy fiber–CA3 synapse. A, B, Characteristic time course of fEPSP changes after an LTP-inducing stimulation protocol in a control slice and in a slice from pro-aggregant TauRD mice. Traces above the diagram depict individual fEPSPs taken before induction of LTP, 25 min after induction and in the presence of DCG IV. C, The summary diagram demonstrates the complete reversal of LTP suppression in the pro-aggregant-OFF group. D, Impaired PTP in slices of the anti-aggregant group is reversed after switch-OFF. mf, Mossy fiber.
Histological analysis of the pro-aggregant OFF mice, which expressed the human TauRD first for 10 months and then switched off for ∼4 months, demonstrated only a moderate reduction in Tau aggregation (Fig. 3; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Numerous Gallyas silver-positive NFTs persist in different brain regions (e.g., hippocampus, subiculum, and somatosensory cortex) of the pro-aggregant OFF mice (Fig. 3B,E,H; supplemental Fig. S2H, available at www.jneurosci.org as supplemental material), whereas no Tau aggregation was seen in negative controls (Fig. 3A,D,G; supplemental S2A,G, available at www.jneurosci.org as supplemental material), anti-aggregant ON mice (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material), and anti-aggregant OFF mice (Fig. 3C,F,I; supplemental S2I, available at www.jneurosci.org as supplemental material). Analogous results were obtained for the neuronal loss, determined by NeuN staining. Pro-aggregant OFF mice revealed impressive neuronal loss in the CA1 and DG region of the hippocampus (Fig. 3K,N; supplemental Fig. S2K, arrows, available at www.jneurosci.org as supplemental material), whereas the wild-type (Fig. 3J,M; supplemental Fig. S2D,J, available at www.jneurosci.org as supplemental material), anti-aggregant ON (supplemental Fig. S2F, available at www.jneurosci.org as supplemental material), and anti-aggregant OFF (Fig. 3L,O; supplemental Fig. S2L, available at www.jneurosci.org as supplemental material) mice did not show neurodegeneration.
Figure 3.
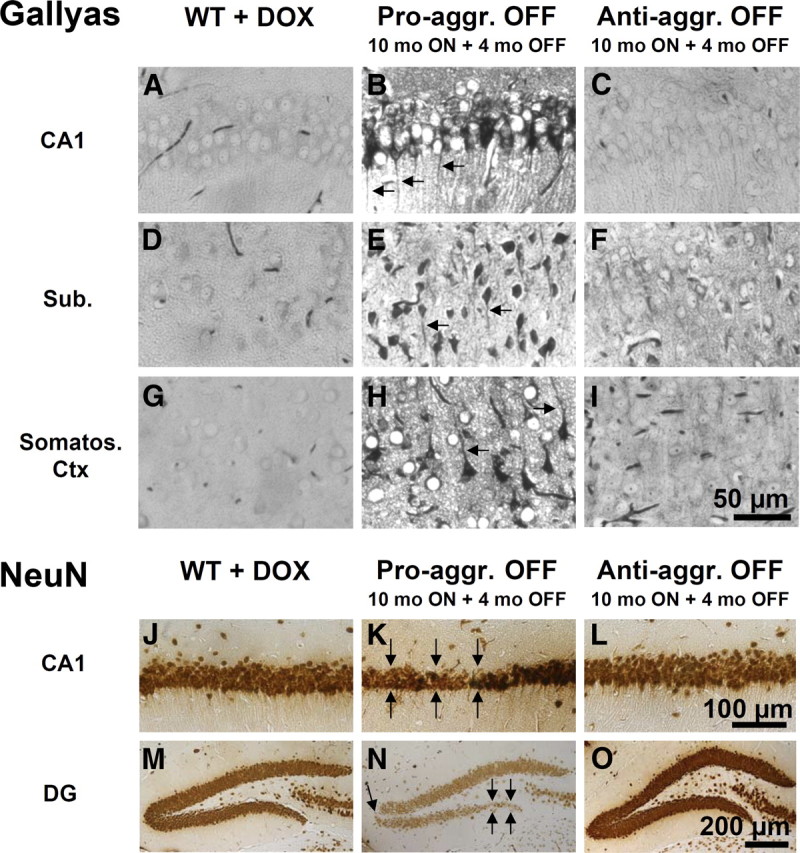
Histology of pro-aggregant and anti-aggregant TauRD mice at 10 months switched ON plus 4 months OFF compared with wild-type mice. Top, Gallyas silver staining for Tau aggregates. Bottom, NeuN staining for neurons. Images B, E, and H illustrate Gallyas-positive neurons in the CA1 region, subiculum, and somatosensoric cortex of pro-aggregant OFF mice compared with wild-type mice (A, D, G) and anti-aggregant OFF mice (C, F, I). Note the missorting of Tau into the apical dendrites of the neurons in pro-aggregant OFF mice (B, E, H, arrows). Images K and N show the neuronal loss in the CA1 region and DG of pro-aggregant OFF mice (arrows) compared with wild-type (J, M) and anti-aggregant OFF (L, O) mice. Note shrinkage of entire pyramidal cell layer in pro-aggregant OFF mice (K) in contrast to wild-type (J) and anti-aggregant OFF (L) mice. Pro-aggr., Pro-aggregant; Anti-aggr., anti-aggregant; WT, wild type; DOX, doxycycline; Sub., subiculum; Somatos. Ctx, somatosensoric cortex. Scale bars: A–I, 50 μm; J–L, 100 μm; M–O, 200 μm.
Biochemical analysis revealed that exogenous TauRD had disappeared completely from the sarcosyl-soluble and -insoluble fractions of pro- and anti-aggregant OFF mice (Fig. 4). The exogenous TauRD disappeared even from the aggregated fraction in the pro-aggregant OFF mice (Fig. 4B, lanes 5 and 6), whereas wild-type, anti-aggregant ON and anti-aggregant OFF mice had no aggregates (Fig. 4B, lanes 1 and 2 and lanes 7 and 8, respectively). This implies that “old” aggregates (generated before switch-off) had turned over and that their content of human TauRD had disappeared and was not replenished after the switch-off. However, since endogenous mouse Tau was still available, it was incorporated into the existing nuclei, thus forming “new” aggregates (Fig. 4B, lanes 5 and 6). As a control, in a parallel cohort of mice, the expression of TauRD was maintained without interruption for the entire period of 10 plus 4 months. Here the histopathological features were similar to the 10 month pro-aggregation mice but had progressed further (supplemental Fig. S2B,E, available at www.jneurosci.org as supplemental material).
In contrast to the histopathological features, there was a clear recovery of synapses in pro-aggregant mice after switch-off, as determined by thin sectioning EM and biochemical analysis of synaptic proteins: compared with controls (100%), synapses decreased by ∼30% after 10 months expression of pro-aggregant TauRD, but only a ∼15% decrease was observed for the case of 10 months ON plus 4 months OFF (Fig. 5A), suggesting that about half of the synapse loss at 10 months had been recovered during the 4 months switch-off period. This was confirmed by biochemical analysis of synaptic proteins (synaptophysin, drebrin, PSD95, NMDAR1) (Fig. 5B) in wild-type mice (14 months old), pro-aggregant TauRD ON mice (14 months continuous expression), and pro-aggregant TauRD OFF mice (10 months ON plus 4 months OFF). The loss of synaptic markers observed after 14 months of TauRD expression (∼40%) was primarily recovered during the 4 months of switch-OFF (Fig. 5B,C). The results imply that the continued expression of the pro-aggregant TauRD species causes a gradual buildup of toxicity in the brain, but they also show that the brain can partially reverse the neuropathology once the toxic protein is switched off.
Figure 5.
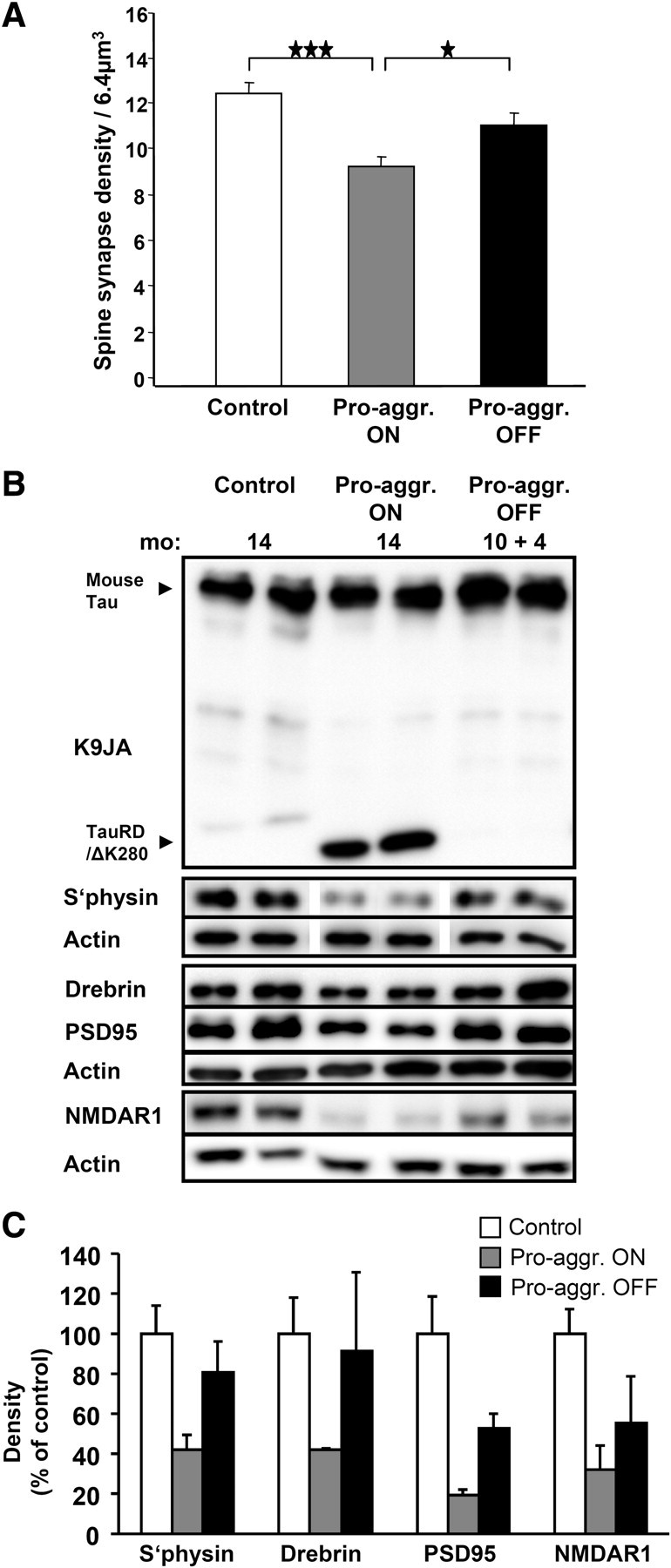
Spine-synapse density versus expression of pro-aggregant TauRD. A, Bars show quantitative evaluation of spine synapses by electron microscopy in the stratum radiatum of the CA1 hippocampal region of control mice (left), after 10 months expression of pro-aggregant TauRD (middle), and after 10 months switch-on plus 4 months switch-off (right). The synapse density decreases after 10 months of TauRD expression to 74% of control. In the case of 10 months switch-on and 4 months off, the spine synapses are rescued to 90% of control, indicating substantial recovery. bar 1/2, ★★★p = 0.000031; bar 2/3, ★p = 0.018; bar 1/3, p = 0.062. B, Western blots showing expression of Tau variants and synaptic proteins in wild-type mice (14 months old), pro-aggregant ON mice (14 months TauRD/ΔK280 expression), and pro-aggregant OFF mice (10 months ON plus 4 months OFF TauRD/ΔK280 expression). Decreased levels of PSD95, drebrin, synaptophysin, and NMDAR1 in pro-aggregant ON mice compared to wild-type mice are shown. Note that the levels of synaptic proteins are partly rescued after the switch off for 4 months. C, Densitometric analysis of the Western blots (B) for the synaptic proteins (synaptophysin, drebrin, PSD95, and NMDAR1), all normalized to β-actin (n = 4). The gray bars indicate the reduction of the synaptic proteins after TauRD/ΔK280 expression in the pro-aggregant ON mice to ∼40% compared with control mice. The black bars show the rescue of synaptic proteins in 4 months switched off pro-aggregant OFF mice to ∼70%, compared with control mice. Significance tests (two-sided t tests) resulted in p values of 0.0176 (synaptophysin), 0.0828 (drebrin), 0.0217 (PSD95), and 0.0133 (NMDAR1) for the differences between wild-type and Pro_ON, resulting in an overall p value of 0.000273 (highly significant) according to Fisher's combined probability test. Similarly, the combined p value for Pro_ON versus Pro_OFF was 0.0319 (significant), whereas the difference between wild-type and Pro-OFF was not significant (combined p value 0.203). Pro-aggr., pro-aggregant; S'physin, synaptophysin. The data in A and C are means ± SEM.
Behavioral analysis after Tau expression and switch-off
Only minor neuromotor changes in transgenic mice
It was of particular interest to test for neuromotor deficiencies because these problems had been reported for earlier generations of Tau-transgenic mice, to the extent that it made cognitive testing impossible (Spittaels et al., 1999; Schindowski et al., 2006). Pro- and anti-aggregant TauRD mice were tested ∼10 months after switching on the transgene expression. At this time, grip strength measures did not differ between genotypes (p > 0.05 for all comparisons) (Table 1), but pro-aggregant TauRD mice showed a lower mean performance on the rotarod (F(2,42) = 6.78; p < 0.01), which was caused bt low values during the first two trials. By the last trial, however, they caught up with the other mice and performed equally well (p > 0.05 for all comparisons) (Table 1). These observations can be explained by the fact that there was no expression of TauRD in the spinal cord, because of the chosen promoter (CaMKIIα), and therefore no Tau-dependent damage to motor neurons. Similarly, there was no difference in spontaneous home cage activity between mutant and control animals (p > 0.05 for all comparisons) (Table 1).
Table 1.
Neuromotor measures
| PRO |
CTRL |
ANTI |
||||
|---|---|---|---|---|---|---|
| ON | OFF | ON | OFF | ON | OFF | |
| Grip strength (mN) | ||||||
| 10 months | ||||||
| Mean | 760 | 758 | 756 | |||
| Last trial | 754 | 753 | 789 | |||
| 14 months | ||||||
| Mean | 783 | 793 | 799 | 764 | 775 | 793 |
| Last trial | 881 | 817 | 870 | 729 | 900 | 954 |
| Rotarod (s, on rod) | ||||||
| 10 months | ||||||
| Mean | 173* | 232 | 209 | |||
| Last trial | 254 | 270 | 262 | |||
| 14 months | ||||||
| Mean | 227 | 211 | 228 | 201 | 208 | 190 |
| Last trial | 276 | 279 | 270 | 300 | 283 | 300 |
| Activity (number of beam crossings) | ||||||
| 10 months | 13,151 | 12,979 | 15,601 | |||
| 14 months | 8717 | 9605 | 7929 | 8562 | 7904 | 9475 |
There are only minor differences in motoric measures between transgenic and nontransgenic mice. Grip strength (in millinewtons) expressed as the mean of 10 successive trials did not differ between three groups (PRO, pro-aggregant; CTRL, control; ANTI, anti-aggregant). On the accelerating rotarod, all groups showed a learning curve across the four consecutive trials. Although pro-aggregant TauRD mice started with a lower performance (*p < 0.01), all groups showed a similar performance on the last trial. Also, total counts during a 23 h recording yielded approximately the same number of beam crossings for all three groups in the home cage activity (p > 0.05 for all comparisons). After switching off half of the mice in each group, no motor differences became noticeable (p > 0.05 for all comparisons).
Learning and memory deficits in pro-aggregant TauRD mice
Spatial learning was assessed in the Morris water maze task that requires mice to learn to find a hidden platform in a circular pool, guided by spatial cues. RM-ANOVAs revealed no group difference (escape latency: F(2,42) = 2.74, p > 0.05; path length: F(2,42) = 2.59, p > 0.05), but there was a major interaction effect between group and day on escape latency and path length (F(8,164) = 3.24, p < 0.01 and F(8,164) = 2.11, p < 0.05, respectively) (Fig. 6A). This indicates that pro-aggregant TauRD mice were capable of learning to find the platform, albeit at a slower rate than control and anti-aggregant mice. Notably, at days 2 and 3 of training, pro-aggregant TauRD mice were much slower in finding the submerged escape platform than control and anti-aggregant mice (pairwise comparisons between pro-aggregant and control mice and between pro- and anti-aggregant mice at both time points, p < 0.01). The swimming velocity in the pro-aggregant TauRD group was not lower than in other groups (p > 0.05 for all comparisons), so we can exclude that the difference in learning rate was attributable to a difference in neuromotor disability. Two days after the acquisition phase, a probe trial revealed no memory-related differences between the groups that finished acquisition training (p > 0.05 for all comparisons). Passive avoidance was tested in the two-compartment, step-through box; the test measures an animal's ability to remember a mild footshock when entering the dark compartment. Whereas pro-aggregant TauRD mice reentered the dark compartment, on average, after 80 s because of impaired memory, almost all anti-aggregant or control mice shied away from the dark compartment, indicating that avoidance learning was normal in these animals (F(2,42) = 7.67; p < 0.001) (Fig. 6D). This test could be performed only once in each group.
Figure 6.
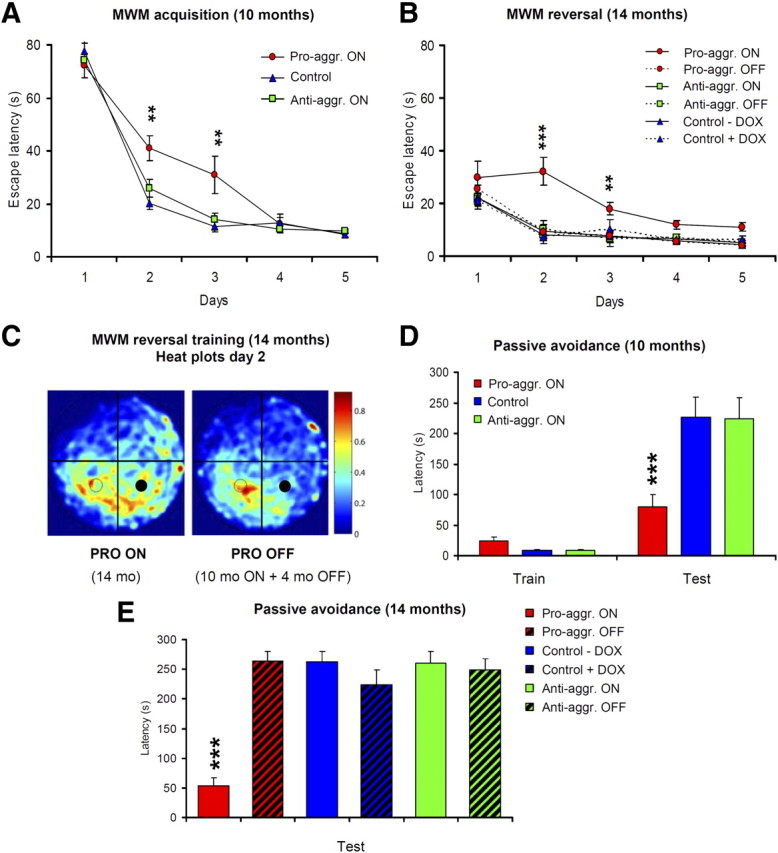
Pro-aggregant TauRD mice show impaired learning in the Morris water maze (MWM) and in the passive avoidance task. MWM acquisition (A, B) and probe trial (C) are shown. A, Escape latencies indicate a slower acquisition in pro-aggregant TauRD mice (red circles) compared with control (blue triangles) and anti-aggregant (green squares) mice. B, Four months later, mice were subjected to a reversal phase in which the platform was placed in another position (right). Pro-aggregant TauRD mice that had been switched on for 10 months and then switched off for 4 months (dashed line) acquired the new position at the control level, whereas pro-aggregant TauRD mice where expression of the transgene was continued were much worse at this task (full line). C, Heat plots for the pro-aggregant TauRD mice on day 2 of the reversal acquisition are shown (dwell frequency is indicated by coloration from red through blue). Pro-aggregant TauRD mice expressing TauRD (left plot) spent more time searching in the former platform quadrant (filled circles) than in the quadrant of the new platform (open circles). After suppression of pro-aggregant TauRD (10 months ON + 4 months OFF; right plot), the mice had a clear preference for the new target quadrant. D, Mean latencies during training and test trial in the passive avoidance task. Switched-ON pro-aggregant TauRD mice were impaired in retention of the single trial task as they reentered the dark compartment much faster during the retention trial compared with anti-aggregant and control mice. E, Mean latencies during the test trial in the passive avoidance task of a second cohort of mice, where the animals were separated into six groups from the beginning of the experiment. Half of the mice received doxycycline for a period of 4 months (pro-aggregant OFF, anti-aggregant OFF, and control plus Dox), whereas the rest of the mice were allowed to continue their TauRD expression. The passive avoidance task demonstrates an impaired retention for the pro-aggregant ON mice, whereas the pro-aggregant OFF mice behave as well as wild-type and anti-aggregant ON and OFF mice. All data are means ± SEM. Asterisks in A and D indicate the significance of differences between PRO-ON versus ANTI-ON and control mice (**p < 0.01; ***p < 0.001). Asterisks in B and E indicate the significance of differences between PRO-ON versus PRO-OFF, ANTI-ON, ANTI-OFF, and control mice (**p < 0.01; ***p < 0.001). Pro-aggr., Pro-aggregant; Anti-aggr., anti-aggregant.
Reversal learning (during which the platform was transferred to another quadrant) was performed in subgroups of animals in which transgene expression was switched off by doxycycline administration for 4 months between the time point of initial testing (∼10 months ON) and final testing (∼14 months ON, or 10 ON plus 4 OFF). Escape latency and path length significantly differed between groups: pro-aggregant TauRD mice that still expressed the transgene were considerably slower to reach the platform on days 2 and 3 compared with the switched off pro-aggregant TauRD group, or the controls, or the anti-aggregant groups (F(2,21) = 6.06 and 5.02; both p < 0.01) (Fig. 6B). Further analysis revealed that pro-aggregant TauRD mice still expressing the transgene spent more time searching the former target quadrant than the quadrant where the platform was now located (F(2,21) = 5.43; p < 0.01) (Fig. 6C). In contrast, switched-off pro-aggregant TauRD mice were now indistinguishable from controls. Again, swimming velocity did not differ between groups (p > 0.05 for all comparisons).
Since the passive avoidance task could be performed only once for each animal, we analyzed a second cohort of mice in which half of the mice received doxycycline right from the beginning of the experiment (∼10 months ON plus 4 months OFF) and the rest of the mice were allowed to continue their transgene expression (∼14 months ON). The results clearly demonstrate that the pro-aggregant ON mice were strongly impaired in learning and memory, whereas the group of pro-aggregant OFF mice behaved like the negative controls and the anti-aggregant ON and OFF mice (Fig. 6E).
Impairment of short-term plasticity and LTP in areas CA3 and CA1
Synaptic transmission and plasticity were examined in the CA1 and CA3 regions of hippocampal slices. The CA3 region, where Tau pathology was particularly pronounced, offers the unique opportunity to directly compare the mossy fiber pathway and the A/C pathway, two excitatory fiber tracts that project onto the same target cell, the CA3 pyramidal neurons, but use a profoundly different mechanism to induce and maintain synaptic plasticity (Nicoll and Schmitz, 2005). fEPSP recordings from stratum lucidum of area CA3 showed that the input/output properties of the mossy fiber–CA3 synapse were not significantly affected in any of the transgenic groups (Fig. 7A) (wild type, n = 12; pro-aggregation, n = 4; pro-aggregation-OFF, n = 9; anti-aggregation, n = 9; anti-aggregation-OFF, n = 7).
Figure 7.

Reduced short-term plasticity at the mossy fiber–CA3 synapse in pro-aggregant TauRD mice. A, Amplitudes of mossy fiber (mf) fEPSPs are plotted as a function of stimulus intensity to determine input–output relationships. B, The top trace depicts mossy fiber fEPSPs in response to four stimuli at 20 Hz recorded from a control slice. The diagram below displays the normalized fEPSP amplitude relative to the amplitude of the first fEPSP. *p < 0.05. C, Strongly reduced frequency facilitation induced by continuous stimulation at 1 Hz for 1 min in pro-aggregant mice. fEPSP amplitude were normalized to baseline value obtained at 0.1 Hz. Only partial recovery was observed in the pro-aggregant-OFF group. Inset, Individual fEPSPs from a control slice before and during frequency facilitation recorded at the same time points as in the normalized diagram. D, Reduction of frequency facilitation in anti-aggregant mice was reversed in the anti-aggregant-OFF group.
As a characteristic and almost defining feature, the mossy fiber–CA3 synapse exhibits strong and ever-increasing facilitation during short trains of repetitive stimulation. Here mossy fiber facilitation was evoked by four stimuli delivered at 20 Hz. To quantify facilitation, the amplitude of fEPSPs was normalized to the first response, and the augmentation of the subsequent responses was plotted relative to the first response (Fig. 7B). Compared with control fEPSPs (n = 18), slices from pro-aggregation mice (n = 10) showed a dramatic impairment of synaptic facilitation (Fig. 7B) that proved fully reversible after expression of the transgene had been switched off (n = 9). Interestingly, also slices from the anti-aggregation group displayed a significant, albeit less profound decrease in facilitation (n = 10) that, again, was fully reversed in the OFF group (n = 7).
A second prominent feature of short-term plasticity at the mossy fiber–CA3 synapse is the so-called frequency facilitation, where continuous stimulation at 1 Hz produces strong facilitation compared with control responses evoked at 0.1 Hz. On returning to control stimulation frequency, the synaptic responses decay quite rapidly to baseline, underscoring the short-lived nature of this phenomenon (Fig. 7C). Expression of pro-aggregant Tau strongly reduced the extent of frequency facilitation (210 ± 17%; n = 10) compared with the control group (346 ± 25%; n = 16; p < 0.001). Cessation of transgene expression restored the initial rising phase of frequency facilitation, consistent with the complete recovery observed in the brief train paradigm (Fig. 7B). However, the plateau phase was significantly lower than that of the control group (232 ± 23%; n = 6; p = 0.016) and did, in fact, not differ from that of the pro-aggregation group (p = 0.13) (Fig. 7C). Interestingly, frequency facilitation was equally diminished in the anti-aggregation group (199 ± 22%; n = 6) when compared with the pro-aggregation group (Fig. 7D) but, unlike the latter group, was fully restored after the transgene expression had been switched off (305 ± 28%; n = 6; p = 0.21 vs control).
To induce LTP, mossy fibers were stimulated at 25 Hz for 5 s. As shown in Figure 8, A and B, this produced an immediate and dramatic rise in synaptic strength [posttetanic potentiation (PTP)] that decayed slowly over a period of about 15–20 min to reach a plateau of stable potentiation (LTP). Suppression of synaptic responses by the group II metabotropic glutamate receptor antagonist DCG IV (2.5 μm), at the end of each experiment, served to document the selective stimulation of the mossy fiber pathway. Both PTP and LTP were significantly reduced in slices from pro-aggregation mice (Fig. 8B,C) (PTP: wild type, 565 ± 28%, n = 14; pro-aggregation, 263 ± 24%, n = 9; p < 0.0001; LTP: wild type, 157 ± 11%; pro-aggregation, 111 ± 8%; p < 0.001). Unlike frequency facilitation, PTP and LTP were recovered in slices from the pro-aggregation-OFF group (PTP, 483 ± 27%; LTP, 135 ± 7%; n = 7; p = 0.119 vs control; p < 0.01 vs pro-aggregation). In slices from anti-aggregation mice, PTP was significantly reduced (313 ± 37%; p < 0.0001), but LTP was indistinguishable from control (159 ± 18%; n = 6). The decrease in PTP was no longer detectable in slices from anti-aggregation-OFF mice (PTP, 550 ± 78%; LTP, 149 ± 5%; n = 6) (Fig. 8D).
As for the mossy fiber pathway, the input–output relationship of the AC pathway was not significantly altered in any of the transgenic groups when compared with control (Fig. 9A) (wild type, n = 9; pro-aggregation, n = 10; pro-aggregation-OFF, n = 7; anti-aggregation, n = 6). On short repetitive stimulation (four stimuli at 20 Hz), facilitation was much less pronounced compared with the mossy fiber synapse, consisting mainly of an augmentation of the second response (PPF) that was not further enhanced by the third and fourth stimulus in the train (Fig. 9B, inset). Across the different groups, we did not detect any significant difference, suggesting that, at this CA3 synapse, short-term plasticity is resistant to Tau aggregation (Fig. 9B) (wild type, n = 16; pro-aggregation, n = 10; pro-aggregation-OFF, n = 7; anti-aggregation, n = 7). With respect to LTP, slices from pro-aggregation mice failed to exhibit any significant potentiation 30 min after induction (Fig. 9C). Notably, PTP was virtually unaffected in the pro-aggregation group, but fEPSP amplitudes decayed then down to baseline, so that no lasting potentiation was measurable 30 min after induction (106 ± 6%; n = 9). In contrast, the theta burst protocol induced robust LTP in control slices (145 ± 6%; n = 9; p < 0.001) as well as in slices from anti-aggregation mice (151 ± 12%; n = 6). Most importantly, the abrogation of LTP in pro-aggregation mice was fully reversible 4 months after termination of transgene expression. In the pro-aggregation-OFF group, LTP amounted to 143 ± 9% (n = 5), which was indistinguishable from control (Fig. 9C).
Figure 9.

Switching off pro-aggregant TauRD expression reverses impairment of LTP at the A/C–CA3 synapse. A, Comparison of input–output relationships between wild-type and transgenic groups did not reveal significant differences. B, Facilitation of fEPSPs by a train of stimuli. The trace above the diagram was recorded in a control slice. The diagram below displays the normalized fEPSP amplitude relative to the amplitude of the first fEPSP. C, Left, Time course of the normalized fEPSP amplitude before and after LTP-inducing stimulation. Note the lack of LTP in the pro-aggregant group and full recovery in the pro-aggregant-OFF group. Right, Superimposed fEPSPs recorded from a control slice (black traces) and from a pro-aggregant slice (red traces) taken at the same time points as in the diagram on the left. WT, Wild type.
Similar to the results from CA3, the fEPSP recordings from area CA1 in hippocampal slices did not reveal any differences in input/output properties for the three groups examined, control and pro- and anti-aggregant mice (Fig. 10A). When PPF, a form of presynaptic short-term plasticity, was tested, we found an increased ratio in anti-aggregant mice at 10 and 20 ms compared with controls (p < 0.05; pairwise comparison) (Fig. 10B). Next, we inspected LTP in the CA1 region, which has been widely used in the investigation of APP/Aβ mice models of AD (Rowan et al., 2003; Shankar et al., 2008). Pro-aggregant TauRD mice did not show any LTP after tetanization. The only noticeable increase in synaptic strength was a PTP of 169 ± 17% (n = 14) 1 min after tetanus. Thereafter, recordings returned to baseline level. Although control mice displayed the expected decremental LTP after a single tetanus (first minute, 185 ± 10%, n = 21; 240 min, 121 ± 13%), potentiation in anti-aggregant mice was even improved (first minute, 213 ± 23%, n = 18; 240 min, 172 ± 23%). Together, this resulted in a significant group difference (RM-ANOVA: F(2,20) = 7.22, p < 0.01) (Fig. 10C). Since an increase in theta burst-induced LTP can be caused by the disinhibition of CA1 neurons (Chapman et al., 1998), we tested whether GABAergic transmission was affected by Tau. However, using Western blot and immunohistochemistry with a GAD65/67 antibody, we found no changes in the number of interneurons (data not shown).
Figure 10.
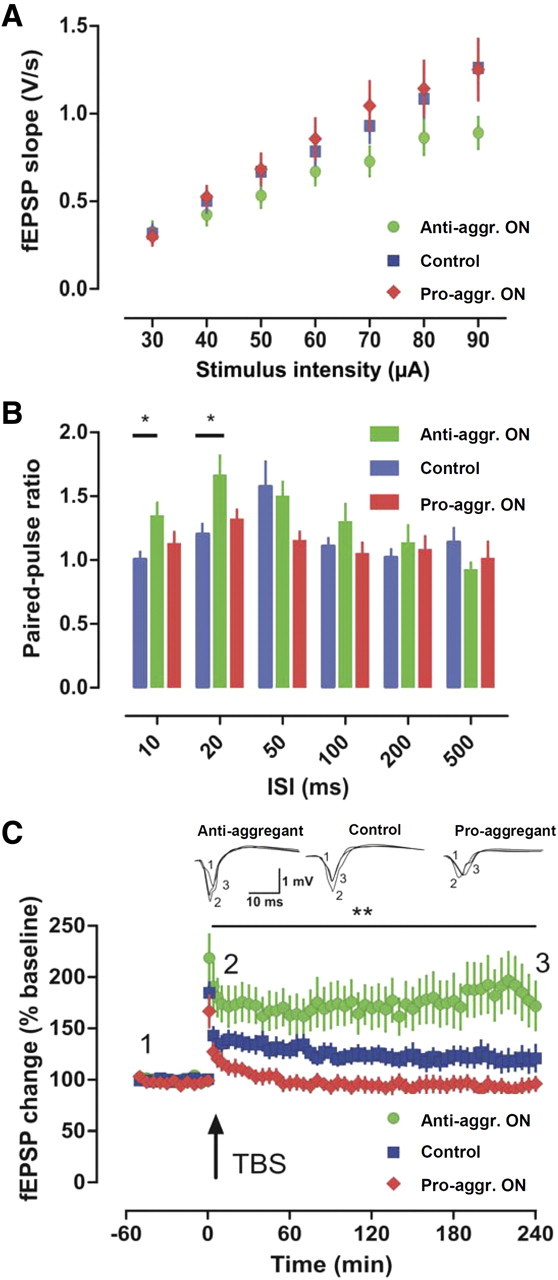
Absence of hippocampal LTP in the CA1 in pro-aggregant TauRD mice. A, Field recordings in the CA1 region of the hippocampus did not reveal any differences in input/output properties (p > 0.05 for all comparisons). B, PPF, a form of short-term plasticity, was altered at the 10 and 20 ms interval in anti-aggregant mice (green bars) compared with control (blue bars; *p < 0.05). C, LTP induced by a single tetanus revealed differences between the genotypes (**p < 0.01). Whereas slices of control mice developed an LTP that slowly returned to baseline during the 4 h recording period, pro-aggregant TauRD mice were completely devoid of LTP. The only potentiation obtained was PTP 1 min after tetanization. Anti-aggregant TauRD mice developed a robust LTP that was even more pronounced than that of controls. Anti-aggr., Anti-aggregant; Pro-aggr., pro-aggregant; ISI, interstimulus interval; TBS, theta burst stimulation.
Discussion
We aimed to characterize effects of regulatable expression of Tau on behavior and LTP in mice, using different forms (pro-aggregant, anti-aggregant) of transgenic TauRD (Mocanu et al., 2008). We examined neural and behavioral plasticity, histopathology, and consequences of switching TauRD on/off. The goal was to distinguish effects of TauRD expression from aggregation and examine reversibility and consequences for LTP, learning, and memory. Previous histopathological and biochemical analysis (Mocanu et al., 2008) revealed the following. (1) Tau pathology correlates with β-structure. Thus, only pro-aggregant TauRD causes aggregation, hyperphosphorylation, and synaptic and neuronal loss. (2) Mouse Tau coaggregates with human TauRD. (3) After switch-off for 6 weeks, human TauRD disappears, but aggregated mouse Tau persists. The present behavioral, electrophysiological, and histopathological analysis revealed that (4) Tau pathology correlates with learning and memory deficits. (5) TauRD transgenes affect synaptic plasticity depending on β-propensity: only pro-aggregant TauRD impairs LTP. (6) Switching pro-aggregant TauRD off for ∼4 months rescues learning/memory and LTP. (7) Synaptic markers and numbers recover, whereas neuronal loss and aggregates persist (mouse Tau only).
What are the implications? The histopathology suggested that aggregation is toxic because only pro-aggregant TauRD generates pathology, but this must be refined by behavioral data: memory and LTP become normal after switching TauRD off, although neuronal loss and aggregation of mouse Tau remain. Thus, toxicity is linked to expression of pro-aggregant TauRD, rather than aggregation. Possible explanations include the following. (1) The aggregation-prone protein may perturb proteostasis by overexerting defenses against protein accumulation, notably proteasomal degradation or macroautophagy which are implicated in Tau clearance (Berger et al., 2006; Dickey et al., 2007; Carrettiero et al., 2009; Wang et al., 2009). (2) Tangles might be tolerable by neurons (Spires-Jones et al., 2008), whereas toxicity might arise from oligomers of pro-aggregant TauRD. Tau oligomers occur in AD brain and correlate with tauopathy (Sahara et al., 2008; Clavaguera et al., 2009). (3) Tau aggregates could interfere with cellular functions, e.g., interactions with RNAs (Kampers et al., 1996), kinases/phosphatases [PP2A (Sontag et al., 1999) and fyn (Bhaskar et al., 2005)], chaperones [hsc70 (Sarkar et al., 2008; Wang et al., 2009) and CHIP (C terminus of Hsp70-interacting protein) (Dickey et al., 2007; Carrettiero et al., 2009)], or motor proteins (Magnani et al., 2007).
One unresolved question is the relationship between changes of Tau (hyperphosphorylation, missorting, aggregation). It is often assumed that phosphorylation triggers other events, e.g., missorting might occur if Tau becomes detached from microtubules by phosphorylation at the KXGS motifs (Thies and Mandelkow,2007). Weighing phosphorylation versus aggregation, toxicity clearly correlates with the competence of TauRD to aggregate, although it lacks most phosphorylation sites. This argues that incipient aggregation triggers phosphorylation of mouse Tau (Fig. 2D2,E2), not vice versa. The trigger could involve activated kinases or inactivated phosphatases (in analogy to effects of a temperature drop) (Planel et al., 2007).
Cognition requires changes in synaptic plasticity, mediated by cytoarchitectural changes (Kandel, 2001). Because neurofibrillary pathology predominates in the hippocampus, memory was evaluated using the hippocampus-dependent Morris water maze and passive avoidance tests (Sutherland and McDonald, 1990; Nakazawa et al., 2004). The pro-aggregant TauRD ON mice failed to remember the stimulus in the passive avoidance test 24 h after an electric shock (Fig. 6D,E), consistent with results on the triple-transgenic mouse model (Billings et al., 2005) and with mice expressing human Tau/R406W (Tatebayashi et al., 2002), indicating impaired associative memory. In the Morris water maze, only pro-aggregant TauRD ON animals exhibited learning difficulties (Fig. 6A--C). The pro-aggregant TauRD ON mice (at 10 months expression) showed longer escape latency (Fig. 6A), a measure of cognitive performance (Graziano et al., 2003). Analogous effects of tauopathy on spatial learning were observed for other AD mouse models (Arendash et al., 2004; Ramsden et al., 2005; Denk and Wade-Martins, 2009). Since pro-aggregant TauRD mice showed only a mild transient neuromotor phenotype (grip strength, rotarod assays) (Table 1), the acquisition impairment cannot be attributed to motor deficits, contrary to mutant mice expressing Tau also in the spinal cord (Ishihara et al., 1999; Spittaels et al., 1999; Lewis et al., 2000; Allen et al., 2002; Zhang et al., 2004; Ikeda et al., 2005). At 14 months pro-aggregant TauRD expression, the mice demonstrated severe impairments in learning a hidden platform and retention at later probe trials (Fig. 6B). Moreover, pro-aggregant TauRD ON mice with continued expression perseverated to the old platform (Fig. 6C). Perseveration is common in AD patients (Pekkala et al., 2008) and may result from postactivation of normally inhibited memory traces (Traykov et al., 2005). This suggests that in the pro-aggregant TauRD ON mice, the memory trace was consolidated, making them unable to adapt via cytoskeletal changes.
Deficiencies in synaptic plasticity in the millisecond to minute range were observed in pro- and anti-aggregant animals, prominently at the CA3–mossy fiber synapse. Remarkably, a decrease in frequency facilitation (Fig. 7C,D) and PTP (Fig. 8C,D) occurred in pro- and anti-aggregation mice to the same extent, whereas PPF was strongly impaired only in pro-aggregant animals (Fig. 7B). The effect is less evident for the A/C synapse in CA3 and for the S/C synapse in CA1 where pro- and anti- aggregant TauRD had no severe effects on short-term plasticity. This depends mainly on presynaptic calcium (Zucker, 1989), whereas mossy fiber short-term plasticity is specifically sensitive to adenosine (Klausnitzer and Manahan-Vaughan, 2008). Both may be disturbed in pro- and anti-aggregant mice.
Spatial learning defects result from defective hippocampal synaptic plasticity, and LTP-like mechanisms subserve cognition (Whitlock et al., 2006). The missorting of Tau from axons to dendrites could influence mRNA transport required for synaptic plasticity (Kanai et al., 1989; Dawson et al., 2001). Plasticity in the CA3 region (where mice show pronounced pathology) is important for contextual learning, memory recall based on pattern completion, and spatial tuning of CA1 neurons (Nakashiba et al., 2008), whereas plasticity of Schaffer–CA1 synapses is implicated in spatial memory and passive avoidance memory (Chen and Tonegawa, 1997; Impey et al., 1998). Notably, we found “classical” NMDA receptor-dependent LTP at Schaffer–CA1 synapses and A/C CA3 synapses (Malenka and Bear, 2004) impaired in pro-aggregant mice (Figs. 9C, 10C), but this was reversed by switching off the transgene. In contrast, anti-aggregant mice displayed LTP at control levels (A/C CA3 synapses) (Fig. 9C) or even a stronger potentiation (CA1 region) (Fig. 10C). Our findings are corroborated by reports of impaired NMDA receptor-dependent LTP in tauopathy models, e.g., in the CA1 region of 6 month 3xTg-AD mice (Oddo et al., 2003) and PS19 mice (Yoshiyama et al., 2007), and in the DG of aged DM-htau mice (Rosenmann et al., 2008). Others did not observe pronounced changes of CA1-LTP in 6-month-old mice (Schindowski et al., 2006) and found deteriorated LTP in 12-month-old htau mice only when potentiation was induced by high-frequency stimulation, not by theta burst (Polydoro et al., 2009). In our study, the impairment was specific for LTP expression in pro-aggregant mice because basic excitability (by input–output relationship) was not affected (Figs. 7A, 9A, 10A).
Surprisingly, anti-aggregant mice displayed enhanced LTP after theta burst stimulation in the stratum radiatum of CA1 (Fig. 10C). Recent findings in hippocampal cell cultures revealed that microtubule plus-ends transiently enter dendritic spines and might enhance synaptic plasticity (Hu et al., 2008; Hoogenraad and Bradke, 2009; Jaworski et al., 2009). The activity-dependent transport mechanism into spines may be altered in anti- and pro-aggregant animals because of TauRD missorting to somatodendritic compartments (Zempel et al., 2010). LTP of the mossy fiber–CA3 synapse, which is presynaptically mediated and NMDA receptor independent (Nicoll and Schmitz, 2005), was decreased after pro- or anti-aggregant TauRD was switched on (Fig. 8C,D). Switching off the transgene rescued LTP to control levels.
From a translational perspective, an important finding of our study is the recovery of memory (Fig. 6B,E), LTP (Figs. 8C, 9C), and synapses (Fig. 5) after switching pro-aggregant TauRD off. This holds for NMDA receptor-dependent and -independent LTP, suggesting that the detrimental effects of TauRD are reversible on both sides of the synapse, once the toxic TauRD disappears. A similar change in paradigm applies to plaques and tangles, once viewed as indestructible but now seen as surprisingly dynamic (Meyer-Luehmann et al., 2008). In mouse models in which Aβ triggers Tau aggregation, the aggregates are reduced after lowering Aβ via immunotherapy (Oddo et al., 2004). In cell models in which pro-aggregant TauRD triggers aggregation, aggregates disappear when expression is switched off (Khlistunova et al., 2006). In apparent contrast, two regulatable mouse models have been reported where Tau expression is controlled by the CaMKII promoter and where Tau aggregates persist even when transgene expression is suppressed (SantaCruz et al., 2005; Mocanu et al., 2008) (Figs. 3B,E,H, 4B). For the Morris water maze assay and the switch-off experiments, SantaCruz and colleagues (Ramsden et al., 2005; SantaCruz et al.,2005) used younger rTg4510 mice than in our study, but in both cases, the cognitive decline is reversible. We assume that the rescue of behavioral deficits in pro-aggregant TauRD OFF mice is linked to the recovery of synapses (Fig. 5A), since neuronal loss still persists (Fig. 3K,N). We suggest two factors for the persistence of tangles, the initial trigger by pro-aggregant TauRD plus mouse Tau, which sustains aggregation once nucleation has occurred, even after amyloidogenic TauRD disappears.
A final comment on implications for Tau-based drug treatments. Several have been proposed, aimed at inhibiting Tau–Tau interactions, hyperphosphorylation, or promoting degradation (Seabrook et al., 2007; Schneider and Mandelkow, 2008). Here we show that behavioral deficits are corrected by removing amyloidogenic Tau. If toxicity were based on interactions of amyloidogenic Tau with other cell components, this might be rescued by enhancing degradation (autophagy, proteasome). Alternatively, if aggregation were the toxic principle, neurons might be detoxified by aggregation inhibitors, and indeed this has been demonstrated in vitro and in cell models (Pickhardt et al., 2007; Congdon et al., 2009).
Footnotes
This work was supported by the Max Planck Society, Deutsche Forschungsgemeinschaft (AL 294/9-1 to C.A.), the Bundesministerium für Bildung und Forschung/Kompetenznetz Degenerative Demenzen program (E.-M.M.), the Breuer Foundation (E.-M.M.), EU-FP7/Memosad (Grant Agreement 2006121 to E.-M.M. and R.D.), Fonds Wetenschappelijk Onderzoek (FWO)-Vlaanderen (Grant G.0327.08 to D.B. and R.D., FWO junior scholarship to A.V.d.J.), the KUL2004 Impulse Program, Interdisciplinair Onderzoek Project 06/004, Onderzoekstoelage 06/23 (D.B.), and a K.U. Leuven fellowship (T.A.). We thank Dr. A. Haemisch and his team at the animal facility at the University of Hamburg Medical School for their continuous help in mouse breeding, Dr. H. Bujard and K. Schoenig (University of Heidelberg, Heidelberg, Germany) for help with inducible transgenic mice, S. Hahn and A. Hofmann for excellent technical assistance, Dr. A. Marx (Max Planck Unit for Structural Molecular Biology, Hamburg, Germany) for advice on statistics, and Dr. J. Decker (Max Planck Unit for Structural Molecular Biology, Hamburg, Germany) for valuable suggestions on this manuscript. We gratefully acknowledge reagents from Dr. E. Kandel (Columbia University, New York, NY; CaMKIIα-tTA activator transgenic mice), Dr. P. Seubert (Elan Pharma, South San Francisco, CA; 12E8 antibody), and Dr. P. Davies (Albert Einstein College, Bronx, NY; MC-1 and PHF-1 antibodies).
References
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendash GW, Lewis J, Leighty R, McGowan E, Cracchiolo JR, Hutton M, Garcia MF. Multi-metric behavioral comparison of APPsw and P301L models for Alzheimer's disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res. 2004;1012:29–34. doi: 10.1016/j.brainres.2004.02.081. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Balschun D, Wolfer DP, Bertocchini F, Barone V, Conti A, Zuschratter W, Missiaen L, Lipp HP, Frey JU, Sorrentino V. Deletion of the ryanodine receptor type 3 (RyR3) impairs forms of synaptic plasticity and spatial learning. EMBO J. 1999;18:5264–5273. doi: 10.1093/emboj/18.19.5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghorn S, Zheng-Fischhöfer Q, Ackmann M, Biernat J, von Bergen M, Mandelkow EM, Mandelkow E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11714–11721. doi: 10.1021/bi000850r. [DOI] [PubMed] [Google Scholar]
- Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, Schmitt I, Wullner U, Evert BO, O'Kane CJ, Rubinsztein DC. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Yen S, Lee G. Disease-related modifications in tau affect the interaction between fyn and tau. J Biol Chem. 2005;280:35119–35125. doi: 10.1074/jbc.M505895200. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Aβ causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Demonstration of amyloid deposits and neurofibrillary changes in whole brain sections. Brain Pathol. 1991;1:213–216. doi: 10.1111/j.1750-3639.1991.tb00661.x. [DOI] [PubMed] [Google Scholar]
- Carrettiero DC, Hernandez I, Neveu P, Papagiannakopoulos T, Kosik KS. The cochaperone BAG2 sweeps paired helical filament- insoluble tau from the microtubule. J Neurosci. 2009;29:2151–2161. doi: 10.1523/JNEUROSCI.4660-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman CA, Perez Y, Lacaille JC. Effects of GABA(A) inhibition on the expression of long-term potentiation in CA1 pyramidal cells are dependent on tetanization parameters. Hippocampus. 1998;8:289–298. doi: 10.1002/(SICI)1098-1063(1998)8:3<289::AID-HIPO10>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Chen C, Tonegawa S. Molecular genetic analysis of synaptic plasticity, activity-dependent neural development, learning, and memory in the mammalian brain. Annu Rev Neurosci. 1997;20:157–184. doi: 10.1146/annurev.neuro.20.1.157. [DOI] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon EE, Figueroa YH, Wang L, Tonuva G, Chang E, Kuret J, Conrad C, Duff KE. Inhibition of tau polymerization with a cyanine dye in two distinct model systems. J Biol Chem. 2009;284:20830–20839. doi: 10.1074/jbc.M109.016089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- Denk F, Wade-Martins R. Knock-out and transgenic mouse models of tauopathies. Neurobiol Aging. 2009;30:1–13. doi: 10.1016/j.neurobiolaging.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Hooge R, Lüllmann-Rauch R, Beckers T, Balschun D, Schwake M, Reiss K, von Figura K, Saftig P. Neurocognitive and psychotiform behavioral alterations and enhanced hippocampal long-term potentiation in transgenic mice displaying neuropathological features of human α-mannosidosis. J Neurosci. 2005;25:6539–6549. doi: 10.1523/JNEUROSCI.0283-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Jr, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt H, Eder M, Frick A, Zieglgänsberger W. Precisely localized LTD in the neocortex revealed by infrared-guided laser stimulation. Science. 1999;286:110–113. doi: 10.1126/science.286.5437.110. [DOI] [PubMed] [Google Scholar]
- Eckermann K, Mocanu MM, Khlistunova I, Biernat J, Nissen A, Hofmann A, Schönig K, Bujard H, Haemisch A, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The beta-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of Alzheimer tauopathy. J Biol Chem. 2007;282:31755–31765. doi: 10.1074/jbc.M705282200. [DOI] [PubMed] [Google Scholar]
- Goode BL, Feinstein SC. Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J Cell Biol. 1994;124:769–782. doi: 10.1083/jcb.124.5.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, Chau M, Denis PE, Feinstein SC. Structural and functional differences between 3-repeat and 4-repeat tau isoforms. Implications for normal tau function and the onset of neurodegenetative disease. J Biol Chem. 2000;275:38182–38189. doi: 10.1074/jbc.M007489200. [DOI] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Studying gene function in eukaryotes by conditional gene inactivation. Annu Rev Genet. 2002;36:153–173. doi: 10.1146/annurev.genet.36.041002.120114. [DOI] [PubMed] [Google Scholar]
- Götz J, Ittner LM. Animal models of Alzheimer's disease and frontotemporal dementia. Nat Rev Neurosci. 2008;9:532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- Götz J, Probst A, Spillantini MG, Schäfer T, Jakes R, Bürki K, Goedert M. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 1995;14:1304–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziano A, Petrosini L, Bartoletti A. Automatic recognition of explorative strategies in the Morris water maze. J Neurosci Methods. 2003;130:33–44. doi: 10.1016/s0165-0270(03)00187-0. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci U S A. 1990;87:5827–5831. doi: 10.1073/pnas.87.15.5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustke N, Trinczek B, Biernat J, Mandelkow EM, Mandelkow E. Domains of tau protein and interactions with microtubules. Biochemistry. 1994;33:9511–9522. doi: 10.1021/bi00198a017. [DOI] [PubMed] [Google Scholar]
- Hoogenraad CC, Bradke F. Control of neuronal polarity and plasticity—a renaissance for microtubules? Trends Cell Biol. 2009;19:669–676. doi: 10.1016/j.tcb.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Hu X, Viesselmann C, Nam S, Merriam E, Dent EW. Activity-dependent dynamic microtubule invasion of dendritic spines. J Neurosci. 2008;28:13094–13105. doi: 10.1523/JNEUROSCI.3074-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Shoji M, Kawarai T, Kawarabayashi T, Matsubara E, Murakami T, Sasaki A, Tomidokoro Y, Ikarashi Y, Kuribara H, Ishiguro K, Hasegawa M, Yen SH, Chishti MA, Harigaya Y, Abe K, Okamoto K, George-Hyslop PS, Westaway D. Accumulation of filamentous tau in the cerebral cortex of human tau R406W transgenic mice. Am J Pathol. 2005;166:521–531. doi: 10.1016/S0002-9440(10)62274-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VMY. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- Jaworski J, Kapitein LC, Gouveia SM, Dortland BR, Wulf PS, Grigoriev I, Camera P, Spangler SA, Di Stefano P, Demmers J, Krugers H, Defilippi P, Akhmanova A, Hoogenraad CC. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron. 2009;61:85–100. doi: 10.1016/j.neuron.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Jaworski T, Dewachter I, Seymour CM, Borghgraef P, Devijver H, Kügler S, Van Leuven F. Alzheimer's disease: old problem, new views from transgenic and viral models. Biochim Biophys Acta. 2010;1802:808–818. doi: 10.1016/j.bbadis.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Kampers T, Friedhoff P, Biernat J, Mandelkow EM. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996;399:344–349. doi: 10.1016/s0014-5793(96)01386-5. [DOI] [PubMed] [Google Scholar]
- Kampers T, Pangalos M, Geerts H, Wiech H, Mandelkow E. Assembly of paired helical filaments from mouse tau: implications for the neurofibrillary pathology in transgenic mouse models for Alzheimer's disease. FEBS Lett. 1999;451:39–44. doi: 10.1016/s0014-5793(99)00522-0. [DOI] [PubMed] [Google Scholar]
- Kanai Y, Takemura R, Oshima T, Mori H, Ihara Y, Yanagisawa M, Masaki T, Hirokawa N. Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. J Cell Biol. 1989;109:1173–1184. doi: 10.1083/jcb.109.3.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. Neuroscience—the molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, Mandelkow E, Mandelkow EM. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006;281:1205–1214. doi: 10.1074/jbc.M507753200. [DOI] [PubMed] [Google Scholar]
- Klausnitzer J, Manahan-Vaughan D. Frequency facilitation at mossy fiber-CA3 synapses of freely behaving rats is regulated by adenosine A1 receptors. J Neurosci. 2008;28:4836–4840. doi: 10.1523/JNEUROSCI.3729-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretz O, Fester L, Wehrenberg U, Zhou L, Brauckmann S, Zhao S, Prange-Kiel J, Naumann T, Jarry H, Frotscher M, Rune GM. Hippocampal synapses depend on hippocampal estrogen synthesis. J Neurosci. 2004;24:5913–5921. doi: 10.1523/JNEUROSCI.5186-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Magnani E, Fan J, Gasparini L, Golding M, Williams M, Schiavo G, Goedert M, Amos LA, Spillantini MG. Interaction of tau protein with the dynactin complex. EMBO J. 2007;26:4546–4554. doi: 10.1038/sj.emboj.7601878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McGowan E, Eriksen J, Hutton M. A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 2006;22:281–289. doi: 10.1016/j.tig.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schönig K, Bujard H, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The potential for β-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008;28:737–748. doi: 10.1523/JNEUROSCI.2824-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissette DA, Parachikova A, Green KN, LaFerla FM. Relevance of transgenic mouse models to human Alzheimer disease. J Biol Chem. 2009;284:6033–6037. doi: 10.1074/jbc.R800030200. [DOI] [PubMed] [Google Scholar]
- Nakashiba T, Young JZ, McHugh TJ, Buhl DL, Tonegawa S. Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science. 2008;319:1260–1264. doi: 10.1126/science.1151120. [DOI] [PubMed] [Google Scholar]
- Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S. NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci. 2004;5:361–372. doi: 10.1038/nrn1385. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Paulson JB, Ramsden M, Forster C, Sherman MA, McGowan E, Ashe KH. Amyloid plaque and neurofibrillary tangle pathology in a regulatable mouse model of Alzheimer's disease. Am J Pathol. 2008;173:762–772. doi: 10.2353/ajpath.2008.080175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekkala S, Albert ML, Spiro A, III, Erkinjuntti T. Perseveration in Alzheimer's disease. Dement Geriatr Cogn Disord. 2008;25:109–114. doi: 10.1159/000112476. [DOI] [PubMed] [Google Scholar]
- Pickhardt M, Larbig G, Khlistunova I, Coksezen A, Meyer B, Mandelkow EM, Schmidt B, Mandelkow E. Phenylthiazolyl-hydrazide and its derivatives are potent inhibitors of tau aggregation and toxicity in vitro and in cells. Biochemistry. 2007;46:10016–10023. doi: 10.1021/bi700878g. [DOI] [PubMed] [Google Scholar]
- Planel E, Richter KE, Nolan CE, Finley JE, Liu L, Wen Y, Krishnamurthy P, Herman M, Wang L, Schachter JB, Nelson RB, Lau LF, Duff KE. Anesthesia leads to tau hyperphosphorylation through inhibition of phosphatase activity by hypothermia. J Neurosci. 2007;27:3090–3097. doi: 10.1523/JNEUROSCI.4854-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polydoro M, Acker CM, Duff K, Castillo PE, Davies P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci. 2009;29:10741–10749. doi: 10.1523/JNEUROSCI.1065-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prange-Kiel J, Rune GM, Leranth C. Median raphe mediates estrogenic effects to the hippocampus in female rats. Eur J Neurosci. 2004;19:309–317. doi: 10.1111/j.0953-816x.2003.03124.x. [DOI] [PubMed] [Google Scholar]
- Preuss U, Biernat J, Mandelkow EM, Mandelkow E. The “jaws” model of tau-microtubule interaction examined in CHO cells. J Cell Sci. 1997;110:789–800. doi: 10.1242/jcs.110.6.789. [DOI] [PubMed] [Google Scholar]
- Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25:10637–10647. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmann H, Grigoriadis N, Eldar-Levy H, Avital A, Rozenstein L, Touloumi O, Behar L, Ben-Hur T, Avraham Y, Berry E, Segal M, Ginzburg I, Abramsky O. A novel transgenic mouse expressing double mutant tau driven by its natural promoter exhibits tauopathy characteristics. Exp Neurol. 2008;212:71–84. doi: 10.1016/j.expneurol.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Rowan MJ, Klyubin I, Cullen WK, Anwyl R. Synaptic plasticity in animal models of early Alzheimer's disease. Philos Trans R Soc Lond B Biol Sci. 2003;358:821–828. doi: 10.1098/rstb.2002.1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara N, Maeda S, Takashima A. Tau oligomerization: a role for tau aggregation intermediates linked to neurodegeneration. Curr Alzheimer Res. 2008;5:591–598. doi: 10.2174/156720508786898442. [DOI] [PubMed] [Google Scholar]
- SantaCruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar M, Kuret J, Lee G. Two motifs within the tau microtubule-binding domain mediate its association with the hsc70 molecular chaperone. J Neurosci Res. 2008;86:2763–2773. doi: 10.1002/jnr.21721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindowski K, Bretteville A, Leroy K, Begard S, Brion JP, Hamdane M, Buee L. Alzheimer's disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Mandelkow E. Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics. 2008;5:443–457. doi: 10.1016/j.nurt.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabrook GR, Ray WJ, Shearman M, Hutton M. Beyond amyloid: the next generation of Alzheimer's disease therapeutics. Mol Interv. 2007;7:261–270. doi: 10.1124/mi.7.5.8. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron. 2008;60:534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Brandt R, Kamibayashi C, Kuret J, White CL, 3rd, Mumby MC, Bloom GS. Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J Biol Chem. 1999;274:25490–25498. doi: 10.1074/jbc.274.36.25490. [DOI] [PubMed] [Google Scholar]
- Spires-Jones TL, de Calignon A, Matsui T, Zehr C, Pitstick R, Wu HY, Osetek JD, Jones PB, Bacskai BJ, Feany MB, Carlson GA, Ashe KH, Lewis J, Hyman BT. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J Neurosci. 2008;28:862–867. doi: 10.1523/JNEUROSCI.3072-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol. 1999;155:2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland RJ, McDonald RJ. Hippocampus, amygdala, and memory deficits in rats. Behav Brain Res. 1990;37:57–79. doi: 10.1016/0166-4328(90)90072-m. [DOI] [PubMed] [Google Scholar]
- Tatebayashi Y, Miyasaka T, Chui DH, Akagi T, Mishima K, Iwasaki K, Fujiwara M, Tanemura K, Murayama M, Ishiguro K, Planel E, Sato S, Hashikawa T, Takashima A. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc Natl Acad Sci U S A. 2002;99:13896–13901. doi: 10.1073/pnas.202205599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by MARK2/Par-1. J Neurosci. 2007;27:2896–2907. doi: 10.1523/JNEUROSCI.4674-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traykov L, Baudic S, Raoux N, Latour F, Rieu D, Smagghe A, Rigaud AS. Patterns of memory impairment and perseverative behavior discriminate early Alzheimer's disease from subcortical vascular dementia. J Neurol Sci. 2005;230:75–79. doi: 10.1016/j.jns.2004.11.006. [DOI] [PubMed] [Google Scholar]
- van Swieten JC, Bronner IF, Azmani A, Severijnen LA, Kamphorst W, Ravid R, Rizzu P, Willemsen R, Heutink P. The DeltaK280 mutation in MAP tau favors exon 10 skipping in vivo. J Neuropathol Exp Neurol. 2007;66:17–25. doi: 10.1097/nen.0b013e31802c39a4. [DOI] [PubMed] [Google Scholar]
- von Bergen M, Barghorn S, Li L, Marx A, Biernat J, Mandelkow E-M, Mandelkow E. Mutations of tau protein in frontotemporal dementia promote aggregation of paired helical filaments by enhancing local beta structure. J Biol Chem. 2001;276:48165–48174. doi: 10.1074/jbc.M105196200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MJ, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Wilsch VW, Behnisch T, Jager T, Reymann KG, Balschun D. When are class I metabotropic glutamate receptors necessary for long- term potentiation? J Neurosci. 1998;18:6071–6080. doi: 10.1523/JNEUROSCI.18-16-06071.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca2+ elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Higuchi M, Yoshiyama Y, Ishihara T, Forman MS, Martinez D, Joyce S, Trojanowski JQ, Lee VM. Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J Neurosci. 2004;24:4657–4667. doi: 10.1523/JNEUROSCI.0797-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]