

Abstract
To reach the open state, the GABAA receptor (GABAAR) is assumed to bind two agonist molecules. Although it is currently believed that GABAAR could also operate in the monoliganded state, the gating properties of singly bound GABAAR are poorly understood and their physiological role is still obscure. In the present study, we characterize for the first time the gating properties of singly bound GABAARs by using a mutagenesis approach and we propose that monoliganded GABAAR contribute in shaping synaptic responses. At saturating GABA concentrations, currents mediated by recombinant GABAARs with a single functional binding site display slow onset, fast deactivation kinetics, and slow rate of desensitization-resensitization. GABAARs with two binding sites activated by brief pulses of subsaturating GABA concentrations (in the range of the GABA concentration profile in the synaptic cleft) could also mediate fast deactivating currents, displaying deactivation kinetics similar to those mediated by GABAARs with a single functional binding site. Model simulations of receptors activated by realistic synaptic GABA waves revealed that a considerable proportion of GABAA receptors open in the monoliganded state during synaptic transmission, therefore contributing in shaping IPSCs.
Introduction
The number of GABA molecules needed to activate GABAA receptors (GABAARs) has been intensively investigated over the three last decades. The first hint that GABAARs possess more than one neurotransmitter binding site came from pioneering studies analyzing the relationship between GABA concentration and GABAAR activation (Constanti, 1979). The determination of GABAAR stoichiometry (Tretter et al., 1997), the characterization of GABAAR binding pocket (Sigel et al., 1992), together with the insight of GABAAR tridimensional organization based on the similarity with the acetylcholine binding protein (AChBP) backbone (Cromer et al., 2002), led to the conclusion that the GABAAR possesses two binding sites at the interface between α and β subunits. However, the physiological relevance of two binding sites on GABAAR is not fully understood. It is currently believed that full receptor activation requires binding of two GABA molecules, although Macdonald et al. (1989) proposed that GABAAR might also be activated in the monoliganded state. This hypothesis was based on the observation that GABAAR mean open time depends on [GABA], being shifted toward short openings at low submicromolar [GABA]. Jones and Westbrook (1995) confirmed Macdonald's theory by showing that currents evoked by short pulses of low GABA concentrations deactivated faster than those evoked by saturating GABA. The explanation for this effect was that brief applications of low [GABA] might activate some GABAARs in the monoliganded state, thus mediating fast-deactivating macroscopic currents due to short-living single channel openings. However, the occurrence of receptor openings in the singly bound state remains a controversial issue. Recently, the observation that brief openings frequently occur at high [GABA] suggested that short openings could be explained without invoking singly bound states leading to an updated kinetic model for GABAARs without the inclusion of monoliganded states (Lema and Auerbach, 2006). By generating concatemers comprising a single mutated β2 subunit, Sigel et al. (1992) demonstrated that GABAARs with a single GABA binding site were functional. Other studies proposed that singly bound states could undergo desensitization (Jones and Westbrook, 1995; Mozrzymas et al., 2003a). In line with this, it has been shown that ambient GABA could absorb receptors into singly bound desensitized states, a mechanism determining reduction of synaptic current amplitude (Overstreet et al., 2000). Although the aforementioned studies have been essential to prove the existence of the monoliganded state of GABAARs, the knowledge of the singly bound state(s) gating features is mostly based on indirect evidence.
In the present study, we characterize for the first time the kinetic properties of currents elicited by the activation of a single binding site of α1β1γ2 receptors by using a mutagenesis approach. Singly bound state currents showed (1) slow rise time (2) fast deactivation kinetics and (3) slow desensitization-resensitization kinetics. Model simulations of GABAAR activated by realistic GABA exposures at the synapse suggested that monoliganded receptors can play a role in shaping GABAergic synaptic currents.
Materials and Methods
Human embryonic kidney 293 cell line, plasmids, and DNA transient transfection.
Human embryonic kidney 293 (HEK293) cells (ATCC-LGC standards) were maintained in DMEM supplemented with 4.5 g/L glucose, 10% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin (Invitrogen) and were transiently cotransfected with DNA encoding for GABAAR subunits α1, β1 and γ2 (1:1:10) (kindly provided by Dr. S. Vicini, Georgetown University, Washington, DC) and/or enhanced green fluorescent protein(EGFP) (pEGFP-N1vector, Clontech) using the Fugene 6 transfection reagent (Roche Applied Science). α1F64C was generated by PCR amino acid substitution of phenylalanine by cysteine in position 64 of the wild-type α1 subunit. α1HA-F64C was obtained by introducing the hemagglutinin (HA) peptide sequence (YPYDVDYA) between amino acid 4 and 5 of the α1F64C sequence. All plasmids were verified by DNA sequencing. Mutated α1 subunits (α1F64C or α1HA-F64C) were cotransfected with the wild-type α1 at 1:5, 1:10 and 1:15 ratios, yielding the same total DNA amount of α1 as in α1β1γ2 transfections (see Fig. 4A). Cells were replated 24 h after transfection onto 18 mm glass coverslips, to avoid the formation of cell clusters. All experiments were performed 48 h after transfection and the inclusion of the γ2 subunit in GABAAR was confirmed by the low GABAAR sensitivity to Zn2+ 1 μm.
Figure 4.

Characterization of α1α1F64Cβ1γ2 receptor gating properties. A, Summary of the dependence of current amplitude (left) and decay kinetics (right) upon the ratio between α1 and α1F64C overexpression. B, The 10–90% rise time of normalized α1α1F64Cβ1γ2 (thick line) and α1β1γ2 (thin line) currents. C, Comparison of the deactivation kinetics between α1α1F64Cβ1γ2 (left) and α1β1γ2 (right) normalized currents. D, Desensitization induced by 100 ms, 10 mm GABA pulse in α1α1F64Cβ1γ2 (left) and α1β1γ2 (right) receptors. E, Desensitization induced by 3000 ms, 10 mm GABA pulse in α1α1F64Cβ1γ2 (left) and α1β1γ2 (right). F, Currents elicited by two consecutive 10 ms pulses spaced by 100 ms from α1α1F64Cβ1γ2 (left) and α1β1γ2 (right) receptors. G, Typical response of α1α1F64Cβ1γ2 (left) and α1β1γ2 (right) currents to long (3000) ms conditioning pulse followed by a short (10 ms) test pulse. H, Summary of recovery from desensitization of paired pulse experiments shown in F. α1α1F64Cβ1γ2 currents (black circles), at the considered time points, showed immediate recovery, while the α1β1γ2 receptors (open circles) displayed biexponential recovery (τfast = 8 ± 0.2 and τslow = 321 ± 21 ms, Afast = 0.36 ± 0.03). I, Summary of recovery from desensitization of paired pulse experiments shown in G. Both α1α1F64Cβ1γ2 currents (black squares) α1β1γ2 currents (open squares) showed biexponential recovery (τw = 27.3 ± 2.7 and τw 8.4 ± 1.1 s, respectively). J, Summary of the dependence of α1β1γ2- and α1α1F64Cβ1γ2 currents deactivation kinetics on [GABA] (black, 1 ms pulse; gray, 20 ms pulse). K, Summary of the dependence of α1β1γ2- and α1α1F64Cβ1γ2 currents onset kinetics on [GABA] (3000 and 20 ms pulses for black and gray, respectively). Each data point results from at least 20 recordings.
Electrophysiological recordings and drug application.
Recordings from excised outside-out patches were performed using the 700B Axopatch amplifier (Molecular Devices). External solution contained (in mm): 145 NaCl, 2 KCl, 2 CaCl2, 2 MgCl2, 10 glucose, and 10 HEPES. Patch pipettes were pulled from borosilicate glass capillaries. Their resistance ranged from 4 to 5 MΩ when filled with intracellular solution containing (in mm): 122 CsCl, 2 CaCl2, 2 MgCl2, 11 BAPTA, 10 HEPES, and 4 Na2ATP (300 mOsm and pH 7.2). Currents were acquired and analyzed using Clampex and Clampfit software (Molecular Devices). GABA was applied by means of an ultrafast perfusion system exploiting a piezoelectric-driven (Physik Instrumente) theta-glass application pipette (Jonas, 1995). The open-tip recordings of the liquid junction potentials revealed that the 10–90% exchange of solution occurred within 100 μs.
Primary antibodies.
Anti-α1 antibody (Alomone Labs) was directed against the N-terminal region of the α1subunit. The rat monoclonal anti-HA antibody (clone 3F10) was purchased from Roche.
Immunocytochemistry and confocal imaging.
Live staining of surface recombinant receptors was performed by incubating live transfected HEK293 cells with the appropriate primary antibody (0.01 mg/ml) in the culture medium at room temperature for 10 min. After fixation with 4% paraformaldehyde for 10 min, cells were incubated with bovine serum albumin (1%) to prevent nonspecific binding and then with the proper Alexa Fluor 647- or Alexa Fluor 546-conjugated secondary antibody (Invitrogen) for 45 min at room temperature. Control experiments without the primary antibody were performed to test fluorescent signals arising from nonspecific binding of the secondary antibody. Coverslips, mounted in DAKO fluorescent mounting medium, were observed using a Leica TCS SP5 laser scanning confocal microscope with excitation lines from 488, 543, and 633 nm lasers (SpectraPhysics) in the sequential mode. Images were acquired using an oil-immersion 63× (numerical aperture 1.4) Plan Apochromat objective and analyzed using Leica LAS AF software and MetaMorph (version 7.5, Molecular Devices).
Subcellular fractionation of membranes.
HEK293 cells were harvested by scraping into ice-cold phosphate buffer, 10 mm, pH 7.4, supplemented with protease inhibitor cocktail (Roche). Hypotonic lysis was allowed in the same buffer for 30 min at 4°C. Samples were centrifuged at 4°C at 10,700 × g for 15 min and the resulting membrane pellet was solubilized in 150 mm NaCl, 10 mm HEPES NaOH, pH 7.4, 1% Triton X-100, and protease inhibitors and clarified of insoluble material by centrifugation.
Immunoprecipitation and immunoblot analysis.
For immunoprecipitation assays, HEK293 cells were cultured in 20 ml flasks and transfected with α1, α1HA-F64C, β1, and γ2L (α1:α1HA-F64C ratio was 1:15) 48 h before the experiments. Membrane fractions, obtained as described above, were incubated with precoupled anti-HA protein A-Sepharose and anti-HA protein G-Sepharose (GE Healthcare) at 4°C for 4 h in binding buffer (10 mm HEPES-NaOH, pH 7.4, 150 mm NaCl) supplemented with 1% Triton X-100. Samples were centrifuged for 5 min at 670 × g. The pellet was washed three times with binding buffer supplemented with 1% Triton X-100 and three times with detergent-free binding buffer. The eluted proteins were then separated by SDS-PAGE and analyzed by immunoblotting. Immunoprecipitates were probed with anti-GABAA receptor α1 antibody, then stripped (30 min at 50°C in 75 mm Tris, pH 6.7, 2% SDS, 100 mm β-mercaptoethanol) and probed with anti-HA antibody. Primary antibodies were revealed by HRP-conjugated secondary antibodies (Sigma), followed by enhanced chemiluminescence (ECL) Plus Western blotting detection system (GE Healthcare) according to the company protocol.
Modeling.
To simulate the GABAAR kinetic behavior, we adopted the Jones and Westbrook (1995) model, which represents the minimal requirement to reproduce the basic features of GABAAR binding and gating. This model assumes opening and desensitized states originating from the both closed singly bound and doubly bound states. Kinetic modeling (see Fig. 5A) was performed with the Channel Lab 2.0 software (developed by S. Traynelis for Synaptosoft), which converted the kinetic model into a set of differential equations and solved them numerically assuming, as the initial condition, that at t = 0 only unbound receptors were present.
Figure 5.
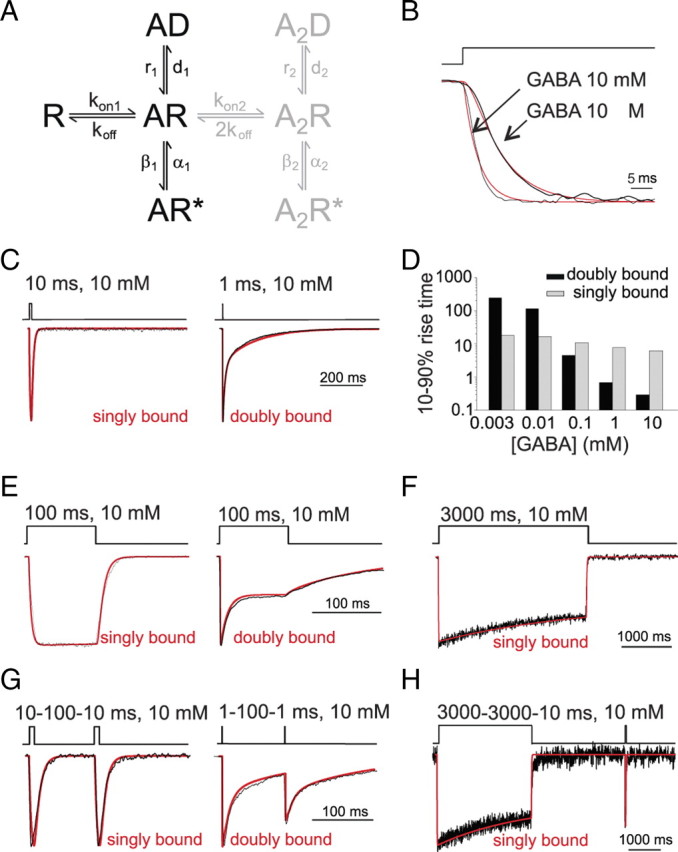
Model simulation of α1β1γ2 (doubly bound) and α1α1F64Cβ1γ2 (singly bound) gating properties. A, Gating scheme for GABAA receptor (Jones and Westbrook, 1995). In the present study, singly bound currents were simulated by using the R, AR, AR*, and AD states (in black). Doubly bound currents were simulated by adding the A2R, A2R* and A2D states (in gray). The rate constants used were: kon1 = 2.877 ± 0.177 ms−1 mm−1, koff = 0.267 ± 0.021 ms−1, β1 = 0.086 ± 0.007 ms−1, α1 = 0.208 ± 0.016 ms−1, d1 = 0.00023 ± 0.00002 ms−1, r1 = 0.00035 ± 0.00003 ms−1, kon2 = 18.29 ms−1 mm−1, β2 = 8.85 ms−1, α2 = 0.42 ms−1, d2 = 1.32 ms−1, r2 = 0.032 ms−1. B, Simulated (red) and experimental (black) onset of currents evoked by saturating and low GABA concentrations (as indicated) from singly bound receptors. C, Simulated (red) and experimental (black) currents showing the deactivation kinetics for singly and doubly bound receptor-mediated currents. D, Summary of simulated 10–90% rise-time dependence on the GABA concentration for singly and doubly bound receptor configurations. E, Simulated (red) desensitization for doubly and singly bound currents superimposed to experimental traces (black). F, Simulated and experimental currents (red and black, respectively), showing slow desensitization onset for singly bound currents. Doubly bound currents are not shown since more doubly bound desensitized state must be included in the model to adequately reproduce desensitization evoked by long GABA pulses. G, Recovery from desensitization using paired pulse protocol for simulated (red) and experimental currents (black). H, Recovery from desensitization of simulated (red) and experimental traces (black) after long conditioning pulses for singly bound currents. Note that in B–H, traces are normalized for better visualization of kinetic differences.
The rate constant optimization procedure was performed by using the NEURON 7.0 Multiple Run fitter module that allows the simultaneous fit of multiple experimental traces. This module implements the Brent's algorithm to optimize and integrate the differential equations using the fast and efficient CVODE variable time step integrator. In the attempt to determine the singly bound rate constants, therefore, for each experiment, we simultaneously fitted currents obtained from three key protocols in the same experiment including: (1) short applications of high GABA doses (10 ms, 10 mm) for resolving 10–90% rise time and the deactivation process; (2) long applications of high GABA doses (3000 ms, 10 mm) and (3) application of low GABA concentration (10 μm). We limited our fitting procedure to three different protocols because of the technical limit of keeping the patch recording stable over time. Simultaneous fit of each experiment (n = 9) generated a set of “singly bound” rate constants. Subsequently, the sets of rate constants obtained from each experiment were averaged, yielding the rate constant values listed in the legend for Figure 5. This procedure also allowed estimation of the rate constants variability [standard deviation of the mean (SDM)] in relation to the experimental variability (see Fig. 5 legend). To test the reliability of the singly bound rate constants in adequately representing receptor activation in the singly bound mode, the same rate constant values were also used to fit experimental protocols not included in rate constant optimization such as: (1) paired pulses protocols; (2) additional GABA low dose (3 mm) and (3) long conditioning pulses followed by a short test pulse. Importantly, the quality of these fits was in the same range of that observed in the traces used for the rate constant optimization. The doubly bound rate constants were determined by simultaneously fitting averaged currents obtained by: (1) short GABA pulses (1 ms, 10 mm); (2) paired pulses (100 ms gap) and (3) low GABA concentration (10 μm) from independent experiments. The rate constant values were in line with our previous studies (Barberis et al., 2007). Note that since doubly bound rate constants were optimized on averaged traces, the values are provided without SDM.
Cooperative binding reaction was introduced by assuming the first binding step ∼6.5 times slower than the second (2.87 and 18.59 ms−1 mm−1, respectively) (Mozrzymas et al., 2003a). Modeling the receptor gating in the singly bound state was performed by considering only the states R, AR, AD, and AR* (see also Fig. 5A).
Model simulations of GABA receptors activated by a neurotransmitter diffusion wave (see Figs. 6, 7) were performed by choosing the 3D free boundary condition solution (Barbour, 2001):
 |
with M the number of neurotransmitter molecules released, α the volume fraction, d the distance from the releasing site, D′ is the diffusion coefficient corrected by the tortuosity (i.e., D′ = D/λ2) and t is the elapsed time from the synaptic release, at t = 0. Equation 1 does not account for GABA neurotransmitter loss following binding to the synaptic receptors and buffering by the transporters. However, a computational investigation (Barbour, 2001) suggests that binding sites into the synaptic cleft negligibly affect the receptor activation. In addition, neurotransmitter buffering by the transporters would mostly affect the GABA concentration outside the synaptic cleft (Bragina et al., 2008). The diffusion coefficient (D) was assumed equal to the one of unhindered glutamine (D = 0.76 μm2/ms) and corrected by the tortuosity coefficient (λ = 1.55) (Barbour, 2001). The volume fraction was α = 0.1, a value smaller than that used in Barbour (2001) (α = 0.21), although recent findings show that due to the very high density at the synapse, smaller volume fraction values are likely to occur (Zuber et al., 2005). Although the law describing the diffusion of neurotransmitter in the synaptic cleft is certainly more complex than that depicted by Equation 1, such 3D diffusion equation well reproduces the peak concentration beneath the releasing site and the submillisecond neurotransmitter clearance from the cleft (see Fig. 6C,D). The concentration of GABA transmitter on the synaptic disk was computed by translating the distance [(d), Eq. 2], to the radial coordinate (r) (see Fig. 6A,B). The opening state of the overall synaptic receptor population was computed by integrating the contribution of the singly and doubly bound open states on the disk. We hypothesized that the receptors were uniformly distributed on the synaptic disk. Then the overall time-dependent opening state, O(t), was obtained by the following:
 |
with Rsyn being the radius of the synapse disk, dA(r) the area element of the annular section at distance r (see Fig. 6B), AR*(r,t) and A2R*(r,t) the singly and doubly bound states at distance r from the center of the disk. Equation 2 was integrated by using a uniform mesh with dR = 0.0125. Smaller integration values did not determine any significant differences. The kinetic model was implemented in NEURON 7.0 (Carnevale and Hines, 2006) and its implementation will be available on the NEURON website.
Figure 6.

Model simulation of GABA diffusion in the synaptic cleft. A, Diagram of a GABAergic synapse. The distance from the releasing site to the synaptic disk is indicated by d (see Materials and Methods). B, Schematic representation of the synaptic disk. The disk area was divided into shells with step dR and the relative contribution of the receptors located in each of them was computed (see Materials and Methods). C, Example of three [GABA] temporal profiles at increasing distances from the disk center (0.2, 0.3 and 0.4 μm) obtained by the simulated release of 4000 GABA molecules. D, GABA concentration spatial distribution at 0.03 ms after release.
Figure 7.

Simulated postsynaptic responses elicited by concentration transients obtained by GABA diffusion in the cleft. A, Simulated postsynaptic responses obtained by release of different GABA molecule number. In black, responses obtained at synapses with synaptic disk radius = 0.1 μm; in red responses obtained at synapses with synaptic disk radius = 0.4 μm. Note that the traces were normalized, to visualize difference in the decay kinetics. B, Open probability of the same traces as in A. C, Summary of the mean time constant (left) and 10–90% rise time (right) dependence upon the number of GABA molecules released at synapses with synaptic disk radius = 0.1 (black), 0.20 (green) and 0.4 (red) μm, respectively. D, Three-dimensional plot summarizing the simulated relative contribution of the singly bound open state at different GABA molecule numbers and synaptic radiuses.
Data analysis.
The current decay was fitted with the function y(t) = ΣAiexp(−t/τi), where Ai are the fractions of respective components (ΣAi = 1) and τi are the time constants. The weighted deactivation time constant was calculated using the formula τw = ΣτiAi. The fractional recovery from desensitization was expressed as r = I2 − Iend/I1 − Iend, where I1 is the first peak amplitude, Iend is the current value immediately before the application of the second pulse, and I2 is the second peak amplitude. In case of long conditioning pulses, the extent of recovery was assessed using the same formula stated above, but Iend corresponded to the value of the current at the end of the conditioning. Dose–response curves were fitted using the equation y = 1/(1 + EC50/[GABA]).
Statistics.
All data points are results of at least 20 different electrophysiological recordings. Values are expressed as means ± SEM. Unless otherwise stated statistical analysis was performed using the paired and unpaired Student's t tests to compare two experimental groups and the one-way ANOVA followed by the Tukey's multiple-comparison test when more than two experimental groups were analyzed.
Results
Dependence of α1β1γ2-mediated current deactivation kinetics on agonist pulse duration
In the attempt to investigate whether α1β1γ2 receptor could be activated in the singly bound configurations we exposed recombinant receptors to ultra fast GABA applications in which the transient agonist exposure would limit GABA binging to one of the two available sites. Patches excised from HEK293 cells expressing α1β1γ2 receptors were exposed to saturating [GABA] with pulses of duration ranging from 1 to 3000 ms. The deactivation time course of currents elicited by brief GABA exposures (1 ms, 10 mm) was fitted by a double exponential function (τfast = 8.2 ± 0.7 and τslow = 126.4 ± 14.6 ms, Afast = 0.58 ± 0.03) yielding a weighted deactivation time constant (τw) of 57.1 ± 6.2 ms (Fig. 1A, top). The increase in the GABA pulse duration led to a slow down of the current deactivation kinetics (Fig. 1A top, B) and the relation between deactivation time constant and pulse duration reached saturation at 1000 ms pulse (τfast = 24.0 ± 0.5 and τslow = 218.3 ± 28.7 ms, Afast = 0.32 ± 0.03, τw = 182.9 ± 20.1 ms, Fig. 1B). As shown by Jones and Westbrook (1995), a crucial determinant for the deactivation kinetics is the relative occupancy of open and desensitized state at the time the agonist is removed, since receptors slowly exiting from desensitization experience multiple transitions in the open state, thus considerably prolonging the deactivation process. During a long GABA pulse, indeed, after the peak occupancy of the open state, receptors are massively absorbed in the desensitized state with consequent slow down of the deactivation kinetics. It has to be pointed out, however, that even the shortest GABA pulse induces some extent of desensitization due to the fact that entry into open and desensitized states advances simultaneously (Mozrzymas et al., 2003a). However, during such brief GABA exposures there is no time for receptors leaving the open state to accumulate into desensitization. For this reason, when considering only doubly bound states, the relative occupancy of open and desensitized states will be proportional to the rate of receptor direct entry into desensitized and open states (d2 and β2, respectively; see Fig. 5A). Under these conditions (when the pulse length is briefer than the time needed to the open and desensitized states to interplay), the occupancy of doubly bound desensitization with respect to the open state is the smallest achievable with consequent fastest current deactivation kinetics. As already proposed by Jones and Westbrook (1995), however, further acceleration of the deactivation kinetics would be possible by taking into account the activation of GABAARs into singly bound state(s) with faster closing rate. To study this issue, we investigated the gating properties of GABAARs activated by short GABA exposures. Relying on the fact that under nonequilibrium conditions, the strength of the pulse is given by both its concentration and duration (C*t), we delivered weak pulses by applying short GABA pulses at subsaturating [GABA]. Currents evoked by ultra-weak GABA pulses (1 μm, 1 ms) were significantly faster than those obtained at saturating doses (at 1 ms pulse; τw = 57.1 ± 6.2 and τw = 17.1 ± 1.2 ms for 10 mm and 1 μm, respectively, p < 0.001, Fig. 1A bottom, B). Interestingly, by reducing GABA concentration and pulse duration, the fast component of the biexponential fit became largely predominant with respect to the slow one. At ultra-weak GABA exposure (1 μm, 1 ms), the deactivation time course was best described by a monoexponential function. In contrast, at long (1000–3000 ms) GABA pulses, the current deactivation time constant was similar at all GABA concentrations tested (1 μm to 10 mm, Fig. 1B). As expected, at long GABA pulses (which allow to reach the current peak), the 10–90% current rise time increased at lower GABA concentrations (0.54 ± 0.1 and 482.5 ± 47.6 ms at 10 and 0.001 mm, respectively, at 3000 ms pulse, p < 0.001, Fig. 1C). On the contrary, at short pulses, the 10–90% rise time was largely dictated by the pulse length since GABA exposure was much shorter than the actual current onset kinetics, in particular at low GABA concentrations (nonequilibrium conditions). However, a moderate increase in the 10–90% rise time (p < 0.05) was observed at decreasing concentrations of GABA delivered with short pulses, probably due to the progressive influence of singly bound states exhibiting slow opening rate. As mentioned above, the capability of GABA to activate GABAARs is given by both the duration and the concentration of the GABA pulse. This is also revealed by studying the dose–response curve at various GABA pulse durations as a function of [GABA] (Fig. 1D) in which short GABA exposures mimicked a decrease in the apparent affinity.
Figure 1.

Dependence of α1β2γ2-mediated current kinetics on GABA concentration and pulse duration. A, Top, Current traces obtained by exposing patches to brief (1 ms) or long (3000 ms) pulses of saturating [GABA]. For reasons of clarity, GABA application protocol is not indicated since the two traces are elicited by different duration pulses. Bottom, Currents evoked by (1 ms) pulses of 10 and 0.001 mm GABA. Note the faster current deactivation at low GABA concentrations. B, Summary of the dependence between GABA pulses duration and weighted current decay time constant obtained at different [GABA]. C, Summary of the dependence of 10–90% rise time of the current elicited by long (black) and short (gray) pulses on GABA concentration and pulse duration. D, Dose dependence of current amplitude at various GABA pulse durations (EC50 = 12.2 ± 1.8 μm at 3000 ms pulses). Each data points results from at least 20 recordings.
α1F64Cβ1γ2 receptors are not functional
Phe64 is a key amino acid for the GABA binding site on the αl subunit (Smith and Olsen, 1994). In the attempt to impair GABA binding to the αl subunit, we screened for possible amino acid substitutions for Phe64. It has been shown that substitution of Leu for αl-Phe64 (α1F64L) strongly decreased the apparent affinity of GABAARs for GABA (Sigel et al., 1992). Amino acid hydrophobicity studies and partial volume quantifications (Wimley and White, 1996) pointed out that, compared with Phe, Leu is less hydrophobic and exhibits smaller steric hindrance. Under the hypothesis that reduced GABA binding to αlF64L was due to the different properties of Leu, we substituted Cys for Phe64 (αlF64C), thus inducing a more dramatic change in both hydrophobicity and steric hindrance. HEK293 cells transfected with αlF64C, along with β1 and γ2 subunits, were probed for αl surface expression by immunocytochemistry with an antibody able to recognize both αl variants. Multicolor confocal microscopy revealed that, similarly to αl, also αlF64C subunit was expressed at the cell surface (Fig. 2A) as confirmed by αl fluorescence line scans in cell-spanning linear regions (Fig. 2B). Once established that α1F64Cβ1γ2 receptors were present at the cell surface, we tested whether these receptors were functional. To this end we exposed cells transfected with α1F64Cβ1γ2 or αlβ1γ2 to saturating [GABA]. Interestingly, no currents were mediated by α1F64Cβ1γ2 receptors, while sizable currents were recorded from αlβ1γ2 receptors (Fig. 2C). To demonstrate that the absence of currents from α1F64Cβ1γ2 was only attributable to impaired GABA binding, we challenged mutant and Wt receptors with pentobarbital, a barbiturate known to directly activate GABAAR also in the absence of binding site for GABA. As expected, pentobarbital (500 μm) elicited sizable currents from both α1F64Cβ1γ2 and α1β1γ2 receptors (Fig. 2D). This evidence further confirms the presence of mutant receptors at the cell surface. Altogether these experiments show that α1F64Cβ1γ2 receptors are expressed at the cell surface and that they are not functional due to impaired GABA binding.
Figure 2.

α1F64Cβ1γ2 receptors expressed at the cell surface are not functional. A, Single plane confocal images of EGFP fluorescence and surface α1 immunolabeling of HEK293 cells transfected with either α1β1γ2 (top) or α1F64Cβ1γ2 (bottom) subunits along with EGFP. Note that the anti-α1 antibody recognizes both α1 and α1F64C. Scale bars, 10 μm (n = 12). B, Line scans of fluorescence intensity of α1 immunolabeling in HEK293 cells transfected with α1β1γ2 (top) or α1F64Cβ1γ2 (bottom) along the white lines (20 μm) reported in A. C, Current traces elicited by applying GABA pulses (1 ms, 10 mm) to patches containing α1β1γ2 (top) and α1F64Cβ1γ2 (bottom) receptors (n = 20). D, Representative current traces elicited by applying pentobarbital (5 s, 500 μm) to patches containing α1β1γ2 (top) and α1F64Cβ1γ2 (bottom) receptors (n = 6). The presence of sizable currents from α1F64Cβ1γ2 indicates that mutant receptors, although not responsive to GABA, are expressed at the cell surface.
α1F64C coassembles with the α1 wild-type subunit
To generate GABAAR with a single binding site, α1 and α1F64C subunits were cotransfected and the possibility that these two subunits could heteromerize within the same receptor was tested by coimmunoprecipitation. Importantly, in these experiments an HA epitope was introduced in α1F64C (α1HA-F64C) between amino acids 4 and 5 of the mature protein (i.e., within the sequence recognized by the anti-α1 antibody), to make the mutant α1 (α1HA-F64C) no longer recognizable by the anti-α1 antibody (Fig. 3A). In this way the anti-α1 antibody exclusively bound to wild-type α1 subunit and the anti-HA antibody selectively recognized α1HA-F64C (Fig. 3A). After immunoprecipitation of membrane fractions of HEK293 cells expressing α1α1HA-F64Cβ1γ2 subunits with the anti-HA antibody, Western blot using the anti-α1 antibody revealed a protein of ∼50 kDa apparent molecular weight corresponding to the α1 subunit (Fig. 3B, top). When the same membrane was stripped and reprobed with the anti-HA antibody, the αlHA-F64C was correctly recognized (Fig. 3B, bottom). Altogether the coimmunoprecipitation assay indicates that αlHA-F64C and α1 are present within the same receptor.
Figure 3.

α1HA-F64C coassembles with α1 subunit in the same GABAAR. A, Single plane confocal images of HEK293 cells overexpressing α1HA-F64Cβ1γ2 receptors and EGFP probed with anti-α1- and anti-HA antibodies as indicated. Scale bar, 10 μm (n = 15). B, Heteromerization of α1 and α1HA-F64C within the same receptor. Membrane fractions of HEK293 cells transfected with α1α1HA-F64Cβ1γ2 (INPUT) were immunoprecipitated with the anti-HA antibody (IP anti-HA) and immunoblotted with the anti-α1 (top) or anti-HA antibody (bottom) (n = 3).
Kinetic properties of GABAAR exhibiting a single functional binding site
In the attempt to study GABAAR with a single binding site, α1F64C subunit was included into α1β1γ2 receptors at different α1:α1F64C ratios. The relative increase of α1F64C amount with respect to α1 led to a marked decrease of the current amplitude and to an acceleration of deactivation kinetics with respect to α1β1γ2-mediated currents (Fig. 4A). At the ratio 1:15 (α1:α1F64C), such dependence reached a plateau (Fig. 4A) suggesting that the cotransfection of α1 and α1F64C at ratio 1:15 together with β1 and γ2 yielded a predominant population of nonfunctional receptors containing two α1F64C subunits and a population of functional receptors containing both α1 and α1F64C. We therefore assumed that at such α1:α1F64C ratio GABA-evoked currents were mediated by α1α1F64Cβ1γ2 receptors with a single functional binding site. The 10–90% rise time of α1α1F64Cβ1γ2 currents evoked at saturating [GABA] was significantly slower than that of α1β1γ2-mediated currents (6.1 ± 0.4 and 0.54 ± 0.1 ms, respectively, p < 0.001, Fig. 4B). Subsequently, we focused on the deactivation kinetics of α1α1F64Cβ1γ2 currents evoked by saturating [GABA]. As currents mediated by α1α1F64Cβ1γ2 display slow rise time, GABA pulse duration was prolonged from 1 to 10 ms to allow the current to reach its peak. The deactivation time course of α1α1F64Cβ1γ2 currents was best described by a monoexponential function showing time constant τ = 9.3 ± 0.5 ms, a value markedly lower than that observed in α1β1γ2 currents (τw = 57.1 ± 6.2 ms, p < 0.001, Fig. 4C). The reduction of the pulse duration from 10 to 3 and 1 ms, left the α1α1F64Cβ1γ2 deactivation kinetics unchanged. Singly bound state desensitization was first studied by delivering long pulses of saturating [GABA]. Interestingly, application of 100 ms, 10 mm GABA did not induce any apparent desensitization on α1α1F64Cβ1γ2-receptors (Fig. 4D). On the contrary, the same protocol induced fast double-exponential apparent desensitization on α1β1γ2-mediated currents (τfast = 6.2 ± 0.2 ms, τslow = 45.5 ± 1.2 ms, Afast = 0.58 ± 0.07) and the ratio between the steady-state and the peak currents was 0.48 ± 0.08. To force the receptor into singly bound desensitized state, we prolonged GABA exposure to 3 s. As shown in Figure 4E, α1α1F64Cβ1γ2 currents displayed monoexponential desensitization onset (τ = 2983.4 ± 193.6 ms) with steady-state/peak current ratio of 0.68 ± 0.05. On the contrary, α1β1γ2 currents desensitized more profoundly (steady-state/peak ratio: 0.28 ± 0.05, p < 0.001, Mann–Whitney test, Fig. 4E) and showed three-exponential desensitization decay (τfast = 7.1 ± 0.8 ms; τmiddle = 101.1 ± 8.6 ms; τslow = 1512.2 ± 87.2 ms; Afast = 0.37 ± 0.02; Amiddle 0.16 ± 0.01). Current responses elicited by two consecutive 10 ms GABA pulses indicated no desensitization accumulation of α1α1F64Cβ1γ2 receptors (Fig. 4F,H). This result is consistent with the lack of apparent desensitization observed in Figure 4D. In contrast, α1β1γ2 receptors activated by paired GABA pulses (1 ms, 10 mm) showed desensitization accumulation and the rate of exit from desensitization was described by a monoexponential recovery curve (Fig. 4F,H). Next, we repeated paired pulse protocols by applying long conditioning pulses (3000 ms) followed by a short test pulse. Under these conditions, α1α1F64Cβ1γ2 receptors showed slower recovery from desensitization with respect to α1β1γ2 receptors (Fig. 4G,I). These data indicate that both desensitization and resensitization occur at lower rate in singly bound with respect to doubly bound. As shown in Figure 4J, the deactivation kinetics of α1α1F64Cβ1γ2 currents was unchanged over [GABA] ranging from 3 μm to 10 mm. In contrast, the 10–90% rise time of α1α1F64Cβ1γ2 currents significantly slowed down at [GABA] < 10 μm, although the dependence of the current onset on the GABA concentration was significantly (p < 0.001) less pronounced than in α1β1γ2 currents (Fig. 4K).
Model simulations of receptor gating
Model simulations were used to provide better quantification of the kinetic behavior of GABAARs activated in the singly bound state. The Jones and Westbrook (1995) model (Fig. 5A) was adopted for the following reasons: (1) it represents the minimal requirement to reproduce the basic GABAAR-mediated currents; (2) it includes monoliganded state capable to open and desensitize; and (3) it has been extensively used to reproduce synaptic-like currents evoked by short agonist exposures. Rate constants in our model were optimized by fitting the simulated currents to the responses obtained with the different tested protocols. The binding reaction process was considered cooperative (Mozrzymas et al., 2003a), to avoid overestimation of singly bound states. Indeed, compared with cooperative binding reaction, sequential noncooperative binding leads to a higher occupancy of the singly bound closed state (AR) with consequent higher probability to perform transitions in the singly bound open state.
Singly bound rate constants were derived from data obtained with GABAARs exhibiting a single functional binding site (α1α1F64Cβ1γ2). The slow rise time at saturating [GABA] (Fig. 4B) together with the fast deactivation kinetics could be explained in terms low efficacy of the singly bound conformational change (β1/α1), although at this stage it cannot be ruled out that unbinding and desensitization rates would also account for the observed kinetic behavior. The 10–90% rise time dose dependence of α1α1F64Cβ1γ2-mediated currents (Fig. 4K) was used to study both the binding rate constant (kon1) and the interplay between the binding process and the opening/closing transitions. The observation that after reducing [GABA] to 3 μm, the rise time was only ∼3 times slower than that recorded at saturating agonist concentrations, however, might be indicative of faster closing rate with respect to the opening rate. Indeed, since the macroscopic rate constant for the opening step is approximated by the sum of the opening and closing rate constants (assuming negligible entry into desensitization at short pulses) (Clements and Westbrook, 1991), at low [GABA], the slow process of entry into the open state would be terminated by fast receptor closing. Under such conditions, therefore, the rise time dependence on GABA concentration would be much less pronounced than in the case of high efficacy opening (such as in the doubly bound state), in line with what we observed. Finally, α1α1F64Cβ1γ2-mediated responses evoked by long agonist application and the paired pulse protocols with long conditioning pulse (Fig. 4D,E,G,I) were informative on the desensitization and resensitization rate constants. Taking into account all the above considerations, we simultaneously fitted the currents obtained at different pulse duration and doses (see Material and Methods) and determined a plausible set of rate constants reproducing the singly and doubly bound current behavior (Fig. 5). (For details on the rate constants optimization procedure see Materials and Methods.) Simulated current onset at saturating [GABA] was 0.3 and 6.2 ms for doubly and singly bound states, respectively (Fig. 5B,D). The simulated deactivation time constant was 8.0 and 53.84 ms for singly and doubly bound configurations, respectively (Fig. 5C). The set of rate constants proposed also reproduced: (1) the lack of fast desensitization at 100 ms pulses; (2) the slow desensitization onset at 3000 ms pulses (τ = 2834 ms) with steady state/peak ratio = 0.7 and (3) the slow recovery from desensitization in paired pulses experiments of long conditioning pulses followed by a test pulses (25% recovery at 3000 ms gap) (Fig. 5E–H).
Desensitization evoked by long saturating pulses was best described by a multiexponential fit, indicating the presence of more than one doubly bound desensitized state (Haas and Macdonald, 1999; Pugh and Raman, 2005). For this reason, the model we adopted did not adequately reproduce doubly bound currents evoked by long (3000 ms) pulses, as well as paired pulse experiments with long conditioning pulses. However, in the present study we did not consider additional slow doubly bound desensitized states as their inclusion in the model did not affect the kinetics of simulated currents evoked by brief agonist exposures.
Model simulations of receptor gating activated by “synaptic” GABA waves
Although the present study shows that GABAAR mediates fast-decaying currents when activated in its monoliganded state, it remains to be established whether the singly bound configuration plays a role at the synapse during synaptic transmission. As stated above, at saturating GABA applications, singly bound open states mediate a small percentage of the total current. This could indicate that synaptic currents, typically elicited by high [GABA] (1–3 mm), would be marginally shaped by monoliganded states of GABAARs. However, it has to be considered that synaptic currents are evoked by extremely short exposures (100 μs) and that, following synaptic vesicle release, [GABA] reaches high values in the vicinity of the release site, but it markedly decreases at the synapse periphery (Fig. 6C,D). It is therefore reasonable to assume that synaptic receptors located at the synapse periphery would be exposed to low concentration/short duration GABA pulses, thus favoring the activation of GABAAR in the monoliganded configuration (Fig. 6C,D). To test this hypothesis, we modeled the behavior of postsynaptic receptors activated by realistic GABA waves diffusing in the synaptic cleft. By using an equation describing three-dimensional diffusion (see Materials and Methods), GABA molecules were assumed to be released in the synaptic space and activate postsynaptic receptors homogeneously distributed within a synaptic disk with a radius ranging from 0.1 to 0.4 μm, Fig. 6A,B). On average, a synaptic vesicle contains 2000–5000 neurotransmitter molecules (Telgkamp et al., 2004). However, in our simulations we considered a range of 1000 to 10,000 released GABA molecules to include the possibility of partial release of the vesicle content and/or low vesicle filling (∼1000 molecules), and release of more than one vesicle (∼10,000 molecules). The peak and time course of the open probability was obtained by integrating the receptor contribution at increasing distances from the disk center (Fig. 6A,B) (see Materials and Methods). The simulated response decay kinetics depended on the number of molecules released (τw = 41.8 and 58.7 ms for 1000 and 10,000 GABA molecules, respectively, with a disk radius of 0.4 μm, Fig. 7A,C). As shown in Figure 7D, this effect is attributable to a higher relative contribution of singly bound states at 1000 with respect to 10,000 GABA molecules released. Interestingly, the dependence of the decay kinetics on GABA molecule number was considerably less pronounced in smaller synapses (τw = 50.6 and 55.6 ms for 1000 and 10,000 GABA molecules, respectively, at 0.1 μm synapses, Fig. 7A,C). This is due to the fact that, in large synapses, receptors located at the periphery of the synaptic disk sense low [GABA] (<200 μm) while, in small synapses, [GABA] peaks at values >1 mm along the synaptic disk radius (Fig. 6C). Thus, the contribution of monoliganded receptors is higher at large synapses with respect to the small ones (Fig. 7D). In line with this, at 0.4 μm synapses, the peak open probability of the simulated responses is considerably lower than that obtained at 0.1 μm synapses (Fig. 7B) indicating that, overall, large synapses are less saturated than small synapses. As expected, for a given synaptic radius, 10–90% current rise time was faster when evoked by a larger number of GABA molecules, as the onset kinetics is proportional to the agonist concentration (10–90% rise-time = 0.56 and 0.29 ms for 1000 and 10,000 GABA molecules, respectively at 0.1 μm synapse radius; Fig. 7C). In addition the 10–90% simulated response rise time depended upon the synaptic disk radius, being it markedly slower at large synapses (0.38 and 1.23 ms for 0.1 at 0.4 μm synapses, respectively, at 2000 released GABA molecules; Fig. 7C). As stated above, large synapses are exposed to an overall lower [GABA] determining slow down of the onset kinetics. Moreover, receptors activating in the singly bound state at the synaptic disk periphery further contribute to slowing down the response rise time since monoliganded state show slow onset rate (Fig. 4B).
Discussion
In the present study we characterized for the first time the kinetic properties of currents mediated by GABAARs activated in the singly bound state(s) by generating a recombinant receptor with a single functional binding site (α1α1F64Cβ1γ2). This represents an advantage with respect to previous works in which monoliganded states were unmasked by applying low [GABA] (Macdonald et al., 1989) or short pulses of low [GABA] (Jones and Westbrook, 1995). The use of concatenated GABA subunits (concatemers) with one impaired binding site has the advantage of providing precise control of both receptor stoichiometry and position of the mutated subunit within the receptor (Baumann et al., 2003). However, the folding and the assembly of these large molecules are difficult to predict. In keeping with this, Baumann et al. (2003) reported different apparent affinities between receptor obtained by transfecting concatemers or individual subunits, indicating that the links between the subunits may alter receptor function. In this study we used independent subunits to generate GABAAR with one functional binding site, although the activation of α1α1F64Cβ1γ2 receptors may not be strictly equivalent to that of α1β1γ2 receptors in the singly bound configuration since it cannot be excluded that the point mutation introduced in the α1 subunit might interfere with receptor gating. Nevertheless, it has to be pointed out, however, that several studies report that alteration of the binding site lead to selective effect on the affinity leaving efficacy unaffected (Fisher, 2004; Wagner et al., 2004). The similar decay kinetics of currents elicited by ultra-weak pulses and that of currents mediated by α1α1F64Cβ1γ2 receptors suggests that the F64C point mutation does not induce dramatic changes in α1α1F64Cβ1γ2 gating properties. At saturating [GABA], α1α1F64Cβ1γ2 currents exhibited fast deactivation kinetics and slow onset. This behavior can be explained by assuming that singly bound open state exhibits both low efficacy (β1/α1) and slow opening rate (α1+b1). Moreover the current onset kinetics can also be accelerated by fast entry into desensitization (Mozrzymas et al., 2003a).
Since at saturating [GABA], singly bound state would mediate ∼2% of the maximal current obtained in the fully liganded state (Baumann et al., 2003) it could be objected that such current component would play a negligible role at the synapse. However, it has to be considered that currents studied by Baumann et al. (2003) were elicited under substantially different conditions from those occurring at the synapse during synaptic transmission. In fact, it has been shown that following fusion of a synaptic vesicle, the neurotransmitter remains within the synaptic cleft for an average time of 100 μs (Mozrzymas et al., 2003b). This implies that postsynaptic receptors are activated in substantial nonequilibrium conditions that favor activation of GABAAR in the monoliganded state. In addition, partial synaptic vesicle fusion through a fusion pore would lead to ultra-weak synaptic exposures (He et al., 2006). Our model simulations show that singly bound states would be predominant at ∼1000 GABA molecules released in large synapses (0.4 μm radius). However, it is difficult to establish to what extent these conditions occur in a physiological context. By considering that a single synaptic vesicle releases on average ∼2500 GABA molecules (Telgkamp et al., 2004; Pugh and Raman, 2005) and that the average synaptic disk radius is ∼0.2 μm (Nusser et al., 1997), the relative contribution of singly bound state to the typical postsynaptic response would be ∼17%. This would determine the speed-up of the deactivation time constant by ∼11% with respect to the time constant obtained at 10,000 GABA molecules (representing a near-saturating GABA exposure). This difference, although relatively mild, might play a role at network level in signal integration and oscillations (Klausberger and Somogyi, 2008). We show in Figure 7B that the peak open probability at large synapses is lower than that at small synapses. It is worth noting that to obtain the actual current mediated by the release of GABA, the peak open probability must be rescaled to the receptor number present at the synapse. Assuming the same receptor density, in 0.4 μm synapses there would be 16 times more receptors with respect to 0.1 μm synapses. Taking into account ∼5 times lower peak open probability at 0.4 μm synapses with respect to 0.1 μm synapses (at the same number of GABA molecule released), 0.4 μm synapses would conduct an ∼3 times larger simulated current than that obtained at 0.1 μm synapses. The decay time course of currents obtained by exposing patches containing native or recombinant receptors to brief GABA pulses is usually slower than that of synaptic currents mediated by the same receptor subtype (Ortinski et al., 2004; Pugh and Raman, 2005). Participation of monoliganded states to mIPSCs could contribute explaining this discrepancy together with different desensitization properties of synaptic receptors (Pugh and Raman, 2005) and/or modulation of synaptic GABAARs by interaction with scaffold proteins (Marchionni et al., 2009) and low intracellular chloride ions concentrations (Houston et al., 2009).
Our simulations show that strong synaptic release (10,000 GABA molecules) at small synapses (0.1 μm radius) elicits responses with near-saturating peak open probability (popen = 0.50) while 1000 GABA molecules released at large synapses (0.4 μm radius) yield open probability ∼50 times lower. Therefore, it could be objected that mIPSCs evoked by low quantum content at large synapses would be highly susceptible to potentiation by positive allosteric modulators of GABAARs and/or increase of neurotransmitter in the synaptic cleft. Benzodiazepines (BDZs), for instance, by increasing binding rate constant of GABAARs (Lavoie and Twyman, 1996) would be expected to massively enhance the mIPSCs amplitude and duration. In contrast, the reported increase of mIPSCs amplitude by BDZs ranges from 25 to 44% (Frerking et al., 1995; Perrais and Ropert, 1999; Mozrzymas et al., 2007). However, in addition to the increase of binding rate constant, BDZs potentiate the GABAARs desensitization (Mozrzymas et al., 2007), determining (1) inhibition of currents evoked by saturating GABA pulses and (2) marked reduction of BDZs-induced peak increase of mIPSCs with respect to that observed at long subsaturating (1 μm) GABA pulses. This prevents the possibility to observe huge increases in the mIPSCs peak amplitude induced by BDZs. Furthermore, using dextran to enhance the peak and duration of GABA in the synaptic cleft by increasing the extracellular medium viscosity, only a 15% increase of mIPSCS peak amplitude was reported (Perrais and Ropert, 2000). However, the real [GABA] increase in the synaptic cleft induced by dextran is difficult to assess. Moreover, by considering the averaged range of synaptic disk size and number of GABA molecules released, the open probability dynamic range would be considerably narrower.
Another important issue is related to the fact that different α subunits have been shown to assemble on the same GABAAR (Benke et al., 2004) generating two different binding sites. This raises the possibility that at short synaptic exposures, GABA would preferentially bind to the site exhibiting higher binding rate constant. In this case, the current percentage mediated by singly bound receptor with respect to the maximal (doubly bound) current is difficult to predict and, theoretically, could be higher than that estimated by Baumann et al. (2003).
Footnotes
This work was supported by EU/FP7 (FOCUS to F.B. and A.B.), Telethon-Italy (GGP10135 to A.B.), Compagnia di San Paolo (progetto Neuroscienze to F.B.), and Telethon-Italy (GGP09134 to F.B.).
We thank Dr. M. Messa for his contribution in biochemistry and Dr. S. Vicini for providing us with the α1, β1, and γ2L plasmids.
References
- Barberis A, Mozrzymas JW, Ortinski PI, Vicini S. Desensitization and binding properties determine distinct alpha1beta2gamma2 and alpha3beta2gamma2 GABA(A) receptor-channel kinetic behavior. Eur J Neurosci. 2007;25:2726–2740. doi: 10.1111/j.1460-9568.2007.05530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B. An evaluation of synapse independence. J Neurosci. 2001;21:7969–7984. doi: 10.1523/JNEUROSCI.21-20-07969.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann SW, Baur R, Sigel E. Individual properties of the two functional agonist sites in GABA(A) receptors. J Neurosci. 2003;23:11158–11166. doi: 10.1523/JNEUROSCI.23-35-11158.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benke D, Fakitsas P, Roggenmoser C, Michel C, Rudolph U, Mohler H. Analysis of the presence and abundance of GABAA receptors containing two different types of alpha subunits in murine brain using point-mutated alpha subunits. J Biol Chem. 2004;279:43654–43660. doi: 10.1074/jbc.M407154200. [DOI] [PubMed] [Google Scholar]
- Bragina L, Marchionni I, Omrani A, Cozzi A, Pellegrini-Giampietro DE, Cherubini E, Conti F. GAT-1 regulates both tonic and phasic GABA(A) receptor-mediated inhibition in the cerebral cortex. J Neurochem. 2008;105:1781–1793. doi: 10.1111/j.1471-4159.2008.05273.x. [DOI] [PubMed] [Google Scholar]
- Carnevale N, Hines M. Cambridge, UK: Cambridge UP; 2006. The NEURON Book. [Google Scholar]
- Clements JD, Westbrook GL. Activation kinetics reveal the number of glutamate and glycine binding sites on the N-methyl-d-aspartate receptor. Neuron. 1991;7:605–613. doi: 10.1016/0896-6273(91)90373-8. [DOI] [PubMed] [Google Scholar]
- Constanti A. The GABA dose/conductance relationship on lobster muscle. J Physiol (Paris) 1979;75:645–649. [PubMed] [Google Scholar]
- Cromer BA, Morton CJ, Parker MW. Anxiety over GABA(A) receptor structure relieved by AChBP. Trends Biochem Sci. 2002;27:280–287. doi: 10.1016/s0968-0004(02)02092-3. [DOI] [PubMed] [Google Scholar]
- Fisher JL. The alpha 1 and alpha 6 subunit subtypes of the mammalian GABA(A) receptor confer distinct channel gating kinetics. J Physiol. 2004;561:433–448. doi: 10.1113/jphysiol.2003.051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerking M, Borges S, Wilson M. Variation in GABA mini amplitude is the consequence of variation in transmitter concentration. Neuron. 1995;15:885–895. doi: 10.1016/0896-6273(95)90179-5. [DOI] [PubMed] [Google Scholar]
- Haas KF, Macdonald RL. GABAA receptor subunit gamma2 and delta subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol. 1999;514:27–45. doi: 10.1111/j.1469-7793.1999.027af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Wu XS, Mohan R, Wu LG. Two modes of fusion pore opening revealed by cell-attached recordings at a synapse. Nature. 2006;444:102–105. doi: 10.1038/nature05250. [DOI] [PubMed] [Google Scholar]
- Houston CM, Bright DP, Sivilotti LG, Beato M, Smart TG. Intracellular chloride ions regulate the time course of GABA-mediated inhibitory synaptic transmission. J Neurosci. 2009;29:10416–10423. doi: 10.1523/JNEUROSCI.1670-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P. Fast application of agonists to isolated membrane patches. In: Sakmann B, Neher E, editors. Single-channel recording. New York: Plenum; 1995. pp. 231–243. [Google Scholar]
- Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie AM, Twyman RE. Direct evidence for diazepam modulation of GABAA receptor microscopic affinity. Neuropharmacology. 1996;35:1383–1392. doi: 10.1016/s0028-3908(96)00077-9. [DOI] [PubMed] [Google Scholar]
- Lema GM, Auerbach A. Modes and models of GABA(A) receptor gating. J Physiol. 2006;572:183–200. doi: 10.1113/jphysiol.2005.099093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Rogers CJ, Twyman RE. Kinetic properties of the GABAA receptor main conductance state of mouse spinal cord neurones in culture. J Physiol. 1989;410:479–499. doi: 10.1113/jphysiol.1989.sp017545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchionni I, Kasap Z, Mozrzymas JW, Sieghart W, Cherubini E, Zacchi P. New insights on the role of gephyrin in regulating both phasic and tonic GABAergic inhibition in rat hippocampal neurons in culture. Neuroscience. 2009;164:552–562. doi: 10.1016/j.neuroscience.2009.07.063. [DOI] [PubMed] [Google Scholar]
- Mozrzymas JW, Barberis A, Mercik K, Zarnowska ED. Binding sites, singly bound states, and conformation coupling shape GABA-evoked currents. J Neurophysiol. 2003a;89:871–883. doi: 10.1152/jn.00951.2002. [DOI] [PubMed] [Google Scholar]
- Mozrzymas JW, Zarnowska ED, Pytel M, Mercik K. Modulation of GABA(A) receptors by hydrogen ions reveals synaptic GABA transient and a crucial role of the desensitization process. J Neurosci. 2003b;23:7981–7992. doi: 10.1523/JNEUROSCI.23-22-07981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozrzymas JW, Wójtowicz T, Piast M, Lebida K, Wyrembek P, Mercik K. GABA transient sets the susceptibility of mIPSCs to modulation by benzodiazepine receptor agonists in rat hippocampal neurons. J Physiol. 2007;585:29–46. doi: 10.1113/jphysiol.2007.143602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Cull-Candy S, Farrant M. Differences in synaptic GABA(A) receptor number underlie variation in GABA mini amplitude. Neuron. 1997;19:697–709. doi: 10.1016/s0896-6273(00)80382-7. [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Lu C, Takagaki K, Fu Z, Vicini S. Expression of distinct alpha subunits of GABAA receptor regulates inhibitory synaptic strength. J Neurophysiol. 2004;92:1718–1727. doi: 10.1152/jn.00243.2004. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Jones MV, Westbrook GL. Slow desensitization regulates the availability of synaptic GABA(A) receptors. J Neurosci. 2000;20:7914–7921. doi: 10.1523/JNEUROSCI.20-21-07914.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrais D, Ropert N. Effect of zolpidem on miniature IPSCs and occupancy of postsynaptic GABAA receptors in central synapses. J Neurosci. 1999;19:578–588. doi: 10.1523/JNEUROSCI.19-02-00578.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrais D, Ropert N. Altering the concentration of GABA in the synaptic cleft potentiates miniature IPSCs in rat occipital cortex. Eur J Neurosci. 2000;12:400–404. doi: 10.1046/j.1460-9568.2000.00957.x. [DOI] [PubMed] [Google Scholar]
- Pugh JR, Raman IM. GABAA receptor kinetics in the cerebellar nuclei: evidence for detection of transmitter from distant release sites. Biophys J. 2005;88:1740–1754. doi: 10.1529/biophysj.104.055814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel E, Baur R, Kellenberger S, Malherbe P. Point mutations affecting antagonist affinity and agonist dependent gating of GABAA receptor channels. EMBO J. 1992;11:2017–2023. doi: 10.1002/j.1460-2075.1992.tb05258.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GB, Olsen RW. Identification of a [3H]muscimol photoaffinity substrate in the bovine gamma-aminobutyric acidA receptor alpha subunit. J Biol Chem. 1994;269:20380–20387. [PubMed] [Google Scholar]
- Telgkamp P, Padgett DE, Ledoux VA, Woolley CS, Raman IM. Maintenance of high-frequency transmission at Purkinje to cerebellar nuclear synapses by spillover from boutons with multiple release sites. Neuron. 2004;41:113–126. doi: 10.1016/s0896-6273(03)00802-x. [DOI] [PubMed] [Google Scholar]
- Tretter V, Ehya N, Fuchs K, Sieghart W. Stoichiometry and assembly of a recombinant GABAA receptor subtype. J Neurosci. 1997;17:2728–2737. doi: 10.1523/JNEUROSCI.17-08-02728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DA, Czajkowski C, Jones MV. An arginine involved in GABA binding and unbinding but not gating of the GABA(A) receptor. J Neurosci. 2004;24:2733–2741. doi: 10.1523/JNEUROSCI.4316-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–848. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- Zuber B, Nikonenko I, Klauser P, Muller D, Dubochet J. The mammalian central nervous synaptic cleft contains a high density of periodically organized complexes. Proc Natl Acad Sci U S A. 2005;102:19192–19197. doi: 10.1073/pnas.0509527102. [DOI] [PMC free article] [PubMed] [Google Scholar]