

Abstract
Ketamine is an important analgesia clinically used for both acute and chronic pain. The acute analgesic effects of ketamine are generally believed to be mediated by the inhibition of NMDA receptors in nociceptive neurons. However, the inhibition of neuronal NMDA receptors cannot fully account for its potent analgesic effects on chronic pain because there is a significant discrepancy between their potencies. The possible effect of ketamine on spinal microglia was first examined because hyperactivation of spinal microglia after nerve injury contributes to neuropathic pain. Optically pure S-ketamine preferentially suppressed the nerve injury-induced development of tactile allodynia and hyperactivation of spinal microglia. S-Ketamine also preferentially inhibited hyperactivation of cultured microglia after treatment with lipopolysaccharide, ATP, or lysophosphatidic acid. We next focused our attention on the Ca2+-activated K+ (KCa) currents in microglia, which are known to induce their hyperactivation and migration. S-Ketamine suppressed both nerve injury-induced large-conductance KCa (BK) currents and 1,3-dihydro-1-[2-hydroxy-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-2H-benzimidazol-2-one (NS1619)-induced BK currents in spinal microglia. Furthermore, the intrathecal administration of charybdotoxin, a KCa channel blocker, significantly inhibited the nerve injury-induced tactile allodynia, the expression of P2X4 receptors, and the synthesis of brain-derived neurotrophic factor in spinal microglia. In contrast, NS1619-induced tactile allodynia was completely inhibited by S-ketamine. These observations strongly suggest that S-ketamine preferentially suppresses the nerve injury-induced hyperactivation and migration of spinal microglia through the blockade of BK channels. Therefore, the preferential inhibition of microglial BK channels in addition to neuronal NMDA receptors may account for the preferential and potent analgesic effects of S-ketamine on neuropathic pain.
Introduction
Ketamine has been used clinically as an acute anesthetic for >40 years, and it also has analgesic effects on chronic pain including neuropathic pain in humans and animals (Parsons et al., 1993; Eichenberger et al., 2008). Racemic ketamine contains the optical isomers, S- and R-ketamine, which show stereoselectivity. S-Ketamine has a three to four times more potent analgesic effect than R-ketamine in humans and rodents (Marietta et al., 1977; Ryder et al., 1978). Furthermore, R-ketamine is associated with a higher occurrence of psychomimetic side effects, such as hallucinations and delusions, than S-ketamine at an equianalgesic dose (Marietta et al., 1977; Ryder et al., 1978) because R-ketamine has a weak affinity for σ receptors, whereas S-ketamine displays almost negligible binding (Hustveit et al., 1995). Therefore, S-ketamine-based pain management has been conducted on the clinical stage (Grande et al., 2008; Huge et al., 2010).
The acute analgesic effects of ketamine are generally believed to be mediated through the blockade of phencyclidine binding site of NMDA receptors of the nociceptive neurons (Salt, 1986; Klepstad et al., 1990). However, it is unlikely that the inhibition of neuronal NMDA receptors can fully account for the potent analgesic effects of ketamine on chronic pain including neuropathic pain. Although S-ketamine binds with twice the affinity to phencyclidine binding site of NMDA receptors compared with R-ketamine (Hustveit et al., 1995), this differential affinity solely cannot explain the four times more potent analgesic effects of S-ketamine on neuropathic pain (Mathisen et al., 1995). Furthermore, several lines of evidence have indicated that various molecules can be targets for the analgesic actions of ketamine, including Ca2+-activated K+ (KCa) channels (Denson and Eaton, 1994; Denson et al., 1994), voltage-activated K+ channels (Schnoebel et al., 2005), and HCN1 channels (X. Chen et al., 2009).
The hyperactivation of spinal microglia after peripheral nerve injury contributes to the development of neuropathic pain (Watkins et al., 2001; Tsuda et al., 2005; Scholz and Woolf, 2007). Chang et al. (2009) reported that ketamine suppresses hyperactivation of cultured microglia stimulated with lipopolysaccharide (LPS). Furthermore, there is emerging evidence that reactive microglia express NMDA receptor subunits (Daulhac et al., 2011; Murugan et al., 2011), thus suggesting that the microglial NMDA receptor is a potential pharmacological target of ketamine. However, electrophysiological studies showed no evidence for the expression of functional NMDA receptors in the cultured microglia (Noda et al., 2000). Therefore, this study focused on the effects of ketamine on KCa currents, which are known to induce hyperactivation (Kaushal et al., 2007) and migration (Schilling et al., 2004) of microglia, because these microglial responses are necessary for the development of neuropathic pain (Abbadie et al., 2003; Tsuda et al., 2003). The present results indicate that S-ketamine preferentially suppresses hyperactivation of spinal microglia after nerve injury through the blockade of large-conductance KCa channels (BK channels) but not NMDA receptors. Therefore, the preferential inhibition of microglial BK channels in addition to neuronal NMDA receptors may account for the preferential and potent analgesic effects of S-ketamine on neuropathic pain.
Materials and Methods
Animals.
The experimental protocol was approved by the Animal Research Committee of Kyushu University. All efforts were made to minimize animal suffering and to reduce the number of animals used. Ten-week-old male C57BL/6N mice and Iba1-EGFP transgenic mice (Hirasawa et al., 2005) were used for electrophysiological and behavioral analyses, respectively. The mice were maintained on a 12 h light/dark cycle (light on at 8:00 A.M.) under conditions of 22–25°C ambient temperature with food and water ad libitum. All mice were handled daily for 5 d before the start of the experiment to minimize their stress reactions to manipulation.
Asymmetric synthesis of S- and R-ketamines.
S-Ketamine with excellent selectivity (99.9%) was prepared by asymmetric synthesis using the sequential enantioselective reduction–rearrangement strategy as reported previously (Yokoyama et al., 2009a,b) (see Fig. 1A). Optically pure R-ketamine and a racemate consisting of equal amounts of S- and R-ketamine were also synthesized (see Fig. 1B).
Figure 1.
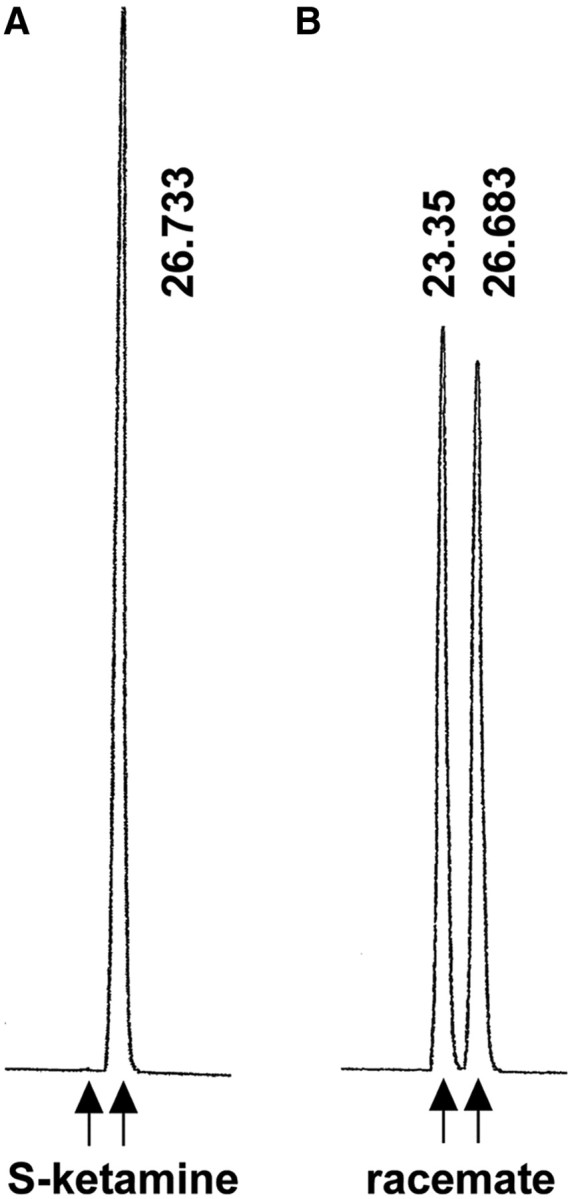
Determination of the enantiopurity of synthetic S-ketamine by HPLC with UV detection (254 nm). A, Asymmetrically synthesized S-ketamine (retention time, 26.733 s) exhibited excellent selectivity (>99.9%). B, The racemate contained equal amounts of S- (retention time, 26.683 s) and R-ketamine (retention time, 23.35 s).
Surgical procedures.
Mice were anesthetized with sodium pentobarbital (50 mg/kg, i.p.). Sterile procedures were used throughout the surgery to prevent infection and to minimize the influence of inflammation. The back of each animal was shaved and cleaned with benzalkonium chloride. A 1 cm incision was made in the middle lumber region (L4 to L5). The L5 transverse process was identified (Rigaud et al., 2008) and carefully removed with bone ronguers. The L4 ventral ramus was carefully isolated and freed from the adjacent nerve, and then the L4 nerve was transected. The incision was washed with saline and closed. Intrathecal administration was performed using a 25 μl Hamilton syringe with a 30 gauge needle according to the methods (Tsuda et al., 2009).
Behavioral analyses.
All mice were habituated to the testing environment for 3 d. All mice were tested for mechanical hypersensitivity of the hindpaw 1 d before and 1 week after spinal nerve transection. The room temperature remained stable at 23°C. The mice were placed in an acrylic cylinder (6 cm diameter) with wire mesh floors and allowed to habituate to the environment for 1 h. The drugs were administrated 1 h before the test trial. Saline (0.2 ml, i.p.; n = 6) or each type of ketamine (20 mg/kg, 0.2 ml, i.p.; n = 6 for each group) and saline (10 μl, i.t.; n = 3) or charybdotoxin (ChTX) (100 nm; 4.3 ng/10 μl, i.t.; n = 4) was administered in the nerve-injured mice. Saline (10 μl, i.t.; n = 3), 1,3-dihydro-1-[2-hydroxy-5-(trifluoromethyl)phenyl]-5-(trifluoromethyl)-2H-benzimidazol-2-one (NS1619) (20 μm; 0.072 μg/10 μl, i.t.; n = 3; and 5555 μm, 20 μg/10 μl, i.t.; n = 3), NS1619 (20 μm; 0.072 μg/10 μl) with S-ketamine (100 μm; 0.27 μg/10 μl, i.t.; n = 4), lysophosphatidic acid (LPA) (1 nmol in 5 μl, i.t.; n = 3), or LPA with ChTX (4.3 ng/10 μl, i.t.; n = 3) were administered to the none-injured mice. Calibrated von Frey filaments (0.02–2.0 g; North Coast Medical) were applied to the midplantar surface of the hindpaw. The 50% paw withdrawal thresholds were calculated using the up-down method (Chaplan et al., 1994). The paw withdrawal threshold was measured on both the right and the left paw for then the intrathecal administration study of the noninjured mice, and the mean was calculated.
Rotarod.
An automated single lane rotarod treadmill (Muromachi; 3 cm diameter drums with grooves to improve the grip) that could be set at an accelerating speed was used as previously described (Hayashi et al., 2008). The starting rotation rate of the rod was 4 rpm and was increased to 40 rpm over 5 min. The mice (n = 6 for each group) were administered saline (intraperitoneally or intratracheally), each type of ketamine (20 mg/kg, i.p.), ChTX (100 nm; 4.3 ng/10 μl, i.t.; n = 3), and NS1619 (20 μm; 0.072 μg/10 μl, i.t.) 1 h before the test trial and then placed on the rotarod apparatus, and the timer was started. The time when the mouse fell from the rod was recorded, with the maximum time being 300 s. No significant difference was observed in any of the groups during the 1 week test period.
Cell culture.
A c-myc-immortalized mouse microglial cell line, MG6 (a kind gift from Dr. H. Kitani, Transgenic Animal Research Center, National Institute of Agrobiological Science, Tsukuba, Japan) (Takenouchi et al., 2005; Nakamichi et al., 2006), was maintained in a growth medium composed of DMEM containing 10% fetal bovine serum (MP Biomedicals) supplemented with 100 μm β-mercaptoethanol, 10 μg/ml insulin, 100 μg/ml streptomycin, and 100 U/ml penicillin in 100 mm Petri dishes (Falcon; BD Biosciences Discovery Labware). Primary cultured microglia were prepared according to previous methods (Tsuda et al., 2003; Hayashi et al., 2006a) and maintained for 10–14 d in Eagle's MEM medium with fetal bovine serum. Floating cells and weakly attached cells on the mixed glial cell layer were isolated by shaking the flask. The resultant cell suspension was then transferred to a Petri dish (Falcon; BD Biosciences Discovery Labware; 1001) and allowed to adhere at 37°C. Unattached cells were removed after 30 min, and microglia were isolated as strongly adhering cells. The purity of microglia was >96% as determined by immunostaining with anti-Iba1 antibody (Wako).
Spinal cord slice preparation.
The animals were anesthetized with sodium pentobarbital (40 mg/kg) 3 d after nerve injury. Ice-cold artificial CSF (ACSF) saturated with 95% O2 and 5% CO2 was perfused through the ascending aorta. ACSF consisted of 124 mm NaCl, 2.5 mm KCl, 1.24 mm KH2PO4, 1.3 mm MgSO4, 2.4 mm CaCl2, 10 mm glucose, and 26 mm NaHCO3. A laminectomy was performed, and the lumber spinal cord was quickly removed and put into ice-cold oxygenated ACSF solution. The pia–arachnoid membrane was carefully peeled off, and a block of the spinal cord from L3 to L5 was embedded in 1% agar. Transverse slices (200 μm thick) were cut from lumber segments L4–L5 with a vibratome (VT1000S; Leica). The slices were then incubated at room temperature for at least 60 min. The slice were placed in a recording chamber and then submerged in and continually perfused with ASCF at a flow rate of 2–3 ml/min.
Electrophysiological recording (slice-patch clamp).
A whole-cell patch-clamp recording was made from the GFP-labeled microglia located at laminae I–II in L4 spinal cord in the slice preparation. GFP-labeled microglia were visually identified by the laser with a wavelength of 488 nm using an upright microscope equipped with a 40× water-immersion objective (Carl Zeiss). The external solution used was ACSF. Patch electrodes were fabricated using a Sutter P-97 (Sutter Instrument) from borosilicate glass (1.5 mm outer diameter, 0.9 mm inner diameter; G-1.5; Narishige). Patch pipettes (2–4 MΩ) were filled with an internal solution containing the following two solutions: (1) the recording for NMDA- and ATP-induced inward currents: 100 mm CsCl, 3 mm Na2ATP, 5 mm HEPES, 1 mm CaCl2, 4 mm MgCl2, 5 mm EGTA, and 10 mm N-methyl-d-glucamine (NMDG) adjusted to pH 7.2 with NMDG; (2) the recording for Ca2+-activated K+ (KCa) currents: 120 mm KCl, 2 mm MgCl2, 10 mm HEPES, 0.1 mm BAPTA adjusted to pH 7.3 with KOH. Patch pipettes were filled with internal solution containing 1% Lucifer yellow for intracellular labeling of recorded microglia and applied to the soma of GFP-labeled microglia. The currents were recorded with an Axopatch 200B patch-clamp amplifier (Molecular Devices). Voltage ramps from −120 to +30 mV were applied for 300 ms to induce KCa currents. The currents signals were filtrated at 3 kHz and digitized at 20 kHz (Digidata 1322A), stored, and analyzed by pCLAMP (Molecular Devices). All electrophysiological recordings were performed at a holding potential of −60 mV. The whole-cell recording mode was established with a brief negative current and pressure pulses after obtaining a cell-attached configuration (1–10 GΩ seal resistance). Recording was started at least 5 min after the rupture of the patch membrane to allow for the stabilization of the intracellular milieu. Microglia showing unstable or large (>50 pA) holding currents were rejected. The series resistance (<30 MΩ) and membrane capacitance were compensated and checked regularly during the recording. Data were discarded when series resistance changed >20% during recording. In some experiments, Lucifer yellow was injected into GFP-labeled microglia with the current injection (−2 nA) at the current-clamp mode. And then the slices were stained with anti-Lucifer antibody (1:50,000; Invitrogen) for 7 d at 4°C.
Electrophysiological recordings (microglial MG6 cell line).
Whole-cell membrane currents were recorded as previously described (Hayashi et al., 2006a). The recordings were performed at room temperature (22–25°C). The external solution contained 130 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, 8 mm d-glucose, pH 7.3 with NaOH. The same internal solution and voltage ramp protocol described above were used for the recording as described above.
Drug application.
All drugs except for ketamine were obtained from Sigma-Aldrich. The test solutions were topically applied using the so-called “Y-tube” solution exchange device (Hayashi et al., 2006a) ∼2 mm away from the cells recorded. Solutions around the recording cells were quickly replaced to test solution using a Y-tube within 500 ms. Test solutions were continually bubbled with 95% O2 and 5% CO2. The solutions were applied for 10 s. The holding currents were not changed by the application of external solutions with a Y-tube. ATP (3 mm), ChTX (100 nm), apamine (1 μm), iberiotoxin (300 nm), tetraethylammonium (TEA) (0.5 mm), NS1619 (20 μm), LPA (1 μm), and ketamine (100 μm) were diluted with external solution. NMDA (0.1–10 mm) and glycine (1 μm), a coagonist of NMDA receptor, were diluted with Mg2+-free external solution.
Immunohistochemistry.
Saline or each type of ketamine was administered to mice (20 mg/kg; n = 3 for each group), and mouse tissues were examined by immunohistochemical analysis as previously described (Hayashi et al., 2008). All mice were deeply anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and perfused transcardially with 0.1 m phosphate buffer (PB), pH 7.4, followed by 4% paraformaldehyde in 0.1 m PB, 0, 1, 3, or 7 d after surgery (n = 3 animals at each time point). The perfused L4 spinal cord segments were dissected and further fixed by immersion in 4% paraformaldehyde overnight at 4°C. Transverse L4 spinal sections (free-floating; 50 μm thick) were prepared by a vibratome (VT1000S; Leica). The floating sections were stained with antibodies for anti-Iba1 (1:10,000; Wako), anti-mature interleukin-1β (mIL-1β) (1:500; Santa Cruz), anti-GFAP (1:5000; Sigma-Aldrich), anti-NeuN (1:5000; Millipore), anti-NR1 (1:1000; Millipore), anti-CD11b (1:800; Serotec), anti-BK-α (1:500; Alomone), anti-brain-derived neurotrophic factor (BDNF) (1:500; Santa Cruz), and anti-P2X4 (1:1000; kindly provided by Dr. Francois Rassendren, Institut de Génomique Fonctionnelle, Montpellier, France) (Ulmann et al., 2008) for 7 d at 4°C with gently agitation. Cultured microglia were incubated with anti-NR1 (1:1000; Millipore) for 2 d at 4°C. The spinal sections and cultured microglia were washed with PBS and incubated with the mixture of secondary antibodies conjugated with Cy3 or Alexa 488 (1:400; Jackson ImmunoResearch) for 3 h at 4°C. The sections were washed with PBS and mounted in the antifading medium Vectashield (Vector Laboratories) and examined with a confocal laser-scanning microscope (CLSM) (LSM510MET; Carl Zeiss). The immunofluorescence (IF) intensity was measured as the average pixel intensity within Iba1-positive cells for mIL-1β immunoreactivity. To calculate the increment rate of immunoreactivity for mIL-1β, the ipsilateral side of immunofluorescence intensity was normalized to contralateral side within the same sections. The immunofluorescence intensity for P2X4 receptors and BDNF within CD11b-positive cells was measured as the average pixel intensity and normalized with sham-operated group. The number of Iba1-positive cells in the spinal dorsal horn was counted and normalized by 0.15 mm2.
Immunoblot analyses.
The MG6 cells were cultured on 100 mm Petri dishes at a density of 5 × 105 cells/ml. For the detection of activated p38 mitogen-activated protein kinase (MAPK), MG6 microglial cells were treated with LPS (100 ng/ml) for 30 min. For the detection of mIL-1β, MG6 microglial cells were (1) primed with LPS (100 ng/ml) for 2 h following replacement to ATP (5 mm) for 1 h or (2) stimulated with LPA (10 μm) for 24 h. MG6 microglia were treated with each type of ketamine or ChTX (100 nm) at 30 min before stimulation with LPS, ATP, or LPA. Each sample was separated by electrophoresis on a 15% SDS-polyacrylamide gel. Proteins on SDS gels were transferred electrophoretically to nitrocellulose membranes. The membranes were washed with PBS and incubated at 4°C overnight under gentle agitation with each primary antibody: anti-p-p38 MAPK (1:1000; Cell Signaling), anti-p38 MAPK (1:1000; Cell Signaling), anti-mIL-1β (1:1000; Santa Cruz), and anti-β-actin (1:1000; Santa Cruz). After washing, the membranes were incubated with horseradish peroxidase (HRP)-labeled anti-goat (1:1000; R&D Systems) or anti-rabbit (1:1000; GE Healthcare) antibody for 1 h at room temperature. Subsequently, the membrane-bound, HRP-labeled antibodies were detected using an enhanced chemiluminescence detection system (ECK lit; GE Healthcare) with an image analyzer (LAS-1000; Fuji Photo Film).
Data analysis.
The data are represented as the mean ± SEM. Statistical analyses of the results were performed with Student's t test, Student's paired t test, or one-way ANOVA with post hoc Tukey's test using the GraphPad Prism software package. A value of p < 0.05 was considered to indicate statistical significance.
Results
Preferential inhibitory effects of S-ketamine on tactile allodynia after nerve injury
The effect of each type of ketamine on the development of tactile allodynia, a hallmark of neuropathic syndrome, was examined. Enantioselectively purified ketamine was synthesized as shown in Figure 1. The paw withdrawal threshold (PWT) to mechanical stimulation of the hindpaw in the saline-treated group was significantly reduced at 3 d after nerve injury and this effect was maintained for up to 7 d, thus indicating the successful development of tactile allodynia after nerve injury. In contrast to the saline-treated group, the systemic administration of each type of ketamine (20 mg/kg) from day 0 to day 7 significantly inhibited the reduction of the PWT after nerve injury. The mean percentage inhibition of S-ketamine for nerve transection-reduced PWT was ∼85%, which was 4.1 and 1.9 times more potent that R-ketamine (21%) or the racemate (44%) (Fig. 2A), respectively. It was also noted that only S-ketamine showed an analgesic effect at 1 and 2 d after nerve injury. Saline or each type of ketamine did not affect the basal activity of mechanical stimulation (Fig. 2B). Motor activity after nerve injury was next examined in the saline-, S- and R-ketamine-, and racemate-treated groups. The time spent on the rod 7 d after surgery was 201.6 ± 31.2 s (saline), 250.2 ± 15.9 s (S-ketamine), 246.7 ± 28.0 s (R-ketamine), and 251.3 ± 20.1 s (racemate), and no significant difference was observed in any of the groups at any of the time points. These results indicate that the potent analgesic effect of S-ketamine is not due to its anesthetic effect.
Figure 2.

The preferential inhibitory effects of S-ketamine on the development of tactile allodynia after nerve injury. A, B, The time course of the paw withdrawal threshold in the ipsilateral (A) and contralateral (B) sides after the systemic administration of saline or each type of ketamine at 20 mg/kg in the mice subjected to nerve transection. Each circle and vertical bar represent the means ± SEM of six independent experiments. The asterisks indicate a statistically significant difference from saline (*p < 0.05; **p < 0.01; ***p < 0.001). The daggers indicate a statistically significant difference from S-ketamine (†p < 0.05; ††p < 0.01; †††p < 0.001).
Preferential inhibitory effect of S-ketamine on hyperactivation of spinal microglia after nerve injury
The finding that only S-ketamine showed an analgesic effect on the early development stage of tactile allodynia after nerve injury strongly suggested that S-ketamine may preferentially inhibit microglial activation, because microglia play a critical role in this stage (Raghavendra et al., 2003; Tsuda et al., 2003; Suter et al., 2009). Therefore, the possible preferential effect of S-ketamine on microglial activation was next examined. Immunohistochemical analyses were conducted on the L4 spinal dorsal horn to investigate the effect of S-ketamine on microglial hyperactivation in the spinal dorsal horn after nerve injury. The level of immunoreactivity for mIL-1β was used as an index of microglial hyperactivation, because reactive microglia, but not ramified microglia, can produce and secrete mIL-1β. The mean IF intensity of mIL-1β in microglia was significantly increased in the ipsilateral side of the spinal dorsal horn 1, 3, and 7 d after nerve injury (Fig. 3A–D,F). The immunoreactivity for mIL-1β on 3 d after nerve injury was mainly localized in Iba1-positive microglia and partially in GFAP-positive astrocyte, but not in NeuN-positive neurons (Fig. 3E). However, the increased immunoreactivity for mIL-1β was mainly localized in astrocytes at 7 d after nerve injury (data not shown). The mean number of Iba1-positive microglia was also significantly increased at the same time (Fig. 3A–D,G). Both the mean expression level of mIL-1β and the mean number of Iba1-positive microglia was significantly lower after treatment with S-ketamine, compared with those of the saline-treated group (Fig. 3B,F,G). However, R-ketamine and the racemate caused significant but relatively weak inhibitory effects on the mean expression level of mIL-1β and the mean number of Iba1-positive microglia (Fig. 3C,D,F,G).
Figure 3.

The preferential inhibitory effects of S-ketamine on the microglial hyperactivation in the spinal dorsal horn after nerve injury. A–D, CLSM images for Iba1 (red), mIL-1β (green), and their merge in the ipsilateral spinal dorsal horn of animals subjected to systemic administration of saline (A), S-ketamine (B), R-ketamine (C), and the racemate (D) at 3 d after nerve injury. Scale bar, 50 μm. E, Localization of mIL-1β (green) in the ipsilateral spinal dorsal horn at 3 d after nerve injury. Scale bar, 10 μm. F, The mean relative IF intensity of mIL-1β within Iba1-positive cells in the spinal dorsal horn of saline- or each type of ketamine-treated mice at 0, 1, 3, and 7 d after nerve injury. Each column and vertical bar represent the mean ± SEM of three independent experiments for each time point. The asterisks indicate a statistically significant difference from saline at day 0 (*p < 0.05; ***p < 0.001). The daggers indicate a statistically significant difference from saline (†p < 0.05; ††p < 0.01; †††p < 0.001). The sharps indicate a statistically significant difference from S-ketamine (#p < 0.05; ###p < 0.001). G, The mean number of Iba1-positive microglia in the ipsilateral and contralateral spinal dorsal horn of saline- or each type of ketamine-treated mice at 0, 1, 3, and 7 d after nerve injury. Each column and vertical bar represent the mean ± SEM of three independent experiments for each time point. The asterisks indicate a statistically significant difference from saline at day 0 (*p < 0.05; ***p < 0.001). The daggers indicate a statistically significant difference from saline (†p < 0.05; ††p < 0.01; †††p < 0.001). The sharps indicate a statistically significant difference between the values (#p < 0.05; ###p < 0.001).
A preferential inhibitory effect of S-ketamine on the hyperactivation of cultured microglia following treatment with LPS, ATP, and LPA
The possible preferential inhibitory effect of S-ketamine on microglial hyperactivation was further examined by immunoblot analyses using p-p38 MAPK and mIL-1β antibody, because reactive spinal microglia produce p-p38 MAPK (Ji et al., 2002; Tsuda et al., 2004) and mIL-1β (Fig. 3A) after nerve injury. The protein band corresponding to p-p38 MAPK was observed in the soluble extracts from MG6 microglial cell 30 min after stimulation with LPS (Fig. 4A,B). The mean amount of p-p38 MAPK was most significantly decreased by treatment with S-ketamine compared with R-ketamine and the racemate (Fig. 4A,B). There is some evidence that ketamine inhibits LPS-induced IL-1β expression in macrophage through the blockade of Toll-like receptor 4 (TLR4)-mediated downstream signaling (T. L. Chen et al., 2009). To exclude that the inhibitory effect of S-ketamine on the microglial activation is due to the blockade of TLR4-mediating signaling, the effects of ketamine on the microglial hyperactivation induced by ATP or LPA, which can induce microglial hyperactivation and tactile allodynia (Inoue et al., 2004, Nakagawa et al., 2007; Ma et al., 2010b), were further examined. The mean amount of mIL-1β induced by treatment with ATP in LPS-primed microglia was most significantly suppressed by treatment with S-ketamine among three types of ketamine (Fig. 4C,D). The mean amount of mIL-1β induced by treatment with LPA was also most significantly suppressed by S-ketamine (Fig. 4E,F). These results strongly suggest that S-ketamine directly and most potently suppresses microglial hyperactivation compared with other species of ketamine.
Figure 4.

Preferential inhibitory effects of S-ketamine on hyperactivation of cultured microglia following stimulation with ATP or LPA. A, C, E, The immunoblot analyses of p-p38 MAPK or mIL-1β in cultured MG6 microglia stimulated with LPS (100 ng/ml) (A), combination of LPS (100 ng/ml) and ATP (5 mm) (C), and LPA (10 μm) (E) in the presence of S- and R-ketamine and racemate. B, D, F, The quantitative analyses of p-p38 MAPK or mIL-1β in the stimulated MG6 microglial cells. Each column and vertical bar represent the mean ± SEM of three independent experiments. The asterisks indicate a statistically significant difference from control group (***p < 0.001). The daggers indicate a statistically significant difference from LPS-, LPS plus ATP-, or LPA-treated group (†††p < 0.001). The sharps indicate a statistically significant difference between the values (##p < 0.01; ###p < 0.001).
The inhibitory effects of S-ketamine on BK currents recorded from microglia
The inhibitory effects of optically pure S-ketamine on microglial hyperactivation were next examined using whole-cell patch-clamp analyses in spinal slices prepared from Iba1-EGFP transgenic mice subjected to nerve injury. In spinal slices, no significant difference was observed in the resting membrane potential between contralateral (−44.2 ± 2.15 mV) and ipsilateral microglia (−37.1 ± 4.29 mV) after nerve injury. The mean value of 3 mm ATP-induced peak currents was 1.50 ± 0.40 pA/pF in contralateral spinal microglia (n = 7 slices from 4 mice) and 3.40 ± 0.99 pA/pF in ipsilateral microglia (n = 5 slices form 3 mice). ATP-induced prolonged currents, which were presumably mediated by P2X4 receptors, were observed in hypertrophic microglia in the ipsilateral side of spinal cord (Fig. 5A), similar as previously reported (Raouf et al., 2007). However, either 0.1 or 1 mm NMDA for 10 s failed to evoke any currents in either contralateral (0.02 ± 0.005 pA/pF; n = 7 slices from 4 mice) or ipsilateral spinal microglia (0.02 ± 0.007 pA/pF; n = 5 slices from 3 mice). In contrast, 10 mm NMDA for 10 s could evoke inward currents in the ipsilateral microglia at 3 d after nerve injury (2.65 ± 0.12 pA/pF; n = 3). The immunoreactivity for NR1 was predominantly found in spinal neurons (Fig. 5C, arrowhead) and partially in spinal microglia (Fig. 5C, arrow). However, the NMDA receptor expressed on spinal microglia is unlikely as a pharmacological target of S-ketamine, because the microglial responses to an extremely high concentration of NMDA (i.e., 10 mm) are considered to be rather experimental artifacts (see Discussion). Morphological analyses of recorded microglia were conducted by intracellular injection of Lucifer yellow. Intracellularly labeled ipsilateral spinal microglia showed hypertrophic morphology after nerve injury (Fig. 5B). However, intracellularly labeled contralateral spinal microglia showed a ramified morphology with small soma and thin and branched processes (Fig. 5B). The effects of ketamine on KCa currents were next examined by voltage-clamp recordings from spinal microglia, because the activation of microglial KCa channels underlies the hyperactivation (Kaushal et al., 2007) and migration (Schilling et al., 2004) of microglia. Ipsilateral spinal microglia exhibited large outward currents (n = 7 slices from 3 mice) in contrast to the contralateral spinal microglia (n = 10 slices from 4 mice) (Fig. 5E,F). Voltage-activated outward rectifier K+ currents of microglia after nerve injury were highly sensitive to 100 nm ChTX, a blocker of both intermediate conductance KCa channels (IK channels) and BK channels (Fig. 5G,J). The voltage-activated outward rectifier K+ currents were partially blocked by iberiotoxin (100 nm), a blocker of BK channels, and fully blocked by a low concentration of TEA (0.5 mm), which is known to selectively inhibit BK channels without affecting either IK or small-conductance SK channels KCa channels (SK channels) (data not shown). However, the nerve injury-induced voltage-activated outward rectifier K+ currents of microglia were not affected by 1 μm apamine, a blocker of SK channels (Fig. 5H,J). Furthermore, immunohistochemical analyses using anti-BK-α antibody revealed that BK channels were expressed in ipsilateral side of spinal microglia (Fig. 5D), which was 1.2-fold higher than contralateral side of spinal microglia. S-Ketamine (100 μm) significantly blocked voltage-activated outward rectifier K+ currents (Fig. 5I,J). These results strongly suggest that nerve injury-induced voltage-activated outward rectifier K+ currents in spinal microglia are mainly mediated by BK channels and S-ketamine significantly suppresses BK currents in reactive microglia.
Figure 5.

The activation of BK channels in spinal microglia on 3 d after nerve injury. A, The typical traces showing NMDA (100 μm)- and ATP (3 mm)-induced inward currents. NMDA did not induce any currents in the contralateral (n = 7) and ipsilateral (n = 5), whereas ATP induced inward currents at the holding potential of −60 mV. NMDA or ATP was applied with Y-tube during the period indicated with a horizontal bar. B, CLSM images for intracellular labeled spinal microglia by injection of Lucifer yellow through the recording pipettes. Scale bar, 10 μm. C, CLSM images for NR1 subunit expression in the ipsilateral side of spinal microglia at 3 d after nerve injury. NR1 immunoreactivity (green) was localized almost in neuron (arrowhead) and partially in microglia (red; arrow). Scale bar, 10 μm. D, CLSM images for BK channels expression in the spinal microglia at 3 d after nerve injury. Microglia (red) express BK channels immunoreactivity (green) using by BK-α antibody. Scale bar, 10 μm. E, The typical traces of voltage ramp (−120 to +30 mV) induced BK currents in the spinal microglia. F, The mean current amplitude induced by the activation of BK channels at +30 mV. Each column and vertical bar represent the mean and SEM of contralateral (n = 7) and ipsilateral (n = 10) side of spinal microglia. The asterisks indicate a statistically significant difference from contralateral (**p < 0.01). G–I, The typical traces shows the effects of ChTX (100 nm) (G), apamine (1 μm) (H), and S-ketamine (100 μm) (I) on BK currents in the ipsilateral side of spinal microglia. J, The mean inhibition rate of BK currents at +30 mV in the presence of ChTX (n = 3), apamine (n = 4), and S-ketamine (n = 3) in the ipsilateral side of spinal microglia. Each column and vertical bar represent the mean ± SEM. The asterisks indicate a statistically significant difference between the values (*p < 0.05; **p < 0.01).
Preferential inhibitory effects of S-ketamine on BK channels in cultured microglia
The effects of optically pure S-ketamine on the activation of BK channels of cultured microglia were examined by voltage-clamp recordings compared with those of R-ketamine and the racemate. As in spinal microglia, inward currents in either MG6 or primary cultured microglia were elicited by 3 mm ATP (n = 4), but not by either 0.1 or 1 mm NMDA (n = 4) (Fig. 6A–C). However, 10 mm NMDA for 10 s evoked inward currents (Fig. 6B) in MG6 and primary cultured microglia, which express NR1 subunit (Fig. 6C). The voltage-activated K+ rectification induced by NS1619 (20 μm), a specific activator of BK channels, was significantly suppressed by S-, R-ketamine and the racemate (Fig. 6D–F). However, the mean inhibitory rate of S-ketamine (n = 6) was significantly higher than R-ketamine (n = 4) or the racemate (n = 4) (Fig. 6I). NS1619-induced BK currents were also observed in GFP-labeled microglia in spinal slices from naive mice, which were significantly suppressed by S-ketamine application with Y-tube system (n = 4) (Fig. 6H). The effects of S-ketamine on LPA-induced BK currents in cultured microglia were further examined. S-Ketamine was found to significantly inhibit LPA (1 μm)-induced BK currents (71.1 ± 2.5%; n = 3) (Fig. 6G,I).
Figure 6.

Preferential inhibitory effects of S-ketamine on BK currents in cultured microglia. A, The typical traces show NMDA (100 μm) and ATP (3 mm) induced inward currents in cultured microglia. NMDA did not induce any currents, whereas ATP induced inward current at holding potential of −60 mV. B, The mean currents of 0.1 (n = 4), 1 (n = 3), and 10 mm (n = 3) NMDA or 3 mm ATP (n = 4) induced currents. Each column and vertical bar represent the mean and SEM. C, Overlaying differential interference contrast (DIC) and fluorescence images with CLSM of NR1 expression (green) in cultured microglia. Scale bar, 10 μm. NMDA (1 mm) did not induce inward currents in primary cultured microglia. D–F, The typical traces show the inhibition of S-ketamine (100 μm) (D), R-ketamine (100 μm) (E), and racemate (100 μm) (F) on NS1619 (20 μm) induced BK currents in cultured microglia. G, The typical traces show the inhibition of S-ketamine (100 μm) on LPA (1 μm) induced BK currents in cultured microglia. H, The typical traces show the inhibition of S-ketamine (100 μm) on NS1619 (20 μm)-induced BK currents in spinal microglia from slice preparation from naive mice. I, The mean inhibition rate of NS1619- or LPA-induced BK currents at +30 mV in the presence of ketamine in the cultured microglia (filled column) and in spinal microglia from slice preparations (open column). NS1619 (NS) + S-ketamine (S), n = 6; NS + R-ketamine (R), n = 3; NS + racemate (rac), n = 4; LPA + S, n = 3; NS + S, n = 5. Each column and vertical bar represent the mean ± SEM. The asterisks indicate a statistically significant difference between the values (*p < 0.05, **p < 0.01, ***p < 0.001).
Possible involvement of BK channels in the development of tactile allodynia
The effects of both NS1619 and ChTX on tactile allodynia were examined to further test the hypothesis that the microglial BK channel is also a molecular target for the analgesic effects of S-ketamine in neuropathic pain. NS1619 showed a biphasic effect on the development of tactile allodynia after nerve injury in a dose-dependent manner (Fig. 7A). The intrathecal administration of NS1619 with a relative low concentration (20 μm; 0.072 μg/10 μl) induced tactile allodynia in the naive mice without affecting motor activity (saline, 294.7 ± 3.1 s; NS1619, 293.7 ± 3.7 s). However, the nerve injury-induced tactile allodynia was significantly suppressed by intrathecal ChTX administration (Fig. 7B), but not by intrathecal apamine administration (data not shown). It was also noted that the intrathecal administration of 0.072 μg of NS1619 induced the morphological alteration of microglia in the spinal dorsal horn as evidenced by large cell bodies with shortened processes (Fig. 7C,D). The simultaneous intrathecal administration of S-ketamine (100 μm; 0.27 μg/10 μl) significantly suppressed NS1619-induced tactile allodynia (Fig. 7A). S-Ketamine also significantly inhibited NS1619-induced morphological alteration of spinal microglia and the increase in the mean number of Iba1-positive microglia (Fig. 7E,G). In contrast, the intrathecal administration of NS1619 with a relative high concentration (5555 μm; 20 μg/10 μl) failed to induce the tactile allodynia in the naive mice (Fig. 7A), but significantly suppressed the nerve injury-induced tactile allodynia (data not shown), as consistent with a previous report (S. R. Chen et al., 2009). However, microglial cell bodies became enlarged and the mean number of Iba1-positive microglia was significantly increased in the spinal dorsal horn of the naive mice after intrathecal administration of 20 μm NS1619 (Fig. 7F,G). However, intrathecal ChTX administration significantly suppressed the nerve injury-induced tactile allodynia (Fig. 7B) and the microglial hyperactivation (Fig. 7H–L) without affecting the motor activity (292.7 ± 4.2 s). Next, the effects of ChTX on the expression of P2X4 receptors and BDNF in spinal microglia after nerve injury were elucidated, because the secretion of BDNF from P2X4 receptor-expressed spinal microglia is a key step for the disinhibition mechanism underlying the initiation of neuropathic pain (Tsuda et al., 2003, 2005; Coull et al., 2005). Nerve injury significantly increased the immunoreactivities for P2X4 in microglia (Fig. 8A–D) and BDNF in both neurons and microglia (Fig. 8E–H). The BDNF production in neurons is thought to be an activity-dependent manner (Hayashi et al., 2006b; Ulmann et al., 2008). Intrathecal ChTX administration significantly suppressed the nerve injury-induced increased immunoreactivities for P2X4 receptors (Fig. 8A–D) or BDNF (Fig. 8E–H) in spinal microglia.
Figure 7.
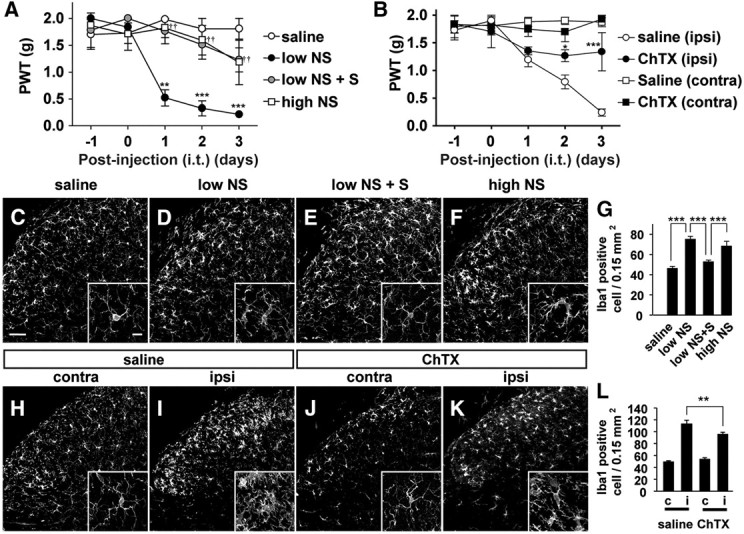
The possible involvement of BK channels in the development of tactile allodynia. A, The time course changes in the PWT after the intrathecal administration of saline, NS1619 with the low concentration (low NS; 20 μm, 0.072 μg/10 μl), combination of low NS with S-ketamine (S, 100 μm; 0.27 μg/10 μl, i.t.), and NS1619 with the high concentration (high NS; 5555 μm, 20 μg/10 μl) in the naive mice. Each circle and vertical bar represent the mean ± SEM. The asterisks indicate a statistically significant difference from saline group (**p < 0.01; ***p < 0.001). The daggers indicate a statistically significant difference between low NS and low NS with S-ketamine (S) (††p < 0.01). B, Effects of the intrathecal administration of saline or ChTX (4.3 ng) on the development of tactile allodynia after nerve injury. Each circle and vertical bar represent the mean ± SEM. The asterisks indicate a statistically significant difference from saline (ipsi) (*p < 0.05; ***p < 0.001). C–F, CLSM images for Iba1-positive spinal microglia in the L4 spinal cord at 3 d after intrathecal treatment with saline (C), low NS (D), combination of low NS and S-ketamine (S) (E), and high NS (F) in the normal mice. Scale bar, 50 μm. The small insets indicate higher magnification images of spinal microglia. Scale bar, 10 μm. G, The mean number of Iba1-positive microglia in the dorsal horn of the intrathecal administration of saline, low NS1619, a combination of low and S-ketamine (S), high NS. Each column and vertical bar represent the mean ± SEM of three independent experiments. The asterisks indicate a statistically significant difference between the values (***p < 0.001). H–K, CLSM images for Iba1-positive spinal microglia in the spinal dorsal horn at 3 d after treatment with intrathecal administration of saline (H, I) or ChTX (J, K) in the mice subjected to nerve injury. L, The effects of the intrathecal administration of ChTX on the mean number of Iba1-positive microglia in the contralateral (c) and ipsilateral (i) spinal dorsal horn of the mice subjected to nerve injury. Each column and vertical bar represent the mean ± SEM of three independent experiments. The asterisks indicate a statistically significant difference between the values (**p < 0.001).
Figure 8.

The possible involvement of BK channels in the nerve injury-induced expression of P2X4 receptors and BDNF in spinal microglia. A–C, CLSM images for P2X4 receptors (A), merged images for P2X4 receptors (green) and CD11b (red), a marker for microglia (B), and their higher magnification images (C) in the dorsal spinal horn at 3 d after nerve injury. D, The relative immunofluorescence intensity of P2X4 receptors in CD11b-positive cells compared with sham group. Scale bars: A, B, 50 μm; C, 10 μm. Each column and vertical bar represent the mean ± SEM of three independent experiments. The asterisks indicate a statistically significant difference from sham group (***p < 0.001). The daggers indicate a significant difference from nerve injury group (†††p < 0.01). E–G, CLSM images for BDNF (E), merged images for BDNF (green) and CD11b (red) (F), and their higher magnification images (G) in the dorsal spinal horn at 3 d after nerve injury. H, The relative immunofluorescence intensity of BDNF receptors in CD11b-positive cells compared with sham group. Scale bars: E, F, 50 μm; G, 10 μm. Each column and vertical bar represent the mean ± SEM of three independent experiments. The asterisks indicate a statistically significant difference from sham group (***p < 0.001). The daggers indicate a significant difference from nerve-injured group (††p < 0.01).
The inhibitory effects of ChTX on LPA-induced microglial hyperactivation and tactile allodynia
LPA is one of the causative factors to induce microglial hyperactivation such as increased expression of BDNF in microglia (Fujita et al., 2008) and subsequent tactile allodynia (Inoue et al., 2004). Therefore, we next examined the effects of ChTX on LPA-induced microglial hyperactivation. The mean production level of mIL-1β in cultured MG6 microglia was significantly increased following treatment with LPA (204.8 ± 23.7%; percentage of control; n = 3). ChTX significantly inhibited the LPA-induced production of mIL-1β in cultured MG6 microglia (100.3 ± 17.6%; percentage of control; n = 3; p < 0.01) (Fig. 9A). Furthermore, a single intrathecal LPA administration induced long-lasting tactile allodynia in the naive mice as reported previously (Fig. 9B) (Inoue et al., 2004). Simultaneous intrathecal administration of ChTX significantly inhibited LPA-induced long-lasting tactile allodynia. These results strongly suggest that BK channels in microglia plays a critical role for their hyperactivation and the subsequent initiation of tactile allodynia.
Figure 9.

The inhibitory effects of ChTX on LPA-induced microglial hyperactivation and tactile allodynia and a schematic representation of the nerve injury-induced reactions of spinal microglia. A, Immunoblot analyses for the LPA (10 μm)-induced increased level of mIL-1β in cultured MG6 microglia in the absence and presence of ChTX (100 μm). B, The time course changes in the PWT after the intrathecal administration of saline, LPA (1 nmol in 5 μl), and combination of LPA with ChTX (100 nm). Each circle and vertical bar represent the mean ± SEM. The asterisks indicate a statistically significant difference from saline group (***p < 0.001). The daggers indicate a statistically significant difference from LPA-treated group (††p < 0.01; †††p < 0.001). C, Schematic representation of a series of sequential signalings in spinal microglia following nerve injury and the pharmacological target of S-ketamine. LPA stimulates the increased synthesis of BDNF in the microglia through upregulation of P2X4 receptors and then enhances nociceptive neurotransmission (1: gray arrow). The current study shows that LPA enhances the production of mIL-1β through the activation of BK channels in the spinal microglia (2: black arrow). Finally, secreted mIL-1β or BDNF itself further enhances the expression levels of P2X4 receptors and the activities of BK channels (3: dashed arrow), leading to the development of neuropathic pain. Therefore, through the inhibition of BK channels, S-ketamine suppresses the nerve injury-induced production of mIL-1β, expression of P2X4 receptors, and BDNF synthesis in the spinal microglia, and finally the development of neuropathic pain (4: open arrow).
Discussion
The present study demonstrated that S-ketamine preferentially inhibited BK channels of spinal microglia and inhibited the development of tactile allodynia after nerve injury. The acute analgesic effects of ketamine are generally believed to be due to the inhibition of neuronal NMDA receptors. IC50 values (in micromolar concentration) of S-ketamine, R-ketamine, and racemate on NMDA receptor-mediated currents in the hippocampal neurons are 0.77 ± 0.1, 1.66 ± 026, and 1.22 ± 1.2, respectively (Y. Hayashi and H. Nakanishi, unpublished data). Therefore, S-ketamine is approximately twice as potent as R-ketamine to inhibit NMDA receptor-mediated currents. However, S-ketamine showed approximately four times more potent analgesic effects on neuropathic pain than R-ketamine in the current study. Furthermore, S-ketamine was found to preferentially inhibit the phosphorylation of p38 MAPK and the production of mIL-1β, key molecules involved in the induction of neuropathic pain (Ji et al., 2002; Tsuda et al., 2004), in cultured microglia after cellular hyperactivation. Therefore, the preferential and potent analgesic effects of S-ketamine on neuropathic pain may be mediated by both neuron and microglia-based mechanisms.
The most likely microglia-based mechanism is that S-ketamine inhibits the microglial hyperactivation through the blockade of NMDA receptors expressed on microglia. However, the expression of functional NMDA receptors in microglia is still a matter of controversy. We detected immunoreactivity for NR1 subunit in both spinal microglia as reported previously (Daulhac et al., 2011; Murugan et al., 2011). However, inward currents were evoked from spinal and cultured microglia by 10 mm NMDA, but not by NMDA with the concentration of 1 mm or less as reported previously (Noda et al., 2000). These results are apparently inconsistent with observations by Tikka and Koistinaho (2001) that 0.5 mm NMDA significantly increases the cell proliferation associated with the activation of p38 MAPK. A possible explanation for the discrepancy is differential application periods of NMDA. In the current study, 1 mm NMDA was applied for 10 s, whereas they applied 0.5 mm NMDA for 30 min or 24 h, suggesting that the total amount of NMDA exposed to microglia in their study is much higher than that in the present study. Therefore, the microglial responses to extremely high concentrations of NMDA in the present as well as a previous study are considered to be rather experimental artifacts. Furthermore, Coderre et al. (2005) reported that the glutamate concentration in spinal cord subjected to neuropathic pain was determined to be up to 131.8 ng/ml. Surprisingly, glutamate levels in sham group (265.7 ng/ml) were twofold higher than in the nerve-injured group; however, the findings were measured at 7 d after nerve injury but not 3 d. Therefore, it is reasonable to consider that the glutamate concentration cannot increase enough to activate microglial NMDA receptors. In addition to NMDA receptors, microglia also express AMPA/kainate (KA) receptors. However, our previous study showed that AMPA induced inward currents in primary cultured microglia only in the presence of cyclothiazide, which blocks desensitization of AMPA-preferring receptors (Noda et al., 2000). In our preliminary experiments, S-ketamine had no effect on the inward current induced by KA in cultured microglia. Therefore, the effect of S-ketamine on microglial glutamate receptors including NMDA and AMPA/KA receptors can be totally excluded.
Several lines of evidence suggested that microglial KCa channels play an important role in migratory activities (Schilling et al., 2004). Furthermore, the activation of microglial KCa channels is also involved in the phosphorylation of p38 MAPK and subsequent production of inflammatory cytokines (Kaushal et al., 2007). Therefore, the current study focused on KCa channels as the molecular substrate of microglia for analgesic effects of S-ketamine, because hyperactivation and migration of spinal microglia are necessary for the development of neuropathic pain (Abbadie et al., 2003; Tsuda et al., 2003). Both IK and BK channels are involved in microglial migratory activity. However, the expression pattern of KCa in microglia depends on experimental conditions. IK channels are preferentially expressed in cultured murine microglia (Schilling et al., 2004; Kaushal et al., 2007), whereas BK channels are observed in microglia under the acute brain slices (Bordey and Spencer, 2003; Schilling and Eder, 2007). In the current study, BK channel blockers including ChTX, iberiotoxin, and a low concentration of TEA suppressed the voltage-activated K+ rectification recorded from spinal microglia after nerve injury. In addition, increased immunoreactivity for BK channels was detected in spinal microglia. The involvement of SK channels, which are not voltage-gated channels (Kaushal et al., 2007), in spinal microglia after nerve injury can be excluded because the resting membrane potential exhibited no significant difference. However, the involvement of IK channels in microglia cannot be totally ruled out because of the limited effect of iberiotoxin. Although further studies will be needed to elucidate the mechanism underlying the inhibitory effect of S-ketamine on BK channels, one possible mechanism is that S-ketamine preferentially interacts with intracellular Ca2+ through acting on Ca2+-binding site of BK channels, which is localized in an intracellular domain of BK channels (Denson et al., 1994).
In in vivo studies, BK channels are inhibited by ChTX (1–4 ng) (Lang et al., 1997; Paton et al., 2001), resulting in enhancement of the neuronal excitability. Therefore, intrathecal ChTX administration was expected to cause an increase in neuronal excitability and enhancement of tactile allodynia. On the contrary, ChTX (4.3 ng) significantly inhibited the development of tactile allodynia and microglial hyperactivation in the spinal dorsal horn after nerve injury. Furthermore, ChTX significantly suppressed nerve injury-induced BDNF synthesis in spinal microglia. These observations strongly suggest that ChTX prevents the disinhibition of spinal neurons caused by BDNF. However, S. R. Chen et al. (2009) reported that NS1619 suppressed the nerve injury-induced tactile allodynia. In contrast, we found that intrathecal NS1619 administration induced tactile allodynia in naive mice. This discrepancy may be due to differential concentrations of NS1619 used in our and a previous study. The concentration of NS1619 (20 μg) used by S. R. Chen et al. (2009) is ∼300 times higher than that used in the current study (0.072 μg). The differential consequences on the pain hypersensitivity of NS1619 with different two concentrations might be due to the differential sensitivities of neuronal and microglial BK channels. The effective dose of NS1619 in vivo was ∼30 μg (Borbouse et al., 2009). Thus, 0.072 μg of NS1619 might induce microglial hyperactivation and allodynia-like behavior without affecting the neuronal excitability. In contrast, 20 μg of NS1619 might spread into both the DRG and the spinal dorsal horn (Ji et al., 2002). The continuous inputs from DRG neurons as well as hyperactivation of spinal microglia are absolutely required for the development of tactile allodynia. In the spinal dorsal horn, 20 μg of NS1619 activates microglia (Fig. 7F,G) but suppresses the neuronal excitability. Furthermore, 20 μg of NS1619 also suppresses the spike generation of DRG neurons (Zhang et al., 2003).
In reactive spinal microglia after nerve injury, increased intracellular Ca2+ in turn activates BK channels, the activities of which keep the membrane potential of cells at hyperpolarized levels. The absence of difference on the resting membrane potential of spinal microglia after nerve injury may be due to the activities of BK channels, which may turn off Ca2+ influx-induced membrane depolarization and sustain the resting membrane potential of the at hyperpolarization levels. Ca2+ influx is necessary for the secretion of IL-1β, whereas membrane depolarization is known to inhibit IL-1β processing and secretion from microglia (Andrei et al., 2004; Stock et al., 2006). Therefore, it is considered that Ca2+ influx and the subsequent activation of BK channels play important roles in IL-1β processing and secretion from reactive microglia. However, the exact mechanism by which the membrane hyperpolarization is required for IL-1β processing and secretion from microglia remains unclear. Several mechanisms might be involved in the elevation of intracellular Ca2+ concentration in microglia. LPA activates store-operated Ca2+ entry (Schilling et al., 2004) or nonselective cation channels in microglia (Möller et al., 2001). Furthermore, prolonged treatment with LPA induces secretion of ATP and expression of P2X4 receptors in microglia (Fujita et al., 2008). ATP-induced fast and slow inward currents recorded from ipsilateral spinal microglia are likely mediated by P2X7 and P2X4 receptors, respectively (Fig. 5A) (Raouf et al., 2007), expressed on reactive spinal microglia after nerve injury (Tsuda et al., 2003; Clark et al., 2010).
LPA biosynthesized de novo in the spinal dorsal horn after nerve injury (Ma et al., 2010a) stimulates microglia to induce the BDNF synthesis through activation of P2X4 receptors. Ca2+ influx mainly through P2X4 receptors and the subsequent activation of BK channels induce the processing and secretion of mIL-1β from reactive microglia. Increased intracellular Ca2+ further activates Ca2+ sensing signaling including p38-MAPK (Trang et al., 2009). Both mIL-1β and BDNF secreted from microglia in turn further enhance the activities of BK channels (Langer et al., 2003; Gao et al., 2010). S-Ketamine strongly inhibited LPA-induced BK currents recorded from cultured microglia. Furthermore, increased expression of P2X4 receptors, BDNF synthesis, and the development of neuropathic pain were significantly inhibited by the blockade of BK channels. Therefore, S-ketamine inhibits the microglial P2X4 receptor-triggered vicious cycle and the subsequent development of neuropathic pain though the inhibition of microglial BK channels (Fig. 9C).
Footnotes
This work was supported by Japan Science and Technology Agency, Core Research for Evolutional Science and Technology, and Grants-in-Aid from Ministry of Education, Culture, Sports, Science, and Technology, Japan. We thank Dr. Francois Rassendren (Institut de Génomique Fonctionnelle, Montpellier, France) for providing the antibody for P2X4 and Dr. Hiroshi Kitani (Transgenic Animal Research Center, National Institute of Agrobiological Science, Tsukuba, Japan) for providing the MG6 microglial cell line.
The authors declare no competing financial interests.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrei C, Margiocco P, Poggi A, Lotti LV, Torrisi MR, Rubartelli A. Phospholipases C and A2 control lysosome-mediated IL-1β secretion: implications for inflammatory processes. Proc Natl Acad Sci U S A. 2004;101:9745–9750. doi: 10.1073/pnas.0308558101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borbouse L, Dick GM, Asano S, Bender SB, Dincer UD, Payne GA, Neeb ZP, Bratz IN, Sturek M, Tune JD. Impaired function of coronary BKCa channels in metabolic syndrome. Am J Physiol Heart Circ Physiol. 2009;297:H1629–H1637. doi: 10.1152/ajpheart.00466.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordey A, Spencer DD. Chemokine modulation of high-conductance Ca2+-sensitive K+ currents in microglia from human hippocampi. Eur J Neurosci. 2003;18:2893–2898. doi: 10.1111/j.1460-9568.2003.03021.x. [DOI] [PubMed] [Google Scholar]
- Chang Y, Lee JJ, Hsieh CY, Hsiao G, Chou DS, Sheu JR. Inhibitory effects of ketamine on lipopolysaccharide-induced microglial activation. Mediators Inflamm. 2009;2009:705379. doi: 10.1155/2009/705379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen SR, Cai YQ, Pan HL. Plasticity and emerging role of BKCa channels in nociceptive control in neuropathic pain. J Neurochem. 2009;110:352–362. doi: 10.1111/j.1471-4159.2009.06138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TL, Chang CC, Lin YL, Ueng YF, Chen RM. Signal-transducing mechanisms of ketamine-caused inhibition of interleukin-1β gene expression in lipopolysaccharide-stimulated murine macrophage-like Raw 264.7 cells. Toxicol Appl Pharmacol. 2009;240:15–25. doi: 10.1016/j.taap.2009.06.013. [DOI] [PubMed] [Google Scholar]
- Chen X, Shu S, Bayliss DA. HCN1 channel subunits are a molecular substrate for hypnotic actions of ketamine. J Neurosci. 2009;29:600–609. doi: 10.1523/JNEUROSCI.3481-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Staniland AA, Marchand F, Kaan TK, McMahon SB, Malcangio M. P2X7-dependent release of interleukin-1β and nociception in the spinal cord following lipopolysaccharide. J Neurosci. 2010;30:573–582. doi: 10.1523/JNEUROSCI.3295-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre TJ, Kumar N, Lefebvre CD, Yu JS. Evidence that gabapentin reduces neuropathic pain by inhibiting the spinal release of glutamate. J Neurochem. 2005;94:1131–1139. doi: 10.1111/j.1471-4159.2005.03263.x. [DOI] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Daulhac L, Maffre V, Mallet C, Etienne M, Privat AM, Kowalski-Chauvel A, Seva C, Fialip J, Eschalier A. Phosphorylation of spinal N-methyl-d-aspartate receptor NR1 subunits by extracellular signal-regulated kinase in the spinal dorsal horn neurons and microglia contributes to diabetes-induced painful neuropathy. Eur J Pain. 2011;15:169.e1–169.e12. doi: 10.1016/j.ejpain.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Denson DD, Eaton DC. Ketamine inhibition of large conductance Ca2+-activated K+ channels is modulated by intracellular Ca2+ Am J Physiol. 1994;267:C1452–C1458. doi: 10.1152/ajpcell.1994.267.5.C1452. [DOI] [PubMed] [Google Scholar]
- Denson DD, Duchatelle P, Eaton DC. The effect of racemic ketamine on the large conductance Ca2+-activated potassium (BK) channels in GH3 cells. Brain Res. 1994;638:61–68. doi: 10.1016/0006-8993(94)90633-5. [DOI] [PubMed] [Google Scholar]
- Eichenberger U, Neff F, Sveticic G, Björgo S, Petersen-Felix S, Arendt-Nielsen L, Curatolo M. Chronic phantom limb pain: the effects of calcitonin, ketamine, and their combination on pain and sensory thresholds. Anesth Analg. 2008;106:1265–1273, table of contents. doi: 10.1213/ane.0b013e3181685014. [DOI] [PubMed] [Google Scholar]
- Fujita R, Ma Y, Ueda H. Lysiphosphatidic acid-induced membrane ruffling and brain-derived neurotrophic factor gene expression are mediated by ATP release in primary microglia. J Neurochem. 2008;107:152–160. doi: 10.1111/j.1471-4159.2008.05599.x. [DOI] [PubMed] [Google Scholar]
- Gao Y, Yang Y, Guan Q, Pang X, Zhang H, Zeng D. IL-1β modulates the Ca2+-activated big-conductance K channels (BK) via reactive oxygen species in cultured rat aorta smooth muscle cells. Mol Cell Biochem. 2010;338:59–68. doi: 10.1007/s11010-009-0338-4. [DOI] [PubMed] [Google Scholar]
- Grande LA, O'Donnell BR, Fitzgibbon DR, Terman GW. Ultra-low dose ketamine and memantine treatment for pain in an opioid-tolerant oncology patient. Anesth Analg. 2008;107:1380–1383. doi: 10.1213/ane.0b013e3181733ddd. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Ishibashi H, Hashimoto K, Nakanishi H. Potentiation of the NMDA receptor-mediated responses through the activation of the glycine site by microglia secreting soluble factors. Glia. 2006a;53:660–668. doi: 10.1002/glia.20322. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Tomimatsu Y, Suzuki H, Yamada J, Wu Z, Yao H, Kagamiishi Y, Tateishi N, Sawada M, Nakanishi H. The intra-arterial injection of microglia protects hippocampal CA1 neurons against global ischemia-induced functional deficits in rats. Neuroscience. 2006b;142:87–96. doi: 10.1016/j.neuroscience.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Yoshida M, Yamato M, Ide T, Wu Z, Ochi-Shindou M, Kanki T, Kang D, Sunagawa K, Tsutsui H, Nakanishi H. Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor A in mice. J Neurosci. 2008;28:8624–8634. doi: 10.1523/JNEUROSCI.1957-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa T, Ohsawa K, Imai Y, Ondo Y, Akazawa C, Uchino S, Kohsaka S. Visualization of microglia in living tissues using Iba1-EGFP transgenic mice. J Neurosci Res. 2005;81:357–362. doi: 10.1002/jnr.20480. [DOI] [PubMed] [Google Scholar]
- Huge V, Lauchart M, Magerl W, Schelling G, Beyer A, Thieme D, Azad SC. Effects of low-dose intranasal S-ketamine in patients with neuropathic pain. Eur J Pain. 2010;14:387–394. doi: 10.1016/j.ejpain.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Hustveit O, Maurset A, Oye I. Interaction of the chiral forms of ketamine with opioid, phencyclidine, sigma and muscarinic receptors. Pharmacol Toxicol. 1995;77:355–359. doi: 10.1111/j.1600-0773.1995.tb01041.x. [DOI] [PubMed] [Google Scholar]
- Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712–718. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglial activation and nitric oxide-dependent neurodegeneration. J Neurosci. 2007;27:234–244. doi: 10.1523/JNEUROSCI.3593-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepstad P, Maurset A, Moberg ER, Oye I. Evidence of a role for NMDA receptors in pain perception. Eur J Pharmacol. 1990;187:513–518. doi: 10.1016/0014-2999(90)90379-k. [DOI] [PubMed] [Google Scholar]
- Lang EJ, Sugihara I, Llinás R. Differential roles of apamin- and charybdotoxin-sensitive K+ conductances in the generation of inferior olive rhythmicity in vivo. J Neurosci. 1997;17:2825–2838. doi: 10.1523/JNEUROSCI.17-08-02825.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer P, Gründer S, Rüsch A. Expression of Ca2+-activated BK channel mRNA and its splice variants in the rat cochlea. J Comp Neurol. 2003;455:198–209. doi: 10.1002/cne.10471. [DOI] [PubMed] [Google Scholar]
- Ma L, Uchida H, Nagai J, Inoue M, Aoki J, Ueda H. Evidence for de novo synthesis of lysophosphatidic acid in the spinal cord through phospholipase A2 and autotoxin in nerve injury-induced neuropathic pain. J Pharmacol Exp Ther. 2010a;333:540–546. doi: 10.1124/jpet.109.164830. [DOI] [PubMed] [Google Scholar]
- Ma L, Nagai J, Ueda H. Microglial activation mediates de novo lysophosphatidic acid production in a model of neuropathic pain. J Neurochem. 2010b;115:643–653. doi: 10.1111/j.1471-4159.2010.06955.x. [DOI] [PubMed] [Google Scholar]
- Marietta MP, WAY WL, Castagnoli N, Jr, Trevor AJ. On the pharmacology of the ketamine enantiomorphs in the rat. J Pharmacol Exp Ther. 1977;202:157–165. [PubMed] [Google Scholar]
- Mathisen LC, Skjelbred P, Skoglund LA, Oye I. Effect of ketamine, an NMDA receptor inhibitor, in acute and chronic orofacial pain. Pain. 1995;61:215–220. doi: 10.1016/0304-3959(94)00170-J. [DOI] [PubMed] [Google Scholar]
- Möller T, Contos JJ, Musante DB, Chun J, Ransom BR. Expression and function of lysophosphatidic acid receptors in cultured rodent microglial cells. J Biol Chem. 2001;276:25946–25952. doi: 10.1074/jbc.M102691200. [DOI] [PubMed] [Google Scholar]
- Murugan M, Sivakumar V, Lu J, Ling EA, Kaur C. Expression of N-methyl-d-aspartate receptor subunits in amoeboid microglia mediates production of nitric oxide via NF-κB signaling pathway and oligodendrocyte cell death in hypoxic postnatal rats. Glia. 2011;59:521–539. doi: 10.1002/glia.21121. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Wakamatsu K, Zhang N, Maeda S, Minami M, Satoh M, Kaneko S. Intrathecal administration of ATP produces long-lasting allodynia in rats: differential mechanism in the phase of the induction and maintenance. Neuroscience. 2007;147:445–455. doi: 10.1016/j.neuroscience.2007.03.045. [DOI] [PubMed] [Google Scholar]
- Nakamichi K, Saiki M, Kitani H, Kuboyama Y, Morimoto K, Takayama-Ito M, Kurane I. Suppressive effect of simvastatin on interferon-β-induced expression of CC chemokine ligand 5 in microglia. Neurosci Lett. 2006;407:205–210. doi: 10.1016/j.neulet.2006.08.044. [DOI] [PubMed] [Google Scholar]
- Noda M, Nakanishi H, Nabekura J, Akaike N. AMPA-kainate subtypes of glutamate receptor in rat cerebral microglia. J Neurosci. 2000;20:251–258. doi: 10.1523/JNEUROSCI.20-01-00251.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons CG, Gruner R, Rozental J, Millar J, Lodge D. Patch clamp studies on the kinetics and selectivity of N-methyl-d-aspartate receptor antagonism by memantine (1-amino-3,5-dimethyladamantan) Neuropharmacology. 1993;32:1337–1350. doi: 10.1016/0028-3908(93)90029-3. [DOI] [PubMed] [Google Scholar]
- Paton JF, Li YW, Schwaber JS. Response properties of baroreceptive NTS neurons. Ann N Y Acad Sci. 2001;940:157–168. doi: 10.1111/j.1749-6632.2001.tb03674.x. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Raouf R, Chabot-Doré AJ, Ase AR, Blais D, Séguéla P. Differential regulation of microglial P2X4 and P2X7 ATP receptors following LPS-induced activation. Neuropharmacology. 2007;53:496–504. doi: 10.1016/j.neuropharm.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Rigaud M, Gemes G, Barabas ME, Chernoff DI, Abram SE, Stucky CL, Hogan QH. Species and strain differences in rodent sciatic nerve anatomy: implications for studies of neuropathic pain. Pain. 2008;136:188–201. doi: 10.1016/j.pain.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryder S, Way WL, Trevor AJ. Comparative pharmacology of the optical isomers of ketamine in mice. Eur J Pharmacol. 1978;49:15–23. doi: 10.1016/0014-2999(78)90217-0. [DOI] [PubMed] [Google Scholar]
- Salt TE. Mediation of thalamic sensory input by both NMDA receptors and non-NMDA receptors. Nature. 1986;322:263–265. doi: 10.1038/322263a0. [DOI] [PubMed] [Google Scholar]
- Schilling T, Eder C. Ion channel expression in resting and activated microglia of hippocampal slices from juvenile mice. Brain Res. 2007;1186:21–28. doi: 10.1016/j.brainres.2007.10.027. [DOI] [PubMed] [Google Scholar]
- Schilling T, Stock C, Schwab A, Eder C. Functional importance of Ca2+-activated K+ channels for lysophosphatidic acid-induced microglial migration. Eur J Neurosci. 2004;19:1469–1474. doi: 10.1111/j.1460-9568.2004.03265.x. [DOI] [PubMed] [Google Scholar]
- Schnoebel R, Wolff M, Peters SC, Bräu ME, Scholz A, Hempelmann G, Olschewski H, Olschewski A. Ketamine impairs excitability in the superficial spinal dorsal horn neurones by blocking sodium and voltage-gated potassium currents. Br J Pharmacol. 2005;146:826–833. doi: 10.1038/sj.bjp.0706385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Stock C, Schilling T, Schwab A, Eder C. Lysophoshatidylcholine stimulates IL-1β release from microglia via a P2X7 receptor-independent mechanism. J Immunol. 2006;177:8560–8568. doi: 10.4049/jimmunol.177.12.8560. [DOI] [PubMed] [Google Scholar]
- Suter MR, Berta T, Gao YJ, Decosterd I, Ji RR. Large A-fiber activity is required for microglial proliferation and p38 MAPK activation in the spinal cord: different effects of resiniferatoxin and bupivacaine on spinal microglial changes after spared nerve injury. Mol Pain. 2009;5:53. doi: 10.1186/1744-8069-5-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenouchi T, Ogihara K, Sato M, Kitani H. Inhibitory effects of U73122 and U73343 on Ca2+ influx and pore formation induced by the activation of P2X7 nucleotide receptors in mouse microglial cell line. Biochim Biophys Acta. 2005;1726:177–186. doi: 10.1016/j.bbagen.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-d-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166:7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neurosci. 2009;29:3518–3528. doi: 10.1523/JNEUROSCI.5714-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature. 2003;424:778–783. doi: 10.1038/nature01786. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia. 2004;45:89–95. doi: 10.1002/glia.10308. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big program from molecules in “small” glia. Trends Neurosci. 2005;28:101–107. doi: 10.1016/j.tins.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Masuda T, Kitano J, Shimoyama H, Tozaki-Saitoh H, Inoue K. IFN-γ receptor signaling mediates spinal microglia activation driving neuropathic pain. Proc Natl Acad Sci U S A. 2009;106:8032–8037. doi: 10.1073/pnas.0810420106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, Buell GN, Reeve AJ, Chessell IP, Rassendren F. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J Neurosci. 2008;28:11263–11268. doi: 10.1523/JNEUROSCI.2308-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Yokoyama R, Matsumoto S, Nomura S, Higaki T, Yokoyama T, Kiyooka S-I. Enantioselective construction of nitrogen-substituted quaternary carbon centers adjacent to the carbonyl group in the cyclohexane ring: first asymmetric synthesis of anesthetic S-ketamine with high selectivity. Tetrahedron. 2009a;65:5181–5191. [Google Scholar]
- Yokoyama T, Yokoyama R, Nomura S, Matsumoto S, Fujiyama R, Kiyooka S-I. Synthesis of S-ketamine via [1,3]-chirality transfer of a stereocenter created by enantioselective aldol reaction. Bull Chem Soc Jpn. 2009b;82:1528–1532. [Google Scholar]
- Zhang XF, Gopalakrishnan M, Shieh CC. Modulation of action potential firing by iberiotoxin and NS1619 in rat dorsal root ganglion neurons. Neuroscience. 2003;122:1003–1011. doi: 10.1016/j.neuroscience.2003.08.035. [DOI] [PubMed] [Google Scholar]