

Abstract
Corticotropin-releasing factor receptor type 1 (CRFR1) plays a major role in the regulation of neuroendocrine and behavioral responses to stress and is considered a key mediator of anxiety behavior. The globus pallidus external (GPe), a main relay center within the basal ganglia that is primarily associated with motor and associative functions, is one of the brain nuclei with the highest levels of CRFR1 expression in the rodent brain. However, the role of CRFR1 in the GPe is yet unknown. In the present study, we used a lentiviral-based system of RNA interference to show that knockdown of CRFR1 mRNA expression in the GPe of adult mice induces a significant increase in anxiety-like behavior, as revealed by the light–dark transfer, open-field, and elevated plus-maze tests. This effect was further confirmed by pharmacological administration of the selective CRFR1 antagonist NBI 30775 (1.75 μg/side) directly into the GPe. In the marble-burying test, blockade of CRFR1 in the GPe increased the percentage of marbles buried and the duration of burying behavior. Additionally, we present evidence suggesting that the enkephalin system is involved in the effect of GPe-CRFR1 on anxiety-like behavior. In contrast to the well established anxiogenic role of CRFR1 in the extended amygdala, our data reveal a novel anxiolytic role for CRFR1 in the GPe.
Introduction
Corticotropin-releasing factor (CRF), originally isolated from the hypothalamus (Vale et al., 1981), represents the final common pathway for the integration of the neuroendocrine stress responses in the brain. Chronic hyperactivation of the CRF system has been linked to stress-related emotional disorders such as anxiety and depression (Holsboer, 1999; Zorrilla and Koob, 2004; Bale, 2005).
CRF mediates physiological activities via activation of CRF receptor type 1 (CRFR1), which is widely expressed in the mammalian brain and pituitary, with high expression levels in the anterior pituitary, cerebral cortex, arcuate nucleus, amygdala, hippocampus, and olfactory bulb. Interestingly, CRFR1 is highly expressed in areas assumed to be primarily involved in sensory information processing and motor function, including the cerebellum, red nucleus, pontine gray, substantia nigra, and subthalamic nucleus; and expression is particularly dense in the globus pallidus external (GPe) (Van Pett et al., 2000).
The GPe is a central component of the basal ganglia circuitry, and contributes to the execution and refinement of movements (Kita, 2007). In addition to its primary role in motor planning and execution, several studies support GPe involvement in emotional behavior (Baumann et al., 1999; Critchley et al., 2001). To date, the specific role of CRFR1 in the GPe is unknown. However, there are some experimental data, as indicated below, suggesting a possible functional stress-related role for CRFR1 in the GPe. In a mouse model of central CRF overexpression, which reveals a number of physiological and autonomic symptoms related to chronic stress, CRFR1 mRNA expression was reduced mainly in the globus pallidus (Korosi et al., 2006). Consistent with this finding, CRF levels were significantly increased in the striatum, the main afferent to the GPe, of 72 h sleep-deprived rats, a model that incorporates multiple stress factors such as isolation, immobility, and general stress (Fadda and Fratta, 1997). In addition, CRF has been shown to stimulate the release of met-enkephalin, an anxiolytic endogenous opioid, in the globus pallidus of the rat via activation of CRFR1 (Sirinathsinghji et al., 1989). In light of these findings, we hypothesized that CRFR1 may mediate the involvement of the GPe in stress responses and emotional behavior.
In this study, we show that the levels of CRFR1 mRNA expression in the GPe are downregulated following exposure to stress. We proceeded to knockdown (KD) CRFR1 expression in the GPe, using a lentiviral vector expressing small interfering RNA targeted against the CRFR1 mRNA (lenti-siCRFR1). Intriguingly, in contrast to the well known anxiolytic effect of CRFR1 ablation (Müller et al., 2003) or CRFR1 KD (Sztainberg et al., 2010) in the limbic system, downregulation of CRFR1 mRNA expression in the GPe significantly increased anxiety-like behavior. This anxiogenic effect was further confirmed using a non-peptide CRFR1-selective antagonist, NBI 30775. In addition, we show that enkephalin expression is downregulated in the GPe of CRFR1 knock-out (KO) mice and that CRFR1 is expressed in a subset of GPe neurons that project to the striatum, together suggesting a possible anxiolytic mechanism by which CRFR1 modulates striatal enkephalin release.
Materials and Methods
Animals.
Adult male C57BL/6J mice (Harlan Laboratories) were used for lentiviral stereotaxic injections, pharmacological studies, and in situ hybridization staining. Adult male mice expressing GFP under the control of CRFR1 promoter (CRFR1-GFP) and CRFR1 KO mice were used for immunostaining experiments. Throughout the experiments, the animals were maintained in a temperature-controlled mouse facility (22 ± 1°C) on a reverse 12 light–dark cycle. Food and water were given ad libitum. All experimental protocols were approved by the Institutional Animal Care and Use Committee of The Weizmann Institute of Science.
Restraint stress.
Restraint stress was induced by putting the mice into a cut 50 ml plastic conical tube for 30 min. Mice were decapitated 1 h following stress initiation. A control group of mice that did not undergo restraint stress were decapitated to establish basal levels.
RNA preparation and real-time PCR.
The GPe of naive mice was bilaterally microdissected, using the Palkovits technique as previously described (Palkovits, 1983), for the quantification of CRFR1 mRNA levels. RNA was extracted using 5-PRIME PerfectPure RNA cell & Tissue kit (5 PRIME). Extracted RNA was treated with DNase to avoid false-positive results caused by DNA contamination. The RNA samples were reverse transcribed using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time PCRs were carried as previously described (Sztainberg et al., 2010).
In situ hybridization and cell counts.
Antisense and sense (control) RNA probes were generated using mouse CRFR1 cDNA and labeled with DIG-11-UTP using a labeling kit from Roche Molecular Biochemicals. In situ hybridization of CRFR1 mRNA was performed with the free-floating section method, as previously reported (Korosi et al., 2006). CRFR1-positive cell nuclei within the GPe and the reticular thalamic nucleus (Rt) were counted on two representative sections per animal from the lenti-siCRFR1 and the control virus group (n = 3 each group).
Lentiviral vector design, production, and validation.
The lenti-shCRFR1 vectors were designed as described previously (Sztainberg et al., 2010). In brief, four different short hairpin RNA (shRNA) target sequences from the open reading frame of the mouse CRFR1 gene were cloned into shRNA expression cassettes driven by the H1 promoter in the p156RRLsinPPTCMV-GFP-PREU3Nhe lentiviral construct (kindly provided by Dr. Inder Verma, the Salk Institute for Biological Studies, La Jolla, CA). The recombinant pseudotyped lentiviral vectors were generated by cotransfection of four plasmids into HEK293T cells, as described earlier (Tiscornia et al., 2006). The ability of lenti-shCRFR1 vectors to knock down CRFR1 expression was assessed by Western blot analysis and functional signaling as described previously (Sztainberg et al., 2010).
Intracerebral injections of lentiviral vectors.
A total of 20 adult (8 weeks) C57BL/6J male mice (Harlan Laboratories) received bilateral stereotaxic injections of lentivirus to the GPe as described previously (Sztainberg et al., 2010). Ten mice received the lenti-siCRFR1 and 10 mice the control virus, which consisted of the same lentiviral construct containing a scramble nonrelevant shRNA sequence. Data of four mice from the lenti-siCRFR1 group and one mouse from the control virus group were excluded because of inaccurate injections. The coordinates used for GPe stereotaxic injection were as follows: AP = −1.34 mm, L = ±3.5 mm, H = −4.75 mm relative to bregma (based on a calibration study indicating these coordinates as leading to the GPe in C57BL/6 strain). Confirmation of the accuracy of the injection site was done by immunostaining using biotinylated anti-GFP antibody.
Cannulation of animals.
A total of 18 adult (8 weeks) C57BL/6J male mice (Harlan Laboratories) were cannulated for the open-field test. An additional group of 18 mice were cannulated for the marble-burying test. Mice were anesthetized with isoflurane and placed in a stereotaxic apparatus (Angle Two Stereotaxic Instruments, myNeurolab, Leica Microsystems). Guide cannulae (28 gauge; 5.5 tubing length below pedestal; Plastics One) were inserted bilaterally into the GPe (AP = −0.2 mm, L = 3.2 mm, H = −3 mm, angle = 20°, relative to bregma). Cannulae were secured with C&B-Metabond kit (Parkell) and Jet acrylic dental cement (Lang Dental Manufacturing). Removable dummy cannulae with tip extending 0.5 mm below the guide cannula were placed to maintain potency until the time of injections. Mice were allowed a minimum of 5 d to recover from surgery before any intracerebral microinjection. After the completion of behavioral testing, mice were overdosed with chloral hydrate 35% and perfused intracardially. The brains were removed, sectioned in the coronal plane in 30 μm slices, and Nissl stained to determine the placement of cannula. Only animals with the tip of the cannula located in the GPe were included in the analysis.
Intracerebral drug administration.
Mice were anesthetized with isoflurane, dummy cannulae were removed, and an injection needle (33 ga) was inserted into the guide cannulae. The injection needle was attached by a polyethylene tube to a 2 μl Hamilton syringe. A total of 9 mice were bilaterally injected with the CRFR1-selective antagonist NBI 30775 (3.5 mg/ml) (a gift from Neurocrine Biosciences) in a volume of 0.5 μl (1.75 μg/side) at a constant rate of 0.12 μl/min. Injections were followed by a waiting period of 3 min to assure that full dose was delivered; a total of nine control mice were bilaterally injected with 0.5 μl vehicle (artificial CSF). Immediately after the injections mice were taken to the test room for 20 min habituation followed by a 10 min session of open-field test. One week after the open-field test, home cage locomotor activity was assessed. Mice were singly housed in standard cages and their basal activity was tracked during 2 consecutive days by an infrared-based automated system (Infra-Mot; TSE Systems). At 10:00 A.M. (dark phase) of the third day, mice were bilaterally injected with the CRFR1-selective antagonist NBI 30775 (1.75 μg/side). Immediately following the injections, mice were taken to the test room for 20 min habituation followed by an additional hour of general locomotor activity tracking.
Behavioral studies.
All behavioral studies were performed during the dark period (7:00 A.M. to 7:00 P.M.). In the CRFR1 knockdown experiment, mice were habituated to the test room for 2 h before any test. The open-field, the light–dark transfer, and the elevated plus-maze tests were used for the assessment of anxiety-like behavior. There were 2 recovery days between the tests. The parameters measured in the three tests were quantified using an automated video tracking system (VideoMot2; TSE Systems). For the assessment of general locomotor activity, mice were singly housed in standard cages and their activity was tracked during 3 consecutive days by an infrared-based automated system (Infra-Mot; TSE Systems GmbH). The open field consists of a Plexiglas box (50 × 50 × 22 cm). The arena was illuminated with 120 lx. Each mouse was placed in the corner of the apparatus to initiate a 10 min test session. The time spent in the center of the arena, the number of entries to the center, the latency to enter the center, and the total distance traveled were measured. The light–dark transfer test consists of a polyvinyl chloride box divided into a black dark compartment (14 × 27 × 26 cm) and a white 1050 lx illuminated light compartment (30 × 27 × 26 cm) connected by a small passage. Mice were placed in the dark zone to initiate a 5 min test session. The time spent in the light zone, the number of entries to the light zone, and the distance traveled in the light zone were measured. The elevated plus-maze apparatus consists of a gray polyvinyl chloride maze, comprised of a central part (5 × 5 cm), two opposing open arms (30.5 × 5 cm), and two opposing closed arms (30.5 × 5 × 15 cm). The apparatus was elevated at a height of 53.5 cm and the open arms were illuminated with 6 lx. Mice were placed in the center, facing an open arm to initiate a 5 min session test. The percentage of time spent in the open arms and the number of entries to the close arms were measured. The marble-burying apparatus consists of a gray polyvinyl chloride box (30 × 27 × 26 cm) containing 5 cm of autoclaved bedding with 20 marbles centrally arranged 4 by 5. Mice were placed in the corner of the apparatus and filmed for 30 min. Videos were scored by counting the number of unburied marbles, the duration of burying behavior, and the total distance traveled.
Immunostaining and densitometry analysis.
Immunostaining for GFP was done using biotinylated anti-GFP raised in rabbit as primary antibody (1:1000; ab69313, Abcam) followed by streptavidin-conjugated Cy2 (1:200; cat. number: 016-220-084, Jackson ImmunoResearch Laboratories). Immunostaining for CRF was done using anti-CRF raised in rabbit as primary antibody (1:000; kindly provided by Wylie Vale, Salk Institute for Biological Studies), and anti-rabbit Cy3 as secondary antibody (1:200; cat. number: 711-165-152, Jackson ImmunoResearch Laboratories). Immunostaining for Ucn1 was done using anti-Ucn1 raised in rabbit as primary antibody (1:1000; kindly provided by Wylie Vale), and anti-rabbit Cy3 as secondary antibody (1:200; cat. number: 711-165-152, Jackson ImmunoResearch Laboratories). Immunostaining for CRFR1 was done using anti-CRFR1/2 (C-20) raised in goat as primary antibody (1:1000; sc-1757, Santa Cruz Biotechnology) followed by an anti-goat biotinylated antibody (1:200, ab6884, Abcam) and further reacted with streptavidin-conjugated Cy3 (1:200, cat. number: 016-160-084, Jackson ImmunoResearch Laboratories). Immunostaining for enkephalin was done using anti-enkephalin antibody raised in rabbit as primary antibody (1:200; AB5026, Millipore Bioscience Research Reagents) and anti-rabbit Cy3 as secondary antibody (1:200; cat. number: 711-165-152, Jackson ImmunoResearch Laboratories). Immunostaining for parvalbumin (PV) was done using anti-PV antibody raised in rabbit as primary antibody (1:1000; PV-25, Swant) and anti-rabbit Cy3 as secondary antibody (1:200; cat. number: 711-165-152, Jackson ImmunoResearch Laboratories).
Digital fluorescent images of GPe and central amygdala (CeA) were collected on a E600 Nikon microscope (Nikon). The mean density enkephalin staining was analyzed using Image-Pro plus 4.1 software (Media Cybernetics) on five representative sections per animal from WT (n = 4) and CRFR1 KO (n = 7) mice.
Data analysis.
Data from each of the behavioral tests were analyzed using MANOVA (multifactor ANOVA with repeated measures) to assess an overall “treatment” effect on all monitored indices; this was followed by independent Student's t test (two tailed) of each index comparing the two “treatment” groups. Real-time PCR results were analyzed by Student's t test (two tailed). In situ hybridization cell count and enkephalin densitometry were analyzed by the Mann–Whitney U test. Heteroscedasticity in all the analyses was controlled by Levene's test for equality of variances.
Results
CRFR1 in the GPe is downregulated by stress and innervated by CRF terminals
In situ hybridization staining for CRFR1 on brain slices of naive mice revealed high levels of CRFR1 mRNA expression in the GPe (Fig. 1A). High levels of CRFR1 expression in the GPe were further confirmed by GFP immunostaining on brain slices obtained from mice expressing GFP under the control of CRFR1 promoter (CRFR1-GFP; Justice et al., 2008; Fig. 1B) and immunostaining of endogenous CRFR1 in WT mice (Fig. 1C). To test our hypothesis that the expression of CRFR1 in the GPe is modulated by stress, we compared GPe-CRFR1 mRNA levels of mice 1 h following restraint stress initiation to unstressed control mice. The GPe of each mouse was bilaterally microdissected, and the level of CRFR1 mRNA expression was analyzed by quantitative real-time PCR. The analysis revealed a significant decrease in CRFR1 mRNA expression in stressed mice compared to control nonstressed mice (t(8) = 3.2, p = 0.012, n = 4–6 for each group) (Fig. 1D).
Figure 1.
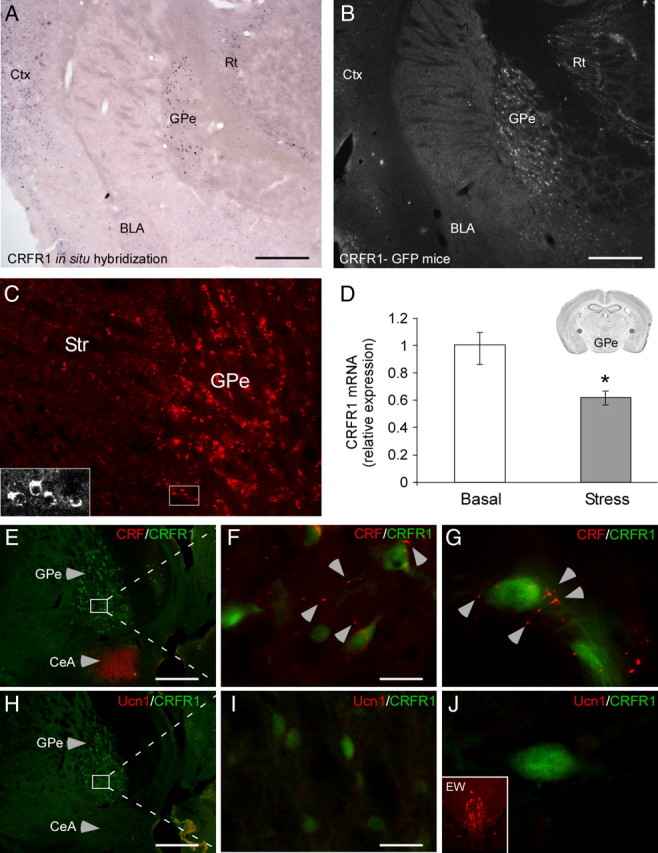
Reduced GPe-CRFR1 mRNA expression in mice exposed to stress and innervation by CRF terminals. A, CRFR1 mRNA expression in the GPe revealed by in situ hybridization staining (scale bar, 500 μm). B, Immunostaining for GFP showing CRFR1-driven GFP expression in the GPe of CRFR1-GFP mouse (scale bar, 500 μm). C, Immunostaining for endogenous CRFR1 in the GPe of a WT mouse. Expression of CRFR1 in the cell soma was confirmed by confocal microscopy. D, Quantitative real-time RT-PCR analysis of CRFR1 mRNA expression in the GPe of nonstressed (basal) and stressed mice (n = 5). Inset, Brain section adapted from the Paxinos and Franklin (2001) mouse brain atlas, showing the site of GPe microdissection. E, H, Sagittal GPe sections of CRFR1-GFP mice coimmunostained for CRF or Ucn1 and CRFR1-driven GFP (scale bar: E, H, 500 μm). F, G, Two additional magnifications reveal CRF terminals in the GPe adjacent to CRFR1-positive neurons (scale bar: F, 30 μm). I, J, In contrast to CRF, no Ucn1-immunoreactive terminals could be detected in the GPe (scale bar: I, 30 μm). J, Inset, Ucn1-positive immunostaining in the Edinger–Westphal (EW) nucleus. Bars represent mean ± SEM, *p < 0.05.
Sagittal brain slices obtained from CRFR1-GFP mice were coimmunostained with anti-GFP and anti-CRF antibodies or anti-GFP and anti-Urocortin 1 (Ucn1) antibodies to elucidate which of the two endogenous ligands of CRFR1 innervate the GPe. Neither CRF- (Fig. 1E) nor Ucn1- (Fig. 1H) positive neuron cell bodies were detected in the GPe. CRF-immunoreactive terminals were clearly observed throughout the GPe (Fig. 1F,G) whereas no Ucn1 immunoreactivity could be detected (Fig. 1I,J), suggesting that CRF is the endogenous CRFR1 ligand present in the GPe.
GPe-specific CRFR1 knockdown or pharmacological blockade increases anxiety-like behavior
To further elucidate the role of CRFR1 in the GPe, we used a lentiviral-based system expressing small interfering RNA against CRFR1, which was recently designed and constructed to knock down CRFR1 (Sztainberg et al., 2010). High-titer lentiviruses expressing shCRFR1 or control viruses (CV) were bilaterally injected into the GPe of C57BL/6 male mice. As the lentiviral construct also contains an enhanced green fluorescent protein (eGFP) reporter, we confirmed the level and distribution of shRNA expression by immunofluorescence staining using an anti-GFP antibody (Fig. 2A,B). KD of CRFR1 mRNA expression in the GPe was further confirmed by in situ hybridization staining (Fig. 2C,D). Mice injected with lenti-shCRFR1 showed a significant (∼60%) reduction in the number of CRFR1 mRNA-expressing cells in the GPe, relative to mice injected with control shRNA viruses (χ2 = 3.97, p = 0.046, n = 3 for each group) (Fig. 2E). No difference was observed in the number of CRFR1-positive cells in the Rt, a CRFR1-expressing nucleus adjacent to the GPe (Fig. 2F–H).
Figure 2.

siRNA-mediated knockdown of CRFR1 mRNA in the GPe. A, Parallel section schemata from the Paxinos and Franklin (2001) mouse brain atlas showing the site where the stereotaxic injection was directed. B, Representative immunofluorescence expression of GFP confirming GPe-specific lentiviral infection. C, D, Representative sections showing specific downregulation of CRFR1 mRNA expression in the GPe of CRFR1 KD mice (D), relative to CV mice (C) detected by in situ hybridization (scale bar, 500 μm). E, Quantification of positive GPe-CRFR1 mRNA-expressing cells of KD mice compared to CV mice, determined by in situ hybridization (n = 3). F, G, Representative sections showing CRFR1 mRNA expression in the Rt of CRFR1 KD mice (G), relative to CV mice (F) detected by in situ hybridization (scale bar, 500 μm). H, Quantification of positive Rt-CRFR1 mRNA-expressing cells of KD mice compared to CV mice, determined by in situ hybridization (n = 3). Bars represent mean ± SEM, *p < 0.05.
To evaluate the effect of GPe-CRFR1 knockdown on anxiety-like behavior, we used three different anxiety paradigms that take advantage of the natural inclination of mice to avoid exposure to predation: the light–dark transfer test, the open-field test, and the elevated plus-maze. In the light–dark transfer test, MANOVA for treatment (CV/KD), indices (time in light zone, entries into light zone, distance in light zone), and their interaction indicated that the treatment had an overall effect (treatment F(1,12) = 10.33; p = 0.007). Furthermore, the interaction treatment × indices was significant (F(2,11) = 5.08, p = 0.027), indicating that the treatment affected the different indices independently. Further t test analysis showed that knockdown of CRFR1 in the GPe increased anxiety, indicated by significant changes in time spent in the light zone (p = 0.019) (Fig. 3A), entries to the light zone (p = 0.028) (Fig. 3B), and distance in the light zone (p = 0.0025) (Fig. 3C). In the open-field test, MANOVA for treatment (CV/KD), indices (time in center, entries into center), and their interaction indicated that the treatment had an overall effect (treatment F(1,12) = 5.28; p = 0.04). Although the interaction treatment × indices was not significant, further t test analysis showed that knockdown of CRFR1 in the GPe increase anxiety, indicated by significant effects in time spent in the center of the arena (p = 0.034) (Fig. 3D) and a strong tendency in the number of visits to the center (p = 0.055) (Fig. 3E). No difference was found in the total distance traveled in the open field between the groups (Fig. 3F). In the elevated plus-maze test, the percentage of time spent in the open arms was decreased in the KD mice, although this difference did not reach statistical significance (p = 0.08) (Fig. 3G). No significant difference was found in the number of entries into the closed arms (Fig. 3H), ruling out the possibility that the observed anxiogenic effect is a result of reduced locomotor activity. In addition, 3 consecutive days of monitoring general home cage locomotor activity showed no significant differences in locomotion between the groups, neither in the light nor in the dark phase (Fig. 3I,J), further confirming that the observed behavioral changes are not due to changes in locomotor activity.
Figure 3.

Knockdown of CRFR1 in the GPe increases anxiety-like behavior. After bilateral stereotaxic injection of a lentiviral vector expressing CRFR1 siRNA to the GPe, mice were tested in a battery of three tests for anxiety-like behavior (n = 6–9). In the light–dark transfer test KD mice displayed a significant decrease in the time spent in the light zone (p = 0.019) (A), the entries into the light zone (p = 0.028) (B), and the distance traveled in the light zone (p = 0.0025) (C) relative to CV mice. In the open-field test, KD mice showed a significant decrease in the time spent in the center of the arena (p = 0.034) (D), and exhibited a tendency toward fewer entries into the center of the arena (p = 0.055) (E). No difference between the groups was found in the total distance traveled (F). In the elevated plus-maze test, KD mice displayed a trend toward decreased percentage of time spent in the open arms (p = 0.08) (G). No difference between the groups was found in the number of entries into the closed arms (H). No difference in general locomotor activity was found between the groups, measured during 3 consecutive days, in the light or the dark phases of the day (I, J). Bars represent mean ± SEM, *p < 0.05, **p < 0.01, ∼p < 0.1.
The anxiogenic effect of CRFR1 knockdown in the GPe was further confirmed by a pharmacological experiment using the CRFR1-selective antagonist NBI 30775. Mice were injected with NBI 30775 or vehicle in the GPe and then tested for anxiety-like behavior in the open-field test. MANOVA for treatment (NBI 30775/vehicle), indices (time in center, entries into center, total distance, latency), and their interaction indicated that the treatment had an overall effect (treatment F(1,18) = 5.91; p = 0.025). Although the interaction treatment × indices was not significant, further t test analysis showed that blocking of CRFR1 in the GPe increased anxiety, indicated by significant effects in time spent in the total distance traveled (p = 0.005) (Fig. 4C), the number of visits to the center (p = 0.04) (Fig. 4D), and a tendency in the latency to enter the center (p = 0.1) (Fig. 4F). No significant difference was found in the time spent in the center of the arena between the groups (Fig. 4E). In addition, no differences between the groups were found in home cage locomotor activity before (Fig. 4G) and at six time points following NBI 30775 or vehicle injection (Fig. 4H). An additional group of mice was used to assess the effect of NBI 30775 in the marble-burying test, where anxiety-like behavior is detected as an “active” response. Mice injected with NBI 30775 in the GPe showed a significant increase in the percentage of marbles buried (t(16) = 2.27, p = 0.037) (Fig. 4I,J) and the duration of burying behavior (t(16) = 2.57, p = 0.02) (Fig. 4K). No significant difference between the groups was found in the total distance traveled in the marble-burying test (Fig. 4L).
Figure 4.

Pharmacological blocking of CRFR1 in the GPe increases anxiety-like behavior. A, B, Representative image of cannula placement and site of injection in the GPe (A) and parallel brain section from the Paxinos and Franklin (2001) mouse brain atlas (B). Blocking of CRFR1 in the GPe (n = 9) resulted in a decrease in the total distance traveled (C), a decrease in the number of entries into the center (D), and an increase in the latency to enter the center of the open-field arena (F). No significant difference between the groups was found in the time spent in the center of the arena (E). No difference in general locomotor activity was found between the groups, measured 2 d before (G), and at 6 time points following NBI 30775 injection (H). In the marble-burying test (n = 8–10), blockade of CRFR1 increased the percentage of marbles buried (I). J, Representative image of the marble-burying test in mouse injected with vehicle (left photo) or NBI 30775 (right photo). K, Blockade of CRFR1 increased the duration of burying behavior. L, No significant difference between the groups was found in the total distance traveled in the marble-burying test. Bars represent mean ± SEM, *p < 0.05, ∼p = 0.1.
Reduced enkephalin expression in the GPe of CRFR1 KO mice
Based on a previous study demonstrating that CRF stimulates the release of the endogenous opioid enkephalin in the globus pallidus via CRFR1 (Sirinathsinghji et al., 1989), we hypothesized that the anxiety-like profile exhibited by KD mice might be explained by a GPe-CRFR1 knockdown-induced modulation of enkephalin release. To confirm this hypothesis, we assessed the GPe enkephalin expression level in CRFR1 KO mice. A densitometry analysis of immunofluorescence staining (Fig. 5A) showed that enkephalin is significantly reduced in the GPe of CRFR1 KO mice compared to their WT littermates (χ2 = 4.32, p = 0.037, n = 4–7 for each group; Fig. 5B). As an internal control, we show that there is no significant difference in enkephalin expression in the CeA (Fig. 5C).
Figure 5.

CRFR1 modulation of enkephalin expression in the GPe. A, Representative images of enkephalin immunostaining in the GPe and CeA of WT and CRFR1 KO mice. B, C, Quantification of enkephalin immunoreactivity in the GPe (B) and the CeA (C) of CRFR1 KO mice compared to WT mice, determined by densitometry analysis of immunofluorescence (n = 4–7). Bars represent mean ± SEM, *p < 0.05.
A subpopulation of CRFR1-immunoreactive cells in the GPe projects to the striatum
The GPe receives most of its enkephalin afferent fibers from the striatal indirect pathway (Haber and Watson, 1983; Gerfen and Young, 1988). In addition, it was previously shown that ∼1/3 of the GPe neurons project back to the striatum and are immunonegative for the Ca2+-binding protein PV (Kita and Kita, 2001). To understand how manipulation of CRFR1 cells in the GPe can affect enkephalin release from the striatum, we further tested whether CRFR1-positive neurons in the GPe are anatomically positioned to modulate the activity of the striatal enkephalin-producing cells. GPe coronal slices from CRFR1-GFP mice were coimmunostained with anti-GFP and anti-PV antibodies (Fig. 6A–F). A cell count analysis revealed that 69.5% of the GPe-CRFR1-positive neurons are positive for PV, and 30.5% of the GPe-CRFR1-positive neurons are PV negative (Fig. 6G), suggesting the existence of a subpopulation of GPe-CRFR1-positive cells that project to the striatum and can affect striatal-enkephalin release to the GPe (Fig. 6H).
Figure 6.

Colocalization of CRFR1 and PV in GPe cells. Immunostaining for PV (A, B), CRFR1-driven GFP (C, D), and their overlay (E, F) in the GPe, at two different magnifications. The white arrow shows a cell coexpressing PV and CRFR1. The blue arrow shows a cell positive for CRFR1 and negative for PV (scale bar: A–F, 250 μm). G, Quantification of CRFR1- and PV-positive cell colocalization presented as percentage of CRFR1-positive cells. H, Proposed model for modulation of enkephalin release from the striatum to the GPe, by GPe-CRFR1-positive neurons.
Discussion
The CRF-CRFR1 system in the limbic brain is thought to play a key role in the pathophysiology of anxiety disorders (Müller and Wurst, 2004). In the present study, we have determined that downregulation of CRFR1 in the GPe of adult mice induces an increase in anxiety-like behavior.
CRF has an important and well established role in the regulation of the HPA axis under basal and stress conditions (Rivier and Vale, 1983; Muglia et al., 1995). In addition, several experiments using animal models have shown an anxiogenic-like behavioral effect of CRF administration, leading to the suggestion that CRF may be involved in the pathophysiology of anxiety-related disorders (Arborelius et al., 1999; Heinrichs and Koob, 2004). Based on the different distribution of the two CRF receptor subtypes in the CNS, CRFR1 is considered to have a key role in mediating the CRF-elicited effects in anxiety (Van Pett et al., 2000). CRFR1 downregulation in the amygdala, either by antisense oligonucleotides (Liebsch et al., 1995) or by lentiviral-mediated small interference RNA (Sztainberg et al., 2010), results in an anxiolytic-like effect. These results are consistent with the behavioral phenotype of both the CRFR1-null mutant mouse (Smith et al., 1998; Timpl et al., 1998) and the limbic-specific conditional CRFR1 knock-out mouse (Müller et al., 2003). Here we found for the first time an anxiogenic effect of CRFR1 downregulation in the GPe, suggesting a novel anxiolytic role for the CRF-CRFR1 system in the basal ganglia.
The basal ganglia, including the striatum and the GP, are best known for their participation in motor planning and execution. However, considerable evidence now supports the notion that the basal ganglia is involved not only in motor control but also in cognitive, emotional, and motivational processes (Calabresi et al., 1997; Graybiel, 2005; Kopell and Greenberg, 2008). We confirmed the previously reported high expression levels of CRFR1 in the GPe (Van Pett et al., 2000; Justice et al., 2008) by in situ hybridization, by immunostaining of GFP on brain slices of transgenic mice expressing GFP under the control of the CRFR1 promoter, and immunostaining of endogenous CRFR1 in WT mice brain slices. Although the C-20 CRFR1/2 antibody has a potential for cross-reactivity with CRFR2, when considering the absence of CRFR2 in the GPe (Van Pett et al., 2000), it is very unlikely that CRFR2 is involved in any of the observed behavioral effects associated with knocking down or pharmacologically blocking CRFR1. Both techniques revealed a dense population of CRFR1-positive cells homogenously distributed in the GPe of adult mice, leading to the hypothesis that CRF acts in the GPe to mediate stress responses and emotional status. To confirm our hypothesis, we assessed CRFR1 mRNA expression levels in the GPe following restraint stress. Compared to naive animals, animals exposed to stress showed a significant downregulation of CRFR1 in the GPe. This result can be interpreted as a stress-induced inhibition of CRFR1 function in the GPe, here proposed to be anxiolytic. Our finding is in accordance with a previous study showing that in a mouse model of central CRF overexpression, which reveals a number of physiological and autonomic symptoms related to chronic stress, CRFR1 mRNA expression is reduced in the globus pallidus (Korosi et al., 2006).
The endogenous ligands of CRFR1 in the mouse brain are CRF and Ucn1 (Reul and Holsboer, 2002). To elucidate which of the ligands innervate the GPe-CRFR1-positive cells, GPe slices of CRFR1-GFP mice were coimmunostained with anti-GFP and anti-CRF antibodies or anti-GFP and anti-Ucn1 antibodies. Neither CRF- nor Ucn1-positive neurons were detected in the GPe. Whereas no Ucn1-immunoreactive terminals could be detected, CRF-immunoreactive terminals were clearly observed throughout the GPe, suggesting that CRF is the endogenous CRFR1 ligand in the GPe. The GPe receives most of its afferent fibers from the striatal indirect pathway coexpressing GABA (Kita and Kitai, 1988) and enkephalin (Haber and Watson, 1983; Gerfen and Young, 1988). In animals that have been pretreated with intracerebroventricular injections of colchicine, we could not find cell bodies with CRF immunoreactivity in the striatum (data not shown), in accordance with previous studies (Keegan et al., 1994; Alon et al., 2009). However, some nerve processes with CRF immunoreactivity could be detected projecting from the central amygdala to the most ventral part of the GPe. Further detailed studies are needed to identify the source of the CRF afferents to the GPe.
To further identify the role of CRFR1 in the GPe, we designed and constructed a lentiviral vector expressing small interference RNA against CRFR1 to genetically downregulate its expression in the GPe. We found that knockdown of CRFR1 in the GPe induced a significant increase in anxiety-like behavior in three different anxiety paradigms. This anxiogenic effect was further confirmed in the open-field and the marble-burying tests by blocking the CRFR1 activity with the selective antagonist NBI 30775. Because the GPe is primarily implicated in motor function, we assessed, after both experiments, the general home cage locomotor activity during 3 consecutive days using an automated system. We found no significant differences in locomotion between KD and CV mice, and between NBI 30775- and vehicle-injected mice, in the light and the dark phases of the day. It is therefore unlikely that the observed anxiogenic effect of GPe-CRFR1-specific knockdown or pharmacological blockade can be attributed to a general decrease in locomotor activity.
In a previous study, it was shown that CRF, when delivered into the GP, induces the release of enkephalin (Sirinathsinghji et al., 1989), an anxiolytic endogenous opioid (König et al., 1996; Kang et al., 2000; Ragnauth et al., 2001) that is released in the GPe by afferent neurons in the striatum. Based on these results, we suggest a mechanism in which reduced CRFR1 expression increases anxiety-like behavior in part by affecting enkephalin release. Indeed, we found that in the absence of CRFR1 in CRFR1 KO mice, the enkephalin expression is specifically downregulated in the GPe. Moreover, we found that ∼30% of the CRFR1-positive cells in the GPe are PV negative, a GPe population that has been shown to project back to the striatum (Kita and Kita, 2001). These findings suggest the existence of a reciprocal loop by which CRFR1-positive cells in the GPe modulate release of enkephalin from the striatum to the GPe, thereby reducing anxiety. However, further detailed tracing studies and functional modulation of the enkephalinergic system in the GPe by CRFR1 activity are needed to evaluate the proposed circuit hypothesis. Finally, considering the high comorbidity between anxiety and basal-ganglia motor disorders (Rosenblatt and Leroi, 2000), and the fact that CRF is extremely decreased in the caudate/putamen of Huntington's disease and Alzheimer patients (Bissette et al., 1985; De Souza et al., 1987), we suggest a new link between motor pathways and stress responsive and emotive pathways, which are integrated in part through the action of CRF and CRFR1 in the GPe.
Footnotes
This work was supported by an FP7 Grant from the European Research Council (#260463); a research grant from the Israel Science Foundation; a research grant from Roberto and Renata Ruhman; a research grant from the Legacy Heritage Biomedical Science Partnership; a research grant from the Israel Ministry of Health; a grant from Mr. and Mrs. Mike Kahn; a research grant from Jorge David Ashkenazi, a research grant from Mr. and Mrs. Barry Wolfe; and a research grant from Nella and Leon Benoziyo Center for Neurosciences. We thank S. Ovadia for his devoted assistance with animal care. We thank Dr. D. E. Grigoriadis (Neurocrine Biosciences, San Diego, CA) for providing the CRFR1 antagonist NBI 30775. A.C. is the incumbent of the Philip Harris and Gerald Ronson Career Development Chair.
References
- Alon T, Zhou L, Pérez CA, Garfield AS, Friedman JM, Heisler LK. Transgenic mice expressing green fluorescent protein under the control of the corticotropin-releasing hormone promoter. Endocrinology. 2009;150:5626–5632. doi: 10.1210/en.2009-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol. 1999;160:1–12. doi: 10.1677/joe.0.1600001. [DOI] [PubMed] [Google Scholar]
- Bale TL. Sensitivity to stress: dysregulation of CRF pathways and disease development. Horm Behav. 2005;48:1–10. doi: 10.1016/j.yhbeh.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Baumann B, Danos P, Krell D, Diekmann S, Leschinger A, Stauch R, Wurthmann C, Bernstein HG, Bogerts B. Reduced volume of limbic system-affiliated basal ganglia in mood disorders: preliminary data from a postmortem study. J Neuropsychiatry Clin Neurosci. 1999;11:71–78. doi: 10.1176/jnp.11.1.71. [DOI] [PubMed] [Google Scholar]
- Bissette G, Reynolds GP, Kilts CD, Widerlöv E, Nemeroff CB. Corticotropin-releasing factor-like immunoreactivity in senile dementia of the Alzheimer type. Reduced cortical and striatal concentrations. JAMA. 1985;254:3067–3069. [PubMed] [Google Scholar]
- Calabresi P, De Murtas M, Bernardi G. The neostriatum beyond the motor function: experimental and clinical evidence. Neuroscience. 1997;78:39–60. doi: 10.1016/s0306-4522(96)00556-8. [DOI] [PubMed] [Google Scholar]
- Critchley HD, Melmed RN, Featherstone E, Mathias CJ, Dolan RJ. Brain activity during biofeedback relaxation: a functional neuroimaging investigation. Brain. 2001;124:1003–1012. doi: 10.1093/brain/124.5.1003. [DOI] [PubMed] [Google Scholar]
- De Souza EB, Whitehouse PJ, Folstein SE, Price DL, Vale WW. Corticotropin-releasing hormone (CRH) is decreased in the basal ganglia in Huntington's disease. Brain Res. 1987;437:355–359. doi: 10.1016/0006-8993(87)91651-9. [DOI] [PubMed] [Google Scholar]
- Fadda P, Fratta W. Stress-induced sleep deprivation modifies corticotropin releasing factor (CRF) levels and CRF binding in rat brain and pituitary. Pharmacol Res. 1997;35:443–446. doi: 10.1006/phrs.1997.0155. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Young WS., 3rd Distribution of striatonigral and striatopallidal peptidergic neurons in both patch and matrix compartments: an in situ hybridization histochemistry and fluorescent retrograde tracing study. Brain Res. 1988;460:161–167. doi: 10.1016/0006-8993(88)91217-6. [DOI] [PubMed] [Google Scholar]
- Graybiel AM. The basal ganglia: learning new tricks and loving it. Curr Opin Neurobiol. 2005;15:638–644. doi: 10.1016/j.conb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- Haber SN, Watson SJ. The comparison between enkephalin-like and dynorphin-like immunoreactivity in both monkey and human globus pallidus and substantia nigra. Life Sci. 1983;33(Suppl 1):33–36. doi: 10.1016/0024-3205(83)90437-x. [DOI] [PubMed] [Google Scholar]
- Heinrichs SC, Koob GF. Corticotropin-releasing factor in brain: a role in activation, arousal, and affect regulation. J Pharmacol Exp Ther. 2004;311:427–440. doi: 10.1124/jpet.103.052092. [DOI] [PubMed] [Google Scholar]
- Holsboer F. The rationale for corticotropin-releasing hormone receptor (CRH-R) antagonists to treat depression and anxiety. J Psychiatr Res. 1999;33:181–214. doi: 10.1016/s0022-3956(98)90056-5. [DOI] [PubMed] [Google Scholar]
- Justice NJ, Yuan ZF, Sawchenko PE, Vale W. Type 1 corticotropin-releasing factor receptor expression reported in BAC transgenic mice: implications for reconciling ligand-receptor mismatch in the central corticotropin-releasing factor system. J Comp Neurol. 2008;511:479–496. doi: 10.1002/cne.21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W, Wilson SP, Wilson MA. Overexpression of proenkephalin in the amygdala potentiates the anxiolytic effects of benzodiazepines. Neuropsychopharmacology. 2000;22:77–88. doi: 10.1016/S0893-133X(99)00090-1. [DOI] [PubMed] [Google Scholar]
- Keegan CE, Herman JP, Karolyi IJ, O'Shea KS, Camper SA, Seasholtz AF. Differential expression of corticotropin-releasing hormone in developing mouse embryos and adult brain. Endocrinology. 1994;134:2547–2555. doi: 10.1210/endo.134.6.8194481. [DOI] [PubMed] [Google Scholar]
- Kita H. Globus pallidus external segment. Prog Brain Res. 2007;160:111–133. doi: 10.1016/S0079-6123(06)60007-1. [DOI] [PubMed] [Google Scholar]
- Kita H, Kita T. Number, origins, and chemical types of rat pallidostriatal projection neurons. J Comp Neurol. 2001;437:438–448. doi: 10.1002/cne.1294. [DOI] [PubMed] [Google Scholar]
- Kita H, Kitai ST. Glutamate decarboxylase immunoreactive neurons in rat neostriatum: their morphological types and populations. Brain Res. 1988;447:346–352. doi: 10.1016/0006-8993(88)91138-9. [DOI] [PubMed] [Google Scholar]
- König M, Zimmer AM, Steiner H, Holmes PV, Crawley JN, Brownstein MJ, Zimmer A. Pain responses, anxiety and aggression in mice deficient in pre-proenkephalin. Nature. 1996;383:535–538. doi: 10.1038/383535a0. [DOI] [PubMed] [Google Scholar]
- Kopell BH, Greenberg BD. Anatomy and physiology of the basal ganglia: implications for DBS in psychiatry. Neurosci Biobehav Rev. 2008;32:408–422. doi: 10.1016/j.neubiorev.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Korosi A, Veening JG, Kozicz T, Henckens M, Dederen J, Groenink L, van der Gugten J, Olivier B, Roubos EW. Distribution and expression of CRF receptor 1 and 2 mRNAs in the CRF over-expressing mouse brain. Brain Res. 2006;1072:46–54. doi: 10.1016/j.brainres.2005.12.034. [DOI] [PubMed] [Google Scholar]
- Liebsch G, Landgraf R, Gerstberger R, Probst JC, Wotjak CT, Engelmann M, Holsboer F, Montkowski A. Chronic infusion of a CRH1 receptor antisense oligodeoxynucleotide into the central nucleus of the amygdala reduced anxiety-related behavior in socially defeated rats. Regul Pept. 1995;59:229–239. doi: 10.1016/0167-0115(95)00099-w. [DOI] [PubMed] [Google Scholar]
- Muglia L, Jacobson L, Dikkes P, Majzoub JA. Corticotropin-releasing hormone deficiency reveals major fetal but not adult glucocorticoid need. Nature. 1995;373:427–432. doi: 10.1038/373427a0. [DOI] [PubMed] [Google Scholar]
- Müller MB, Wurst W. Getting closer to affective disorders: the role of CRH receptor systems. Trends Mol Med. 2004;10:409–415. doi: 10.1016/j.molmed.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Müller MB, Zimmermann S, Sillaber I, Hagemeyer TP, Deussing JM, Timpl P, Kormann MS, Droste SK, Kühn R, Reul JM, Holsboer F, Wurst W. Limbic corticotropin-releasing hormone receptor 1 mediates anxiety-related behavior and hormonal adaptation to stress. Nat Neurosci. 2003;6:1100–1107. doi: 10.1038/nn1123. [DOI] [PubMed] [Google Scholar]
- Palkovits M. Punch sampling biopsy technique. Methods Enzymol. 1983;103:368–376. doi: 10.1016/s0076-6879(83)03025-6. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KB. The mouse brain in stereotaxic coordinates. 2nd edition. San Diego: Academic; 2001. [Google Scholar]
- Ragnauth A, Schuller A, Morgan M, Chan J, Ogawa S, Pintar J, Bodnar RJ, Pfaff DW. Female preproenkephalin-knockout mice display altered emotional responses. Proc Natl Acad Sci U S A. 2001;98:1958–1963. doi: 10.1073/pnas.041598498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reul JM, Holsboer F. Corticotropin-releasing factor receptors 1 and 2 in anxiety and depression. Curr Opin Pharmacol. 2002;2:23–33. doi: 10.1016/s1471-4892(01)00117-5. [DOI] [PubMed] [Google Scholar]
- Rivier C, Vale W. Modulation of stress-induced ACTH release by corticotropin-releasing factor, catecholamines and vasopressin. Nature. 1983;305:325–327. doi: 10.1038/305325a0. [DOI] [PubMed] [Google Scholar]
- Rosenblatt A, Leroi I. Neuropsychiatry of Huntington's disease and other basal ganglia disorders. Psychosomatics. 2000;41:24–30. doi: 10.1016/S0033-3182(00)71170-4. [DOI] [PubMed] [Google Scholar]
- Sirinathsinghji DJ, Nikolarakis KE, Herz A. Corticotropin-releasing factor stimulates the release of methionine-enkephalin and dynorphin from the neostriatum and globus pallidus of the rat: in vitro and in vivo studies. Brain Res. 1989;490:276–291. doi: 10.1016/0006-8993(89)90245-x. [DOI] [PubMed] [Google Scholar]
- Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee KF. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron. 1998;20:1093–1102. doi: 10.1016/s0896-6273(00)80491-2. [DOI] [PubMed] [Google Scholar]
- Sztainberg Y, Kuperman Y, Tsoory M, Lebow M, Chen A. The anxiolytic effect of environmental enrichment is mediated via amygdalar CRF receptor type 1. Mol Psychiatry. 2010;15:905–917. doi: 10.1038/mp.2009.151. [DOI] [PubMed] [Google Scholar]
- Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19:162–166. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, Verma IM. Production and purification of lentiviral vectors. Nat Protoc. 2006;1:241–245. doi: 10.1038/nprot.2006.37. [DOI] [PubMed] [Google Scholar]
- Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt JC, Chan RK, Li HY, Arias C, Prins GS, Perrin M, Vale W, Sawchenko PE. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Zorrilla EP, Koob GF. The therapeutic potential of CRF1 antagonists for anxiety. Expert Opin Investig Drugs. 2004;13:799–828. doi: 10.1517/13543784.13.7.799. [DOI] [PubMed] [Google Scholar]