

Abstract
Rubinstein-Taybi syndrome (RSTS) is an inheritable disease associated with mutations in the gene encoding the CREB (cAMP response element-binding protein)-binding protein (CBP) and characterized by growth impairment, learning disabilities, and distinctive facial and skeletal features. Studies in mouse models for RSTS first suggested a direct role for CBP and histone acetylation in cognition and memory. Here, we took advantage of the genetic tools for generating mice in which the CBP gene is specifically deleted in postmitotic principal neurons of the forebrain to investigate the consequences of the loss of CBP in the adult brain. In contrast to the conventional CBP knock-out mice, which exhibit very early embryonic lethality, postnatal forebrain-restricted CBP mutants were viable and displayed no overt abnormalities. We identified the dimer of histones H2A and H2B as the preferred substrate of the histone acetyltransferase domain of CBP. Surprisingly, the loss of CBP and subsequent histone hypoacetylation had a very modest impact in the expression of a number of immediate early genes and did not affect neuronal viability. In addition, the behavioral characterization of these mice dissociated embryonic and postnatal deficits caused by impaired CBP function, narrowed down the anatomical substrate of specific behavioral defects, and confirmed the special sensitivity of object recognition memory to CBP deficiency. Overall, our study provides novel insights into RSTS etiology and clarifies some of the standing questions concerning the role of CBP and histone acetylation in activity-driven gene expression, memory formation, and neurodegeneration.
Introduction
The Rubinstein-Taybi syndrome (RSTS) is a rare inheritable disease associated with mutations in either one of two related genes encoding transcriptional coactivators with lysine acetyltransferase activity (KAT) [previously referred to as histone acetyltransferase activity, or HAT (Allis et al., 2007)]: the cAMP-response element binding protein (CREB)-binding protein (CBP), whose gene (crebbp) is mutated in ∼60% of the patients (Petrij et al., 1995) and p300, paralogous to CBP and responsible for ∼3% of the cases (Roelfsema et al., 2005). Children with RSTS display distinctive anatomical features, such as hypertelorism, prominent nose, and broad thumbs and first toes (Wiley et al., 2003), experience difficulties in planning and executing motor acts, and have a short attention span and low intelligence level (Gotts and Liemohn, 1977; Hennekam et al., 1992).
Recent studies in RSTS mouse models have provided important new insights into the etiology of this condition and demonstrated a good correlation between reduced KAT activity and deficits in synaptic plasticity and memory (Bourtchouladze et al., 2003; Alarcón et al., 2004; Korzus et al., 2004; Wood et al., 2005, 2006; Viosca et al., 2010). Moreover, because intellectual abilities rely on the ability to store and retrieve information and memory storage is known to depend on de novo gene expression, it has been proposed that the reduced histone acetylation in CBP-deficient mice may cause the misregulation of the transcription of loci important in neural plasticity and thus contribute to the observed cognitive impairments (Barco, 2007). Although RSTS is not a neurodegenerative disease and no cell loss has been reported in the different mouse models for this disorder, several lines of evidence indicate that CBP and histone acetylation may also play a role in the pathogenesis of diverse neurodegenerative diseases (Saha and Pahan, 2006). Another interesting notion emerging from studies on RSTS mouse models, in particular of the reversal of phenotypical traits after pharmacological treatment with inhibitors of histone deacetylases (HDACi), is that the cognitive and neurological deficits associated to RSTS may have two distinct components: one originated during the development of the CNS, and a second one resulting from the continued requirement of the enzymatic activity of CBP throughout life (Alarcón et al., 2004; Korzus et al., 2004).
Previous loss-of-function approaches to investigate the role of CBP in neuronal gene expression and survival were constrained by the early embryonic lethality of CBP knock-out mice and the partial or uncertain level of inhibition of CBP function in heterozygous and transgenic lines (Barco, 2007). Here, we address some of the important questions raised in previous studies, such as the substrate specificity of CBP and its role in neuronal survival and activity-driven gene expression through the characterization of a conditional knock-out mouse strain in which CBP expression is specifically lost in postmitotic forebrain neurons. We also investigate the behavioral deficits associated with CBP deficiency once the impact of developmental defects has been eliminated or at least primarily reduced.
Materials and Methods
Animals and treatments.
The generation of both CBPf/f (Zhang et al., 2004) and CaMKIIα–cre (CKIIcre) (Tsien et al., 1996) mice has been described previously. Mice were maintained and bred under standard conditions, consistent with Spanish and European regulations and approved by the Institutional Animal Care and Use Committee. The genetic background of all mice was C57BL/6J. Analyses were conducted in 2- to 4-month-old animals, except for the cell death analyses, in which a second group of 8-month-old animals was also examined. Sodium butyrate (NaBu) (1.2 mg/kg in PBS) was administered by intraperitoneal injection. Vehicle-treated mice received an injection of PBS. Sampling was performed 30 min later, before the decay of the transient increase of acetylation (data not shown). Exposure to novelty consisted of placing an individual animal in a white Plexiglas square box (50 × 50 × 30 cm) containing plastic tubings and small objects/toys of different shapes, colors, and sizes for 1 h. Control animals were maintained in their standard cages. Animals were rapidly killed to minimize the possible induction of immediate early genes (IEG) by stress.
Plasmids and transient transfection of HEK293 cells.
Standard manipulations of Escherichia coli, cell culture, proteins, and nucleic acids were performed essentially as described (Ausubel et al., 1999). For transfection experiments in HEK293 cells, CBP and p300 coding sequences were cloned in pMM400 vector (Mayford et al., 1996) that contain a tet-responsive element and a minimal cytomegalovirus promoter, which allowed inducible expression of the protein of interest. These plasmids were cotransfected with either pTet–Off or pEGFP–N3 (Invitrogen). The tTA protein expressed from pTet–Off strongly activates transcription of CBP and p300 genes in the absence of doxycycline. HEK293 cells were grown in 90% DMEM supplemented and 10% fetal calf serum supplemented with 2 mm glutamine and penicillin/streptomycin (100 U/ml to 100 μg/ml) (Invitrogen). HEK293 cells were transfected with Lipofectamine 2000 (Invitrogen) following the instructions of the manufacturer.
Western blot and immunohistochemistry.
Nissl staining, immunohistochemistry, and Western blot analysis were performed as previously described (López de Armentia et al., 2007; Sanchís-Segura et al., 2009). Microdissected CA1 subregions were used as source of protein extracts. For immunohistochemistry, mice were anesthetized, perfused with 4% paraformaldehyde, and postfixed overnight, and 50 μm sections were obtained using a vibratome. Antibodies used in the present study were as follows: α-CBP C1 and c-fos antibodies (Santa Cruz Biotechnology), α-H2B and α-H3 (Millipore Corporation), α-neuronal-specific nuclear protein (NeuN) (Millipore Corporation), α-GFAP, α-MAP2, α-synaptophysin, α-actin, and biotinylated and HRP-conjugated α-mouse secondary antibodies (Sigma-Aldrich), HRP-conjugated α-rabbit secondary antibody (Cell Signaling Technology), and Alexa Fluor 488, 555, and 594 secondary antibodies (Invitrogen). The different α-acetyl-histone antibodies were locally produced (Sanchís-Segura et al., 2009) except for the acetylated lysine-specific antibodies (Millipore Corporation). Cell counting and length measurements were performed using NIH ImageJ software in images obtained with confocal microscopy in single stacks or in a conventional epifluorescence microscope. For each animal, two brain slices were examined, and the resulting values were averaged. For the quantification of CBP, c-fos, and acetylhistones immunostainings, the signal intensities for single individual nuclei in a frame (0.375 × 0.375 mm, confocal microscopy) were obtained and normalized by the median of intensities to scale the values between frames.
PCR, reverse transcription, and quantitative PCR.
To obtain genomic DNA, the tissues were treated with 100 μl of 25 mm NaOH and 0.2 mm EDTA, pH 12, for 20 min at 95°C and then buffered with an equal volume of 1 m Tris-HCl, pH 5. PCR were set up using GoTaq DNA polymerase (Promega): a final volume reaction of 25 μl contained 1:20 dilutions of DNA, 5× Green buffer (Promega), 1.5 mm MgCl2, 0.2 mm dNTP mix, 0.5 mm each primer, and 0.625 U of GoTaq DNA polymerase. PCR was performed for 30 cycles (1 min at 95°C, 1 min at 54°C, 1 min at 72°C). The sequences of the primers Z38 and Z40 were as described previously (Zhang et al., 2004).
In reverse transcription (RT)-PCR experiments, total RNA from whole hippocampus, microdissected CA1 subregion, cortex, striatum and cerebellum was purified using TRI reagent (Sigma) according to the instructions of the manufacturer. One microgram was reverse transcribed using RevertAid First Strand cDNA Synthesis kit (Fermentas). Then, conventional PCR were performed using Pfu DNA polymerase (Fermentas): a final volume reaction of 25 μl contained 50 ng of cDNA, 10× buffer with 20 mm MgSO4, 0.2 mm dNTP mix, 0.5 mm each primer, and 0.625 U of Pfu DNA polymerase. PCR was performed for 35 cycles (45 s at 95°C, 45 s at 55°C, 1 min at 72°C). The sequences (from 5′ to 3′) of the primers were as follows: 5′ CREB binding domain (CBD), CGGCTCCATGGGTGCAGGGCAACAGAATGCCA; E8 reverse (E8rev), AAGGAGGCGCTGCTGTAGGTAT. Quantitative RT-PCRs (qRT-PCR) were performed in an Applied Biosystems 7300 real-time PCR unit using SYBR GreenER mix (Invitrogen) and primers specific for CBP, p300, Arc, c-fos, Npas4, Egr1, Nr4a1, FosB, Dusp1, and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). All primer sequences are available on request. Each independent sample was assayed in duplicate and normalized using GAPDH levels.
Behavior.
For all behavioral tasks, adult mutant and control littermate males were used. The SHIRPA primary screen, open field, elevated plus maze, and fear conditioning tasks were performed as described previously (Viosca et al., 2009, 2010). To study motor coordination and learning, mice were trained on a rotarod (Ugo Basile) at a constant speed (4 rpm) four times per day during 2 d. On the testing day, the rotarod was set to increase its speed 1 rpm every 16 s starting at 4 rpm, and the interval to fall off was measured. For the analysis of circadian locomotor activity, each mouse was individually housed in a PhenoTyper cage (45 × 45 × 55 cm, length × width × height; Noldus Information Technology) during 5 d and 4 nights with water and food available ad libitum. All cages contained a top unit with a digital video camera and infrared lights, sawdust on the floor, and an immobile shelter. Data were acquired with EthoVision video-tracking software (Noldus Information Technology) for 3 consecutive days after 36 h of habituation. To evaluate nociception and sensitivity to thermal stimulus, mice were placed in the test chamber of a hotplate (IITC Life Science) set at 54°C, and the latency until the animal exhibited heat pain behavior was recorded. All mice were tested three times with 1 h interval between each measurement. In the novel object recognition memory task, mice were habituated to an open blue cylinder for 2 consecutive days (10 min/d). During training (third day), the mice explored for 20 min two identical objects, and for testing (fourth day), the animals were presented for 10 min to one of the training objects and to a novel object. The familiar and the novel objects differed in shape, color, and texture and were located in the same positions in which the objects were located during the training session. The use of different objects as novel or familiar, as well as the relative position of the novel object, was balanced between genotypes. Time spent exploring the objects in both the training and testing sessions was measured with video-tracking system (SMART; Panlab SL). The discrimination index (DI) was determined using the following formula: DI = 100 × (time exploring A − time exploring B)/(time exploring A + time exploring B). Spatial learning in the Morris water maze (MWM) was assessed in a circular tank (170 cm diameter) filled with opaque white water as described previously (Viosca et al., 2009). Mice were monitored throughout the training and testing sessions with a video-tracking system (Panlab SL). A platform of 10 cm diameter was submerged 1 cm below the water surface in the center of the target quadrant. The training protocol, both when the platform was visible (3 d: V1 to V3) and hidden (8 d: H1 to H8), consisted of four trials per day with a 45 min intertrial interval. If the mice did not find the platform after 120 s, they were gently guided to it. Mice were transferred to their cages only after remaining on the platform for at least 10 s. Memory retention trials of 60 s were performed at the beginning of day H5 (probe trial 1) and 24 h after concluding the training on day H8 (probe trial 2). Statistical analyses used ANOVAs, and means ± SEM are presented in the figures. The experimenter was blind to genotypes, and the result of the PCR-based genotyping was provided as a factor for statistical analysis of the behavioral data once the battery of tasks was concluded.
Results
CBP depletion in principal forebrain neurons
To explore the consequences of the neuronal elimination of CBP in adult brain function bypassing the embryonic lethality caused by CBP depletion, we produced mice bearing both a floxed crebbp allele (CBPf) in homozygosity (Zhang et al., 2004) and the transgene CaMKIIα–cre, which drives the expression of cre recombinase in postmitotic principal neurons of the forebrain (Tsien et al., 1996). This genetic manipulation results in mice that express a truncated CBP protein (CBPstop523) that lacks KAT activity (amino acids 1300–1700) and is also unable to interact with a number of transcription factors involved in neuronal function, such as CREB, c-fos, jun, nuclear factor-κB, and others, because critical domains (KIX, CH3) involved in these interactions are encoded by exons downstream of the deletion (Chan and La Thangue, 2001).
Previous work has demonstrated that both CBPf/f mice (Zhang et al., 2004) and CaMKIIα–cre transgenic mice (Tsien et al., 1996) are indistinguishable from wild-type mice. CaMKIIα–cre/CBPf/f mice, from now on referred as CKIIcre/CBPf/f, have a normal lifespan and show no overt anatomical or behavioral abnormality (Table 1). These initial observations suggest that, although CBP is essential during embryonic development, it is largely dispensable for the survival of postmitotic neurons. As expected, given that the mutation is restricted to postmitotic neurons, CKIIcre/CBPf/f mice do not present the facial dismorphia and skeletal defects observed in heterozygous mutants (Tanaka et al., 2000; Viosca et al., 2010). Body weight and size were also similar between genotypes, and even CKIIcre/CBPf/f mice were slightly larger, indicating that the growth impairment observed in heterozygous mice does not have a neurological origin or depends on neuronal populations not affected in CKIIcre/CBPf/f mice.
Table 1.
Neurological characterization of CKIIcre/CBPf/f and control littermates
| CBPf/f (n = 9) | CKIIcre/CBPf/f (n = 10) | p | |
|---|---|---|---|
| Weight (g) | 28.6 ± 0.94 | 30.2 ± 0.53 | 0.16 |
| Body length (mm) | 90 (90–96.5) | 97.5 (93.75–100) | 0.02 |
| Body position | 3 (2–3.25) | 3 (2–3) | 0.54 |
| Spontaneous activity | 2 (1–2) | 2 (1–2) | 0.65 |
| Respiration rate | 2 (2–2) | 2 (2–2) | 1.00 |
| Tremor | 0 (0–0) | 0 (0–0) | 1.00 |
| Barbering | 0 (0–0) | 0 (0–1) | 0.36 |
| Urination | 0 (0–0) | 0 (0–0) | 1.00 |
| Defecation | 0.5 (0–1.25) | 0 (0–0) | 0.08 |
| Transfer arousal | 4 (2–4.25) | 4 (1–4) | 0.39 |
| Fear | 1 (0.75–1) | 1 (1–1) | 0.15 |
| Palpebral closure | 0 (0–0) | 0 (0–0) | 1.00 |
| Piloerection | 0 (0–0) | 0 (0–0) | 1.00 |
| Gait | 0 (0–0) | 0 (0–1) | 0.13 |
| Pelvic elevation | 3 (2.75–3) | 3 (2–3) | 0.65 |
| Tail elevation | 1 (1–1) | 1 (1–1) | 0.32 |
| Touch scape | 2 (2–2) | 2 (1–2) | 0.16 |
| Positional passivity | 0 (0–0) | 0 (0–0.25) | 0.54 |
| Trunk curl | 0 (0–0) | 0 (0–0) | 1.00 |
| Feet clasping | 1 (1–1) | 1 (1–1) | 1.00 |
| Limb grasping | 1 (1–1) | 1 (1–1) | 1.00 |
| Visual placing | 2 (2–2) | 2 (2–2) | 1.00 |
| Negative geotaxis | 0 (0–0) | 0 (0–0) | 0.32 |
| Grip strength | 3 (3–3) | 3 (3–3) | 0.32 |
| Pinna reflex | 1 (0.75–2) | 1.5 (0.75–2) | 0.52 |
| Corneal reflex | 2 (2–2) | 2 (2–2) | 0.32 |
| Toe pinch | 0.5 (0–3) | 1.5 (0–2.25) | 0.78 |
| Pole latency (up) | 2 (0–6.25) | 2 (0–6.75) | 1.00 |
| Pole latency (down) | 1 (0–4.75) | 2 (0–3) | 0.90 |
| Wire maneuver | 2 (1–3) | 1.5 (0.75–2) | 0.16 |
| Righting reflex | 0 (0–0) | 0 (0–0) | 1.00 |
| Contact righting reflex | 1 (1–1) | 1 (1–1) | 1.00 |
| Irritability | 0 (0–0) | 0 (0–0) | 0.32 |
| Vibrissae | 1 (0–1) | 1 (1–1) | 0.28 |
| Skin color | 1 (1–1) | 1 (1–1.25) | 0.54 |
| Limb tone | 1 (1–1) | 1 (1–1) | 1.00 |
| Abdominal tone | 1 (1–2) | 1 (1–1.25) | 0.24 |
| Lacrimation | 1 (1–1) | 1 (1–1) | 1.00 |
| Provoked biting | 1 (0–1) | 0.5 (0–1) | 0.37 |
| Aggression | 0 (0–0) | 0 (0–0) | 1.00 |
| Vocalization | 0 (0–1) | 0 (0–0.25) | 0.34 |
| Seizures | 0 (0–0) | 0 (0–0) | 1.0 |
| Hotplate | 12.7 ± 0.66 | 11.9 ± 0.53 | 0.39 |
Data are expressed as either mean ± SEM or median, followed by the interquartile range in parentheses. p values are calculated using t test for data expressed as mean ± SEM or Mann–Whitney test for data expressed as median, followed by the interquartile range.
We confirmed cre-mediated recombination of the crebbp locus in genomic DNA samples. In agreement with the expected pattern of cre recombinase expression, exon-specific PCR reactions (Fig. 1A) demonstrated the extensive elimination of exon 7 in genomic DNA from the hippocampus, cortex, and striatum of CKIIcre/CBPf/f mice (Fig. 1B). In contrast, we did not observe any recombination in cerebellum in which the calcium/calmodulin-dependent kinase IIα (CaMKIIα) promoter is not active (Fig. 1B). The elimination of exon 7 caused the production of a shorter CBP transcript in the tissues expressing the cre recombinase (Fig. 1C). qRT-PCR analyses revealed that the amount of full-length CBP transcripts was reduced in the same brain regions showing recombination in the crebbp locus (Fig. 1D, Ex7-8 bars). The reduction of full-length CBP transcripts was especially severe in the cortex and in the hippocampus (∼71 and ∼35%, respectively). As expected, this reduction did not reach 100%, because the genetic manipulation eliminates CBP expression only in principal neurons, whereas glial cells, interneurons, and other neural types maintain normal expression of the crebbp gene. In addition, not all the neurons in a given region expressed the cre recombinase (see below). Interestingly, exon 7-lacking transcripts seemed to be unstable, because we observed a significant reduction of CBP transcripts also when the qRT-PCR was targeted to sequences downstream of the deletion (Fig. 1D, 3′ bars).
Figure 1.

Forebrain-specific recombination of the crebbp locus. A, Scheme of the crebbp locus showing the loxP sites (thick arrows) surrounding exon 7. Primers for checking recombination at the genomic DNA [named Z38 and Z40 (Zhang et al., 2004)] and transcript levels (named 5′CBD and E8rev) are also depicted. B, The recombination of genomic DNA (band marked as “recomb”) was detected only in genomic DNA from cre-expressing tissues (Ctx, cortex; Hpc, hippocampus; Str, striatum) but not in the cerebellum (Cb) or the tail. MW, Molecular weight markers. C, Exon-specific RT-PCR using 5′CBD and E8rev primers identifies two different transcript species with the expected sizes of 370 and 267 bp, confirming the deletion of exon 7 in the cortex but not in the cerebellum. D, Quantitative RT-PCR of RNA from CKIIcre/CBPf/f and control littermates (n = 4 per genotype) using a primer pair specific for full-length CBP transcripts (E7fwd and E8rev) or a pair of primers located 3′ in the KAT domain (3′). E, Immunohistochemistry analysis of CBP demonstrates that the expression of cre recombinase driven by the CaMKIIα--cre transgene is scattered over the cortex and hippocampus. CBP loss is also appreciable at the amygdala but not in the cerebellum in which cre recombinase is not expressed. The different cortical layers (I–VI) and other anatomical landmarks are indicated. CA1, CA1 subregion; CA3, CA3 subregion; DG, dentate gyrus; LA, lateral amygdala; BA, basal amygdala; Str, striatum; ML, molecular layer; GL, granular layer. Scale bar, 100 μm. F, Higher-magnification image of the CA1 subregion demonstrating the existence of two distinct populations of CA1 pyramidal neurons: CBP positive and CBP negative. Scale bar, 50 μm. G, Left, Quantification of the CBP-positive cells in different brain regions. Right, DAPI staining showed that a similar number of cells were analyzed in both groups of animals. CBPf/f, n = 2; CKIIcre/CBPf/f, n = 3. Data are expressed as mean ± SEM. *p < 0.01; **p < 0.001.
Immunohistochemistry experiments using a CBP-specific antibody confirmed the neuron-specific reduction of CBP in different forebrain regions (Fig. 1E). In the hippocampus, the staining revealed a scattered pattern for the loss of CBP in the pyramidal cell layer of the CA1 subfield (Fig. 1F). The elimination of CBP was observed in ∼90% of the CA1 pyramidal neurons and in ∼40% of the granular cells of the dentate gyrus (DG); no loss of CBP was observed in the CA3 subfield (Fig. 1G). The pattern of CBP elimination in the cortex also showed interesting asymmetries. We observed a general reduction of CBP throughout the cortex (Fig. 1E), but layers 2 and 3 seemed more severely affected (∼70% of the cells; data not shown) than the other layers. The loss of CBP also affected most principal neurons in the amygdala formation, whereas immunostaining in the cerebellum was similar in CKIIcre/CBPf/f and control mice. Because of the prominent reduction of CBP in the CA1 subfield of the hippocampus, we focused our analyses primarily in this area.
CBP loss causes a strong reduction of neuronal histone acetylation
The reduction of CBP levels (and consequently of its KAT activity) has been associated with histone hypoacetylation. In previous analyses of cbp+/− mice, we detected a significant reduction of neuronal histone H2B acetylation (Alarcón et al., 2004), but the neurons of these animals still maintained a functional crebbp allele. Neurons with disruption in both alleles represent a better approach to clarify the contribution of the activity of CBP to the acetylation of each one of the four nucleosome histones and to determine the substrate preference of this enzyme in neurons.
Western blot analyses in the CA1 subregion revealed a significant (∼50%) reduction in the acetylation of bulk histones H2B and H2A and a trend indicating also reduced acetylation of histones H3 and H4 (Fig. 2A). To gain additional detail, we used immunohistochemistry staining and confocal microscopy and took advantage of the scattered pattern of CBP loss. This circumstance allowed the direct comparison of neighboring CBP-expressing and CBP-lacking neurons in the same field of view. We found that the same cells lacking CBP also showed a dramatic reduction in the acetylation of the four nucleosome histones. This reduction was especially severe in the case of histones H2B and H2A, although the acetylation of histones H3 and H4 was also significantly reduced (Fig. 2B,C). The same pattern, pronounced decrease of H2A/H2B acetylation and more modest reduction of H3/H4, was also observed in cortical and dentate gyrus neurons (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The analysis of the correlation between CBP expression and histone acetylation levels in individual cells indicates that the KAT activity of CBP contributes to the acetylation state of the four nucleosome histones but is especially important in determining the acetylation level of histones H2A and H2B (Pearson's correlation coefficient of 0.78 for AcH2A, 0.79 for AcH2B, 0.62 for AcH3, and 0.69 for AcH4). Interestingly, intraperitoneal injection of the HDACi sodium butyrate was able to restore and even to increase the histone acetylation levels in the brain of CKIIcre/CBPf/f mice, indicating that other cellular KAT activities also have these histones as substrates (Fig. 3 and results not shown).
Figure 2.
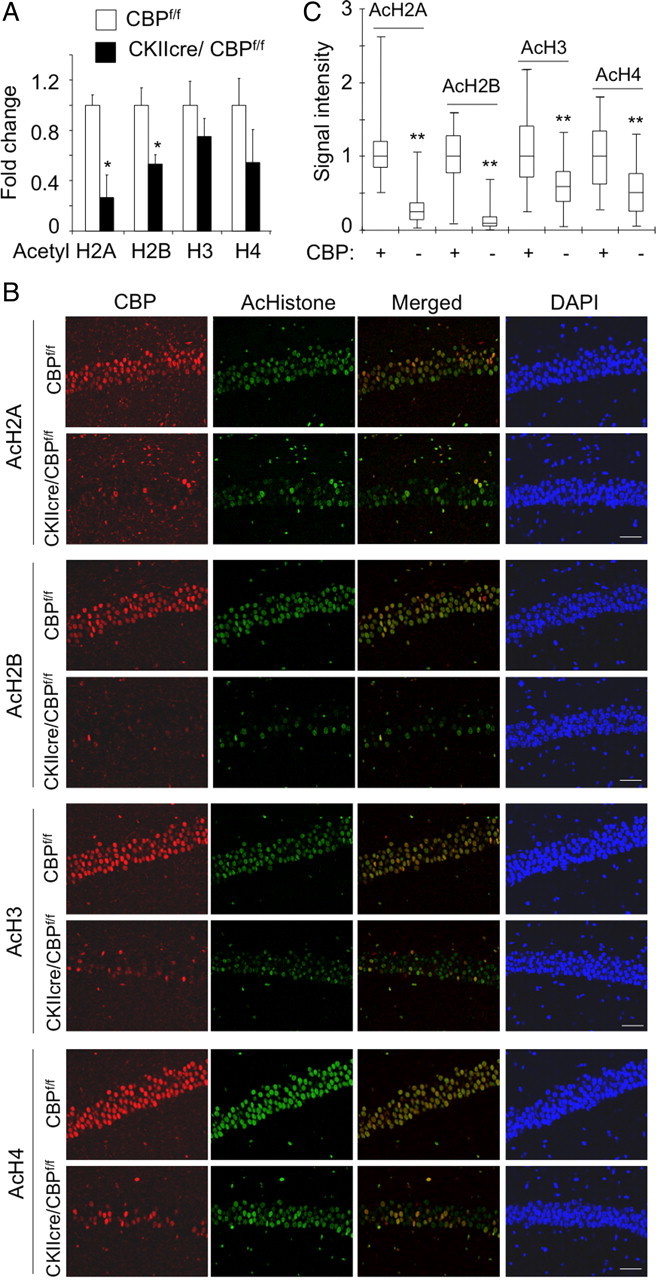
Strong reduction of histone acetylation in CKIIcre/CBPf/f mice. A, Quantification of the results of Western blots of CA1 subregion protein extracts from CKIIcre/CBPf/f mice and control littermates using α-β-actin, α-H2B and α-H3 (non-acetylated), α-AcH2B, α-AcH2A, α-AcH3, and α-AcH4 antibodies (n = 6 for CBPf/f and n = 5 for CKIIcre/CBPf/f). Data are expressed as mean ± SEM. See representative blot results in Figure 3. We included samples from mice injected with NaBu or vehicle (PBS) as a control (results not shown). B, Representative images of the cellular layer in the CA1 subfield of CKIIcre/CBPf/f mice and control littermates. Sagittal brain sections were immunostained with antibodies against the acetylated forms of histones H2A, H2B, H3, and H4 (green fluorescence) and against CBP (red fluorescence). Mutant mice showed important reduction in AcH2A and AcH2B followed by AcH3 and AcH4. Note the single-cell correlation between levels of CBP and acetylation. Scale bar, 50 μm. C, The quantification of immunohistochemistry signal in individual neurons revealed that in the CA1 subfield of CKIIcre/CBPf/f mice the CBP+ cells maintain higher levels of histone acetylation than CBP− cells. The difference is especially dramatic in the case of AcH2B and AcH2A. *p < 0.01; **p < 0.001.
Figure 3.

CBP-dependent histone acetylation deficits can be reversed with sodium butyrate. A, Representative result of Western blot analyses of CA1 subfield protein extracts from CKIIcre/CBPf/f mice and control littermates injected with sodium butyrate (NB) or PBS using α-β-actin, α-H2B and α-H3 (non-acetylated), α-AcH2B, α-AcH2A, α-AcH3, and α-AcH4 antibodies. B, Quantification of the Western blots against AcH2B and AcH3 in hippocampal extracts from CBPf/f and CKIIcre/CBPf/f mice treated with vehicle (V, solid bars) or sodium butyrate (NB, hatched bars). Data are expressed as mean ± SEM. n = 4 for both genotypes. *p < 0.05; **p < 0.01; #p < 0.01 in the comparison between genotypes. C, Representative images of the cellular layer in the CA1 subfield of CKIIcre/CBPf/f mice and control littermates treated with NaBu or PBS and stained with α-AcH2B. Scale bar, 50 μm.
To confirm the apparent preference of KAT activity by CBP for histones H2A and H2B versus H3 and H4, we overexpressed CBP in HEK293 cells and performed Western blots for each of the four acetylated histones. In agreement with our observations in knock-out mice, the overexpression of CBP affected more the acetylation state of histones H2B and H2A than that of H3 and H4 (Fig. 4A,B). In the case of H2B acetylation, we also explored the specificity for the different lysine (K) residues in the N-terminus tail. CBP did not show a clear specificity and caused the hyperacetylation of K5, K12, K15, and K20 (Fig. 4C,D). Interestingly, although the paralogous KAT p300 exhibited the same specificity in these in vitro assays, it cannot compensate the loss of CBP in neurons in vivo.
Figure 4.

Substrate specificity of CBP. A, Western blot analysis of acetylation for the four nucleosome histones in extracts of HEK293 cells transfected either with a plasmid encoding CBP (C) or p300 (P) or with an empty vector (E). Hyperacetylation was only observed in AcH2A and AcH2B when cotransfected with the plasmid pTet–Off but not with an unrelated vector (pGFP) that not expressed the required tTA to achieve maximum levels of KAT protein expression. CBP and p300 showed similar substrate specificity. B, Quantification of Western blot results in A. C, Substrate specificity of CBP in H2B acetylation. Western blot analysis of acetylation for the four lysine residues at the N-terminal tail of histone H2B in extract of HEK293 cells transfected either with a plasmid encoding CBP or p300 or with an empty vector. D, Quantification of the data in C. Data are expressed as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Overall, these experiments identify CBP as the main KAT activity responsible for the steady-state level of acetylation of the H2A/H2B dimers but also suggest that there are other KAT activities present in the cell that have these histones as substrate when HDACis disrupt the acetylation/deacetylation balance.
The elimination of CBP and ensuing histone hypoacetylation is not critical for neuronal viability
The reduction of CBP activity and histone acetylation has been proposed to have a causal role in neurodegenerative diseases; therefore, we explored in CKIIcre/CBPf/f mice whether the neuronal elimination of CBP was associated with neuronal death in the adult brain.
We did not observe any reduction in brain weight (values in 8-month-old mice: CBPf/f, 0.37 ± 0.01 g; CKIIcre/CBPf/f, 0.39 ± 0.01 g; n = 4 for both genotypes) nor apparent loss of neurons in the brain areas showing extensive ablation of CBP neither at the time of the analyses presented above (2- to 3-month-old mice, 4′,6′-diamidino-2-phenylindole (DAPI) panels in Fig. 2 and data not shown) nor 5 months later (8-month-old mice in Fig. 5A). Thus, the thickness of the cell layers in the CA1 and DG subfields were similar in mutant and control mice (Fig. 5B), and both the relative thickness of the cortical layers and the number of cortical neurons were maintained in CKIIcre/CBPf/f mice despite the strong differences in histone acetylation and CBP expression (Fig. 5C and results not shown). Moreover, Neurosilver staining did not reveal cell death or tissue damage (Fig. 5D). The immunostaining with anti-GFAP and anti-NeuN antibodies to detect astrogliosis and the loss of neurons, respectively, did not reveal any difference between the two genotypes (Fig. 5E) [NeuN-positive (NeuN+) cells/mm2: CBPf/f, 1130.14 ± 50.11; CKIIcre/CBPf/f, 1148.97 ± 48.06; GFAP+ cells/mm2: CBPf/f, 452.05 ± 21.41; CKIIcre/CBPf/f, 466.61 ± 37.63; n = 4 for both genotypes]. Immunostaining for the dendritic marker MAP2 and the synaptic marker synaptophysin (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) also supported the anatomical integrity of the hippocampus in CKIIcre/CBPf/f mice.
Figure 5.

Loss of CBP does not cause neuronal death. A, Nissl staining of brain sagittal sections of an 8-month-old CKIIcre/CBPf/f mouse and a control (CBPf/f) littermate did not show gross abnormalities. Scale bar, 150 μm. B, The thickness of the CA1 and DG (upper and lower blades) were not significantly reduced in CKIIcre/CBPf/f mice (n = 4 per genotype). C, The thickness of cortical layers and the number of cortical neurons (data not shown) were also not affected (n = 4 per genotype). Up, Cortical upper layers (II–III); Low, cortical lower layers (IV–VI). PtA cortex, Parietal associative cortex. D, Neurosilver staining in brain slices from CKIIcre/CBPf/f and control mice did not reveal any overt difference and dark cells (i.e., degenerating neuron) were not detected in either genotype. Scale bar, 50 μm. E, Representative images of the cellular layer in the CA1 subfield of CKIIcre/CBPf/f and control mice in slices immunostained with markers for mature neurons (NeuN) or glial cells (GFAP) and counterstained with DAPI (not shown). Scale bar, 50 μm. F, qRT-PCR quantification of p300 mRNA in the hippocampus (Hpc) and cortex of CKIIcre/CBPf/f mice and control littermates (n = 4 per genotype). Similar results were obtained with microdissected CA1 subfield samples (data not shown).
As mentioned previously, neurons contain a second KAT enzyme highly homologous to CBP, p300, which has been also associated with RSTS and has similar substrate preferences (Fig. 4). We tested whether CBP loss caused a compensatory upregulation of p300, which might prevent neuronal death. qRT-PCR assay for p300 transcripts did not reveal significant differences between CKIIcre/CBPf/f and control littermates, indicating that the overexpression of p300 does not compensate for the reduction of CBP (Fig. 5F).
Expression and novelty induction of IEGs in CBP-lacking hippocampal neurons
Both functions of CBP, as KAT and as transcriptional coactivator, have been linked to activity-driven gene expression. We therefore investigated whether CBP loss blocked the hippocampal induction of IEGs in response to novelty, a process likely required for the consolidation of hippocampal-dependent memory (Lisman and Grace, 2005; Moncada and Viola, 2007). To address this question, we analyzed the induction of seven representative IEGs [fos, npas4, arc, dusp1, egr1 (also known as zif268), nr4a1 (also known as nur77), and fosB] in response to 1 h of novelty exposure (NE) (Sheng et al., 1990; Patterson et al., 1992). qRT-PCR analyses of hippocampal samples showed a clear induction for all these genes in response to NE but did not reveal any change depending on genotype (Fig. 6A). Because this result was unexpected and there was a trend indicating a possible reduction in transcript level for some of these genes, we repeated the experiment with an independent group of mice in which we microdissected the CA1 subfield, a region in which CBP depletion was particularly extensive. qRT-PCR analyses of CA1 samples revealed a more clear trend toward a reduction of the induction of some of these IEGs in CKIIcre/CBPf/f mice (Fig. 6B). However, the two-way ANOVA of CA1 qRT-PCR data did not reveal significant interaction between genotype and condition (home cage vs novelty) for the expression of any of these IEGs, although the expression of egr1, dusp1, and nr4a1 showed a significant genotype effect (Table 2). The multivariate ANOVA (MANOVA) of transcript levels for this group of genes did not show significant genotype effect or interaction with condition (Table 2). Overall, these analyses demonstrate that the expression of some of IEGs is slightly reduced in the hippocampus of CKIIcre/CBPf/f mice, but the ability for induction in response to novelty is still essentially preserved in the absence of CBP.
Figure 6.

Induction of IEGs by novelty in CKIIcre/CBPf/f mice. A, qRT-PCR of indicated IEGs in hippocampal samples from CKIIcre/CBPf/f and control siblings after exploration of a novel environment for 1 h (NE) or kept in their home cages (HC) (n = 4, for all conditions). B, Quantitative RT-PCR of the same IEGs in microdissected CA1 subregions of CKIIcre/CBPf/f mice and control siblings after exploration of a novel environment for 1 h (n values: CBPf/f HC, 6; CBPf/f NE, 7; CKIIcre/CBPf/f HC, 5; and CKIIcre/CBPf/f NE, 7). *p < 0.05 (two-way ANOVA genotype effect; for additional details, see Table 2). C, c-fos immunostaining of the CA1 subfield did not reveal changes in c-fos induction between CKIIcre/CBPf/f mice and control siblings (2–3 mice per group). Scale bar, 25 μm.
Table 2.
Summary of two-way ANOVA and MANOVA of qRT-PCR data for IEG expression in microdissected CA1 subregions
| Two-way ANOVA |
p value |
||
|---|---|---|---|
| Condition | Genotype | Condition × genotype | |
| arc | <0.001 | 0.21 | 0.26 |
| fos | <0.001 | 0.19 | 0.23 |
| npas4 | <0.001 | 0.15 | 0.53 |
| egr1 | <0.001 | 0.003 | 0.21 |
| nr4a1 | <0.001 | 0.007 | 0.25 |
| fosB | <0.001 | 0.19 | 0.29 |
| dusp1 | <0.001 | 0.027 | 0.45 |
| MANOVA | <0.001 | 0.25 | 0.97 |
Significant (p < 0.05) effects are indicated in bold.
To explore further this lack of effect, we examined in greater detail the expression of c-fos, an IEG previously shown to depend on CBP function (Korzus et al., 2004) but whose induction was unaffected in CKIIcre/CBPf/f mice, using immunohistological techniques. As reported previously, c-fos induction on novelty was detected in a large number of CA1 pyramidal neurons and in a few scattered granule cells of the dentate gyrus (Hess et al., 1995; Wallace et al., 1995; Chawla et al., 2005) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Again, we took advantage of the scattered pattern of CBP loss and concurrent reduction of histone acetylation to compare the induction of IEG in CBP-positive and CBP-negative CA1 pyramidal neurons of a given animal. The double immunostaining of brain sections with antibodies against c-fos and CBP revealed that a similar number of cells responded to the novelty paradigm in CKIIcre/CBPf/f mice and control littermates (percentage c-fos+ cells: CBPf/f, 33.54 ± 4.15; CKIIcre/CBPf/f, 30.54 ± 3.47). Moreover, c-fos induction was observed in both CBP-positive and CBP-negative cells in the CA1 subfield of CKIIcre/CBPf/f mice [percentage c-fos+ cells: among CBP+ cells, 19.35 ± 7.60; among CBP-negative (CBP−) cells, 32.41 ± 5.32]. No correlation, either positive or negative, was found between the novelty-triggered induction of c-fos expression and CBP-dependent histone acetylation in individual pyramidal cells at the CA1 subregion (Pearson's correlation coefficient in CKIIcre/CBPf/f mice = −0.02).
Behavioral deficits in CKIIcre/CBPf/f mice
Previous behavioral studies in several CBP-deficient strains (heterozygous or transgenic) have demonstrated that the reduction of CBP function causes long-term memory deficits and other behavioral impairments (Barco, 2007). However, these studies have left some important open questions such as the identification of the anatomical substrate for these deficits and the distinction between developmental and adult functions of CBP. We therefore investigated the behavioral phenotype of CKIIcre/CBPf/f mice to gain insight into these issues.
We first subjected CKIIcre/CBPf/f mice to a comprehensive battery of basal neurological tests, the SHIRPA modified screen (Viosca et al., 2009, 2010). No differences were observed in basic reflexes, strength, muscle tone, and other parameters evaluated in this primary screen except for the slight increase in body length commented above (Table 1). Next, we examined the behavior of CKIIcre/CBPf/f mice in an open field, a test for anxiety and basic locomotor and exploratory behavior. Ambulatory distance in a 30 min test (CBPf/f, 59.63 ± 3.49 m; CKIIcre/CBPf/f, 64.72 ± 4.00 m; t(17) = 0.95; p = 0.36), percentage of distance in the center of the arena (CBPf/f, 6.05 ± 0.75; CKIIcre/CBPf/f, 5.92 ± 0.88; t(17) = 0.10; p = 0.92), which is a common measure of anxiety, and other parameters such as stereotypic time, resting time, and vertical activity were similar for both genotypes. We exposed the animal to the same box in three additional sessions. This caused habituation in wild types, and the total traveled distance decreased between sessions (Fig. 7A) (CBPf/f, F(2,16)Day = 6.61; p = 0.01) but not in CKIIcre/CBPf/f mice (Fig. 7A) (CKIIcre/CBPf/f, F(1.1,10.5)Day = 3.62; p = 0.08). Locomotor activity was also assessed using the automatic system Phenotyper for a period of 72 h (Fig. 7B). Both CKIIcre/CBPf/f mice and their control littermates exhibited more activity during the dark phase of the light cycle (t test dependent samples, t(9) = 7.94, p = 0.00 and t(8) = 10.97, p = 0.00, respectively). CKIIcre/CBPf/f mice showed more diurnal activity than their control siblings (F(1,17)genotype = 31.01; p < 0.001). We also studied these mice in the elevated plus maze, a task in which mice face a conflict between their tendency to explore new environments and their innate aversion to open spaces. Both CKIIcre/CBPf/f and control mice showed a similar number of entries and spent the same amount of time in the open arms (Fig. 7C) (t(17) = 0.28; p = 0.78; results not shown). Together with the observations in the open field, these parameters indicate normal levels of anxiety.
Figure 7.

Locomotor activity, anxiety, learning, and memory in CKIIcre/CBPf/f mice. A, CKIIcre/CBPf/f mice initially behave as control mice in an open field (see Results), but a modest difference in locomotor habituation was observed when the mice were exposed to the same arena during 3 consecutive days (CBPf/f, n = 9 and CKIIcre/CBPf/f, n = 10). B, CKIIcre/CBPf/f mice showed more activity in the Phenotyper boxes during the light phase of the day. C, CKIIcre/CBPf/f mice showed normal anxiety as measured in the elevated plus maze. OA, Open arm; CA, closed arm. D, CKIIcre/CBPf/f mice showed impaired memory for object recognition when tested 24 h after training. E, No significant differences between genotypes were observed during training in both the visible platform (V1–V3) and the hidden platform tasks (H1–H8). F, Spatial memory was assessed in two probe trials (P1 and P2), and no significant difference was observed in the percentage of time spent in the target quadrant (TQ) or in the number of annulus crossings. G, Normal freezing in contextual and cued conditioning 24 h and 1 week after training (CBPf/f, n = 9 and CKIIcre/CBPf/f, n = 10). PS, Preshock; Cx, context test; PT, pretone; Tn, tone test.
We examined next the animals' performance in an accelerated paradigm for the rotarod task, a motor coordination and learning task in which cbp+/− mice were impaired (Alarcón et al., 2004). Interestingly, we did not observe any impairment in CKIIcre/CBPf/f mice (latency to fall: CBPf/f, 275.5 ± 11.1 s; CKIIcre/CBPf/f, 275.3 ± 17.4 s; t test, t(17) = −0.57; p = 0.99). Also, we found no difference between the two genotypes in the hotplate test, a test to measure nociception and sensitivity to thermal stimulus (Table 1).
Next, we examined the performance of CKIIcre/CBPf/f mice in a novel object recognition memory task, a non-aversive memory task that relies on the natural exploratory behavior of mice and that has consistently revealed memory deficits in all the CBP-deficient strains analyzed so far (Barco, 2007). As expected, control mice spent more time exploring the novel object during testing (Fig. 7D) (CBPf/f vs 0, t(8) = 5.05; p = 0.001). In contrast, CKIIcre/CBPf/f mutants spent similar time exploring the novel and the old object (Fig. 7G) (CKIIcre/CBPf/f vs 0, t(9) = 1.25; p = 0.24), indicating that, like other CBP-deficient strains previously analyzed, CKIIcre/CBPf/f mutants have impaired long-term recognition memory.
To continue with the analysis of cognitive abilities, we examined the performance of CKIIcre/CBPf/f mice in spatial navigation using the MWM. In agreement with preceding studies on cbp+/− mice, we did not detect significant differences between groups in neither the visible nor the hidden platform tasks. The path lengths and the latencies to find the platform declined with similar slope over training in mutant and control littermates (Fig. 7E and results not shown). The probe trials performed on days 5 and 8 did not reveal any statistical difference in the performance associated with the genotype (Fig. 7F). In agreement with our observations in the rotarod supporting normal motor performance, we found that, conversely to heterozygous mice (Alarcón et al., 2004), these mutants did not swim slower than their control littermates (F(1,17)genotype = 0.73, p = 0.40; F(7,119)genotype × session = 0.30, p = 0.95).
Finally, we assessed CKIIcre/CBPf/f mice in two fear conditioning tasks that measure the capability of the mouse to form associations between an aversive stimulus (mild footshock) and neutral environmental cues, such as the spatial context in the case of contextual fear conditioning, and a tone delivered simultaneously to the shock in the case of the cued fear conditioning. Conversely to our previous observations in cbp+/− mice, but in agreement with previous findings in transgenic mice expressing a truncated CBP variant driven by the CaMKIIα promoter (Korzus et al., 2004), CKIIcre/CBPf/f mutants showed the same level of freezing as control mice in both tasks (Fig. 7G).
Discussion
The role of epigenetic mechanisms of regulation of gene expression in cognition has attracted great interest in recent years (Hsieh and Gage, 2005; Levenson and Sweatt, 2005; Tsankova et al., 2007; Borrelli et al., 2008; Zocchi and Sassone-Corsi, 2010). Our study represents an important contribution to this rapidly progressing field because the analysis of brain-specific crebbp knock-outs did not only provide novel insight into RSTS etiology, it also clarified important open questions concerning the substrate preferences of CBP and the role of CBP and histone acetylation in neurodegeneration, activity-driven gene expression, and memory formation.
CBP loss and reduced histone acetylation do not interfere with neuronal viability: implications for neurodegenerative diseases
A number of studies have shown that the pathogenesis of Alzheimer's disease (AD) and Huntington disease (HD) is accompanied by the selective downregulation of genes involved in neuroplasticity and suggested that transcriptional deregulation may represent an early mechanism of pathology in both diseases (T. Shimohata et al., 2000a,b; M. Shimohata et al., 2005; Obrietan and Hoyt, 2004; Fischer et al., 2007; Palop et al., 2007). Moreover, in the case of HD, the deregulation of gene transcription has been specifically linked to the sequestration of CBP in huntingtin aggregates (McCampbell et al., 2000; Nucifora et al., 2001; Steffan et al., 2001). Independent studies have shown that the administration of HDACis attenuates neuronal loss in animal models for HD (Steffan et al., 2001; Ferrante et al., 2003; Hockly et al., 2003), AD (Fischer et al., 2007), and other neurodegenerative diseases (Minamiyama et al., 2004; Mai et al., 2009). Based on both lines of evidence, it has been proposed that epigenetic changes in the chromatin play a role in neurodegeneration and that targeting the balance between histone acetylation and deacetylation may hold great potential for neuroprotection and neurorestoration (Fischer et al., 2007; Chuang et al., 2009; Mai et al., 2009). The evidence presented here indicates that the relationship between histone acetylation and neurodegeneration is more complex than expected. CKIIcre/CBPf/f mice exhibit a dramatic reduction of histone acetylation, particularly of histones H2A and H2B, and this does not seem to affect neuronal viability, indicating that sustained low levels of histone acetylation do not necessarily cause neuronal death. In agreement with this view, no neurodegenerative process has been ever reported in RSTS patients or in animal models for this condition (Wiley et al., 2003; Barco, 2007).
How to explain the apparent discrepancy between the absence of neurodegeneration in mice lacking CBP and the beneficial effects of HDACi? Maybe promoting histone acetylation is beneficial under cellular stress conditions but does not affect neuronal viability in normal situations. Alternatively, the biologically relevant targets of these pathological conditions may be H3/H4 and cellular KAT activities other than CBP, whereas the reduction of the acetylation of H2A and H2B are mere side effects. However, the fact that H3 and H4 acetylation is also reduced in CKIIcre/CBPf/f mice argues against this possibility. One might also consider that the requirement for CBP is not cell autonomous, and CBP-positive neurons adjacent to CBP-negative neurons might prevent their death through, for example, the release of neurotrophic factors. It is also possible that >8 months are needed to see the deleterious consequences of CBP elimination because AD and HD are two slowly progressing diseases. Finally, our results also open the possibility that the beneficial effects of HDACi are not related to histone acetylation but to other non-histone substrates of HDAC enzymes, such as transcription factors or cytoskeleton proteins. Nevertheless, the lysine acetylome consists on hundreds of substrates involved in numerous functions (Choudhary et al., 2009).
CBP-dependent histone acetylation and activity-driven gene expression
Studies in cbp+/− mice have demonstrated that CBP deficiency causes the reduction of histone H2B acetylation (Alarcón et al., 2004). This seminal finding was rather surprising, because biochemical assays and studies in cultured cells had identified histone H2B as a substrate for the KAT activity of CBP but did not reveal this level of specificity (McManus and Hendzel, 2003; Kouzarides, 2007). The experiments presented here extend the analysis of the substrate specificity of CBP in vivo in the CNS. Our results suggest that the balance of KAT and HDAC activities in hippocampal neurons makes the acetylation of the N-terminal tail of histones H2A and H2B, especially the second one, more sensitive to changes in CBP activity than those of histone H3 and H4. Our experiments with NaBu also indicate that, although the KAT activity of CBP seems to be primarily responsible for the basal acetylation state of H2A and H2B, other KAT activities with this specificity, such as p300, become active when the steady-state acetylation/deacetylation balance is disrupted by HDACis.
The pair H2A/H2B forms dimers, whereas H3/H4 forms tetramers. Both structures have different affinities for the DNA and play different roles on chromatin structure and transcription (Kouzarides, 2007). Although the role of H2A and H2B acetylation in transcription has been, in general, less studied than for H3 and H4, the acetylation of H2A/H2B dimers is thought to facilitate their release from the chromatin (Ito et al., 2000). The presence of acetylated H2B seems to be a feature of only the most active genes and has been associated with the maintenance of the overall transcriptional competence of specific loci (Myers et al., 2003). In fact, a recent study identified AcH2B(K5) as one of the posttranslational histone posttranslational modification more strongly associated with gene expression levels (Karlić et al., 2010). Our results seem to indicate that this may be a consequence rather than a cause of the high transcriptional activity and suggest that, although the acetylation of H2A/H2B may enhance transcription, it is not strictly necessary for activity-driven gene induction. Our results also indicate that CBP is not an essential coactivator for the induction of the IEGs analyzed in this work, despite the fact that the promoters of these genes contain the DNA binding motifs for the binding of activity-regulated transcription factors known to interact with CBP. This conclusion challenges the proposed, but still unconfirmed, critical role of CBP and activity-driven histone acetylation in memory-related transcription, but it is in good agreement with previous studies in cbp+/− mice after kainate-induced seizures (Alarcón et al., 2004) and with recent observations in primary mouse embryonic fibroblasts, demonstrating that the elimination of both CBP and p300 does not prevent the induction of IEGs by forskolin (Kasper et al., 2010).
Novel insights into RSTS mental impairment: anatomical substrates and dissociation of developmental and postnatal deficits
The behavioral analysis of CKIIcre/CBPf/f mice has revealed interesting deficits, such as slightly decreased locomotor habituation and object recognition memory impairment. However, these deficits were more modest than expected considering the early lethality of the conventional knock-out mice and the importance of the functions allocated to this protein. A factor to consider in the interpretation of cognitive deficits in CKIIcre/CBPf/f mice (or the lack of them) is the mosaic pattern of CBP ablation. Although this feature has been very useful in our correlative studies on individual cells, it represents a caveat for behavioral experiments because we cannot discard that the remaining CBP+ cells in hippocampus, amygdala, or other structures are sufficient to support the assessed forms of memory.
Important information can be extracted nevertheless from the comparison between CBP-deficient strains (see comparison of previous RSTS models by Josselyn, 2005; Barco, 2007). For example, some noncognitive impairments, such as the reduced swimming speed or the poor rotarod performance observed in heterozygous mice but absent in CKIIcre/CBPf/f mice, are likely developmental or caused by CBP deficiency in cell types not targeted in the forebrain-specific knock-outs, such as the Purkinje of granular cells in the cerebellum. Also, we confirmed the particular sensitivity of object recognition memory to the reduction of CBP function and identified the hippocampus and cortical areas as the most likely anatomical substrate for this cognitive impairment. The impaired expression of the activity-regulated genes egr1, nr4a1, and dusp1 in the hippocampus of CKIIcre/CBPf/f mice might contribute to this memory impairment because slight changes of nr4a1 induction have been associated to the memory-enhancing effects of the HDACi trichostatin A (Vecsey et al., 2007). Spatial navigation and fear conditioning tasks have produced less consistent results in RSTS mouse models. Both tasks did not show any impairment in forebrain-restricted knock-outs, indicating either that the small remaining population of CBP-positive neurons is sufficient to sustain these memories or that CBP function in the postnatal brain is not critical for these forms of learning, which is in agreement with previous observations in other forebrain-specific CBP-deficient strains (Korzus et al., 2004). Interestingly, it has been shown recently that acetylation of K12 in histone H4 may mediate gene induction in the contextual fear conditioning paradigm (Peleg et al., 2010). Although the main KAT activity underlying this posttranslational modification has not been yet identified, it is plausible that specific KAT activities are required for different learning tasks and memory processes in different brain areas. Additional studies are necessary to determine the still obscure function of histone acetylation in cognition and mental retardation.
Footnotes
Research at the laboratory of A.B. is supported by Spanish Ministry of Science and Innovation Grants BFU2008-00611, CSD2007-00023, and SAF2008-03194-E (part of the coordinated ERA-Net NEURON Project Epitherapy) and grants from Fundación Ramón Areces and Fundació La Marató de TV3 063510. L.M.V. has a Ramón y Cajal contract given by the Spanish Ministry of Science and Innovation. We thank Eva Benito, Eloisa Herrera, and José López-Atalaya for critical reading of this manuscript and helpful advice. We also thank Raffaela Tulino for technical assistance.
The authors declare no competing financial interests.
References
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/− mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004;42:947–959. doi: 10.1016/j.neuron.2004.05.021. [DOI] [PubMed] [Google Scholar]
- Allis CD, Berger SL, Cote J, Dent S, Jenuwien T, Kouzarides T, Pillus L, Reinberg D, Shi Y, Shiekhattar R, Shilatifard A, Workman J, Zhang Y. New nomenclature for chromatin-modifying enzymes. Cell. 2007;131:633–636. doi: 10.1016/j.cell.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Short protocols in molecular biology. Ed 4. New York: Wiley; 1999. [Google Scholar]
- Barco A. The Rubinstein-Taybi syndrome: modeling mental impairment in the mouse. Genes Brain Behav. 2007;6(Suppl 1):32–39. doi: 10.1111/j.1601-183X.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchouladze R, Lidge R, Catapano R, Stanley J, Gossweiler S, Romashko D, Scott R, Tully T. A mouse model of Rubinstein-Taybi syndrome: defective long-term memory is ameliorated by inhibitors of phosphodiesterase 4. Proc Natl Acad Sci U S A. 2003;100:10518–10522. doi: 10.1073/pnas.1834280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–2373. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- Chawla MK, Guzowski JF, Ramirez-Amaya V, Lipa P, Hoffman KL, Marriott LK, Worley PF, McNaughton BL, Barnes CA. Sparse, environmentally selective expression of Arc RNA in the upper blade of the rodent fascia dentata by brief spatial experience. Hippocampus. 2005;15:579–586. doi: 10.1002/hipo.20091. [DOI] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447:178–182. doi: 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Gotts EE, Liemohn WP. Behavioral characteristics of three children with the broad thumb-hallux (Rubinstein-Taybi) syndrome. Biol Psychiatry. 1977;12:413–423. [PubMed] [Google Scholar]
- Hennekam RC, Baselier AC, Beyaert E, Bos A, Blok JB, Jansma HB, Thorbecke-Nilsen VV, Veerman H. Psychological and speech studies in Rubinstein-Taybi syndrome. Am J Ment Retard. 1992;96:645–660. [PubMed] [Google Scholar]
- Hess US, Lynch G, Gall CM. Regional patterns of c-fos mRNA expression in rat hippocampus following exploration of a novel environment versus performance of a well-learned discrimination. J Neurosci. 1995;15:7796–7809. doi: 10.1523/JNEUROSCI.15-12-07796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, Gage FH. Chromatin remodeling in neural development and plasticity. Curr Opin Cell Biol. 2005;17:664–671. doi: 10.1016/j.ceb.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Ito T, Ikehara T, Nakagawa T, Kraus WL, Muramatsu M. p300-mediated acetylation facilitates the transfer of histone H2A-H2B dimers from nucleosomes to a histone chaperone. Genes Dev. 2000;14:1899–1907. [PMC free article] [PubMed] [Google Scholar]
- Josselyn SA. What's right with my mouse model? New insights into the molecular and cellular basis of cognition from mouse models of Rubinstein-Taybi Syndrome. Learn Mem. 2005;12:80–83. doi: 10.1101/lm.93505. [DOI] [PubMed] [Google Scholar]
- Karlić R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 2010;107:2926–2931. doi: 10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper LH, Lerach S, Wang J, Wu S, Jeevan T, Brindle PK. CBP/p300 double null cells reveal effect of coactivator level and diversity on CREB transactivation. EMBO J. 2010;29:3660–3672. doi: 10.1038/emboj.2010.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron. 2004;42:961–972. doi: 10.1016/j.neuron.2004.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Levenson JM, Sweatt JD. Epigenetic mechanisms in memory formation. Nat Rev Neurosci. 2005;6:108–118. doi: 10.1038/nrn1604. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- López de Armentia M, Jancic D, Olivares R, Alarcón JM, Kandel ER, Barco A. cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J Neurosci. 2007;27:13909–13918. doi: 10.1523/JNEUROSCI.3850-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai A, Rotili D, Valente S, Kazantsev AG. Histone deacetylase inhibitors and neurodegenerative disorders: holding the promise. Curr Pharm Des. 2009;15:3940–3957. doi: 10.2174/138161209789649349. [DOI] [PubMed] [Google Scholar]
- Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH. CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet. 2000;9:2197–2202. doi: 10.1093/hmg/9.14.2197. [DOI] [PubMed] [Google Scholar]
- McManus KJ, Hendzel MJ. Quantitative analysis of CBP- and P300-induced histone acetylations in vivo using native chromatin. Mol Cell Biol. 2003;23:7611–7627. doi: 10.1128/MCB.23.21.7611-7627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Sobue G. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2004;13:1183–1192. doi: 10.1093/hmg/ddh131. [DOI] [PubMed] [Google Scholar]
- Moncada D, Viola H. Induction of long-term memory by exposure to novelty requires protein synthesis: evidence for a behavioral tagging. J Neurosci. 2007;27:7476–7481. doi: 10.1523/JNEUROSCI.1083-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers FA, Chong W, Evans DR, Thorne AW, Crane-Robinson C. Acetylation of histone H2B mirrors that of H4 and H3 at the chicken beta-globin locus but not at housekeeping genes. J Biol Chem. 2003;278:36315–36322. doi: 10.1074/jbc.M305822200. [DOI] [PubMed] [Google Scholar]
- Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Hoyt KR. CRE-mediated transcription is increased in Huntington's disease transgenic mice. J Neurosci. 2004;24:791–796. doi: 10.1523/JNEUROSCI.3493-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Grover LM, Schwartzkroin PA, Bothwell M. Neurotrophin expression in rat hippocampal slices: a stimulus paradigm inducing LTP in CA1 evokes increases in BDNF and NT-3 mRNAs. Neuron. 1992;9:1081–1088. doi: 10.1016/0896-6273(92)90067-n. [DOI] [PubMed] [Google Scholar]
- Peleg S, Sananbenesi F, Zovoilis A, Burkhardt S, Bahari-Javan S, Agis-Balboa RC, Cota P, Wittnam JL, Gogol-Doering A, Opitz L, Salinas-Riester G, Dettenhofer M, Kang H, Farinelli L, Chen W, Fischer A. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 2010;328:753–756. doi: 10.1126/science.1186088. [DOI] [PubMed] [Google Scholar]
- Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, Breuning MH. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- Roelfsema JH, White SJ, Ariyürek Y, Bartholdi D, Niedrist D, Papadia F, Bacino CA, den Dunnen JT, van Ommen GJ, Breuning MH, Hennekam RC, Peters DJ. Genetic heterogeneity in Rubinstein-Taybi syndrome: mutations in both the CBP and EP300 genes cause disease. Am J Hum Genet. 2005;76:572–580. doi: 10.1086/429130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006;13:539–550. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchís-Segura C, López-Atalaya JP, Barco A. Selective boosting of transcriptional and behavioral responses to drugs of abuse by histone deacetylase inhibition. Neuropsychopharmacology. 2009;34:2642–2654. doi: 10.1038/npp.2009.125. [DOI] [PubMed] [Google Scholar]
- Sheng M, McFadden G, Greenberg ME. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron. 1990;4:571–582. doi: 10.1016/0896-6273(90)90115-v. [DOI] [PubMed] [Google Scholar]
- Shimohata M, Shimohata T, Igarashi S, Naruse S, Tsuji S. Interference of CREB-dependent transcriptional activation by expanded polyglutamine stretches: augmentation of transcriptional activation as a potential therapeutic strategy for polyglutamine diseases. J Neurochem. 2005;93:654–663. doi: 10.1111/j.1471-4159.2005.03060.x. [DOI] [PubMed] [Google Scholar]
- Shimohata T, Onodera O, Tsuji S. Interaction of expanded polyglutamine stretches with nuclear transcription factors leads to aberrant transcriptional regulation in polyglutamine diseases. Neuropathology. 2000a;20:326–333. doi: 10.1046/j.1440-1789.2000.00350.x. [DOI] [PubMed] [Google Scholar]
- Shimohata T, Nakajima T, Yamada M, Uchida C, Onodera O, Naruse S, Kimura T, Koide R, Nozaki K, Sano Y, Ishiguro H, Sakoe K, Ooshima T, Sato A, Ikeuchi T, Oyake M, Sato T, Aoyagi Y, Hozumi I, Nagatsu T, Takiyama Y, Nishizawa M, Goto J, Kanazawa I, Davidson I, Tanese N, Takahashi H, Tsuji S. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat Genet. 2000b;26:29–36. doi: 10.1038/79139. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Naruse I, Hongo T, Xu M, Nakahata T, Maekawa T, Ishii S. Extensive brain hemorrhage and embryonic lethality in a mouse null mutant of CREB-binding protein. Mech Dev. 2000;95:133–145. doi: 10.1016/s0925-4773(00)00360-9. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viosca J, Schuhmacher AJ, Guerra C, Barco A. Germline expression of H-Ras(G12V) causes neurological deficits associated to Costello syndrome. Genes Brain Behav. 2009;8:60–71. doi: 10.1111/j.1601-183X.2008.00443.x. [DOI] [PubMed] [Google Scholar]
- Viosca J, López-Atalaya JP, Olivares R, Eckner R, Barco A. Syndromic features and mild cognitive impairment in mice with genetic reduction on p300 activity: differential contribution of p300 and CBP to Rubinstein-Taybi syndrome etiology. Neurobiol Dis. 2010;37:186–194. doi: 10.1016/j.nbd.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Wallace CS, Withers GS, Weiler IJ, George JM, Clayton DF, Greenough WT. Correspondence between sites of NGFI-A induction and sites of morphological plasticity following exposure to environmental complexity. Brain Res Mol Brain Res. 1995;32:211–220. doi: 10.1016/0169-328x(95)00076-5. [DOI] [PubMed] [Google Scholar]
- Wiley S, Swayne S, Rubinstein JH, Lanphear NE, Stevens CA. Rubinstein-Taybi syndrome medical guidelines. Am J Med Genet A. 2003;119A:101–110. doi: 10.1002/ajmg.a.10009. [DOI] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Attner MA, Oliveira AM, Brindle PK, Abel T. A transcription factor-binding domain of the coactivator CBP is essential for long-term memory and the expression of specific target genes. Learn Mem. 2006;13:609–617. doi: 10.1101/lm.213906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Hofmann C, Casanova E, Schütz G, Lutz B. Generation of a conditional allele of the CBP gene in mouse. Genesis. 2004;40:82–89. doi: 10.1002/gene.20068. [DOI] [PubMed] [Google Scholar]
- Zocchi L, Sassone-Corsi P. Joining the dots: from chromatin remodeling to neuronal plasticity. Curr Opin Neurobiol. 2010;20:432–440. doi: 10.1016/j.conb.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]