

Abstract
In Kv1.1, single point mutants found below the channel activation gate at residue V408 are associated with human episodic ataxia type-1, and impair channel function by accelerating decay of outward current during periods of membrane depolarization and channel opening. This decay is usually attributed to C-type inactivation, but here we provide evidence that this is not the case. Using voltage-clamp fluorimetry in Xenopus oocytes, and single-channel patch clamp in mouse ltk− cells, of the homologous Shaker channel (with the equivalent mutation V478A), we have determined that the mutation may cause current decay through a local effect at the activation gate, by destabilizing channel opening. We demonstrate that the effect of the mutant is similar to that of trapped 4-aminopyridine in antagonizing channel opening, as the mutation and 10 mm 4-AP had similar, nonadditive effects on fluorescence recorded from the voltage-sensitive S4 helix. We propose a model where the Kv1.1 activation gate fails to enter a stabilized open conformation, from which the channel would normally C-type inactivate. Instead, the lower pore lining helix is able to enter an activated-not-open conformation during depolarization. These results provide an understanding of the molecular etiology underlying episodic ataxia type-1 due to V408A, as well as biophysical insights into the links between the potassium channel activation gate, the voltage sensor and the selectivity filter.
Introduction
Episodic ataxia type-1 (EA-1) is an inherited neurological movement disorder that produces continuous myokymia and episodes of uncontrolled contractions in skeletal muscle (VanDyke et al., 1975). EA-1 is caused by mutations in the potassium channel gene KCNA1, which encodes the Shaker-related channel Kv1.1 (Browne et al., 1994). One important mutation locus is at site V408, which is in the ion permeation pathway. When expressed in heterologous systems, V408A (Adelman et al., 1995; D'Adamo et al., 1998) and V408L (Demos et al., 2009) show accelerated decay of outward potassium current during prolonged depolarization of the membrane potential. However, the molecular mechanism by which mutations at this site lead to faster current decay is unclear.
Normally, the Kv1 channel pore is gated by two distinct mechanisms, found at opposite ends of the transmembrane conduit. The inner “activation” gate consists of a narrowing of the ion passage at a conserved Pro-Val-Pro bundle crossing sequence in the intracellular aspect of the S6 helix in the four channel subunits (Liu et al., 1997), one helical turn extracellular to V408. Activation gate movement is coupled to translocations of the channel's voltage sensing apparatus by interactions between the S4-S5 linker and S6 (Lu et al., 2002; Soler-Llavina et al., 2006). Depolarization of the membrane potential causes outward and rotational movements of the charged S4 membrane helix, allowing the bundle crossing to open to the flux of potassium ions (for review, see Bezanilla, 2000). The outer “inactivation” gate is contained within the channel's externally located selectivity filter, and is responsible for the high preference of the channel to conduct potassium (Doyle et al., 1998; Zhou et al., 2001b). Prolonged depolarizations cause a series of conformational rearrangements within the selectivity filter, hindering its ability to coordinate the conduction of potassium ions (Baukrowitz and Yellen, 1995), which leads to a characteristic decay in ionic currents. Although current decay normally is controlled mainly by outer pore inactivation in Kv1 channels, it remains unclear as to which of the above two gating mechanisms is being altered by V408A.
To address the question of how alanine substitution at V408 enhances current decay in Kv1.1, we used voltage-clamp fluorimetry, as well as single-channel patch-clamp electrophysiology, to monitor protein conformational rearrangements during membrane depolarization. Voltage-clamp fluorimetry is a technique capable of visualizing discrete structural reorientations within a protein while simultaneously recording ionic currents (Mannuzzu et al., 1996; Cha and Bezanilla, 1997). We used the Shaker potassium channel, the Drosophila melanogaster homolog of Kv1.1, as a model for Kv1 behavior, due to its functional similarity to the Kv1.1 channel, and the well established relationships of fluorescence signals from the Shaker A359C reporter site with conformational changes associated with both activation and inactivation processes (Claydon et al., 2007). Our findings rule out enhanced constriction of the outer pore by V408A and support a mechanism for V408A in which the activation gate of mutant channels can enter an “activated-not-open” conformation during depolarization, which accelerates the macroscopic rate of current decay during depolarization.
Materials and Methods
Construct cloning and oocyte preparation.
A Shaker IR clone (consisting of wild-type Shaker B with N-type inactivation gate residues 6–46 deleted) was a gift from Carol Deutsch (Jefferson Medical College, Philadelphia, PA), and the Kv1.1 clone was purchased from Origene. Both clones were excised from their original plasmids by PCR amplification and enzymatic treatment. Oligonucleotides complementary to the cDNA 5′ and 3′ ends, also possessing recognition sequences for the HindIII and EcoRI restriction enzymes, were designed and synthesized by Integrated DNA Technologies. PCR amplifications were then performed, using the Phusion PCR kit (New England Biolabs), as recommended by the manufacturer. Linear PCR products were then digested using HindIII and EcoRI enzymes (New England Biolabs), and ligated into a similarly digested pBluescriptII SK+ vector, from Fermentas using T4 ligase (New England Biolabs). To generate single site mutations, mutagenic primers containing modified sequences encoding the desired mutations and overlapping the site of interest were designed and synthesized at Integrated DNA Technologies. A two-step overlapping PCR protocol was performed using the Phusion system, to generate mutations, as described by the manufacturer. For RNA translation, constructs were linearized using SacII (New England Biolabs). RNA was generated using the T7 mMessage mMachine kit (Ambion), then purified using LiCl purification.
Stage V–VI oocytes were surgically excised from anesthetized female Xenopus laevis frogs, and maintained in oocyte Ringer's (OR2) solution, containing (in mm): 82.5 NaCl, 2.5 KCl, 5 HEPES, 1 MgCl2, and adjusted to pH 7.4 with NaOH (all chemicals were from Sigma-Aldrich, unless otherwise noted). Before injection, oocytes were digested in type I collagenase (Invitrogen) in OR2 to remove their follicular layers, then transferred to OR3 solution, consisting of Leibovitz's Medium (Invitrogen), 50% v/v in water, supplemented with 5 ml each of glutamine and gentamycin (Invitrogen), and 15 mm HEPES, and adjusted to pH 7.4 with NaOH. Oocytes were each injected with 5–25 ng of RNA and incubated in OR3 at 19°C. Recordings were performed 24–48 h postinjection. All procedures involving live animals conformed with the guidelines established by the Canadian Council for Animal Care and the University of British Columbia Committee on Animal Care.
Two-electrode voltage-clamp electrophysiology and fluorimetry.
Equipment and techniques for two-electrode voltage-clamp electrophysiology and voltage-clamp fluorimetry were as described by Claydon et al. (2007). ND96, our default recording solution, contained, in mm: 96 NaCl, 3 KCl, 10 HEPES, 1 MgCl2, 2 CaCl2, and was adjusted to pH 7.4 with NaOH. A modified version with elevated external potassium contained, in mm: 99 KCl, 10 HEPES, 1 MgCl2, 2 CaCl2, and was adjusted to pH 7.4 with NaOH. For 4-aminopyridine recordings, ND96 was supplemented with the indicated concentration of 4-AP. Recording pipettes contained 3 m KCl. Tetramethylrhodamine maleimide (TMRM) was used as a fluorophore, and was applied to oocytes immediately before recording, as described by Claydon et al. (2007).
Single-channel patch-clamp recordings.
Mouse ltk−cells were transiently transfected with 0.5 μg of Shaker IR or Shaker IR V478A DNA along with 1 μg of GFP DNA using LipofectAMINE 2000 (Invitrogen). All recordings were made at room temperature 24–48 h after transfection. Single-channel currents were recorded in the cell-attached patch configuration with an Axopatch 200B patch-clamp amplifier and pClamp8 software (Molecular Devices Inc.). Patch electrodes were fabricated using thin-walled borosilicate glass (World Precision Instruments) and coated with Sylgard (Dow Corning). Electrode resistances were between 10 and 25 MΩ. The single-channel currents were low-pass filtered at 2 kHz (−3 dB, 4-pole Bessel filter) and sampled at 10 kHz. No junction potential correction was done on data acquired in the cell-attached recording.
For single-channel patch-clamp recordings, the patch pipettes contained (in mm): 135 NaCl, 5 KCl, 10 HEPES, 1 MgCl2, 1 CaCl2, and was adjusted to pH 7.4 with NaOH. The bath solution contained (in mm): 135 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, and was adjusted to pH 7.4 with KOH.
Data analysis.
Single-channel records were analyzed with pClamp10 software (Molecular Devices Inc.) after digital filtering at 1 kHz unless stated otherwise. Given a combined analog and digital filter frequency of 894 Hz, the rise time (Tr) of the system was calculated to be 0.37 ms using the formula 0.3321/fc, where fc represents the overall combined analog and digital filter cutoff frequency in Hz (Colquhoun and Sakmann, 1985). Half-amplitude threshold analysis (Colquhoun and Sakmann, 1985) was used to detect events longer than 2 Tr and generate idealized records from which dwell time histograms and ensemble time courses were constructed. Histograms were plotted with a logarithmic time axis. Events were logarithmically binned and subject to square root transformation (Sigworth and Sine, 1987) and normalization for comparison. Open dwell time and burst duration histograms were fit with Gaussian distributions to yield two or three mean time constants, calculated using the method outlined by Colquhoun and Sakmann (1985). n = 3 for Shaker IR and Shaker IR V478A.
Off-line data analysis was performed using pClamp10 (Molecular Devices), Origin 8.0 (OriginLab Corporation), and GraphPad Prism 4.0. Boltzmann curves were fit using the equation: y = 1/(1 + e(V1/2 − V)/k), where y is the conductance or fluorescence amplitude normalized with respect to maximal conductance or fluorescence amplitude, V1/2 is the half-activation potential, V is the test voltage, and k is the slope factor, all in millivolts.
All data are presented as means ± SEM. All comparisons used one-way ANOVA. Statistical significance was assessed using the post hoc Bonferroni test. p < 0.05 was used as a threshold of significance. To facilitate comparisons of A359C fluorescence to track voltage sensor return (which decayed as double exponentials), weighted time constants were generated following the method of Lacroix et al. (2011). Following fitting of double exponential curves using pClamp10, a weighted average was generated for each dataset by multiplying each τ value by its corresponding amplitude, then dividing by the sum of both amplitudes. All datasets represent results of at least 6 cells from 3 separate experiments, except single-channel records, where n = 3 cells. Figure 8 (see below) was generated using the PyMOL Molecular Graphics System, Version 1.3, Schrödinger, LLC.
Figure 8.

EA-1 mutations may weaken channel open state stability. A, Intracellular regions of the S5 and S6 helices from two adjacent subunits in the Kv1.2-2.1 chimera (PDB ID: 2R9R) are shown (red and green coloration denote separate subunits). Key residues (all within the S6 helices) are highlighted in spherical representation, including V478 from the first subunit and A471, L472 and P475 from the second subunit (residue numbering as in Shaker IR). B, Substitution of V478 for alanine results in the absence of both γ-carbons normally present with a valine side chain.
Results
A mutation in the lower S6 accelerates current decay in Shaker IR and hKv1.1
To investigate the functional effects of a known disease mutation at Kv1.1 V408, we injected Xenopus oocytes with mRNA encoding wild-type Kv1.1 or the mutant Kv1.1 V408A. As mentioned in the introduction, we also injected the Drosophila melanogaster Kv1.1 homolog, Shaker IR, and Shaker IR V478A, equivalent to V408A in Kv1.1 (Fig. 1A, asterisk). When 35 s depolarizing pulses to +20 mV were applied to these oocytes, channels activated rapidly, then underwent a slow, time-dependent decay in current, which is usually attributed to C-type inactivation (Fig. 1B,C). These traces were fit with single exponentials for comparison of the time constants of current decay (Fig. 1E). In V408A mutants, the Kv1.1 channel currents decayed significantly faster compared with wild-type. Current decay in Shaker IR V478A was also accelerated, suggesting a likely shared mechanism between Kv1.1 and Shaker IR. This is possibly because C-type inactivation occurs more quickly in Shaker IR due to a more inactivation compatible residue at site T449 (López-Barneo et al., 1993). To examine this possibility, T449 in Shaker IR was substituted with tyrosine, which specifically slows C-type inactivation (López-Barneo et al., 1993), and also matches the identity in Kv1.1 (Fig. 1A, +). Current decay in Shaker IR T449Y was slowed (Fig. 1D) compared with Shaker IR, whereas in the V478A version of the T449Y Shaker IR channel, current decay remained fast and comparable to Shaker IR V478A and Kv1.1 V408A (Fig. 1E). These data establish that Shaker IR is a suitable channel in which to study EA-1 mutations. The absence of modulation of C-type inactivation rate by T449Y in the background of the V478A mutant also suggests that the current decay rate following channel opening in V478A mutants may not be related to modulation of C-type inactivation, but may in fact reflect a separate underlying mechanism.
Figure 1.

A valine to alanine mutation accelerates current decay in Kv1.1 and Shaker channels. A, An alignment of the selectivity filter and S6 pore lining regions in Shaker and Kv1.1 demonstrates 93% amino acid conservation across the region shown. * denotes site of EA-1 mutant V408A (in Kv1.1) and the equivalent mutant V478A in Shaker. + denotes a Shaker site “T449” (Kv1.1 Y379), a poorly conserved site involved in modulation of C-type inactivation. B–D, Currents during pulses to +60 mV from a holding potential of −80 mV are shown for Kv1.1 and Kv1.1 V408A (B), Shaker IR and Shaker IR V478A (C), and Shaker IR T449Y and Shaker IR T449Y V478A (D). E, Mean time constants from fits to traces in B–D. Measured mean τ values were as follows: 24.1 ± 0.6 s (Kv1.1 wt), 4.89 ± 0.42 s (Kv1.1 V408A); 9.52 ± 0.76 s (Shaker IR), 6.30 ± 0.30 s (Shaker IR V478A); 13.7 ± 1.3 s (Shaker IR T449Y), 5.08 ± 0.35 s (Shaker IR T449Y V478A). Comparisons were made using one-way ANOVA with Bonferroni post hoc tests for significance, **p < 0.01 or ***p < 0.001. Horizontal scale bars denote 5 s.
Pore opening, but not voltage-sensing by the S4 helix, is shifted to depolarized membrane potentials by the V478A mutation
Given that channel current decay in both mutant and wild-type channels is dependent on the channel being in its open state, we also investigated the voltage dependence of activation and of movement in the S4 helix, representing the primary charge carrying component of the voltage sensor. Previous work has demonstrated the sensitivity of activation gate stability to mutations at site V478 (Hackos et al., 2002; Maylie et al., 2002; Yifrach and MacKinnon, 2002; Kitaguchi et al., 2004), so we considered that functional effects of mutation on channel behavior may be linked to disruptions in structure in the lower S6 activation gate region. When oocytes expressing Kv1.1 or Kv1.1 V408A (Fig. 2A) or Shaker IR and Shaker IR V478A (Fig. 2C, top) were pulsed to a series of depolarizing voltages, in 10 mV steps from a −80 mV holding potential, and the resulting current amplitudes were divided by potential gradient and normalized to generate conductance–voltage relationships [G(V)], a significant rightward shift could be observed in both Kv1.1 (Fig. 2B), and Shaker IR (Fig. 2D, filled symbols).
Figure 2.
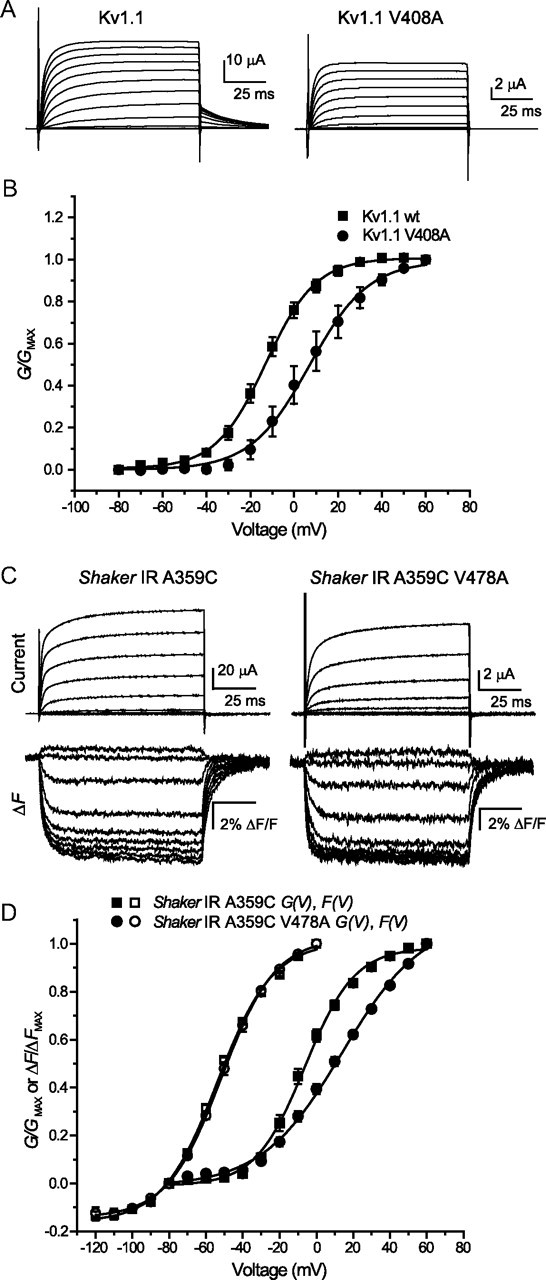
EA-1 mutants shift the voltage dependence of activation, but not of voltage sensor movement. A, Currents recorded in response to pulses from −80 to +60 mV, in 10 mV steps from a holding potential of −80 mV, in Kv1.1 and Kv1.1V408A. B, Steady-state conductance values calculated from currents in A are normalized and plotted against voltage, then fit with two state Boltzmann curves. Values of V1/2 and k for the G(V) curve of Kv1.1 were −12.7 ± 2.3 mV and 10.3 ± 0.6 mV, respectively, while for Kv1.1 V408A they were +5.49 ± 4.62 mV and +12.1 ± 1.1 mV. C, A protocol similar to the one shown in A is used to elicit outward currents from Shaker IR A359C and Shaker IR A359C V478A, while concurrently recording fluorescence from A359C modified by TMRM dye. For clarity, 20 mV steps are shown, for potentials from −100 through 60 mV. D, Steady-state conductance values and fluorescence signals from C are normalized and plotted against voltage. Conductance–voltage (G–V) curves are fit with single Boltzmann functions from −80 to +60 mV, whereas F(V) curves are fit from −120 to 0 mV. Values of V1/2 and k for the F(V) curve of Shaker IR A359C were −53.7 ± 1.1 mV and 15.0 ± 0.6 mV, respectively, while for F(V) of Shaker IR A359C V478A they were −51.6 ± 1.4 mV and 14.7 ± 0.9 mV, respectively. Values of V1/2 and k for the G(V) curve of Shaker IR A359C were −7.07 ± 2.57 mV and 13.7 ± 1.1 mV, respectively, while for G(V) of Shaker IR A359C V478A they were +9.60 ± 2.76 mV and 21.6 ± 0.7 mV, respectively. Where error bars are invisible they are contained within the data points.
While we were unable to record fluorescence traces from Kv1.1 channels, possibly due to the absence of requisite residues within the external linkers (Sørensen et al., 2000), the similarity between the effects of V to A mutants on Shaker IR and Kv1.1 voltage- and time-dependent gating events suggested that Shaker IR could be an adequate model channel for use in voltage-clamp fluorimetry. We chose to use the well characterized Shaker IR A359C channels (whose G–V relationships are unchanged from channels without the fluorescence reporter mutant—data not shown). When these channels are tagged with TMRM, and subjected to voltage-clamp fluorimetry, a series of voltage-dependent deflections can be recorded (Fig. 2C, bottom). The faster of these represents fast S4 movements and is the functional correlate of activation gating charge transfer “qON” (Cha and Bezanilla, 1997), which primarily takes place at potentials negative to 0 mV, the point to which these values were normalized (Fig. 2D, open squares). The Boltzmann curve fit to the voltage dependence of these fluorescence deflections (F–V), in keeping with properties of Kv channel gating currents, covers voltages at more negative potentials with respect to the conductance (Cha and Bezanilla, 1997; Claydon et al., 2007). When the same technique was applied to Shaker IR A359C V478A (Fig. 2C, bottom), the Boltzmann fit of the generated F(V) relationship overlaid well with the curve from Shaker IR A359C (Fig. 2D, open circles), suggesting that changes in voltage sensing are not responsible for the positive shift in G(V) observed with the V478A mutation.
Slower movements of the voltage sensor associated with outer pore constriction in Shaker IR A359C are attenuated by V478A mutation
Being that C-type inactivation is correlated with slower movements of the voltage sensor, which can be observed during longer periods of depolarization (Claydon et al., 2007), we next compared fluorescence dynamics between the V478A mutant and wild-type channel. We used fluorescence constructs possessing the Kv1.1 residue tyrosine at the T449 site (Fig. 1). Shaker IR A359C T449Y and Shaker IR A359C T449Y V478A were pulsed to a series of depolarizing potentials from a holding potential of −80 mV (Fig. 3A,B). A second component of A359C fluorescence, apparent during long periods of channel opening, is known to be associated with the onset of C-type inactivation (Claydon et al., 2007), though it appears to occur on a faster time scale than current decay in non-mutant channels, perhaps indicating that it reports on a voltage sensor reorientation preceding selectivity filter constriction (see discussion). We noticed that, while the rate of current decay was appreciably accelerated at all potentials by V478A (Fig. 3B,C), the secondary component of fluorescence was unexpectedly slowed (Fig. 3B,D) and strongly reduced in amplitude. This is inconsistent with an acceleration of secondary voltage sensor movement or C-type inactivation, as the amplitude of this slow component is expected to reflect the contribution of C-type inactivation to current decay. The reduction in fluorescence decay rate and amplitude more likely implied that a process distinct from C-type inactivation may underlie the effects of V478A mutants on current decay.
Figure 3.

Opposite effects on kinetics of Shaker IR current decay and A359C secondary fluorescence quenching resulting from mutation V478A. A, Outward currents recorded in response to a series of 20 s pulses to voltages from −20 to +40 mV, in 20 mV steps, in Shaker IR A359C T449Y, and a sample fluorescence trace recorded simultaneously with current at +40 mV. B, Currents and fluorescence recorded from Shaker IR A359C T449Y V478A in response to the same protocols as in A. Gray lines in A and B represent single exponential fits. Fluorescence traces are normalized to the amplitudes of their fast components for comparison of relative slow fluorescence amplitudes. C, Mean time constants for current traces in A and B. D, Mean time constants fit to current and fluorescence at +40 mV for constructs in A and B.
V478 mutation and 4-aminopyridine similarly reduce the amplitude of slow movements of the voltage sensor
We next considered the possibility that disruptions in the final transition of the channel activation pathway, or in coupling of the voltage sensor to activation, rather than acceleration of C-type inactivation, may be responsible for the faster current decay in V478A mutants, and could explain the unexpected decrease of slow fluorescence amplitude in Figure 3B. Such an effect might also account for the shift in activation voltage dependence and the apparent uncoupling from voltage sensor movement as tracked by the F(V) of A359C fluorescence, as shown in Figure 2. Normally, the concerted opening transition from the “activated-not-open” state to the open state is tightly coupled to S4 movement (Smith-Maxwell et al., 1998; Ledwell and Aldrich, 1999). As before, we examined the slow component of Shaker IR A359C fluorescence, as emission spectra from this construct can report on a slow conformational change of the S4 helix that accompanies C-type inactivation. Furthermore, we introduced 4-AP as an additional tool to study activation gate dynamics. 4-AP is known to enter the inner pore of open channels and bind within the inner vestibule, blocking the permeation pathway and “pulling” the activation gate closed (McCormack et al., 1994; Armstrong and Loboda, 2001). This has the effect of trapping 4-AP inside the activation gate and antagonizing the concerted opening transition, thereby maintaining the channel in the “activated-not-open” state (del Camino et al., 2005). As a consequence, C-type inactivation, which takes place primarily from the open state, is largely prevented.
When Shaker IR A359C was subjected to 10 s pulses to +60 mV (Fig. 4A), the slowly inactivating ionic current was accompanied by a slow decay in fluorescence intensity after the initial rapid fluorescence signal, as observed previously (Fig. 3). When oocytes were bathed in 10 mm 4-AP, potassium current was significantly blocked, C-type inactivation was prevented, and the normalized amplitude of the second fluorescence component was considerably diminished (Fig. 4C). When the same experiment was performed on Shaker IR A359C V478A, a decaying outward current was observed, but the slow fluorescence signal was already small (Fig. 4B), so that when 4-AP was applied, although current amplitude was strongly decreased, the slow fluorescence amplitude was unchanged (Fig. 4C). When the mutation T449Y was introduced to Shaker IR A359C, slow fluorescence amplitude was diminished from Shaker IR A359C (Fig. 4C), and following 4-AP application, fluorescence amplitude resembled that from Shaker IR A359C with 4-AP or Shaker IR A359C V478A (Fig. 4C). Finally, when both T449Y and V478A were present together, slow fluorescence amplitude was even further reduced, but unchanged following addition of 4-AP (Fig. 4C). Because Shaker channels in the presence of the V478A mutation exhibited the same effects on slow fluorescence decay as wild-type channels with 4-AP, and yet the Shaker IR V478A channel remained capable of conducting current, the effect of V478A on current decay must take place after the channel activation gate has opened. However, the absence of slow fluorescence suggests that current decay in Shaker IR V478A takes place before, or in lieu of, secondary movements of the voltage sensor associated with C-type inactivation. Therefore, it's plausible that current decay with the V478A mutation may in fact be due to perturbation of the activation gate region, promoting bundle crossing closing during depolarization and preventing secondary S4 movement. Consistently, 4-AP, which is known to associate with greatest preference to the activated-not-open state of the channel (Armstrong and Loboda, 2001; del Camino et al., 2005), binds with an ∼20 times greater potency to Shaker IR A359C V478A compared with Shaker IR A359C when subjected to repetitive pulse protocols. Shaker IR A359C V478A had an IC50 of 48 μm in 4-AP, compared with Shaker IR A359C with an IC50 of 868 μm (data not shown). The increased potency is consistent with a reduction in exposure of the open state following 4-AP binding in the presence of V478A, leading to a stronger 4-AP trapping effect (McCormack et al., 1994; Armstrong and Loboda, 2001). In concert with results using voltage-clamp fluorimetry, this finding suggests that local perturbations in the lower S6 may underlie the changes in current decay rate.
Figure 4.

V478A mutants and 4-aminopyridine similarly prevent C-type inactivation by destabilization of the channel open state. A, B, Currents (top) and fluorescence from Shaker IR A359C (A) and Shaker IR A359C V478A (B), in ND96 (black) and in ND96 + 10 mm 4-aminopyridine (gray), in response to pulses from +60 mV from −80 mV. Slow fluorescence deflection amplitudes are normalized to the amplitudes of the rapid fluorescence components of each signal (amplitudes were measured between points denoted by arrows). C, Normalized amplitudes of slow fluorescence signals from Shaker IR A359C and A359C V478A, and Shaker IR A359C T449Y and A359C T449Y V478A. Fluorescence amplitudes of the fast component were normalized to a value of 1, then slow amplitudes were quantified by normalization to the fast components from the same traces, in ND96 (dark gray) and 10 mm 4-AP (light gray). D, Fluorescence traces from Shaker IR A359C in the presence of ND96 (black) and ND96 containing 99 mm (gray). E, Fluorescence traces from Shaker IR A359C V478A in the presence of ND96 (black) and ND96 containing 99 mm external K+ (gray). F, Slow fluorescence amplitudes recorded from channels in the presence of ND96 (dark gray) and ND96 with 99 mm external K+ (light gray) are normalized to fast amplitudes within the same traces, and then plotted against each other for comparison. Comparisons were made using one-way ANOVA with Bonferroni post hoc tests for significance, ***p < 0.001 or p > 0.05 (NS, not significantly different).
Raising the concentration of extracellular K+ is known to be a potent way of inhibiting C-type inactivation (López-Barneo et al., 1993; Levy and Deutsch, 1996), therefore we examined its effects on the slow component of fluorescence in Shaker IR channels with and without the V478A mutations. As seen previously in the 4-AP experiments, long pulses in Shaker IR A359C demonstrated a large, slow, secondary component of fluorescence, which can be greatly reduced by increasing external K+ concentration from 3 to 99 mm (Fig. 4D). However, no effect of increasing external K+ concentration was observed on the slow ΔF signals from V478A mutant channels (Fig. 4E). This suggests that the secondary voltage sensor environmental changes in the outer pore region that accompany C-type inactivation during longer periods of depolarization are absent in the V478A mutation (Fig. 4F), and that the extent of C-type inactivation is reduced in these channels.
Facilitation of S4 segment remobilization by the pore mutant and 4-AP during membrane potential repolarization
To further understand the behavior of the activation gate and voltage sensors of Shaker IR V478A during long depolarizing stimuli, we examined voltage sensor return during repolarization. The process of C-type inactivation is functionally associated with an energetic stabilization of the gating charges of S4 in an outward position, known as charge immobilization (Fedida et al., 1996; Olcese et al., 1997) or relaxation (Villalba-Galea et al., 2008), such that, upon repolarization of the membrane potential, the return of the voltage sensor to its resting position within the closed channel is slowed. If, as we suspect, the extent of C-type inactivation is reduced in channels with the V478A mutation, then charge immobilization would occur to a lesser extent compared with the channel lacking the pore mutation.
To examine charge immobilization, channels were subjected to long pulses to +60 mV, then returned to a series of voltages from 0 to −120 mV in 10 mV steps (Fig. 5A). Fluorescence amplitudes were measured 50 ms following the start of repolarization pulses, when any fast S4 movements would likely have occurred, but slower ones reflecting charge immobilization would not have. Energetic stabilization of S4 in its outward position would be expected to negatively shift the voltage dependence of S4 to its resting conformation (Olcese et al., 1997) compared with the F(V) relationship observed during channel activation (Fig. 2D). The voltage dependence of the changes in Shaker IR A359C fluorescence intensity, when measured in this way, was hyperpolarized by ∼40 mV (Fig. 5B,D), and, expectedly, the addition of 10 mm 4-AP largely attenuated the shift. When Shaker IR A359C V478A was subjected to the same experimental protocol, the voltage dependence of A359C fluorescence during repolarization overlaid well with the trace of Shaker IR A359C in the presence of 4-AP. This is consistent with the idea that the mutation and 4-AP both antagonize the stability of the open state (Fig. 5C,D). When 4-AP was subsequently added to Shaker IR A359C V478A, a slight additional rightward shift of the Boltzmann curve was seen, aligning it with the F(V) for Shaker IR A359C during activation, and indicating that charge immobilization had been fully prevented by this combination.
Figure 5.

V478A mutation prevents S4 charge immobilization in Shaker IR. A, Sample current (top) from a voltage protocol (bottom) to measure fluorescence immobilization (for explanation see text). Arrows indicate the period over which fluorescence signals in B and C were recorded. B, Fluorescence traces recorded during repolarization to the indicated potentials in Shaker IR A359C, in ND96 (top) and in ND96 with 10 mm 4-AP (bottom). Arrows indicate where ΔF/Fmax was measured for D. C, Fluorescence traces recorded during repolarization to the indicated potentials for Shaker IR A359C V478A. D, Fluorescence amplitudes 50 ms after start of repolarizing voltage pulses in B and C plotted against voltage and fit with single Boltzmann functions, in ND96 (filled symbols) and 10 mm 4-AP (open symbols). V1/2 values of Boltzmann fits for Shaker IR A359C fluorescence were −98.9 ± 1.5 mV with 0 mm 4-AP and −60.7 ± 1.5 mV with 10 mm 4-AP. V1/2 values of Boltzmann fits for Shaker IR A359C V478A fluorescence were −65.6 ± 1.4 mV with 0 mm 4-AP and −55.1 ± 2.1 mV with 10 mm 4-AP. A Boltzmann function fit to Shaker IR A359C fluorescence during activation pulses (from Fig. 2) is overlaid in gray (V1/2 = −53.7 ± 1.1 mV). E, Time constants fit at all potentials to repolarization fluorescence traces for Shaker IR A359C and A359C V478A. τ values were significantly different at all displayed potentials where they could be fit, p < 0.05.
To chart the time dependence of voltage sensor return, the fluorescence traces were fit to weighted time constants, in a method similar to one used previously with gating currents (Lacroix et al., 2011). We observed that at all potentials, the time constant of fluorescence return was accelerated by V478A mutants compared with Shaker IR A359C (Fig. 5E), further consistent with an attenuation of conformational changes in S4 in the presence of V478A.
A stable channel open state is prevented by V478A mutation
Data acquired using voltage-clamp fluorimetry suggest that a destabilization of the open state at depolarized potentials may be responsible for the acceleration of current decay in Shaker IR V478A. A possible explanation is that channels do not reach a stabilized conducting state following opening, in which the voltage sensors have entered their relaxed states, instead allowing the activation gate to “slip” closed even while the voltage sensors are in their activated conformation. If this were the case, we would expect that in measurements of currents from single channels, the open dwell time during depolarization, a measure of open state stability, would be affected by V478A. To investigate this, we expressed wild-type and mutant channels in ltk− cells, and studied them using single-channel cell-attached patch clamp. Patches expressing a single Shaker IR channel or single Shaker IR V478A channel were subjected to repeated 8 s pulses to +60 mV from a holding potential of −80 mV (Fig. 6A,B). To quantify the effects of the mutation on current decay, we compared the number of sweeps ending with the channel closed, and consistent with the acceleration of current decay in the V478A mutation, found that 69.9 ± 0.03% of sweeps ended with the channel in a closed state in Shaker IR V478A compared with 54.7 ± 0.02% in Shaker IR (p < 0.01). Inset views allow for closer examination of flickering (bottom), which was further subjected to burst analysis. It appears that the V478A mutant shows extensive rapid closing events during bursts such that the full open level is rarely observed, at the sampling rate and filter frequency that were used.
Figure 6.

Shaker IR V478A has altered cell-attached single-channel properties. A, Representative sweeps of Shaker IR currents elicited from 8 s pulses to +60 mV from a holding potential of −80 mV, as shown. Bottom shows a higher time resolution view of the area indicated by the dotted lines. B, Representative sweeps of Shaker IR V478A currents, with a higher time resolution view in the bottom.
Dwell times of individual opening events were measured, and plotted against occurrence frequencies normalized to the maximum value. A significant reduction in open dwell time was found in Shaker IR V478A compared with Shaker IR (Fig. 7A). While Shaker IR could be fit with three Gaussian functions, the fit corresponding to the largest time constant, τ3, was abolished by V478A. Thus, sustained openings longer than 10 ms were removed in the presence of V478A. In addition, burst durations (rapid opening behavior between closings longer than 5 ms) were binned and plotted against normalized frequency, then fit with Gaussian functions to determine mean burst durations (Fig. 7B). Here, the distribution was also fit as the sum of three Gaussian distributions, and the largest of these was not seen with V478A mutation. These data suggest a less stable open state in the presence of V478A. Conversely, no significant effects of V478A were observed in closed times between high-frequency opening events (Fig. 7C) or in relative frequency of closing events longer than 5 ms (Fig. 7D). Together, these findings are consistent with fluorescence data, which suggest that a stabilized open conformation of the channel, associated with transfer of the S4 helix into its relaxed position and longer periods of activation gate opening, is being prevented by the EA-1 associated mutation V478A.
Figure 7.

Shaker IR V478A does not enter a long-lasting open state and has shorter bursts than Shaker IR. A, Frequency histogram of opening event durations normalized to maximum frequency and binned logarithmically by duration for currents from Shaker IR (black) and Shaker IR V478A (gray). Gaussian distributions fit to the peaks had mean time constants, for Shaker IR, of τ1 = 0.8 ± 0.1 ms, τ2 = 1.3 ± 0.1 ms, τ3 = 4.7 ± 0.9 ms, and for Shaker IR V478A, of τ1 = 0.8 ± 0.1 ms and τ2 = 1.4 ± 0.3 ms. B, Normalized frequency histogram of burst duration (single bursts delimited by closed intervals longer than 5 ms). Gaussian distributions fit to the peaks had mean time constants, for Shaker IR, of τ1 = 3.4 ± 0.1 ms, τ2 = 25.2 ± 4.0 ms, τ3 = 169.0 ± 15.5 ms, and for Shaker IR V478A, of τ1 = 9.9 ± 0.6 ms and τ2 = 98.3 ± 16.4 ms. C, Normalized frequency histogram of intraburst durations. Single Gaussian fits (data not shown) had mean time constants of 1.1 ± 0.1 ms and 1.3 ± 0.1 ms for Shaker IR and Shaker IR V478A, respectively. D, Normalized frequency histogram of interburst durations (denoted as closing events longer than 5 ms). These curves were not fit with Gaussian functions.
Discussion
Insights into atypical gating in an EA-1 mutant Kv1 channel from voltage-clamp fluorimetry and single-channel patch clamp
In this paper, we have investigated how EA-1 associated mutations in the lower S6 of a Shaker family channel can lead to the increased rate of decay of outward current amplitude, as a reduction in Kv1.1 current amplitude in cerebellar basket cells is known to underlie the onset of disease (Herson et al., 2003). We have established using voltage-clamp fluorimetry and single-channel patch-clamp electrophysiology that this decay likely results from destabilization of the open state accelerating closing of the activation gate at depolarized potentials. Several lines of evidence support this conclusion. While current decay is significantly faster in Shaker IR V478A and Kv1.1 V408A compared with their non-mutant forms, a slowly decaying fluorescence signal from Shaker IR A359C, which is known to accompany C-type inactivation (Claydon et al., 2007), is slowed and greatly reduced in amplitude by the mutation V478A. Furthermore, two known antagonists of C-type inactivation, mutation at site T449 (López-Barneo et al., 1993), and elevated external K+ (Levy and Deutsch, 1996), while producing significant effects on current decay and slow fluorescence amplitude in non-mutant channels, had no discernible effects on V478A mutant current or fluorescence.
In addition, the potency of 4-aminopyridine was greatly increased in the presence of V478A, consistent with a local disruption in the lower S6. Slow fluorescence signals from Shaker IR A359C were attenuated by 4-AP, and this effect was mimicked by V478A, with no additional effect of 4-AP, suggesting that both effects likely take place in the activation gate. Finally, the strong reduction in gating charge immobilization with V478A, which normally occurs during prolonged depolarization and is linked with the development of C-type inactivation (Olcese et al., 1997), indicates that the accelerated current decay seen with V478A cannot be explained by an increased rate and contribution of slow C-type inactivation.
In concert with fluorescence experiments, we have used patch-clamp electrophysiology to probe the effects of V478A on single-channel opening behavior. We observed a reduction in mean open time during flickering, as well as shortening of burst durations themselves. These findings most likely reflect a destabilization of the open state, causing local perturbations in conformation that could allow channels to close during depolarizations. This finding is similar to a proposed mechanism of current decay during long periods of activation proposed in spHCN channels (Shin et al., 2004) and in Kv4.2 (Dougherty et al., 2008), and is consistent with the finding that multiple pathways of current decay exist in other Kv1 channels, as was shown in Kv1.3 (Marom and Levitan, 1994). Though it represents a different conclusion, this result also appears to be consistent with observations of a shorter mean open time in single-channel analysis of homotetramers of Kv1.1 V408A as well as Kv1.1-Kv1.2 heterotetramers possessing the same mutation (D'Adamo et al., 1999).
Interestingly, the valine examined in this study is known to form part of the binding site for compatible N termini involved in fast inactivation (Zhou et al., 2001a). Indeed, impaired binding has been observed in Kv1.1-Kv1.4 tetramers (Imbrici et al., 2006), as well as in Kv1.1 coexpressed with a Kvβ1.1 subunit (Maylie et al., 2002), and entry into the open state is a known requirement for peptide binding (Hoshi et al., 1990; Demo and Yellen, 1991). An intriguing implication of our findings is that a reduction in the time spent in the open state, as a consequence of destabilization by the V to A mutation, may in fact underlie the decrease in the extent of N-type inactivation observed in mutants at this site in Kv1.1.
A proposed mechanism for normal and disease-causing channel gating in Shaker channels
We have used slow secondary quenching of the Shaker A359C fluorophore to track secondary movements of the S4 helix following opening. A plausible explanation for the slow quenching phenomenon observed in A359C during long pulses is that it reflects a reorientation of the S4 with respect to the outer pore leading to its increased stability in that position (Loots and Isacoff, 1998; Loots and Isacoff, 2000), which may represent a mode shift in channel gating (Batulan et al., 2010; Haddad and Blunck, 2011), and favor the onset of C-type inactivation. Recent work suggests this is likely due to a relaxation of the voltage sensor, which is associated with, but not obligatorily linked to C-type inactivation (Villalba-Galea et al., 2008; Batulan et al., 2010; Lacroix et al., 2011). Consistent with this, our data using the T449Y mutant in Shaker IR A359C to antagonize inactivation suggests that, while it is likely that charge immobilization and C-type inactivation are in fact tightly linked, the former appears capable in some conditions of taking place at a faster rate than the latter (Fig. 3). As such, the S4 helix may enter into a stable outward conformation with respect to the outer pore (i.e., the relaxed state), leading to longer duration opening events in wild-type channels but not those possessing the EA-1 mutation. As C-type inactivation occurs preferentially from the open state under normal conditions, stabilization would probably favor the onset of C-type inactivation, whereas disruption of stability could lead to closing during periods of depolarization, and actually reduce the extent to which C-type inactivation can occur.
Based on the findings of this study, we propose a mechanism by which prolonged depolarizations may lead to conformational changes within the pore and voltage sensor and lead to C-type inactivation, and how the EA-1 mutation Kv1.1 V408A and its Shaker homolog V478A may contribute to the process. When the membrane becomes depolarized at positive potentials, the voltage sensors move to their active positions, and the lower S6 is pulled to its open position. Normally in Kv1 channels, the lower S6 can then transition between the first open and the stabilized open state. In the Kv1.2-2.1 crystal structure (Long et al., 2007), an apparent hydrophobic pocket exists in this region which we propose may allow contacts between the side chains of V478, and A471, L472 and P475 of an adjacent subunit to stabilize the lower S6 (Fig. 8A), and reduce the mechanical load on the S4 helix allowing it to undergo a mode shift (Haddad and Blunck, 2011) to a relaxed position. This conformation then facilitates entry into a stable quiescent state where the selectivity filter has become constricted (C-type inactivation). Conversely, when the mutation V478A is present, the entry to a stabilized open state is prevented as evidenced by single-channel records, which may be due to the absence of stabilizing hydrophobic interactions between S6 helices of adjacent subunits (Fig. 8B). Under these conditions, the “mechanical load” of the open activation gate can no longer be transferred from the S4 to the pore. As such, as the S4 attempts to adopt its relaxed state, the S6 is able to slip into its closed conformation, blocking the flow of current. Mutational analysis of hydrophobic interactions behind the activation gate would help to establish the properties of a putative “stabilized open state.”
Summary
In summary, we have established a mechanism by which mutations at Kv1.1 V408A leads to an increase in the rate of current decay. Our results suggest that the phenotype results from instability of the open state, causing the activation gate to close at depolarized potentials, with implications for future strategies for development of treatments. These results also demonstrate the utility of the voltage-clamp fluorimetry methodology to determine the diverse biophysical mechanisms underlying pathophysiology caused by genetic mutations. Using a combination of approaches, we have demonstrated that the established phenotype of neurological genetic disease episodic ataxia type-1 can result from a distinct closing phenomenon in the channel activation gate, introduced or accelerated by mutations within the permeation pore, rather than modulation of existing mechanisms.
Footnotes
This project was supported by grants from the Heart and Stroke Foundation of British Columbia and the Yukon (D.F., E.A.A.), and the Canadian Institutes of Health Research (D.F.). E.A.A. is supported by a Canada Research Chair. We appreciate the assistance of Ka Kee Salazar in performing site-directed mutagenesis. We thank Damiano Angoli for technical advice and assistance in generating preliminary data for this paper.
The authors declare no competing financial interests.
References
- Adelman JP, Bond CT, Pessia M, Maylie J. Episodic ataxia results from voltage-dependent potassium channels with altered functions. Neuron. 1995;15:1449–1454. doi: 10.1016/0896-6273(95)90022-5. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, Loboda A. A model for 4-aminopyridine action on K channels: Similarities to tetraethylammonium ion action. Biophys J. 2001;81:895–904. doi: 10.1016/S0006-3495(01)75749-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batulan Z, Haddad GA, Blunck R. An intersubunit interaction between S4–S5 linker and S6 is responsible for the slow off-gating component in shaker K+ channels. J Biol Chem. 2010;285:14005–14019. doi: 10.1074/jbc.M109.097717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev. 2000;80:555–592. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- Browne DL, Gancher ST, Nutt JG, Brunt ER, Smith EA, Kramer P, Litt M. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet. 1994;8:136–140. doi: 10.1038/ng1094-136. [DOI] [PubMed] [Google Scholar]
- Cha A, Bezanilla F. Characterizing voltage-dependent conformational changes in the shaker K+ channel with fluorescence. Neuron. 1997;19:1127–1140. doi: 10.1016/s0896-6273(00)80403-1. [DOI] [PubMed] [Google Scholar]
- Claydon TW, Vaid M, Rezazadeh S, Kehl SJ, Fedida D. 4-aminopyridine prevents the conformational changes associated with p/c-type inactivation in shaker channels. J Pharmacol Exp Ther. 2007;320:162–172. doi: 10.1124/jpet.106.110411. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sakmann B. Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate. J Physiol. 1985;369:501–557. doi: 10.1113/jphysiol.1985.sp015912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Adamo MC, Liu Z, Adelman JP, Maylie J, Pessia M. Episodic ataxia type-1 mutations in the hKv1.1 cytoplasmic pore region alter the gating properties of the channel. EMBO J. 1998;17:1200–1207. doi: 10.1093/emboj/17.5.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Adamo MC, Imbrici P, Sponcichetti F, Pessia M. Mutations in the KCNA1 gene associated with episodic ataxia type-1 syndrome impair heteromeric voltage-gated K(+) channel function. FASEB J. 1999;13:1335–1345. doi: 10.1096/fasebj.13.11.1335. [DOI] [PubMed] [Google Scholar]
- del Camino D, Kanevsky M, Yellen G. Status of the intracellular gate in the activated-not-open state of shaker K+ channels. J Gen Physiol. 2005;126:419–428. doi: 10.1085/jgp.200509385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demo SD, Yellen G. The inactivation gate of the shaker K+ channel behaves like an open-channel blocker. Neuron. 1991;7:743–753. doi: 10.1016/0896-6273(91)90277-7. [DOI] [PubMed] [Google Scholar]
- Demos MK, Macri V, Farrell K, Nelson TN, Chapman K, Accili E, Armstrong L. A novel KCNA1 mutation associated with global delay and persistent cerebellar dysfunction. Mov Disord. 2009;24:778–782. doi: 10.1002/mds.22467. [DOI] [PubMed] [Google Scholar]
- Dougherty K, De Santiago-Castillo JA, Covarrubias M. Gating charge immobilization in Kv4.2 channels: the basis of closed-state inactivation. J Gen Physiol. 2008;131:257–273. doi: 10.1085/jgp.200709938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Fedida D, Bouchard R, Chen FS. Slow gating charge immobilization in the human potassium channel Kv1.5 and its prevention by 4-aminopyridine. J Physiol. 1996;494:377–387. doi: 10.1113/jphysiol.1996.sp021499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackos DH, Chang TH, Swartz KJ. Scanning the intracellular S6 activation gate in the shaker K+ channel. J Gen Physiol. 2002;119:521–532. doi: 10.1085/jgp.20028569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad GA, Blunck R. Mode shift of the voltage sensors in shaker K+ channels is caused by energetic coupling to the pore domain. J Gen Physiol. 2011;137:455–472. doi: 10.1085/jgp.201010573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herson PS, Virk M, Rustay NR, Bond CT, Crabbe JC, Adelman JP, Maylie J. A mouse model of episodic ataxia type-1. Nat Neurosci. 2003;6:378–383. doi: 10.1038/nn1025. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Imbrici P, D'Adamo MC, Kullmann DM, Pessia M. Episodic ataxia type 1 mutations in the KCNA1 gene impair the fast inactivation properties of the human potassium channels Kv1.4-1.1/Kvbeta1.1 and Kv1.4-1.1/Kvbeta1.2. Eur J Neurosci. 2006;24:3073–3083. doi: 10.1111/j.1460-9568.2006.05186.x. [DOI] [PubMed] [Google Scholar]
- Kitaguchi T, Sukhareva M, Swartz KJ. Stabilizing the closed S6 gate in the shaker kv channel through modification of a hydrophobic seal. J Gen Physiol. 2004;124:319–332. doi: 10.1085/jgp.200409098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacroix JJ, Labro AJ, Bezanilla F. Properties of deactivation gating currents in shaker channels. Biophys J. 2011;100:L28–30. doi: 10.1016/j.bpj.2011.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledwell JL, Aldrich RW. Mutations in the S4 region isolate the final voltage-dependent cooperative step in potassium channel activation. J Gen Physiol. 1999;113:389–414. doi: 10.1085/jgp.113.3.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DI, Deutsch C. Recovery from C-type inactivation is modulated by extracellular potassium. Biophys J. 1996;70:798–805. doi: 10.1016/S0006-3495(96)79619-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Holmgren M, Jurman ME, Yellen G. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 1997;19:175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Loots E, Isacoff EY. Protein rearrangements underlying slow inactivation of the shaker K+ channel. J Gen Physiol. 1998;112:377–389. doi: 10.1085/jgp.112.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loots E, Isacoff EY. Molecular coupling of S4 to a K(+) channel's slow inactivation gate. J Gen Physiol. 2000;116:623–636. doi: 10.1085/jgp.116.5.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Lu Z, Klem AM, Ramu Y. Coupling between voltage sensors and activation gate in voltage-gated K+ channels. J Gen Physiol. 2002;120:663–676. doi: 10.1085/jgp.20028696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannuzzu LM, Moronne MM, Isacoff EY. Direct physical measure of conformational rearrangement underlying potassium channel gating. Science. 1996;271:213–216. doi: 10.1126/science.271.5246.213. [DOI] [PubMed] [Google Scholar]
- Marom S, Levitan IB. State-dependent inactivation of the Kv3 potassium channel. Biophys J. 1994;67:579–589. doi: 10.1016/S0006-3495(94)80517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maylie B, Bissonnette E, Virk M, Adelman JP, Maylie JG. Episodic ataxia type 1 mutations in the human Kv1.1 potassium channel alter hKvbeta 1-induced N-type inactivation. J Neurosci. 2002;22:4786–4793. doi: 10.1523/JNEUROSCI.22-12-04786.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack K, Joiner WJ, Heinemann SH. A characterization of the activating structural rearrangements in voltage-dependent shaker K+ channels. Neuron. 1994;12:301–315. doi: 10.1016/0896-6273(94)90273-9. [DOI] [PubMed] [Google Scholar]
- Olcese R, Latorre R, Toro L, Bezanilla F, Stefani E. Correlation between charge movement and ionic current during slow inactivation in shaker K+ channels. J Gen Physiol. 1997;110:579–589. doi: 10.1085/jgp.110.5.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin KS, Maertens C, Proenza C, Rothberg BS, Yellen G. Inactivation in HCN channels results from reclosure of the activation gate: desensitization to voltage. Neuron. 2004;41:737–744. doi: 10.1016/s0896-6273(04)00083-2. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ, Sine SM. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Maxwell CJ, Ledwell JL, Aldrich RW. Role of the S4 in cooperativity of voltage-dependent potassium channel activation. J Gen Physiol. 1998;111:399–420. doi: 10.1085/jgp.111.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soler-Llavina GJ, Chang TH, Swartz KJ. Functional interactions at the interface between voltage-sensing and pore domains in the shaker K(v) channel. Neuron. 2006;52:623–634. doi: 10.1016/j.neuron.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Cha A, Latorre R, Rosenman E, Bezanilla F. Deletion of the S3–S4 linker in the shaker potassium channel reveals two quenching groups near the outside of S4. J Gen Physiol. 2000;115:209–222. doi: 10.1085/jgp.115.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDyke DH, Griggs RC, Murphy MJ, Goldstein MN. Hereditary myokymia and periodic ataxia. J Neurol Sci. 1975;25:109–118. doi: 10.1016/0022-510x(75)90191-4. [DOI] [PubMed] [Google Scholar]
- Villalba-Galea CA, Sandtner W, Starace DM, Bezanilla F. S4-based voltage sensors have three major conformations. Proc Natl Acad Sci U S A. 2008;105:17600–17607. doi: 10.1073/pnas.0807387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yifrach O, MacKinnon R. Energetics of pore opening in a voltage-gated K(+) channel. Cell. 2002;111:231–239. doi: 10.1016/s0092-8674(02)01013-9. [DOI] [PubMed] [Google Scholar]
- Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001a;411:657–661. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-fab complex at 2.0 A resolution. Nature. 2001b;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]