

Abstract
Neurons in spinal dorsal horn lamina I play a pivotal role for nociception that critically depends on a proper balance between excitatory and inhibitory inputs. Any modification in synaptic strength may challenge this delicate balance. Long-term potentiation (LTP) at glutamatergic synapses between nociceptive C-fibers and lamina I neurons is an intensively studied cellular model of pain amplification. In contrast, nothing is presently known about long-term changes of synaptic strength at inhibitory synapses in the spinal dorsal horn. Using a spinal cord–dorsal root slice preparation from rats, we show that conditioning stimulation of primary afferent fibers with a stimulating protocol that induces LTP at C-fiber synapses also triggered LTP at GABAergic synapses (LTPGABA). This LTPGABA was heterosynaptic in nature and was mediated by activation of group I metabotropic glutamate receptors. Opening of ionotropic glutamate receptor channels of the AMPA/KA or NMDA subtype was not required for LTPGABA. Paired-pulse ratio, coefficient of variation, and miniature IPSCs analysis revealed that LTPGABA was expressed presynaptically. Nitric oxide as a retrograde messenger signal mediated this increase of GABA release at spinal inhibitory synapses. This novel form of synaptic plasticity in spinal nociceptive circuits may be an essential mechanism to maintain the relative balance between excitation and inhibition and to improve the signal-to-noise ratio in nociceptive pathways.
Introduction
Neurons in lamina I of the spinal dorsal horn are critically involved in the integration of nociceptive information that is relayed to the brain (Todd, 2010). Lamina I neurons receive monosynaptic glutamatergic input from Aδ- and C-fiber nociceptors and are under permanent GABAergic control by interneurons (Yoshimura and Nishi, 1995; Seagrove et al., 2004; Dahlhaus et al., 2005).
An appropriate GABAergic inhibition in superficial spinal dorsal horn is fundamental for normal nociception, as intrathecal application of GABAA receptor antagonists induces signs of severe pain in animals (Yaksh, 1989; Ishikawa et al., 2000). Low-threshold sensory input to lamina I neurons, which is absent under normal conditions, is unmasked upon blockade of GABAergic inhibition (Baba et al., 2003; Torsney and MacDermott, 2006; Schoffnegger et al., 2008). Consistently, bicuculline enhances low threshold-mediated action potential firing of lamina I neurons in vivo (Seagrove et al., 2004). In addition, changes in spinal GABAergic inhibition have been shown to play important roles in the manifestation of neuropathic and inflammatory pain states (Moore et al., 2002; Coull et al., 2003; Poisbeau et al., 2005). Conversely, thermal hyperalgesia and tactile allodynia in neuropathic animals can be reversed by GABA receptor agonist applied intrathecally (Malan et al., 2002).
Activity-dependent forms of plasticity at spinal excitatory synapses are believed to underlie diverse forms of pain amplification (Sandkühler, 2009). Long-term potentiation (LTP) at glutamatergic synapses between nociceptive nerve fibers and spinal dorsal horn neurons has been studied intensively (Randic et al., 1993; Ikeda et al., 2003, 2006; Drdla et al., 2009). Perceptual correlates of LTP in nociceptive pathways have been demonstrated in humans (Klein et al., 2004), and behavioral correlates exist in animals (Zhang et al., 2005). In striking contrast, nothing is presently known about long-term changes of synaptic efficacy at spinal inhibitory synapses. Long-term changes of GABAergic inhibition of spinal lamina I neurons could significantly shift the balance between inhibitory and excitatory drive and may also have an impact on the induction of plasticity at excitatory synapses, as described for other brain regions (Chen et al., 2010).
Here we identified a novel form of heterosynaptic LTP at GABAergic synapses on spinal lamina I neurons that is triggered by activity in primary afferent nerve fibers. We found that convergent monosynaptic Aδ- or C-fiber afferent input was required for the induction of heterosynaptic LTP at GABAergic terminals that was mediated by group I metabotropic glutamate receptors (mGluRs) and nitric oxide (NO) as a retrograde messenger.
Materials and Methods
Spinal cord slice preparation.
Young male Sprague Dawley rats (aged 25–35 d) were deeply anesthetized with isoflurane and killed by decapitation. A laminectomy was performed and the spinal cord was quickly removed into ice-cold incubation solution consisting of (in mm): 95 NaCl, 1.8 KCl, 1.2 KH2PO4, 0.5 CaCl2, 7 MgSO4, 26 NaHCO3, 15 glucose, and 50 sucrose, oxygenated with 95% O2/5% CO2, pH 7.4, measured osmolarity 310 – 320 mOsm · l−1. The dura mater and the ventral roots were removed. Parasagittal slices with attached dorsal roots (10–15 mm long) were cut at 400–600 μm thickness using a vibrating microslicer (DTK-1000, Dosaka) and kept in the incubation solution at 33°C for at least 30 min. After this, the slices were stored in the same solution at room temperature (20–24°C).
All experiments were in accordance with directive 2010/63/EU of the European Parliament and the council of the European Union.
Whole-cell patch-clamp recordings from spinal lamina I neurons.
A single slice was placed in the recording chamber where it was continuously superfused at a rate of 3–4 ml · min−1 with oxygenated extracellular solution, which was identical to the incubation solution except for the following (in mm): 127 NaCl, 24 CaCl2, 1.3 MgSO4, 0 sucrose. Recordings were made at 32 ± 1°C. Neurons of the dorsal horn were visualized with an Olympus BX 51WI (Olympus Optical) microscope equipped with Dodt infrared optics (Dodt et al., 1999). Only neurons at a distance of <20 μm from the dorsal white/gray matter border were considered as being lamina I neurons and thus used for experiments.
GABAergic IPSCs were recorded at a holding potential of 0 mV using an Axopatch 200B patch-clamp amplifier and the pCLAMP 9 software package (both Molecular Devices). No correction for the liquid junction potential was made. Signals were low-pass filtered at 2–10 kHz, sampled at 10–20 kHz, and analyzed offline using pCLAMP 9. Patch pipettes (2–5 MΩ) from borosilicate glass were filled with a cesium-based intracellular solution composed of the following (in mm): 120 CsMeSO3, 20 TEA-Cl, 2 MgCl2, 2 Na2ATP, 10 EGTA, 0.5 NaGTP, 10 HEPES, pH 7.28, with CsOH; measured osmolarity was 300 mOsm · l−1.
IPSCs were pharmacologically isolated by bath application of antagonists to glycinergic and glutamatergic ionotropic transmission [1 μm strychnine, 50 μm d-(−)-2-amino-5-phosphonopentanoic acid (d-AP5), and 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX)]. A patch pipette filled with extracellular solution was placed within 20–40 μm from the cell body of the patched neuron, and monosynaptic IPSCs were evoked by single monopolar square pulse stimulation. Test pulses of 0.1 ms duration were given at intervals of 15 s. At the end of all experiments, bicuculline (10 μm) was bath applied and completely blocked IPSCs, confirming that they were mediated by GABAA receptors.
Conditioning high-frequency stimulation (HFS, 100 pulses at 100 Hz repeated 3 times at 10 s intervals) was applied to attached dorsal roots at C-fiber intensity (5 mA) and 0.1 ms pulse width via a suction electrode with an isolated current stimulator (A360, World Precision Instruments). When HFS was applied, neurons were kept in voltage-clamp mode at 0 mV. To test whether the neurons received monosynaptic input from the attached dorsal root, all drugs were washed out at the end of the experiment. The neurons were then voltage clamped to a holding potential of −70 mV and EPSCs were evoked by stimulating the attached dorsal root via the suction electrode. Evoked EPSCs were classified as Aδ-fiber-evoked when the calculated conduction velocity was above 1.5 m · s−1 and as C-fiber-evoked when the conduction velocity was below 0.5 m · s−1. Monosynaptic input was identified by the absence of failures in response to 10 stimuli at 10 Hz (Aδ-fiber input) or 1 Hz (for C-fiber input) stimulation of dorsal roots and a jitter in response latencies <10% of the response delay. In all experiments only one neuron per spinal cord slice was used.
GABAergic miniature IPSCs (mIPSCs) were recorded in presence of lidocaine (2 mm) to block action potential-dependent effects. Lidocaine applied to the bath solution completely blocked action potential firing of spinal dorsal horn neurons induced by postsynaptic depolarization and monosynaptic EPSCs evoked by stimulation of attached dorsal roots within 5 min. Monosynaptic EPSCs completely recovered after wash-out of lidocaine for 15 min (n = 3; data not shown).
Series resistance was monitored throughout the experiment. Neurons were discarded if the series resistance changed by >10% during the experiment.
Analysis.
IPSCs were analyzed offline using Clampfit 10. Synaptic strength was quantified by measuring the peak amplitude of monosynaptic IPSCs. The mean amplitude of 20 IPSCs evoked by test stimuli served as a control. Two pulses were given with an interstimulus interval of 50 ms. The paired-pulse ratio (PPR) was determined by dividing the amplitude of the second IPSC by the amplitude of the first IPSC and averaged over 5 min epochs of 20 IPSCs. The squared coefficient of variation CV2 (CV−2 = mean2/variance) was calculated for 5 min epochs of 20 IPSCs and normalized to the control period.
Means of IPSC amplitudes, PPR, and CV−2 are reported for 5 min of control compared with the 5 min period from 25 to 30 min after HFS or wash-out of (S)-3,5-dihydroxyphenylglycine (DHPG) or as otherwise stated in the text.
Individual mIPSCs were counted and analyzed offline using MiniAnalysis software (Synaptosoft). Each mIPSC event was visually accepted or rejected based upon the rise and decay times after an automatic screening by the software.
Statistical analysis of the data was performed using SigmaStat 3.1 (SPSS). Results are given as means ± 1 SEM. Statistical comparison of mean values were determined using Student's paired t test for normally distributed data or Wilcoxon signed rank test for non-normally distributed data. The critical value for statistical significance was set at p < 0.05 (*), p < 0.01 (**).
Drugs.
All drugs were added to the bath solution at known concentrations. Drugs used were bicuculline (Fluka); 2-diethylamino-N-(2,6-dimethylphenyl)acetamide (lidocaine; Alexis); N-ω-nitro-l-arginine methyl ester (l-NAME), CNQX, and GDP-βS and strychnine (all Sigma Aldrich); DHPG; 2-methyl-6-(phenylethynyl)pyridine (MPEP), and 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxyl-3-oxide (cPTIO; all Tocris Bioscience); (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385), S-nitroso-N-acetyl-dl-penicillamin (SNAP), and d-AP5 (all Ascent Scientific).
Drugs were applied for at least 20 min before HFS or DHPG application or as otherwise stated in the text. Control drug-free experiments were interleaved with experiments using drug application.
Results
Conditioning stimulation of small primary afferent nerve fibers induces LTP at GABAergic synapses
We performed whole-cell patch-clamp recordings from spinal lamina I neurons. To isolate monosynaptic GABAergic IPSCs, recordings were made in the presence of ionotropic glutamate (CNQX, d-AP5) and glycine (strychnine) receptor antagonists. IPSCs were evoked by focal stimulation in the superficial dorsal horn (Fig. 1A). Neurons were kept at a holding potential of 0 mV with a cesium-based internal solution. Under these conditions IPSCs appeared as outward currents and were completely blocked by bath application of bicuculline. These IPSCs were thus mediated by GABAA receptors (Fig. 1B).
Figure 1.
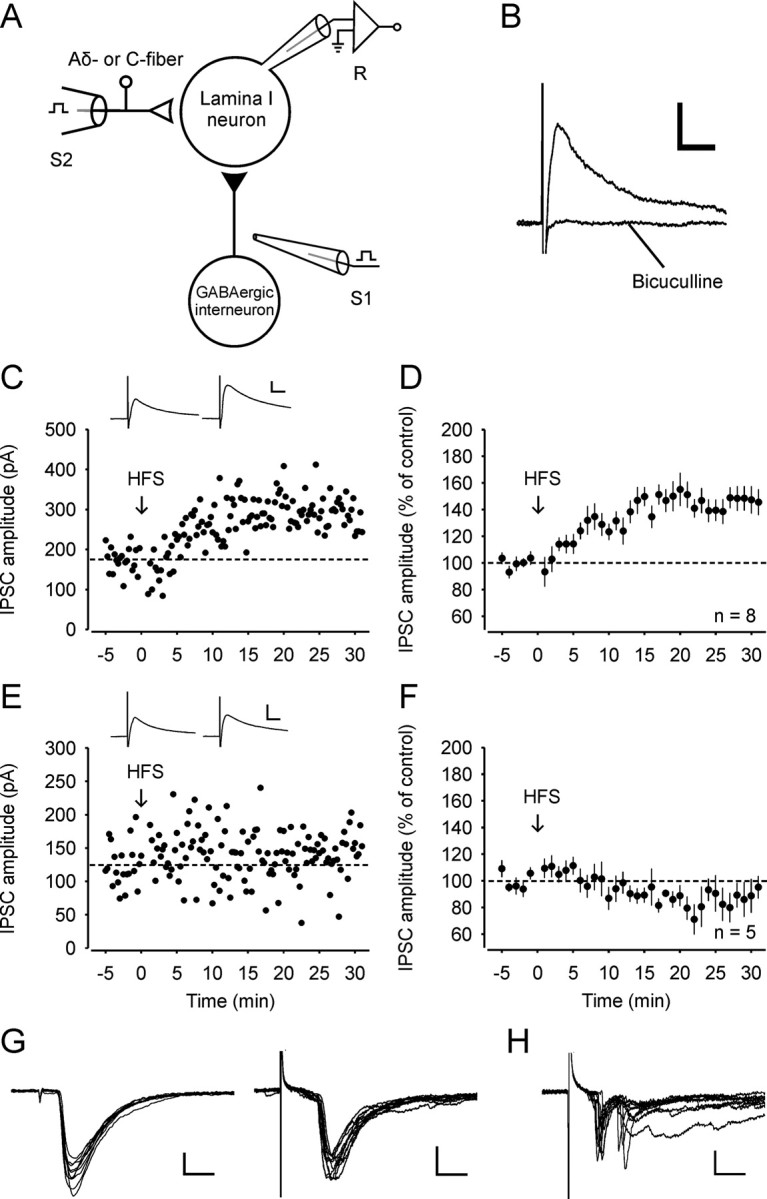
LTP at GABAergic synapses with spinal lamina I neurons that receive monosynaptic glutamatergic input from primary afferent Aδ-fibers or C-fibers. A, Schematic illustration of stimulating and recording electrodes. Monosynaptic IPSCs were evoked by a focal stimulation electrode (S1) placed medially to the recorded lamina I neuron (R). An attached dorsal root was stimulated via a suction electrode (S2) that was used for conditioning stimulation and for the identification of glutamatergic input. Black triangle, Inhibitory synapse; open triangle, excitatory synapse. B, Typical example of a monosynaptic IPSC recorded in the presence of CNQX, d-AP5, and strychnine. The IPSC was abolished by bicuculline. C, Representative example of LTPGABA in a neuron that received monosynaptic afferent input from Aδ-fibers. HFS was applied at the time point zero (arrow). CNQX, d-AP5, and strychnine were present in the bath solution throughout the recording period. Monosynaptic Aδ-fiber input was verified at the end of the experiment. The dotted lines in this and in all other graphs indicate the mean IPSC amplitudes during the control period. Inset: Average of 20 IPSCs 0–5 min before and 25–30 min after conditioning stimulation. D, Average time course of IPSCs recorded from all eight spinal lamina I neurons that received monosynaptic Aδ-fiber or C-fiber input. Each point was averaged from four IPSCs and normalized to the control period of 20 IPSCs. E, Example of an experiment where HFS did not affect IPSC amplitudes. The neuron received polysynaptic but no monosynaptic input from C-fibers. F, Time course of IPSCs from five neurons that did not receive any monosynaptic input. G, Examples of monosynaptic Aδ-fiber (elicited at a frequency of 10 Hz and stimulation intensity of 120 μA; left) and monosynaptic C-fiber input (evoked at 1 Hz and 3.2 mA; right). H, Example of polysynaptic input from dorsal root afferents (stimulated at 1 Hz and 5 mA). In G and H glutamatergic EPSCs were evoked by stimulation of attached dorsal roots at a holding potential of −70 mV at the end of the experiments. Ten EPSCs are superimposed in each example. Calibration: 10 ms, 100 pA (B, C, and E); 2.5 ms, 100 pA (left) and 10 ms, 100 pA (right) (G); 10 ms, 50 pA (H).
We established a stable baseline of IPSCs evoked by focal stimulation for 5 min. We then applied conditioning high-frequency stimulation to the attached dorsal root via a suction electrode (Fig. 1A). HFS induced LTP of the GABAergic IPSC amplitudes (LTPGABA) in 8 out of 13 lamina I neurons tested (to 148 ± 9% of control; p < 0.001; Fig. 1C,D). We next evaluated the glutamatergic afferent input to the neurons under study. All drugs were washed out for at least 10 min at the end of the experiment. The neurons were then clamped to a holding potential of −70 mV, and synaptic input from attached dorsal root was assessed. Monosynaptic EPSCs were identified as described previously (Dahlhaus et al., 2005). Analysis of the data revealed that all eight neurons in which LTPGABA was induced received either monosynaptic Aδ-fiber or monosynaptic C-fiber input from attached dorsal roots (Fig. 1G). The remaining five neurons, in which HFS was ineffective (88 ± 11% of control; Fig. 1E,F), received polysynaptic but no monosynaptic input from the attached dorsal roots in the slice preparation (Fig. 1H).
LTPGABA is mediated by group I metabotropic glutamate receptors
We next explored the mechanisms that trigger LTPGABA. During conditioning stimulation, ionotropic glutamate receptors were blocked by specific antagonists applied to the bath solution. We thus hypothesized that group I mGluRs, which are highly expressed in the superficial dorsal horn (Jia et al., 1999), may be activated during HFS and mediate LTPGABA. To test this hypothesis we performed experiments in the presence of 100 μm LY367385 and 30 μm MPEP, which are selective antagonists of mGluR subtypes 1 and 5, respectively. Only neurons that received either monosynaptic Aδ-fiber or C-fiber input were used for further analysis. During bath application of the group I mGluR antagonists, induction of LTPGABA was abolished in all neurons tested (92 ± 13% of control; n = 5; Fig. 2A,B), indicating that activation of group I mGluRs is necessary for the induction of LTPGABA. We next tested whether activation of group I mGluRs is also sufficient for the induction of LTPGABA. Bath application of the selective group I mGluR agonist DHPG (50 μm) for 10 min induced a transient enhancement of the IPSC amplitudes followed by a long-lasting potentiation (to 136 ± 12% of control; 30 min after wash-out, n = 17, p < 0.001; Fig. 2C,D). The DHPG-induced LTPGABA remained undiminished for the remaining of the recording periods up to 50 min after wash-out (Fig. 2C). Superficial dorsal horn neurons express high levels of group I mGluRs on their somata and also on the terminals of GABAergic neurons (Jia et al., 1999; Tao et al., 2000). To examine, whether postsynaptic mGluR signaling is involved in DHPG-induced LTPGABA, we added the G-protein inhibitor GDP-βS (0.5 mm) to the recording pipette solution. Bath application of DHPG induced a transient increase of the IPSC amplitudes, but the long-lasting potentiation was completely blocked (to 85 ± 12% of control, 30 min after wash-out of DHPG; n = 6; Fig. 2E,F). These results demonstrate that DHPG-induced long-lasting potentiation requires activation of postsynaptic G-proteins.
Figure 2.

LTPGABA is mediated by group I metabotropic glutamate receptors. A, Example of an experiment where HFS was delivered in the presence of mGluR1 and mGluR5 antagonists MPEP (30 μm) and LY367385 (100 μm), respectively. The neuron received monosynaptic C-fiber input. B, Average of five experiments where group I mGluR antagonists were applied during HFS. All neurons received either monosynaptic Aδ-fiber or C-fiber input. C, Example of an experiment where the specific group I mGluR agonist DHPG (50 μm) was applied to the bath solution and potentiated the IPSC amplitudes. D, Average time course of IPSCs recorded from 17 spinal lamina I neurons where DHPG was bath-applied. E, Example of an experiment where DHPG was bath applied when GDP-βS (0.5 mm) was included in the pipette solution to inhibit postsynaptic G-proteins. F, Average of six experiments where GDP-βS, included in the pipette solution, abolished DHPG-induced LTPGABA. Insets, Average of 20 IPSCs 0–5 min before and 25–30 min after conditioning stimulation (A) or 25–30 min after wash-out of DHPG (C and E). Calibration: 10 ms, 100 pA.
LTPGABA is expressed presynaptically
To determine the locus of the expression of LTPGABA, paired-pulse stimulation experiments were performed. After induction of LTPGABA by HFS, the paired-pulse ratio decreased (to 0.75 ± 0.08 of control value; n = 8, p < 0.05; Fig. 3A). The decrease in PPR is consistent with a presynaptic expression of LTPGABA. In neurons that did not display LTPGABA, no significant changes of the PPR were observed (p > 0.05; n = 5; Fig. 3B). In experiments where HFS-induced LTPGABA was blocked by group I mGluR antagonists, the PPR remained unchanged (p > 0.05; n = 5; Fig. 3B). We next asked whether DHPG-induced LTPGABA is also expressed presynaptically. Indeed, DHPG-induced LTPGABA was associated with a decrease in the PPR (to 0.83 ± 0.05 of control value; n = 10; p < 0.01; Fig. 3B). The presynaptic expression of LTPGABA was further confirmed by changes in the CV−2, where CV is the coefficient of variation. LTPGABA was accompanied by an increase of the CV−2 (to 2.27 ± 0.45 of control value; n = 8; p < 0.05; Fig. 3C). Likewise, DHPG-induced LTPGABA was also associated with an increase of the CV−2 (to 2.55 ± 0.47 of control value; n = 17; p < 0.05; Fig. 3D). No change of the CV−2 was observed, however, in neurons that did not express LTPGABA, either because they had no monosynaptic input from dorsal roots or because group I mGluRs were blocked (Fig. 3D).
Figure 3.

Presynaptic expression of LTPGABA. A, HFS-induced LTPGABA is accompanied by a decrease in the PPR. Synaptic currents were evoked by two stimuli with an interpulse interval of 50 ms. PPR of 20 IPSCs were averaged in 5 min blocks, normalized to the control period, and plotted against time. Inset: Average of 20 IPSCs 0–5 min (black) before and 25–30 min (gray) after HFS are superimposed. Calibration, 25 ms, 50 pA. B, Summary graph of the change in PPR 25–30 min after HFS or DHPG wash-out. No change in PPR was observed in neurons without monosynaptic afferent input (No mono; n = 5) or in neurons with monosynaptic input but when HFS was delivered in presence of group I mGluR antagonists (MPEP + LY367385; n = 5). DHPG-induced (DHPG; n = 10) LTPGABA was also accompanied by a decrease of the PPR. C, LTPGABA is associated with an increase of the CV−2. Normalized data are plotted in 5 min blocks against time. Inset: Example of variability of 20 IPSCs 0–5 min before and 25–30 min after HFS. Calibration: 20 ms, 100 pA. D, Summary showing the effects on the CV−2 under various conditions. Labeling is the same as in B. All neurons included in A and C received either monosynaptic Aδ-fiber or C-fiber input from attached dorsal roots.
To further investigate whether transmitter release is enhanced at GABAergic synapses after conditioning HFS, miniature IPSCs were recorded in presence of lidocaine (2 mm) to block action potential-dependent effects. These mIPSCs were completely blocked by bicuculline, confirming that they were mediated by GABAA receptors (n = 5, data not shown). A stable baseline of mIPSCs was first recorded for 5 min as a control, followed by a wash-out period of lidocaine for 15 min. Then HFS was applied to the attached dorsal root and lidocaine was subsequently washed in (Fig. 4A). The frequency of mIPSCs 25–30 min after HFS was significantly increased in neurons that received monosynaptic input from the attached dorsal root (from 233 ± 82 to 565 ± 289 events in 5 min; n = 6; p < 0.05; Fig. 4B,C,D). The amplitude of mIPSCs was not altered after conditioning HFS (baseline 17 ± 1 pA vs 17 ± 1 pA at 25–30 min after HFS; p > 0.05; Fig. 4 B,C,D). Neither the frequency nor the amplitude of mIPSCs was affected in three neurons that did not receive monosynaptic input from attached dorsal roots (data not shown).
Figure 4.

HFS increases the frequency but not the amplitude of mIPSCs in spinal lamina I neurons. A, Experimental design for the recording of mIPSCs. LIDO, Lidocaine. B, Representative example of mIPSCs recorded from a spinal lamina I neuron that received monosynaptic Aδ-fiber input from attached dorsal root under baseline conditions and 25–30 min after HFS. Calibration: 100 ms, 20 pA. C, The cumulative probability curve of inter-event intervals for mIPSCs from the same neuron as that in A was shifted 25–30 min after HFS compared to baseline, indicating an enhanced transmitter release. In contrast, no change of the cumulative probability curve of amplitudes of mIPSCs was observed. A total of 118 events for baseline and 300 events for 25–30 min after HFS were plotted. D, Summary of the change in frequency and amplitude of mIPSCs from six neurons that received monosynaptic glutamatergic input from primary afferent Aδ-fibers or C-fibers.
Collectively, the data indicate that LTPGABA is due to a persistent increase in the release of GABA. The postsynaptic induction and the presynaptic expression of LTPGABA suggest that a retrograde messenger is involved.
Nitric oxide signaling mediates LTPGABA
NO is a potential retrograde messenger in the superficial dorsal horn where NO synthase (NOS) is expressed (Saito et al., 1994). We thus tested whether NO is required for the induction of LTPGABA. Bath application of the NO scavenger cPTIO (30 μm) completely prevented the induction of LTPGABA. Compared to baseline, IPSC amplitudes 25–30 min after HFS were significantly depressed (to 83 ± 8.0% of control; n = 5; p < 0.01; all neurons received either monosynaptic Aδ-fiber or monosynaptic C-fiber input; Fig. 5A,B). cPTIO is a membrane-impermeant scavenger of NO, suggesting that NO must diffuse through the extracellular space to exert its action on GABAergic terminals. We then tested whether NO is also required for DHPG-induced LTPGABA. When DHPG was applied to the bath solution in the presence of the NOS inhibitor l-NAME (100 μm), LTPGABA was not only fully blocked but converted into a long-term depression (LTD) of the IPSC amplitudes (to 73 ± 17% of control; n = 7; p < 0.01; Fig. 5C,D).
Figure 5.

Induction of LTPGABA on spinal lamina I neurons requires NO signaling. A, Example of an experiment where HFS was applied in the presence of the NO scavenger cPTIO (30 μm). The neuron received monosynaptic C-fiber input. Inset: Average of 20 IPSCs 0–5 min before and 25–30 min after conditioning stimulation. B, Average of five experiments where HFS was delivered in presence of cPTIO and failed to trigger LTPGABA. All neurons received either monosynaptic Aδ-fiber or C-fiber input from attached dorsal roots. C, Example of an experiment where DHPG was bath applied in presence of the NOS inhibitor l-NAME (100 μm). Wash-in of the group I mGluR agonist induced a long-lasting depression instead of a potentiation of the IPSC amplitudes. Inset: Average of 20 IPSCs 0–5 min before bath application of DHPG and 25–30 min after wash-out of the inducing agonist. D, Average of seven experiments where DHPG was applied in presence of l -NAME. Calibration: 10 ms, 50 pA (A); 10 ms, 100 pA (C).
Next we tested whether NO is sufficient to enhance GABA release. Bath application of the NO donor SNAP (200 μm) resulted in an increase of the IPSC amplitudes (to 166 ± 17% of control; n = 5; p < 0.001; 25–30 min after wash-in; Fig. 6A,B). The SNAP-induced potentiation was accompanied by a decrease of the PPR (to 0.80 ± 0.03 of the control value; n = 5; p < 0.01; Fig. 6C) and an increase of the CV−2 (to 3.46 ± 0.52 of the control value; n = 5; p < 0.05; Fig. 6D), consistent with an increase in the release probability of GABA.
Figure 6.

The NO donor SNAP enhances GABAergic IPSCs in spinal lamina I neurons and occludes HFS-induced LTPGABA. A, Example of an experiment where the NO donor SNAP (200 μm) was applied to the bath solution and potentiated the IPSC amplitudes. Inset: Average of 20 IPSCs 0–5 min before and 25–30 min after application of SNAP. B, Average time course of all five experiments where SNAP was applied. C, D, The SNAP-induced potentiation of IPSCs was accompanied by a decrease of the PPR (C) and an increase of the CV−2 (D), indicating an increase of GABA release. E, Pretreatment with the NO donor SNAP blocks LTPGABA induced by conditioning HFS. An example of IPSCs recorded from a spinal lamina I neuron that received monosynaptic Aδ-fiber input from an attached dorsal root is shown. The slice was incubated with SNAP for 30 min before the whole-cell configuration. Inset: Average of 20 IPSCs 0–5 min before and 25–30 min after conditioning stimulation. F, Average of five experiments where slices were pretreated with SNAP. All neurons received monosynaptic glutamatergic input from primary afferent Aδ-fibers or C-fibers. Calibration: 10 ms, 100 pA (A); 10 ms, 50 pA (E).
We also asked whether SNAP-induced potentiation occludes LTPGABA. When slices were pretreated with SNAP for at least 30 min before the whole-cell configuration was achieved, HFS failed to induce LTPGABA. Compared to control, the amplitudes of IPSCs 25–30 min after HFS were significantly depressed (to 83 ± 14.0% of control; n = 5; p < 0.05; all neurons received either monosynaptic Aδ- or monosynaptic C-fiber input; Fig. 6E,F). This suggests that SNAP-induced potentiation shares signaling pathways with HFS-induced potentiation at spinal GABAergic synapses and further supports the belief that NO signaling is required for LTPGABA.
Discussion
The present study revealed a novel form of synaptic plasticity in the spinal dorsal horn that was induced by conditioning stimulation of primary afferent Aδ- or C-fibers. The heterosynaptic LTP at GABAergic synapses, which was induced postsynaptically by group I mGluRs, expressed presynaptically and involved NO as a retrograde messenger (Fig. 7). This is the first report to demonstrate the following: (1) activity-dependent plasticity at inhibitory synapses in the spinal cord; and (2) a heterosynaptic form of LTP in spinal dorsal horn neurons.
Figure 7.

Schematic diagram of heterosynaptic LTP at GABAergic synapses with spinal lamina I neurons that receive monosynaptic glutamatergic input from primary afferent Aδ-fibers or C-fibers. Glutamate is released upon high-frequency conditioning stimulation and activates group I metabotropic glutamate receptors on spinal lamina I neurons. Nitric oxide is released via a postsynaptic, G-protein-dependent mechanism and enhances transmitter release at GABAergic synapses converging onto the same neuron.
LTPGABA in the superficial dorsal horn is heterosynaptic in nature
We applied the conditioning HFS to the dorsal root while spinal ionotropic glutamate receptors were blocked by selective antagonists in the bath solution. Thus, glutamate released from the terminals of primary afferent nerve fibers by the conditioning stimulation could not excite any postsynaptic neuron. Consequently, all disynaptic and polysynaptic pathways were blocked. Glutamate required for the activation of mGluRs could then derive from three sources only: (1) directly from monosynaptic primary afferent input converging onto the neurons under study; (2) from primary afferents contacting other spinal neurons via spillover; or (3) from ambient levels of glutamate, e.g., released from glial cells. The fact that LTPGABA could be triggered exclusively in those neurons that received monosynaptic input from Aδ-fibers or C-fibers suggests that any spillover of glutamate from primary afferents or ambient glutamate was not sufficient for the induction of LTPGABA. The most direct explanation of the present data is that glutamate was released from primary afferents and activated postsynaptic metabotropic glutamate receptors on spinal lamina I neurons, which then triggered LTPGABA by a heterosynaptic mechanism (Fig. 7).
LTPGABA in spinal dorsal horn as described here shares some fundamental features with previously described LTPGABA at synapses in the ventral tegmental area (Nugent et al., 2007). Both forms of synaptic plasticity are triggered by high-frequency conditioning stimulation of glutamatergic synapses; they are heterosynaptic in nature, expressed presynaptically, and involve NO as a retrograde messenger. Both forms of LTPGABA also differ in important aspects. While LTPGABA in the ventral tegmental area requires activation of NMDA receptors (Nugent et al., 2007), LTPGABA in the spinal dorsal horn was independent of NMDA receptors but instead required activation of group I mGluRs.
GABA and glycine can be co-released from the same synaptic vesicle at spinal inhibitory synapses (Jonas et al., 1998). If glycine and GABA were co-released from the same vesicles in the present study, presynaptically expressed LTP should then also apply to glycinergic transmission. Co-release of glycine and GABA has been shown to occur mainly in the immature spinal cord. Mixed GABAA/glycine receptor-mediated postsynaptic events become less frequent during maturation and disappear in spinal dorsal horn neurons by postnatal day 23 (Keller et al., 2001). Significant co-release may thus not to be expected in 25- to 35-d-old rats used in the present study. Furthermore, GABAergic and glycinergic spinal synapses are regulated differently under physiological conditions (Choi et al., 2009; Yowtak et al., 2011) and by peripheral nerve injury (Moore et al., 2002; Coull et al., 2003), suggesting a transmitter-specific regulation of synaptic release. It will be interesting to explore in future experiments if and how transmission at spinal glycinergic synapses might also be regulated by a heterosynaptic mechanism.
Group I mGluR activation is necessary and sufficient for presynaptically expressed LTPGABA
In the superficial spinal dorsal horn group I mGluRs are expressed on the somata of spinal neurons and on the terminals of GABAergic neurons (Jia et al., 1999; Tao et al., 2000). Here, induction of LTPGABA by HFS was prevented when mGluR1 and mGluR5 receptors were blocked. Activation of group I mGluRs by bath application of DHPG induced LTPGABA, and this required postsynaptic G-protein coupling. These results demonstrate that activation of postsynaptic G-protein-coupled receptors is not only necessary but also sufficient for the induction of LTPGABA in spinal lamina I neurons.
Activation of group I mGluRs leads to either LTP (Anwyl, 2009) or LTD (Lüscher and Huber, 2010) at excitatory synapses in various areas of the brain. The expression of synaptic plasticity can be presynaptic or postsynaptic, depending upon the type of synapse under study. At GABAergic synapses, activation of group I mGluRs mainly leads to presynaptically expressed LTD (Chevaleyre and Castillo, 2003; Heifets et al., 2008; Pan et al., 2008). In the only previous study where activation of group I mGluRs was necessary for the induction of LTP at GABAergic synapses on CA1 pyramidal cells, the expression was postsynaptic (Patenaude et al., 2003). Interestingly, we now provide evidence that LTP at GABAergic synapses with spinal lamina I neurons is expressed presynaptically. LTPGABA induced either by conditioning HFS of primary afferents or by bath application of DHPG was associated with a decrease of the PPR and an increase in the CV−2. Additionally, the frequency but not the amplitude of mIPSCs was increased by conditioning HFS. Together, these findings are consistent with a presynaptic expression of LTPGABA. Our results thus revealed a previously unrecognized presynaptic regulation of GABAergic transmission triggered by activation of postsynaptic group I mGluRs.
Nitric oxide enhances GABA release in the superficial spinal dorsal horn
NO is a prime candidate for a retrograde messenger in the spinal dorsal horn (Meller and Gebhart, 1993). Its functions for nociception are, however, still not fully understood. Inhibition of NO synthesis in the spinal cord reduces inflammatory and neuropathic pain, suggesting an overall pronociceptive effect of NO (Meller et al., 1992; Malmberg and Yaksh 1993). Spinal application of NO donors or cGMP analogs revealed conflicting results as pronociceptive and antinociceptive effects have been observed (Sousa and Prado, 2001; Tegeder et al., 2002; Schmidtko et al., 2008). The effect of NO on the spontaneous activity of spinal neurons varies with respect to the lamina location. In spinal laminae I and II, but not in lamina X, NO reduces the frequency of action potential discharges in a substantial fraction of neurons (Pehl and Schmid, 1997). Here we report that bath application of a NO donor enhanced GABAergic transmission at spinal lamina I neurons and that NO signaling was necessary for the induction of LTPGABA. The NO-sensitive guanylate cyclase is found in presynaptic terminals of spinal GABAergic synapses (Ding and Weinberg, 2006). Activation of presynaptic guanylate cyclase has been shown to control NO-dependent LTP not only at GABAergic synapses but also at glutamatergic synapses (Zhuo et al., 1994; Nugent et al., 2007). In fact, we have shown previously that NO signaling mediates one type of LTP at glutamatergic synapses between C-fibers and spinal lamina I projection neurons. NO was required for LTP induced by conditioning low-frequency stimulation (at 2 Hz), but not for LTP induced by HFS (100 Hz) (Ikeda et al., 2006). The latter conditioning stimulus was also used in the present study. The signaling pathways of NO-dependent, presynaptically expressed LTP at GABAergic and glutamatergic synapses may thus overlap, while the induction protocols are different. Our results further substantiate the emerging role of NO for plasticity at inhibitory synapses (Makara et al., 2007; Nugent et al., 2007; Niehaus et al., 2010).
Potential functional consequences of LTPGABA in nociceptive pathways
Previous work indicates that most neurons located in lamina I of the spinal cord, being either excitatory or inhibitory, receive monosynaptic input from either Aδ-fibers and/or C-fibers (Yoshimura and Jessell, 1990; Dahlhaus et al., 2005; Yasaka et al., 2007; Pinto et al., 2010), and many also express group I mGluRs (Alvarez et al., 2000). In the present study, we recorded from unidentified lamina I neurons and showed that LTPGABA could be induced in all neurons that received monosynaptic input from small primary afferents. In contrast, the same conditioning stimulation triggers LTP only at a subset of glutamatergic C-fiber synapses of lamina I neurons projecting to the parabrachial area (Ikeda et al., 2006). Thus, an enhancement of inhibition due to LTPGABA at many GABAergic synapses at lamina I neurons and parallel LTP at synapses of selected C-fibers with spinal projection neurons leads to an enhanced contrast between the afferent signal and the background “noise.” Thereby, heterosynaptic LTPGABA in lamina I may function as a filter for improving novelty detection and for enhancing the signal-to-noise ratio in spinal nociceptive pathways.
Inhibitory systems in spinal dorsal horn may serve at least five principle functions (Sandkühler, 2009, 2012) that would all be strengthened by LTPGABA. (1) Attenuation of nociceptive responses to an appropriate level to prevent hyperalgesia. (2) Muting of nociceptive neurons in the absence of any sensory stimulus to prevent spontaneous pain. (3) Functionally separating pathways for different sensory modalities. This is required for avoiding any crosstalk between low threshold sensory input such as touch and the nociceptive system. A cross-talk would otherwise lead to touch-evoked pain (allodynia). (4) Limiting the spread of excitation to somatotopically adequate areas of the spinal dorsal horn. A failure of this function of inhibitory spinal systems would lead to spreading sensations, including secondary hyperalgesia. (5) The prevention of LTP induction at synapses of nociceptive afferent nerve fibers. LTPGABA likely has a considerable impact on the inducibility of synaptic plasticity in the nociceptive system of the spinal dorsal horn. Enhanced GABAergic inhibition blocks both the induction and the maintenance of LTP of C-fiber-evoked field potentials in spinal dorsal horn in vivo (Hu et al., 2006). When inhibition via GABAA receptors is fully blocked, conditioning stimulation, which normally induces LTD, now leads to LTP induction (Miletic and Miletic, 2001). Likewise, blocking spinal inhibition during conditioning stimulation of dorsal root afferents converts depression of long-lasting excitatory optical responses in the superficial dorsal horn to facilitation (Ikeda et al., 2000). A globally enhanced GABAergic inhibition due to LTPGABA is therefore expected to hinder any subsequent LTP induction at glutamatergic synapses converging onto the same postsynaptic dorsal horn neurons.
In summary, the present study identified a novel mechanism through which spinal nociception can be modulated in an activity-dependent manner. It will be interesting to explore whether this form of synaptic plasticity is modified under inflammatory or neuropathic pain conditions.
Footnotes
This project was supported by the Austrian Science Fund (FWF Grant P19367) and by the Anniversary Fund (Jubilaeumsfond) of the Austrian National Bank to J.S.
The authors declare no competing financial interests.
References
- Alvarez FJ, Villalba RM, Carr PA, Grandes P, Somohano PM. Differential distribution of metabotropic glutamate receptors 1a, 1b, and 5 in the rat spinal cord. J Comp Neurol. 2000;422:464–487. doi: 10.1002/1096-9861(20000703)422:3<464::aid-cne11>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology. 2009;56:735–740. doi: 10.1016/j.neuropharm.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Baba H, Ji RR, Kohno T, Moore KA, Ataka T, Wakai A, Okamoto M, Woolf CJ. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Mol Cell Neurosci. 2003;24:818–830. doi: 10.1016/s1044-7431(03)00236-7. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Zhang M, Yin DM, Wen L, Ting A, Wang P, Lu YS, Zhu XH, Li SJ, Wu CY, Wang XM, Lai C, Xiong WC, Mei L, Gao TM. ErbB4 in parvalbumin-positive interneurons is critical for neuregulin 1 regulation of long-term potentiation. Proc Natl Acad Sci U S A. 2010;107:21818–21823. doi: 10.1073/pnas.1010669107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Choi IS, Nakamura M, Cho JH, Park HM, Kim SJ, Kim J, Lee JJ, Choi BJ, Jang IS. Cyclic AMP-mediated long-term facilitation of glycinergic transmission in developing spinal dorsal horn neurons. J Neurochem. 2009;110:1695–1706. doi: 10.1111/j.1471-4159.2009.06275.x. [DOI] [PubMed] [Google Scholar]
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sík A, De Koninck P, De Koninck Y. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 2003;424:938–942. doi: 10.1038/nature01868. [DOI] [PubMed] [Google Scholar]
- Dahlhaus A, Ruscheweyh R, Sandkühler J. Synaptic input of rat spinal lamina I projection and unidentified neurons in vitro. J Physiol. 2005;566:355–368. doi: 10.1113/jphysiol.2005.088567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JD, Weinberg RJ. Localization of soluble guanylyl cyclase in the superficial dorsal horn. J Comp Neurol. 2006;495:668–678. doi: 10.1002/cne.20901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodt H, Eder M, Frick A, Zieglgänsberger W. Precisely localized LTD in the neocortex revealed by infrared-guided laser stimulation. Science. 1999;286:110–113. doi: 10.1126/science.286.5437.110. [DOI] [PubMed] [Google Scholar]
- Drdla R, Gassner M, Gingl E, Sandkühler J. Induction of synaptic long-term potentiation after opioid withdrawal. Science. 2009;325:207–210. doi: 10.1126/science.1171759. [DOI] [PubMed] [Google Scholar]
- Heifets BD, Chevaleyre V, Castillo PE. Interneuron activity controls endocannabinoid-mediated presynaptic plasticity through calcineurin. Proc Natl Acad Sci U S A. 2008;105:10250–10255. doi: 10.1073/pnas.0711880105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XD, Ge YX, Hu NW, Zhang HM, Zhou LJ, Zhang T, Li WM, Han YF, Liu XG. Diazepam inhibits the induction and maintenance of LTP of C-fiber evoked field potentials in spinal dorsal horn of rats. Neuropharmacology. 2006;50:238–244. doi: 10.1016/j.neuropharm.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Asai T, Murase K. Robust changes of afferent-induced excitation in the rat spinal dorsal horn after conditioning high-frequency stimulation. J Neurophysiol. 2000;83:2412–2420. doi: 10.1152/jn.2000.83.4.2412. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Heinke B, Ruscheweyh R, Sandkühler J. Synaptic plasticity in spinal lamina I projection neurons that mediate hyperalgesia. Science. 2003;299:1237–1240. doi: 10.1126/science.1080659. [DOI] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jäger T, Sandkühler J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Marsala M, Sakabe T, Yaksh TL. Characterization of spinal amino acid release and touch-evoked allodynia produced by spinal glycine or GABAA receptor antagonist. Neuroscience. 2000;95:781–786. doi: 10.1016/s0306-4522(99)00461-3. [DOI] [PubMed] [Google Scholar]
- Jia H, Rustioni A, Valtschanoff JG. Metabotropic glutamate receptors in superficial laminae of the rat dorsal horn. J Comp Neurol. 1999;410:627–642. [PubMed] [Google Scholar]
- Jonas P, Bischofberger J, Sandkühler J. Corelease of two fast neurotransmitters at a central synapse. Science. 1998;281:419–424. doi: 10.1126/science.281.5375.419. [DOI] [PubMed] [Google Scholar]
- Keller AF, Coull JAM, Chéry N, Poisbeau P, De Koninck Y. Region-specific developmental specialization of GABA-glycine cosynapses in laminas I-II of the rat spinal dorsal horn. J Neurosci. 2001;21:7871–7880. doi: 10.1523/JNEUROSCI.21-20-07871.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein T, Magerl W, Hopf HC, Sandkühler J, Treede RD. Perceptual correlates of nociceptive long-term potentiation and long-term depression in humans. J Neurosci. 2004;24:964–971. doi: 10.1523/JNEUROSCI.1222-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makara JK, Katona I, Nyíri G, Németh B, Ledent C, Watanabe M, de Vente J, Freund TF, Hájos N. Involvement of nitric oxide in depolarization-induced suppression of inhibition in hippocampal pyramidal cells during activation of cholinergic receptors. J Neurosci. 2007;27:10211–10222. doi: 10.1523/JNEUROSCI.2104-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malan TP, Mata HP, Porreca F. Spinal GABAA and GABAB receptor pharmacology in a rat model of neuropathic pain. Anesthesiology. 2002;96:1161–1167. doi: 10.1097/00000542-200205000-00020. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Yaksh TL. Spinal nitric oxide synthesis inhibition blocks NMDA-induced thermal hyperalgesia and produces antinociception in the formalin test in rats. Pain. 1993;54:291–300. doi: 10.1016/0304-3959(93)90028-N. [DOI] [PubMed] [Google Scholar]
- Meller ST, Gebhart GF. Nitric oxide (NO) and nociceptive processing in the spinal cord. Pain. 1993;52:127–136. doi: 10.1016/0304-3959(93)90124-8. [DOI] [PubMed] [Google Scholar]
- Meller ST, Pechman PS, Gebhart GF, Maves TJ. Nitric oxide mediates the thermal hyperalgesia produced in a model of neuropathic pain in the rat. Neuroscience. 1992;50:7–10. doi: 10.1016/0306-4522(92)90377-e. [DOI] [PubMed] [Google Scholar]
- Miletic G, Miletic V. Contribution of GABA-A receptors to metaplasticity in the spinal dorsal horn. Pain. 2001;90:157–162. doi: 10.1016/s0304-3959(00)00398-5. [DOI] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ. Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci. 2002;22:6724–6731. doi: 10.1523/JNEUROSCI.22-15-06724.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus JL, Murali M, Kauer JA. Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur J Neurosci. 2010;32:108–117. doi: 10.1111/j.1460-9568.2010.07256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007;446:1086–1090. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- Pan B, Hillard CJ, Liu QS. D2 dopamine receptor activation facilitates endocannabinoid-mediated long-term synaptic depression of GABAergic synaptic transmission in midbrain dopamine neurons via cAMP-protein kinase A signaling. J Neurosci. 2008;28:14018–14030. doi: 10.1523/JNEUROSCI.4035-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patenaude C, Chapman CA, Bertrand S, Congar P, Lacaille JC. GABAB receptor- and metabotropic glutamate receptor-dependent cooperative long-term potentiation of rat hippocampal GABAA synaptic transmission. J Physiol. 2003;553:155–167. doi: 10.1113/jphysiol.2003.049015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehl U, Schmid HA. Electrophysiological responses of neurons in the rat spinal cord to nitric oxide. Neuroscience. 1997;77:563–573. doi: 10.1016/s0306-4522(96)00495-2. [DOI] [PubMed] [Google Scholar]
- Pinto V, Szûcs P, Lima D, Safronov BV. Multisegmental Aδ- and C-fiber input to neurons in lamina I and the lateral spinal nucleus. J Neurosci. 2010;30:2384–2395. doi: 10.1523/JNEUROSCI.3445-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poisbeau P, Patte-Mensah C, Keller AF, Barrot M, Breton JD, Luis-Delgado OE, Freund-Mercier MJ, Mensah-Nyagan AG, Schlichter R. Inflammatory pain upregulates spinal inhibition via endogenous neurosteroid production. J Neurosci. 2005;25:11768–11776. doi: 10.1523/JNEUROSCI.3841-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randić M, Jiang MC, Cerne R. Long-term potentiation and long-term depression of primary afferent neurotransmission in the rat spinal cord. J Neurosci. 1993;13:5228–5241. doi: 10.1523/JNEUROSCI.13-12-05228.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito S, Kidd GJ, Trapp BD, Dawson TM, Bredt DS, Wilson DA, Traystman RJ, Snyder SH, Hanley DF. Rat spinal cord neurons contain nitric oxide synthase. Neuroscience. 1994;59:447–456. doi: 10.1016/0306-4522(94)90608-4. [DOI] [PubMed] [Google Scholar]
- Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev. 2009;89:707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- Sandkühler J. Spinal plasticity and pain. In: Koltzenburg M, McMahon SB, editors. Wall and Melzack's textbook of pain. Ed 6. Philadelphia: Elsevier Churchill Livingstone; 2012. In press. [Google Scholar]
- Schmidtko A, Gao W, Sausbier M, Rauhmeier I, Sausbier U, Niederberger E, Scholich K, Huber A, Neuhuber W, Allescher HD, Hofmann F, Tegeder I, Ruth P, Geisslinger G. Cysteine-rich protein 2, a novel downstream effector of cGMP/cGMP-dependent protein kinase I-mediated persistent inflammatory pain. J Neurosci. 2008;28:1320–1330. doi: 10.1523/JNEUROSCI.5037-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoffnegger D, Ruscheweyh R, Sandkühler J. Spread of excitation across modality borders in spinal dorsal horn of neuropathic rats. Pain. 2008;135:300–310. doi: 10.1016/j.pain.2007.12.016. [DOI] [PubMed] [Google Scholar]
- Seagrove LC, Suzuki R, Dickenson AH. Electrophysiological characterisations of rat lamina I dorsal horn neurones and the involvement of excitatory amino acid receptors. Pain. 2004;108:76–87. doi: 10.1016/j.pain.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Sousa AM, Prado WA. The dual effect of a nitric oxide donor in nociception. Brain Res. 2001;897:9–19. doi: 10.1016/s0006-8993(01)01995-3. [DOI] [PubMed] [Google Scholar]
- Tao YX, Li YQ, Zhao ZQ, Johns RA. Synaptic relationship of the neurons containing a metabotropic glutamate receptor, mGluR5, with nociceptive primary afferent and GABAergic terminals in rat spinal superficial laminae. Brain Res. 2000;875:138–143. doi: 10.1016/s0006-8993(00)02608-1. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Schmidtko A, Niederberger E, Ruth P, Geisslinger G. Dual effects of spinally delivered 8-bromo-cyclic guanosine mono-phosphate (8-bromo-cGMP) in formalin-induced nociception in rats. Neurosci Lett. 2002;332:146–150. doi: 10.1016/s0304-3940(02)00938-2. [DOI] [PubMed] [Google Scholar]
- Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. 2010;11:823–836. doi: 10.1038/nrn2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torsney C, MacDermott AB. Disinhibition opens the gate to pathological pain signaling in superficial neurokinin 1 receptor-expressing neurons in rat spinal cord. J Neurosci. 2006;26:1833–1843. doi: 10.1523/JNEUROSCI.4584-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989;37:111–123. doi: 10.1016/0304-3959(89)90160-7. [DOI] [PubMed] [Google Scholar]
- Yasaka T, Kato G, Furue H, Rashid MH, Sonohata M, Tamae A, Murata Y, Masuko S, Yoshimura M. Cell-type-specific excitatory and inhibitory circuits involving primary afferents in the substantia gelatinosa of the rat spinal dorsal horn in vitro. J Physiol. 2007;581:603–618. doi: 10.1113/jphysiol.2006.123919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura M, Jessell T. Amino acid-mediated EPSPs at primary afferent synapses with substantia gelatinosa neurones in the rat spinal cord. J Physiol. 1990;430:315–335. doi: 10.1113/jphysiol.1990.sp018293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura M, Nishi S. Primary afferent-evoked glycine- and GABA-mediated IPSPs in substantia gelatinosa neurones in the rat spinal cord in vitro. J Physiol. 1995;482:29–38. doi: 10.1113/jphysiol.1995.sp020497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yowtak J, Lee KY, Kim HY, Wang J, Kim HK, Chung K, Chung JM. Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. Pain. 2011;152:844–852. doi: 10.1016/j.pain.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XC, Zhang Y-Q, Zhao ZQ. Involvement of nitric oxide in long-term potentiation of spinal nociceptive responses in rats. Neuroreport. 2005;16:1197–1201. doi: 10.1097/00001756-200508010-00013. [DOI] [PubMed] [Google Scholar]
- Zhuo M, Hu Y, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]