

Abstract
Phosphorylation plays a central role in the dynamic regulation of the processing of the amyloid precursor protein (APP) and the production of amyloid-β (Aβ), one of the clinically most important factors that determine the onset of Alzheimer's disease (AD). This has led to the hypothesis that aberrant Aβ production associated with AD results from regulatory defects in signal transduction. However, conflicting findings have raised a debate over the identity of the signaling pathway that controls APP metabolism. Here, we demonstrate that activation of the c-Jun N-terminal protein kinase (JNK) is essential for mediating the apoptotic response of neurons to Aβ. Furthermore, we discovered that the functional loss of JNK signaling in neurons significantly decreased the number of amyloid plaques present in the brain of mice carrying familial AD-linked mutant genes. This correlated with a reduction in Aβ production. Biochemical analyses indicate that the phosphorylation of APP at threonine 668 by JNK is required for γ-mediated cleavage of the C-terminal fragment of APP produced by β-secretase. Overall, this study provides genetic evidence that JNK signaling is required for the formation of amyloid plaques in vivo. Therefore, inhibition of increased JNK activity associated with aging or with a pathological condition constitutes a potential strategy for the treatment of AD.
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by the presence of extracellular amyloid plaques in the brain formed by the progressive deposition of amyloid-β (Aβ). Aβ is a 4 kDa peptide that derives from the sequential cleavage of the amyloid precursor protein (APP) by β- and γ-secretases (Mattson, 2004). Mutations in genes associated with familial forms of AD (FAD) result in elevated production of Aβ that precedes disease pathology (Hardy, 1997; Bertram et al., 2010). These findings have led to the idea that the accumulation of Aβ is a primary cause of AD. Aβ neurotoxicity is mediated, at least in part, by enhancing the levels of reactive oxygen species (ROS) (Behl et al., 1994). Furthermore, a positive-feedback loop by which oxidative stress produced following the interaction of Aβ with the receptor for advanced glycation endproducts (RAGE) increases RAGE expression, thereby enhancing Aβ neurotoxicity, has been suggested (Yan et al., 1996).
Activation of the proapoptotic c-Jun N-terminal protein kinase (JNK) signaling pathway constitutes one possible mechanism by which ROS produced by the accumulation of Aβ impairs synaptic function and leads to neuronal loss. This is mostly demonstrated by the observation that Aβ-induced neuronal death is significantly reduced in cortical neurons lacking JNK expression (Morishima et al., 2001). JNK is a member of the mitogen-activated protein kinase (MAPK) family that phosphorylates and activates transcription factors of the AP-1 (activator protein-1) family, including c-Jun and ATF2, in response to various stresses (Davis, 2000). Analogous to other MAPKs, JNK is activated by phosphorylation at threonine (Thr) and tyrosine (Tyr) residues by two dual-specificity MAPK kinases (MKK4 and MKK7) (Wang et al., 2007a).
Consistent with a role of JNK signaling in mediating Aβ toxicity (Morishima et al., 2001), recent studies have demonstrated the neuroprotective effect of inhibiting JNK activity in rodent AD models (Braithwaite et al., 2010; Ramin et al., 2011). Furthermore, JNK-mediated APP phosphorylation at Thr (T) 668 has been proposed as a mechanism to prevent APP degradation and increase Aβ production in a neuroglioma cell line (Colombo et al., 2009). The importance of this finding is highlighted by evidence that T668 phosphorylation is increased in human AD brain (Lee et al., 2003). However, the demonstration that the cyclin-dependent kinase 5 (Cdk5) is responsible for phosphorylating APP at T668 in naturally degenerating CAD cells, a CNS-derived neuronal cell line used for studying neuronal cell biology and pathology, has challenged the idea that JNK is implicated in controlling APP metabolism under pathological conditions (Muresan and Muresan, 2007). In contrast, this study provided evidence that JNK phosphorylates APP to control its transport in distal neurites under normal conditions.
Conflicting findings regarding the role of JNK signaling in regulating APP processing may be attributed to differences in experimental conditions and emphasize the importance of using animal models to identify physiologically relevant regulatory mechanisms. Therefore, we decided to test the effect of the loss of JNK activity in the brain of mice that overexpress FAD-linked mutant genes to elucidate the requirement of JNK signaling in AD pathology.
Materials and Methods
Mice.
The mouse strains were maintained in a pathogen-free facility at the University of Manchester. All animal procedures were performed under license in accordance with the United Kingdom Home Office Animals (Scientific Procedures) Act (1986) and institutional guidelines.
Genotype determination of mice and tissues.
Offspring carrying flox alleles, creERT2, APP, and PS1 transgenes were identified by PCR on tail and brain tissue DNA, as previously described (Jankowsky et al., 2004; Wang et al., 2007b).
Tissue culture.
Primary cultures of cortical neurons were prepared from the cerebral cortices of 17-d-old embryos (E17) and cultured on poly-ornithine-precoated six-well plates in Neurobasal media containing B27 supplement, 1% penicillin/streptomycin, and 1% glutamine, as previously described (Dajas-Bailador et al., 2008). Cells carrying the creERT2 transgene were identified by PCRs on genomic DNA extracted from the embryos. Four days after plating, 50% of the media was replaced by fresh Neurobasal media containing B27 supplement minus AO, 1% penicillin/streptomycin, and 1% glutamine. Seven days after plating, neurons were incubated in 4-hydroxytamoxifen (4-OHT) (0.1 μm) diluted in 95% ethanol to induce gene deletion. Three days later, neurons were treated with Aβ42 (200 nm; American Peptide). Aβ42 solubilized in DMSO was diluted in physiological buffer before being added onto the cells. Where indicated, the cells were pretreated for 30 min with zVAD (10 μm; R&D Systems) or N-acetylcysteine (NAC) (1 mm; Sigma-Aldrich), or for 1 h with a neutralizing anti-RAGE antibody (50 μg/ml; R&D Systems). In Figure 5, neurons were used 7 d (Fig. 5B,C) and in Figure 6 neurons were used 11 d (Fig. 6B) after the addition of 4-OHT.
Figure 5.

MKK4 and MKK7 are required for APP phosphorylation at T668. A, mkk4/7fl/fl littermates carrying the AD disease genes with or without CaMKII-creERT2 were injected 1 month after birth with tamoxifen to produce 3- and 5-month (M)-old animals lacking MKK4 and MKK7 expression in the brain (mkk4/7Δbr) and suitable controls (mkk4/7wt). i, Endogenous JNK activity in brain extracts was measured by protein kinase assay. Radioactivity incorporated in the GST-c-Jun substrate was quantified by PhosphorImager. The autoradiography represents two distinct animals per genotype per time point. The data correspond to the mean ± SE (N = 4 animals). ii, Protein lysates were analyzed by immunoblot with specific antibodies. The data represent two distinct animals per genotype per time point. B, C, Cortical neurons were obtained from mkk4/7fl/fl mice carrying the APPswe/PS1dE9 transgene with (+) or without (−) CaMKII-creERT2 and treated with 4-OHT after 7 DIV to produce mkk4/7−/− neurons and suitable controls (mkk4/7+/+). Neurons were used 7 d after the addition of 4-OHT. Where indicated, the cells were treated for 2.5 h with DAPT (1 μm; Sigma-Aldrich) before being harvested. Cell lysates were analyzed by immunoblot using specific antibodies (B, Ci). The data are representative of two experiments. Cell survival was measured by MTT assay (ii). The amount of Aβ42 in cell lysates was measured by ELISA (Invitrogen) (iii). The data correspond to the mean ± SD (N = 2).
Figure 6.

The amyloidogenic cleavage of endogenous APP requires JNK signaling. A, Brains were obtained from 3- and 5-month (M)-old mkk4/7Δbr mice and control (mkk4/7wt) littermate. B, Cortical neurons lacking MKK4 and MKK7 (mkk4/7−/−) and suitable controls (mkk4/7+/+) were maintained in culture for 14 or 18 d. Lysates were analyzed by immunoblot using an antibody against APP (i). The amount of Aβ42 was measured by ELISA (IBL International) (ii). The data correspond to the mean ± SD (N = 2).
Preparation of cell lysates and immunoblot analysis.
Proteins were extracted from cells in Triton lysis buffer (Wang et al., 2007b). Twenty to 50 μg proteins were subjected to immunoblot analysis following SDS-PAGE with antibodies against c-Jun (9165), JNK1/2 (Santa Cruz; sc-571), MKK4 (3346), MKK7 (4172), act caspase 3 (9664), APP (Covance; SIG-39152-200), P-T668-APP (BioSource International; 443364), RAGE (Santa Cruz; sc-8230), or amino acids 1–17 of human Aβ (6E10; Sigma-Aldrich) to detect CTFβ. Antibodies were purchased from Cell Signaling Technology unless indicated otherwise. Immune complexes were detected by enhanced chemiluminescence with anti-goat, anti-rabbit, or anti-mouse IgG coupled to horseradish peroxidase as the secondary antibody.
Protein kinase assay.
Endogenous JNK activity was measured in cell lysates in the presence of [γ-32P]ATP following precipitation with glutathione S-transferase (GST)-c-Jun (Wang et al., 2007b). The radioactivity incorporated into the recombinant protein was quantitated after SDS-PAGE by PhosphorImager analysis.
Immunofluorescence.
Neurons were fixed in 4% paraformaldehyde (PFA) before being incubated with a specific antibody to MAP2 (Sigma-Aldrich; 4542) or to RAGE (Santa Cruz; sc-8230). Immune complexes were detected with secondary antibody conjugated to fluorescein or Texas Red (Jackson ImmunoResearch). Nuclei were stained with DAPI (5 μg/ml). Fluorescence images were viewed with an Olympus wide-field microscope.
Death assays.
Cell survival was quantified by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Caspase activity was measured by spectrofluorometer using the DEVD-AMC caspase 3-specific fluorogenic substrate (Alexis Biochemicals).
ELISA.
Intracellular concentration of Aβ42 was measured using a specific sandwich-type ELISA (human Aβ42 kit; Invitrogen; KHB3441; or mouse/rat Aβ42 kit; IBL International; 27721) according to the manufacturer's instruction.
Histological and immunohistochemical analyses.
Mice were anesthetized and perfused with 0.9% saline, followed by 4% PFA. The 30 μm cryosections of brain were blocked in PBS containing 10% goat serum and 0.1% Triton X-100 for 1 h at room temperature before being incubated overnight at 4°C with primary antibodies to MKK4 (1:100; BD Biosciences Pharmingen), MKK7 (1:100; Cell Signaling), or Aβ42 (1:500; Alpha Diagnostic). The following day, the slides were rinsed in PBS and incubated at room temperature for 1 h with secondary biotinylated anti-rabbit antibody. The slides were processed using the ABC detection kit (Vector Laboratories). The presence of the antigens was revealed using the DAB (diaminobenzidine) (brown) peroxidase substrate kit (Vector Laboratories).
Electron microscopy.
Brains were fixed with 2.5% PFA and 0.1% glutaraldehyde in phosphate buffer, and then dissected and small blocks of the cerebral cortex, hippocampus, and entorhinal cortex were postfixed with 1% osmium tetraoxide, dehydrated in methanol, and embedded in Epon. Semithin sections were stained with toluidine blue, and selected ultrathin sections, with uranyl acetate and lead citrate.
Results
The deletion of mkk4 and mkk7 protects cortical neurons from Aβ-induced apoptosis
Mice lacking mkk4 or mkk7 die before birth (Wang et al., 2007a). To circumvent this early lethality and thereby permit the study of the physiological function of JNK signaling in AD pathology, we developed novel mouse models in which specific exons of the mkk4 and mkk7 genes were flanked by LoxP sites [referred to as the flox (fl) allele] (Wang et al., 2007b; A. Hübner and R. J. Davis, unpublished observation). These sites, which do not interfere with the normal expression of the genes, constitute a binding domain for the DNA recombinase Cre. Homozygous mkk4fl and/or mkk7fl mice were mated with a transgenic line expressing Cre fused to a mutated form of the ligand binding domain of the estrogen receptor (ERT2) (Hayashi and McMahon, 2002). The ERT2 moiety ensures the cytoplasmic sequestration of Cre. Cortical neurons were prepared from the embryos and cultured for 7 d before being incubated with 4-OHT to trigger the nuclear translocation of Cre where it specifically recombines the fl alleles.
Immunoblot analysis confirmed that the inactivation of the mkk4 and mkk7 genes only occurred in homozygous flox neurons carrying the creERT2 transgene (Fig. 1A). The loss of MKK4 and/or MKK7 in matured neurons did not cause any obvious change in cell morphology (Fig. 1B,E). In subsequent experiments, cortical neurons expressing or not Cre and treated with 4-OHT will be referred to as (−/−) and (+/+) cells, respectively.
Figure 1.

The loss of MKK4 and MKK7 protects neurons against Aβ42-induced apoptosis. Cortical neurons displaying homozygous fl mutation for the mkk4 and/or mkk7 alleles and expressing (+) or not (−) CreERT2 were incubated for 72 h with 0.1 μm 4-OHT before being treated with Aβ42 (200 nm) for the indicated times. Where indicated, the cells were pretreated with zVAD (10 μm). A, Protein lysates were analyzed by immunoblot with antibodies against MKK4, MKK7, and JNK. B, E, Immunofluorescence was performed with a specific antibody to MAP2. The immune complex was detected with a secondary antibody conjugated to fluorescein (green). DNA was stained with DAPI (blue). Scale bar, 10 μm. C, Caspase 3 activity was measured by caspase assay. D, Cell survival was measured by MTT assay. The data correspond to the mean ± SE (N = 3).
To determine the functional consequence of the loss of MKK4 and/or MKK7 expression, the viability of mkk4/7+/+, mkk4−/−, mkk7−/−, and mkk4/7−/− neurons treated with Aβ42 was examined. Bright-field microscopy indicated that mkk4/7+/+ neurons underwent extensive cell death after Aβ42 stimulation. The loss of axon integrity, as demonstrated by the disappearance of MAP2 staining, correlated with the detection of fragmented nuclei characteristic of apoptotic cells (Fig. 1B). Consistently, caspase 3 activity was increased in neurons treated with Aβ42 (Fig. 1C). Aβ42-induced neuronal death was prevented by incubating the cells with zVAD, a well characterized caspase inhibitor (Fig. 1D). Similarly, Aβ42 induced the apoptotic death of mkk4−/− and mkk7−/− neurons (Fig. 1E). In contrast, mkk4/7−/− neurons remained viable (Fig. 1D,E) and did not display increased caspase 3 activity (Fig. 1C) following Aβ42 stimulation.
Consistent with the requirement of JNK to mediate Aβ-induced apoptosis (Morishima et al., 2001), Aβ42 treatment increased JNK activity in mkk4/7+/+ neurons with a maximum at 1 h (Fig. 2A). This was prevented following the loss of MKK4 and MKK7 (Fig. 2A) and significantly blocked in neurons pretreated with the antioxidant NAC (Fig. 2B). Impaired JNK activity in mkk4/7−/− neurons correlated with low level of c-Jun expression under both basal and stimulated conditions, supporting the evidence that JNK increases the transcriptional activity of c-Jun and the ability of c-Jun to control its own promoter (Davis, 2000) (Fig. 2C). Furthermore, immunoblot analysis confirmed the proteolytic cleavage of caspase 3 in mkk4/7+/+, but not in mkk4/7−/−, neurons stimulated with Aβ42 (Fig. 2C). In addition, mkk4 and mkk7 gene deletion prevented Aβ42-mediated increased RAGE expression (Fig. 2D). The functional importance of this finding is demonstrated by the ability of a neutralizing RAGE antibody to block JNK activation (Fig. 2E) and increased caspase 3 activity (Fig. 2F) in mkk4/7+/+ neurons incubated with Aβ42.
Figure 2.
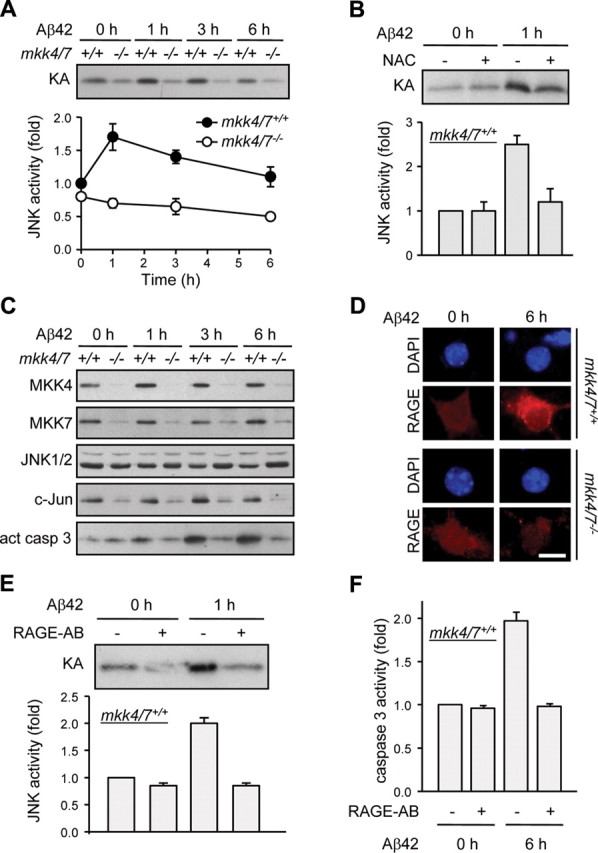
MKK4 and MKK7 are required for Aβ-induced JNK activation. Cortical neurons were incubated with Aβ42 (200 nm) for the indicated times 3 d after 4-OHT treatment to induce gene deletion. Where indicated, the cells were pretreated with NAC (1 mm) or incubated in presence of a neutralizing RAGE antibody (RAGE-AB) (50 μg/ml) before stimulation. A, B, E, Endogenous JNK activity was measured by protein kinase assay. Radioactivity incorporated in the GST-c-Jun substrate was quantified by PhosphorImager. Values are expressed as fold of untreated mkk4/7+/+ sample. The data correspond to the mean ± SE (N = 3) (A, B) or ± SD (N = 2) (E). C, Protein lysates were analyzed by immunoblot with antibodies against MKK4, MKK7, JNK1/2, c-Jun, and active (act) caspase 3. The data are representative of three independent experiments. D, Immunofluorescence was performed with a specific antibody to RAGE. The immune complex was detected with a secondary antibody conjugated to Texas Red (red). DNA was stained with DAPI (blue). Scale bar, 10 μm. F, Caspase 3 activity was measured by caspase assay. The data correspond to the mean ± SE (N = 3).
Overall, these studies provide genetic evidence that MKK4 and MKK7 are both required to mediate the apoptotic response of neurons by activating JNK following Aβ–RAGE interaction and the production of ROS.
Inactivation of MKK4 and MKK7 in the adult forebrain
To determine the physiological significance of the loss of active JNK in AD pathology, we created a mouse model in which the expression of MKK4 and MKK7 could be specifically abolished in neurons in the adult forebrain. This was achieved by crossing the mkk4/7fl/fl mice with a transgenic line expressing CreERT2 under the control of the Ca2+/calmodulin-dependent protein kinase II (CaMKII) gene promoter (Erdmann et al., 2007). One-month-old mkk4/7fl/fl littermates carrying or not the CaMKII-creERT2 transgene were injected intraperitoneally every day for 5 d with a nontoxic amount of tamoxifen (200 μg) to induce Cre activation and the neuronal specific deletion of the fl alleles.
Immunostaining of brain sections with antibodies to MKK4 and MKK7 showed that MKK4 was highly expressed in the cerebral cortex, while MKK7 was mostly detected in the CA2 and CA3 regions of the hippocampus (Fig. 3A). The inactivation of the mkk4 and mkk7 genes only occurred in the brain of mkk4/7fl/fl mice expressing Cre and injected with tamoxifen (Fig. 3A). This was confirmed by immunoblot analysis of brain extracts (Fig. 3B). Similar expression of MKK4 and MKK7 in the cerebellum of mkk4/7fl/fl and (CaMKII-creERT2)mkk4/7fl/fl mice injected with tamoxifen demonstrated the selective ablation of the proteins in specific areas of the brain (Fig. 3A). JNK activity was decreased by ∼80% following the deletion of the mkk4 and mkk7 genes (Fig. 3C). Consistent with our previous results in cell cultures (Fig. 2), this resulted in impaired c-Jun expression (Fig. 3B). In subsequent experiments, mkk4/7fl/fl and (CaMKII-creERT2)mkk4/7fl/fl mice injected with tamoxifen will be referred to as mkk4/7wt and mkk4/7Δbr animals, respectively.
Figure 3.

Inactivation of MKK4 and MKK7 in the adult forebrain. One-month-old mkk4/7fl/fl mice carrying (+) or not (−) the CaMKIIcreERT2 transgene were injected with tamoxifen to induce Cre-mediated recombination of the fl alleles. A, One month later, brain sections were analyzed by immunohistochemistry using antibodies against MKK4 or MKK7. Scale bar, 10 μm. HC, Hippocampus; DG, dentate gyrus. B, Protein lysates were analyzed by immunoblot with specific antibodies. The data are representative of two independent experiments. C, Endogenous JNK activity was measured by protein kinase assay. Radioactivity incorporated in the GST-c-Jun substrate was quantified by PhosphorImager. Values are expressed as fold of maximum. The data correspond to the mean ± SD (N = 2 animals).
The loss of MKK4 and MKK7 prevents amyloid plaque formation
To establish the requirement of JNK signaling in the onset and in the progression of AD, we examined the effect of the loss of MKK4 and MKK7 in the brain of mice that overexpress APP carrying the Swedish mutation (APPswe) and presenilin 1 lacking exon 9 (PS1dE9) (Jankowsky et al., 2004). APPswe is a better substrate for β-secretase than α-secretase (Citron et al., 1992), while PS1dE9 confers γ-secretase a constitutive activity. The deletion of exon 9 also results in a serine-to-cysteine substitution that alters the specificity of γ-secretase to favor the production of Aβ42 relative to the less pathogenic Aβ40 (Perez-Tur et al., 1995). As expected, expression of the APPswePS1dE9 transgene in mkk4/7wt mice resulted in amyloid deposition as soon as 3 months of age with a progressive increase in plaque number at 5 months (Fig. 4A,B). The amount of Aβ42 produced in the brain increased with a similar kinetics (Fig. 4C). Electron microscopy confirmed the presence of a large number of clustered dystrophic neurites filled with altered mitochondria and residual bodies around amyloid deposits characteristic of AD pathology (Fig. 4D).
Figure 4.
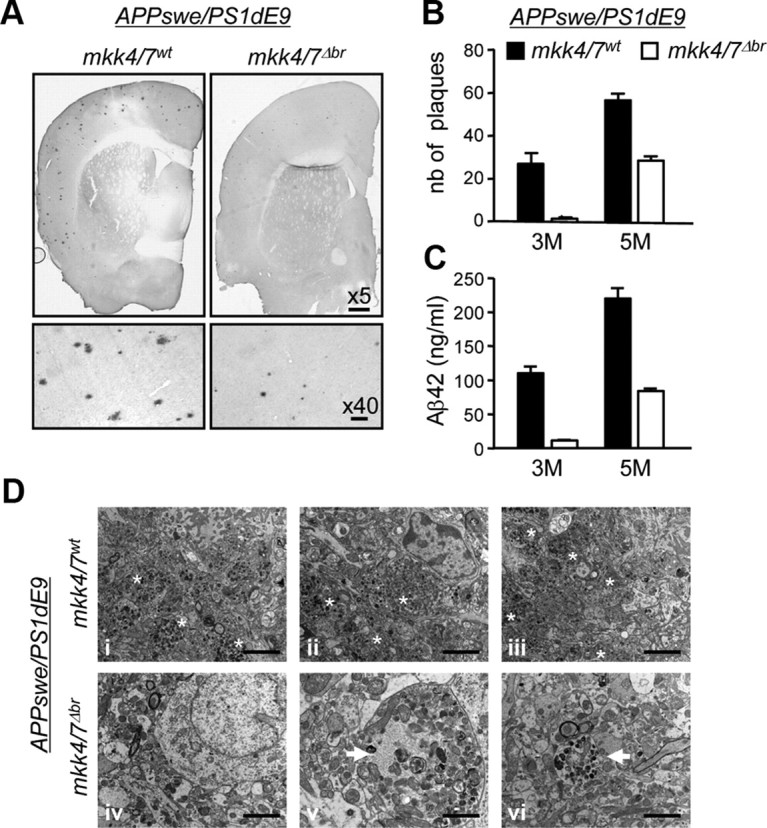
The loss of MKK4 and MKK7 prevents the formation of amyloid plaques. A breeding program was set up to create a mouse model harboring homozygous mkk4fl and mkkk7fl alleles together with a transgene encoding APPswe and PS1dE9, with or without CaMKII-creERT2. Littermates were injected 1 month after birth with tamoxifen, to produce animals lacking MKK4 and MKK7 expression in the brain (mkk4/7Δbr) and suitable controls (mkk4/7wt). A, The brains of 5-month-old mice were analyzed by immunohistochemistry using an antibody to Aβ42. Scale bars: for 5×, 200 μm; 40×, 20 μm. B, The number (nb) of plaques was counted in serial coronal brain sections at 3 and 5 months (M) after birth. C, The amount of Aβ42 was measured by ELISA (Invitrogen). The data correspond to the mean ± SE (N = 3 animals). D, Brain sections were analyzed by EM. The images are from three distinct animals per genotype (i–iii, mkk4/7wt; iv–vi, mkk4/7Δbr). Dystrophic neurites filled with altered mitochondria and residual bodies are clustered around amyloid deposits in mkk4/7wt mice (asterisks), whereas rare isolated dystrophic neurites (arrows) are detected in mkk4/7Δbr animals. Scale bars: i–iv, 2 μm; v, 1 μm; vi, 1.5 μm.
In contrast, the number of plaques was greatly reduced in the absence of MKK4 and MKK7 (Fig. 4A,B). In fact, plaques that did develop in the brain of the mkk4/7Δbr mice carrying the APPswe/PS1dE9 transgene were found in areas positive for MKK4 and MKK7 expression and JNK phosphorylation. This was expected since the deletion of fl genes following tamoxifen-induced Cre does not occur with 100% efficiency. Consistent with a reduction in Aβ deposition, the level of Aβ42 was significantly decreased following the loss of MKK4 and MKK7 (Fig. 4C). Furthermore, MKK4/7-deficient brains displayed a marked decrease of the number of dystrophic neurite clusters and only a few isolated neurites with residual bodies (Fig. 4D). Together, these results demonstrate that active JNK is required for the formation of amyloid plaques in vivo.
The loss of MKK4 and MKK7 prevents the phosphorylation of APP at T668
To understand the mechanism by which JNK signaling increases Aβ42 production, we investigated the effect of the loss of MKK4 and MKK7 expression on APP phosphorylation at T668, a site known to be phosphorylated by JNK (Standen et al., 2001) and implicated in the amyloidogenic cleavage of APP (Lee et al., 2003). In addition, phosphorylation of APP at T668 has been shown to be increased in human AD brain (Lee et al., 2003). Consistent with our previous data (Figs. 2A,D, 3C), the loss of MKK4 and MKK7 in the brain of mice expressing the APPswe/PS1dE9 transgene caused a reduction in RAGE expression and a significant decrease in JNK activity (Fig. 5Ai). This prevented the phosphorylation of APP at T668 (Fig. 5Aii). The low level of phospho (P)-T668-APP correlated with the accumulation of the full-length (FL) protein as well as a C-terminal fragment (CTF) produced after the cleavage of APP by α or β secretases (Fig. 5Aii). Further analyses using an antibody raised against amino acids 1–17 of human Aβ (6E10) confirmed that mkk4 and mkk7 gene deletion increased CTFβ (Fig. 5Aii).
The effect of mkk4 and mkk7 gene deletion on APP processing was tested in cortical neurons expressing the APPswe/PS1dE9 transgene, after 14 d in culture. Consistent with our in vivo data, the loss of MKK4 and MKK7 following Cre activation prevented APP phosphorylation at T668, causing an accumulation of APP and CTFβ (Fig. 5B). A similar phenotypic abnormality has been observed in cortical neurons defective in PS1 or expressing loss-of-function variants (Naruse et al., 1998). Therefore, we hypothesized that mkk4 and mkk7 gene deletion increased APP level by preventing γ-mediated cleavage of CTF. To test this hypothesis, we examined the effect of DAPT, a known inhibitor of γ-secretase. The results show that incubation of mkk4/7+/+ neurons with DAPT increased APP and CTF levels (Fig. 5Ci), as well as cell survival (Fig. 5Cii), to a similar extent as that associated with the loss of MKK4 and MKK7. Consistent with the observation that neurons lacking MKK4 and MKK7 are less sensitive to the expression of the APPswe/PS1dE9 transgene than wild-type cells, the level of Aβ42 was significantly lower in mkk4/7−/− than in mkk4/7+/+ neurons (Fig. 5Ciii). In addition, we found that the brain of 5-month-old mkk4/7Δbr mice and mkk4/7−/− neurons cultured for 18 d displayed higher levels of APP and CTF compared with wild-type (wt or +/+) samples (Fig. 6Ai,Bi). Consistently, the loss of MKK4 and MKK7 prevented increased production of Aβ42 in aging brains and neurons (Fig. 6Aii,Bii).
Together, these results provide strong evidence that increased JNK activity downstream of MKK4 and MKK7 is required for the amyloidogenic cleavage of mutant, but also endogenous, APP and Aβ42 production.
Discussion
In this study, we demonstrated that the loss of active JNK associated with the absence of both MKK4 and MKK7 protects neurons against Aβ-induced toxicity. Furthermore, we discovered that JNK signaling is required for amyloid plaque formation in vivo. Together, these results provide the first genetic demonstration that activation of JNK triggers the cascade of event that leads to AD pathology.
Phosphorylation plays a central role in the dynamic regulation of APP processing (Gandy and Greengard, 1994). This has led to the hypothesis that aberrant Aβ production associated with AD results from regulatory defects in signal transduction. Our results support this idea by providing a genetic link between amyloid plaque formation and increased JNK activity in neurons via the amplification of a positive-feedback loop associated with the phosphorylation of APP at T668 (Fig. 7). However, a previous study showed that, while JNK phosphorylates APP to control its transport in distal neurites under normal conditions, Cdk5 is responsible for phosphorylating APP in naturally degenerating CAD cells overexpressing or not APP (Muresan and Muresan, 2007). Furthermore, the role of T668 phosphorylation in promoting the amyloidogenic cleavage of APP (Lee et al., 2003; Vingtdeux et al., 2005) has been disputed by evidence that a knock-in mouse model in which T668 was replaced by an alanine residue displayed normal level of Aβ (Sano et al., 2006). Similarly, there is an ongoing debate over the role of phospho-T668 in regulating APP metabolism as a consequence of modulating its ability to interact with its binding partners. For example, Pin1 is a peptidyl-prolyl isomerase that binds the phosphorylated T668 motif of APP, leading to increased Aβ production (Akiyama et al., 2005). However, a role of Pin1 in promoting the nonamyloidogenic processing of APP has also been described previously (Pastorino et al., 2006). In addition, one study suggests that the interaction of Fe65 with the nonphosphorylated form of APP stabilizes APP and inhibits Aβ production (Ando et al., 2001), while others have found that overexpression of Fe65 promotes the production of Aβ (Taru and Suzuki, 2009).
Figure 7.

Increased JNK activity induces amyloid plaque formation and neuronal demise. Under normal conditions, Aβ is a relatively minor product in the brain because the nonamyloidogenic processing of APP by α- and γ-secretases prevails. α-secretase-mediated cleavage of APP generates secreted (s) APPα and CTFα. Processing of CTFα by γ secretase produces p3 and an APP intracellular domain (AICD). However, defects in Aβ clearance, associated with aging or a pathological condition, lead to a rise in basal Aβ level. Aβ interacts with RAGE to produce ROS and activates JNK via MKK4 and MKK7. Active JNK phophorylates AP1 transcription factors to regulate gene expression (i), and APP at T668 to enhance the amyloidogenic cleavage of APP (ii). CTFβ generated by β-secretase is processed by γ-secretase to produce a phosphorylated form of AICD, which can translocate to the nucleus (Chang et al., 2006), and more Aβ, which further stimulates JNK activity. Sustained high-level JNK activity in the brain induces caspase activation (iii) causing synaptic dysfunction (D'Amelio et al., 2011) and/or neuronal death. A key therapeutic strategy may be to interfere with the amplification of positive-feedback loops that shifts the balance toward the amyloidogenic pathway leading to increased Aβ42 production and AD pathology. sec, secretase.
Overall, these discrepancies may be attributed to differences in experimental conditions (wild-type vs mutant APP; endogenous vs ectopic expression; neurons in culture vs animal model). For example, T668 phosphorylation prevents γ-mediated cleavage of wild-type APP (Feyt et al., 2007), while APPswe mutant requires to be phosphorylated at T668 before being cleaved by γ-secretase (Vingtdeux et al., 2005). In contrast, the Swedish mutation that enhances the affinity of APP for β-secretase may bypass the requirement of T668 phosphorylation, which has been proposed to facilitate the cleavage of APP by β-secretase in primary cortical neurons (Lee et al., 2003). Consequently, the functional disruption of JNK signaling may affect β-mediated cleavage of wild-type APP, but not APPswe mutant. This is consistent with the detection of CTFβ in the brain of APPswe/PS1dE9mkk4/7Δbr mice. Together, these apparent conflicting findings support the hypothesis that APP metabolism is regulated by distinct signaling mechanisms during normal aging and in AD condition in which mutant or wild-type forms of APP are often expressed above physiological levels.
Therefore, although we show that JNK signaling is implicated in the processing of endogenous APP, it will be important to determine the requirement of MKK4 and MKK7 in regulating the cleavage of overexpressed wild-type APP. This will help to clarify the specific requirement of T668 phosphorylation in controlling β cleavage of APP. Together, this information will enable us to predict (1) the usefulness of JNK inhibitors to treat late-onset AD patients that do not display the Swedish mutation and (2) take into account the side effects of therapeutic intervention based on inhibiting JNK activity by considering the physiological role of APP phosphorylation at T668 in neurite extension, a process known to be dependent on the JIP-3/JNK pathway (Muresan and Muresan, 2005). Another key question will be to examine the physiological role of JNK signaling in mediating amyloid toxicity in vivo. These issues still need to be addressed before the therapeutic implication of inhibiting JNK activity for treating human AD becomes clear.
Footnotes
This work was supported by a scholarship from the Medical Research Council (S.M.) and by a grant from the Alzheimer's Research United Kingdom (C.T.). We thank A. Whitmarsh (University of Manchester, Manchester, UK) for critically reviewing this work, and the technicians at the animal facility for looking after the mice.
The authors declare no competing financial interests.
References
- Akiyama H, Shin RW, Uchida C, Kitamoto T, Uchida T. Pin1 promotes production of Alzheimer's amyloid beta from beta-cleaved amyloid precursor protein. Biochem Biophys Res Commun. 2005;336:521–529. doi: 10.1016/j.bbrc.2005.08.130. [DOI] [PubMed] [Google Scholar]
- Ando K, Iijima KI, Elliott JI, Kirino Y, Suzuki T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of beta-amyloid. J Biol Chem. 2001;276:40353–40361. doi: 10.1074/jbc.M104059200. [DOI] [PubMed] [Google Scholar]
- Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid beta protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Braithwaite SP, Schmid RS, He DN, Sung ML, Cho S, Resnick L, Monaghan MM, Hirst WD, Essrich C, Reinhart PH, Lo DC. Inhibition of c-Jun kinase provides neuroprotection in a model of Alzheimer's disease. Neurobiol Dis. 2010;39:311–317. doi: 10.1016/j.nbd.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KA, Kim HS, Ha TY, Ha JW, Shin KY, Jeong YH, Lee JP, Park CH, Kim S, Baik TK, Suh YH. Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol Cell Biol. 2006;26:4327–4338. doi: 10.1128/MCB.02393-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Colombo A, Bastone A, Ploia C, Sclip A, Salmona M, Forloni G, Borsello T. JNK regulates APP cleavage and degradation in a model of Alzheimer's disease. Neurobiol Dis. 2009;33:518–525. doi: 10.1016/j.nbd.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Jones EV, Whitmarsh AJ. The JIP1 scaffold protein regulates axonal development in cortical neurons. Curr Biol. 2008;18:221–226. doi: 10.1016/j.cub.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L, Moreno S, Bacci A, Ammassari-Teule M, Marie H, Cecconi F. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat Neurosci. 2011;14:69–76. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Erdmann G, Schütz G, Berger S. Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci. 2007;8:63–72. doi: 10.1186/1471-2202-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feyt C, Pierrot N, Tasiaux B, Van Hees J, Kienlen-Campard P, Courtoy PJ, Octave JN. Phosphorylation of APP695 at Thr668 decreases gamma-cleavage and extracellular Abeta. Biochem Biophys Res Commun. 2007;357:1004–1010. doi: 10.1016/j.bbrc.2007.04.036. [DOI] [PubMed] [Google Scholar]
- Gandy S, Greengard P. Regulated cleavage of the Alzheimer amyloid precursor protein: molecular and cellular basis. Biochimie. 1994;76:300–303. doi: 10.1016/0300-9084(94)90162-7. [DOI] [PubMed] [Google Scholar]
- Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154–159. doi: 10.1016/s0166-2236(96)01030-2. [DOI] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305–318. doi: 10.1006/dbio.2002.0597. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kao SC, Lemere CA, Xia W, Tseng HC, Zhou Y, Neve R, Ahlijanian MK, Tsai LH. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163:83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME. β-Amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21:7551–7560. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muresan Z, Muresan V. c-Jun NH2-terminal kinase-interacting protein-3 facilitates phosphorylation and controls localization of amyloid-β precursor protein. J Neurosci. 2005;25:3741–3751. doi: 10.1523/JNEUROSCI.0152-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muresan Z, Muresan V. The amyloid-beta precursor protein is phosphorylated via distinct pathways during differentiation, mitosis, stress, and degeneration. Mol Biol Cell. 2007;18:3835–3844. doi: 10.1091/mbc.E06-07-0625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naruse S, Thinakaran G, Luo JJ, Kusiak JW, Tomita T, Iwatsubo T, Qian X, Ginty DD, Price DL, Borchelt DR, Wong PC, Sisodia SS. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- Perez-Tur J, Froelich S, Prihar G, Crook R, Baker M, Duff K, Wragg M, Busfield F, Lendon C, Clark RF, Roques P, Fuldner RA, Johnston J, Cowburn R, Forsell C, Axelman K, Lilius L, Houlden H, Karran E, Roberts GW, et al. A mutation in Alzheimer's disease destroying a splice acceptor site in the presenilin-1 gene. Neuroreport. 1995;7:297–301. [PubMed] [Google Scholar]
- Ramin M, Azizi P, Motamedi F, Haghparast A, Khodagholi F. Inhibition of JNK phosphorylation reverses memory deficit induced by β-amyloid (1–42) associated with decrease of apoptotic factors. Behav Brain Res. 2011;217:424–431. doi: 10.1016/j.bbr.2010.11.017. [DOI] [PubMed] [Google Scholar]
- Sano Y, Nakaya T, Pedrini S, Takeda S, Iijima-Ando K, Iijima K, Mathews PM, Itohara S, Gandy S, Suzuki T. Physiological mouse brain Abeta levels are not related to the phosphorylation state of threonine-668 of Alzheimer's APP. PLoS One. 2006;1:e51. doi: 10.1371/journal.pone.0000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standen CL, Brownlees J, Grierson AJ, Kesavapany S, Lau KF, McLoughlin DM, Miller CC. Phosphorylation of Thr668 in the cytoplasmic domain of the Alzheimer's disease amyloid precursor protein by stress-activated protein kinase 1b (Jun N-terminal kinase-3) J Neurochem. 2001;76:316–320. doi: 10.1046/j.1471-4159.2001.00102.x. [DOI] [PubMed] [Google Scholar]
- Taru H, Suzuki T. Regulation of the physiological function and metabolism of AbetaPP by AbetaPP binding proteins. J Alzheimers Dis. 2009;18:253–265. doi: 10.3233/JAD-2009-1148. [DOI] [PubMed] [Google Scholar]
- Vingtdeux V, Hamdane M, Gompel M, Bégard S, Drobecq H, Ghestem A, Grosjean ME, Kostanjevecki V, Grognet P, Vanmechelen E, Buée L, Delacourte A, Sergeant N. Phosphorylation of amyloid precursor carboxy-terminal fragments enhances their processing by a gamma-secretase-dependent mechanism. Neurobiol Dis. 2005;20:625–637. doi: 10.1016/j.nbd.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Wang X, Destrument A, Tournier C. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim Biophys Acta. 2007a;1773:1349–1357. doi: 10.1016/j.bbamcr.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Wang X, Nadarajah B, Robinson AC, McColl BW, Jin JW, Dajas-Bailador F, Boot-Handford RP, Tournier C. Mitogen-activated protein kinase kinase 4 is an essential activator of the c-Jun N-terminal protein kinase during brain development. Mol Cell Biol. 2007b;27:7935–7946. doi: 10.1128/MCB.00226-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]