

Abstract
Changes in function of voltage-gated sodium channels in nociceptive primary sensory neurons participate in the development of peripheral hyperexcitability that occurs in neuropathic and inflammatory chronic pain conditions. Among them, the tetrodotoxin-resistant (TTX-R) sodium channel Nav1.8, primarily expressed by small- and medium-sized dorsal root ganglion (DRG) neurons, substantially contributes to the upstroke of action potential in these neurons. Compelling evidence also revealed that the chemokine CCL2 plays a critical role in chronic pain facilitation via its binding to CCR2 receptors. In this study, we therefore investigated the effects of CCL2 on the density and kinetic properties of TTX-R Nav1.8 currents in acutely small/medium dissociated lumbar DRG neurons from naive adult rats. Whole-cell patch-clamp recordings demonstrated that CCL2 concentration-dependently increased TTX-resistant Nav1.8 current densities in both small- and medium-diameter sensory neurons. Incubation with CCL2 also shifted the activation and steady-state inactivation curves of Nav1.8 in a hyperpolarizing direction in small sensory neurons. No change in the activation and inactivation kinetics was, however, observed in medium-sized nociceptive neurons. Our electrophysiological recordings also demonstrated that the selective CCR2 antagonist INCB3344 [N-[2-[[(3S,4S)-1-E4-(1,3-benzodioxol-5-yl)-4-hydroxycyclohexyl]-4-ethoxy-3-pyrrolidinyl]amino]-2-oxoethyl]-3-(trifluoromethyl)benzamide] blocks the potentiation of Nav1.8 currents by CCL2 in a concentration-dependent manner. Furthermore, the enhancement in Nav1.8 currents was prevented by pretreatment with pertussis toxin (PTX) or gallein (a Gβγ inhibitor), indicating the involvement of Gβγ released from PTX-sensitive Gi/o-proteins in the cross talk between CCR2 and Nav1.8. Together, our data clearly demonstrate that CCL2 may excite primary sensory neurons by acting on the biophysical properties of Nav1.8 currents via a CCR2/Gβγ-dependent mechanism.
Introduction
In sensory primary afferent neurons, voltage-gated sodium channels (VGSCs) are thought to play a critical role in the pathogenesis of chronic pain conditions (Amir et al., 2006; Dib-Hajj et al., 2010). Changes in the expression, trafficking, and function of VGSC have been particularly discussed to explain the neuronal hyperexcitability observed in the painful phenotype associated with nerve injury or peripheral inflammation (Docherty and Farmer, 2009; Swanwick et al., 2010). Several lines of evidence suggest that the tetrodotoxin-resistant (TTX-R) sodium channel Nav1.8 mostly contributes to the enhanced excitability and spontaneous ectopic discharges occurring in small- and medium-sized dorsal root ganglion (DRG) neurons (Devor, 2006; Cummins et al., 2007; Dib-Hajj et al., 2009a). The enhanced responsiveness of these Nav1.8-positive neurons thus leads to the development of hyperalgesia, allodynia, and ongoing spontaneous pain (Dib-Hajj et al., 2010; Lampert et al., 2010).
To date, several inflammatory mediators acting through G-protein-coupled receptors (GPCRs), including PGE2, adenosine, and serotonin, have been shown to sensitize Nav1.8 sodium channels and therefore to significantly increase the excitability of the nociceptor (Priest, 2009; Lampert et al., 2010). In addition to their well established role in the immune system, chemokines were also recently described to orchestrate the cellular response during the initiation/maintenance of chronic pain (Rittner and Brack, 2006; White et al., 2009; Gao and Ji, 2010). Among them, the chemokine CCL2 was shown to play a key role in spinal nociceptive processing, notably by sensitizing primary afferent neurons (Gosselin et al., 2008; White et al., 2009; White and Miller, 2010; Rostène et al., 2011). Acting via the binding to its cognate GPCR CCR2, intrathecal delivery of exogenous CCL2 was found to promote pain hypersensitivity (Tanaka et al., 2004; Dansereau et al., 2008). Accordingly, CCL2-overexpressing or CCR2-deficient mice exhibited altered nociceptive behaviors to thermal, chemical, and mechanical stimuli (Abbadie et al., 2003; Menetski et al., 2007; Zhang et al., 2007). In addition, the use of CCR2 receptor antagonists or blocking antibodies successfully inhibited nociceptive signaling (Bhangoo et al., 2007, 2009; Dansereau et al., 2008; Serrano et al., 2010; Struthers and Pasternak, 2010; Van Steenwinckel et al., 2011). Similar to pain neuromodulators (Rostène et al., 2007), CCL2 is stored in large dense core vesicles known to contain pronociceptive-related peptides [substance P and CGRP (calcitonin gene-related peptide)], is released in a calcium-dependent manner from DRG neuronal cell bodies and terminal nerve endings, and directly excites primary nociceptive neurons by autocrine/paracrine processes (Oh et al., 2001; White et al., 2005; Sun et al., 2006; Dansereau et al., 2008; Jung et al., 2008; Van Steenwinckel et al., 2011).
Thus, the aim of the present study was to investigate whether the mechanism of CCL2-induced sensory neuron excitation might in part arise from the enhanced activation of TTX-R sodium channel Nav1.8 currents. For this purpose, we examined the changes induced by CCL2 on the density and biophysical properties of Nav1.8 currents in acutely dissociated small- and medium-sized DRG neurons from naive adult rats using whole-cell patch-clamp recordings. Additional electrophysiological studies were conducted to determine whether CCR2 receptor antagonist [N-[2-[[(3S,4S)-1-E4-(1,3-benzodioxol-5-yl)-4-hydroxycyclohexyl]-4-ethoxy-3-pyrrolidinyl]amino]-2-oxoethyl]-3-(trifluoromethyl)benzamide (INCB3344)] could inhibit the cross talk between CCR2 and Nav1.8 (Brodmerkel et al., 2005). Finally, in light of the association of CCR2 with Gi/o-protein subunits (Bajetto et al., 2002), we further evaluated the molecular basis of this functional interaction by exploring the effects of signaling inhibitors on Nav1.8 currents evoked by CCR2 activation.
Materials and Methods
Animals.
Adult male Sprague Dawley rats (250–300 g) were housed two per cage in a climate-controlled room on a 12 h light/dark cycle with water and food ad libitum. All experimental procedures were approved by the Animal Care and Use Committee of the Université de Sherbrooke and were in accordance with the guidelines provided by the Canadian Council of Animal Care.
Acute culture of DRG neurons.
DRG neurons from adult rats were acutely isolated as described previously (Zhou et al., 2003). Briefly, rats were exposed to CO2 and decapitated. The bilateral L4–L5 DRGs were isolated, quickly unsheathed in calcium/magnesium-free PBS with glucose (1.6 m), and all adherent connective tissue was removed. Isolated DRGs were then enzymatically digested with collagenase A for 90 min (1 mg/ml; Roche Diagnostics) in PBS containing glucose, and then for 30 min with trypsin (0.25%) (Invitrogen). Subsequently, DRG neurons were mechanically dissociated by repeated trituration using a fine polished Pasteur pipette in culture medium containing 1:1 DMEM and Ham's F12 medium, supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin (100 U/ml)/streptomycin (0.1 mg/ml). Isolated neurons were gently centrifuged (50 × g for 3 min), plated on poly-d-lysine/laminin-coated glass coverslips, and incubated at 37°C in a humidified 95% air/5% CO2 atmosphere. All of the electrophysiological recordings or immunocytochemical experiments were performed within 14–20 h, a sufficient time to allow the cells to adhere to the plate. For the immunocytochemical detection, DRG neurons were fixed for 15 min with 4% paraformaldehyde (PFA) for further immunostaining. During the brief period of culture, DRG neurons displayed only short axonal processes (<10 μm), thus facilitating the voltage-clamp studies, and minimizing changes in electrical properties that can occur in long-term culture.
Immunofluorescence labeling.
For tissue sections, rats were deeply anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and perfused transaortically with saline, followed by 4% PFA containing 0.8% picric acid in 0.1 m PBS. Lumbar DRGs were rapidly dissected and postfixed in the same fixative overnight at 4°C. DRGs were then cryoprotected in 30% sucrose and frozen 1 min at −40°C in isopentane. DRGs were sliced using a cryostat in 12 μm (Jung CM 3100; Leica Microsystems) sections, and then mounted on Superfrost slides. To simultaneously detect CCR2 and Nav1.8, tissue sections and cultured DRG neurons were processed for dual immunofluorescence labeling. Specimens were incubated in 0.1 m PBS containing 3% NGS and 0.1% Triton X-100 for 2 h, and then treated with a rabbit CCR2 antibody for 1 h (Gosselin et al., 2005). After rinsing three times with 0.1 m PBS, sections were incubated with a biotin-conjugated goat anti-rabbit antibody (1:1000; Vector Laboratories) for 1 h and finally revealed by incubation with streptavidin-Alexa Fluor 488 (1:1000; Invitrogen). Subsequently, cultured rat DRG neurons and L4–L6 DRG sections were incubated with a goat anti-rabbit IgG (Vector; 1:100) for 1 h to completely block sites of primary CCR2 antibody. Specimens were then treated with rabbit anti-Nav1.8 (1:500; Alomone Labs) and stained with Alexa Fluor 555-conjugated goat anti-rabbit (1:1000; Invitrogen). Specimens were then washed and mounted with Aquamount. We performed primary and secondary antibody control experiments for each combination of primary and secondary antibodies, to evaluate nonspecific secondary antibody binding and cross-reactivity between primary and secondary antibodies. Experiments performed in the absence of primary and/or secondary antibody revealed an absence of immunofluorescence labeling (data not shown). Double-labeled sections were analyzed by light microscopy using a Zeiss fluorescence microscope (Carl Zeiss) fitted with highly discriminating filters. Pictures were taken using a digital camera (AxioCam HRC; Carl Zeiss) connected to an image acquisition software (AxioVision; Carl Zeiss).
cAMP assay.
Acutely dissociated DRG neurons were dispensed into poly-d-lysine-coated 24-well plates (2 × 105 cells per well). After 16 h, cells were preincubated for 2 h at 37°C in 200 μl of Krebs'–Ringer's solution (10 mm HEPES, 118 mm NaCl, 2.5 mm CaCl2, 4.75 mm KCl, 1.2 mm MgSO4 1.2 mm KH2PO4, 24 mm NaHCO3, 11.1 mm glucose, and 0.3% BSA) in the presence or not of the Gi inhibitor, pertussis toxin (PTX) (250 ng/ml; Sigma-Aldrich). The medium was then replaced by 200 μl of Krebs'–Ringer's solution containing 0.1 mm 3-isobutyl-1-methylxanthine (IBMX) (Sigma-Aldrich), a nonspecific cyclic nucleotide phosphodiesterase inhibitor, and the cells were incubated for 15 min at 37°C. Then, 200 μl of a solution containing CCL2 (at a final concentration of 100 nm) were added to the medium, and the cells were incubated for another 30 min at 37°C. Basal levels were determined in parallel by incubating cells in the absence of CCL2 for 30 min at 37°C in 400 μl of Krebs'–Ringer's solution containing 0.1 mm IBMX. The reaction was stopped by replacing the incubation medium with 100 μl of 0.1N HCl for 20 min at room temperature. Next, the cells were scraped and centrifuged for 10 min at 10,000 rpm. Collected supernatants were then processed directly for cAMP determination by using the cAMP Direct Immunoassay kit (Abcam; ab65355), as recommended by the manufacturer.
Whole-cell patch-clamp recordings.
Sodium currents were recorded from single, small- to medium-sized DRG neurons in the whole-cell patch-clamp configuration 14–20 h after dissociation and plating, using an Axopatch 200B amplifier (Molecular Devices). All experiments were performed at room temperature (21–23°C). The intracellular recording electrodes were made from borosilicate glass capillary tubes (Warner Instruments), pulled using a two-step vertical micropipette puller P83 (Narashige), and heat-polished on a microforge (Narashige).
The pipette solution contained the following (in mm): 10 NaCl, 140 CsCl, 10 EGTA, 1 MgCl2, 2 Na2ATP, 10 HEPES; pH was adjusted to 7.2 by CsOH. Osmolarity was adjusted to 310 mOsm/L with sucrose. Pipettes had a resistance of 2–4 MΩ when filled with the pipette solution. After formation of a tight seal (>1 GΩ), whole-cell access was obtained by rupturing the membrane, and membrane resistance and capacitance (Cm) were determined. Capacity transients were cancelled using computer-controlled circuitry and series resistance was compensated (>85%) in all experiments. The external solution contained the following (in mm): 35 NaCl, 65 NMDG-Cl, 30 tetraethylammonium (TEA)-Cl, 0.1 CaCl2, 0.1 CoCl2, 5 MgCl2, 10 HEPES, and 10 glucose, pH adjusted at 7.4 by NaOH and osmolarity adjusted to 300 mOsm/L. The TEA-Cl and CoCl2 were used to inhibit endogenous K+ and Ca2+ currents, respectively. The external sodium concentration was reduced to 35 mm to maintain adequate clamp of currents and to ensure that the particular current under study was stable. Note that the amplitude of Na+ currents was not affected over the time period used to record the effects of the different drugs.
Using a whole-cell configuration, TTX-R Nav1.8 currents were isolated by prepulse inactivation as described previously (MeLean et al., 1988; Roy and Narahashi, 1992; Cummins and Waxman, 1997). Briefly, standard current–voltage (I–V) families were constructed using a holding potential of −120 mV with 500 ms prepulses to −50 mV before each depolarization to inactivate the fast TTX-sensitive (TTX-S) currents. Thus, standard I–V curves were obtained by the application of a series of test pulses to voltages that ranged from −70 to +40 mV in 10 mV increments after the prepulse inactivation protocol (see Fig. 2). The voltage dependence of inactivation was measured by applying a double-pulse protocol consisting of a 500 ms conditioning potential (−120 to −10 mV, 5 mV increments) followed by a fixed test pulse (−10 mV, 50 ms). The current amplitude (I) was normalized to the maximum control current amplitude (Imax).
Figure 2.

Isolation of TTX-resistant Nav1.8 currents in small-sized rat sensory neurons. Whole-cell voltage-clamp current traces of Nav1.8 in small-diameter sensory neurons recorded following a 30 min incubation with 100 nm CCL2 in the absence (n = 11) or presence of INCB3344 (100 nm; n = 7), PTX (250 ng/ml; n = 9), or gallein (20 μm; n = 6). Representative I–V curves of currents are determined using the pulse protocol indicated in the inset.
Drugs.
CCL2 (PeproTech) was applied at two different concentrations (10 and 100 nm) to the chamber for 30 min before patch-clamp recordings. Cultured DRG neurons were also treated with 100 nm CCL2 in the presence of increasing concentrations (1–100 nm) of the CCR2 antagonist INCB3344 (Global Pfizer; Pure Substance Program). DRG cells were also pretreated overnight with PTX (250 ng/ml) or with gallein, a specific Gβγ dimer signaling inhibitor (20 μm; Tocris Bioscience).
Data analysis and statistics.
The peak inward current values at each potential were plotted to generate I–V curves. Conductance (G) was determined as I/(Vm − Vrev), where I is the current, Vm is the potential at which current is evoked, and Vrev is the reversal potential of the current. Activation was fitted with the following Boltzmann equation: G = Gmax/[1 + exp[(V1/2 − Vm)/k]], where Vm is the test pulse voltage potential at which current is evoked, Gmax is the calculated maximal conductance, V1/2 is the potential of half-activation or inactivation, and k is the slope factor. The normalized curves were fitted using a Boltzmann distribution equation as follows: I = Imax/[1 + exp[(V1/2 − Vm)/k]], where Imax is the peak sodium current elicited after the most hyperpolarized prepulse, Vm is the preconditioning pulse potential, V1/2 is the half-maximal sodium current, and k is the slope factor. INCB3344 dose–response curve was obtained by bracketing the responses to different INCB3344 concentrations (1–100 nm).
Recording data were acquired using a Digidata 1440 A series interface (Molecular Devices) digitized at 10 kHz, low-pass filtered at 2 kHz, and captured using pClamp software (version 10.2; Molecular Devices). For current density measurements, the currents were divided by the cell capacitance (Cm) as read from the amplifier. Cells were selected for recording based on whole-cell capacitance (Cm < 30 pF corresponding to small neurons, <25 μm; Cm < 55 pF corresponding to medium neurons, <25–32 μm). The offset potential was zeroed before patching the cells and leakage current was digitally subtracted on-line using hyperpolarizing potentials, applied after the test pulse.
Data are expressed as means ± SEM. Statistical analyses were performed using Student's t test or one-way ANOVA followed by Dunnett's posttest. A value of p < 0.05 was considered statistically significant. Curves were fitted using Origin software (OriginLab Corporation), and graphs were plotted using GraphPad, version 5.d (GraphPad Software). Final composites were processed using Deneba's Canvas 10.5 imaging software (Deneba Software).
Results
Colocalization of Nav1.8 and CCR2 in cultured rat sensory neurons and DRG tissue
To determine whether Nav1.8 was present within the same sensory neuronal population as CCR2, dual immunofluorescence labeling was performed on dissociated primary afferent neurons and on DRG sections from naive rats (Fig. 1). Dual immunostaining revealed that Nav1.8 and CCR2 colocalized extensively over small- to medium-sized sensory neurons (Fig. 1A–F). At higher magnification, the distribution patterns for both Nav1.8 and CCR2 were entirely restricted to the periphery of the neuron somata (Fig. 1D–F). Accordingly, fluorescence microscopy performed on DRG tissue sections revealed that numerous Nav1.8-positive small and medium sensory neurons were also immunoreactive for CCR2 (Fig. 1G–I). In addition, CCR2 and Nav1.8 immunoreactivities were detected in axonal fibers in DRG sections, suggesting anterograde transport of both proteins to nerve terminals (data not shown). The presence of CCR2 receptors on Nav1.8-positive neurons led us to investigate, by electrophysiological approaches, whether the CCR2 ligand CCL2 could modulate the TTX-resistant Nav1.8 currents.
Figure 1.

Cellular distribution of Nav1.8 and CCR2 in rat DRG neurons. Immunofluorescence staining of CCR2 (green) (A, D) and Nav1.8 (red) (B, E) on acutely dissociated primary afferent neurons. The merged images show dually labeled small- and medium-sized sensory neurons (yellow) (C, F). Note that CCR2 and Nav1.8 are mainly detected at the periphery of the cell. Immunohistochemical labeling performed on DRG sections from naive rats (G–I). Numerous small- to medium-diameter DRG neurons display both Nav1.8 and CCR2 immunoreactivities. Scale bars, 20 μm.
CCL2 enhances Nav1.8 currents in small primary sensory neurons
Several protocols exist to isolate TTX-R from TTX-S sodium currents. Here, we applied a prepulse inactivation protocol with a 500 ms step of −50 mV to isolate the slow-inactivating TTX-R sodium currents (MeLean et al., 1988; Roy and Narahashi, 1992; Cummins and Waxman, 1997). Furthermore, the membrane potential was held at −70 mV to inhibit Nav1.9 currents, leaving the Nav1.8 current intact (Rush et al., 2005; Cang et al., 2009). The family of Nav1.8 sodium currents was generated with a voltage-clamp protocol by applying a range of potentials between −70 and +40 mV for 100 ms (Fig. 2). Using a whole-cell patch-clamp configuration, we therefore examined the effects of CCL2 on the density and kinetic properties of TTX-R Nav1.8 currents in acutely dissociated lumbar DRG neurons. Nav1.8 current amplitude was increased in small neurons (C-fibers) following an incubation of 30 min with 100 nm CCL2 (Fig. 2). Under these conditions, current–voltage (I–V) analysis revealed that the mean peak current amplitude was significantly higher in CCL2-treated (−99.13 ± 2.77 pA/pF; n = 11) than in control DRG neurons (−53.30 ± 2.15 pA/pF; n = 13) (Fig. 3A,B; ***p < 0.001). The peak current density was also affected significantly at the lowest concentration of CCL2 (10 nm) (−63.55 ± 12.26 pA/pF; n = 4; *p < 0.05; Fig. 3A,B).
Figure 3.

Nav1.8 currents are increased in small sensory neurons following exposure to CCL2. A, Current–voltage relationships of Nav1.8 were determined before (black circles) and after application of 10 nm (gray squares) or 100 nm (gray triangles) of CCL2. B, Peak Nav1.8 current densities were significantly enhanced in the presence of 10 and 100 nm CCL2 (*p < 0.05 and ***p < 0.001, respectively, vs control; Student's t test; n = 6–13). Error bars indicate SEM. C, D, CCL2 shifted the activation (C) and steady-state inactivation (D) curves in a hyperpolarizing direction. The midpoint values of the activation (V1/2act) and inactivation (V1/2inact) curves are illustrated in Table 1.
Acute application of 100 nm CCL2 also shifted the activation and steady-state inactivation curves of Nav1.8 in a hyperpolarizing direction (Fig. 3C,D). The midpoint of activation (V1/2act) was significantly decreased in CCL2-treated neurons compared with control neurons (−18.74 ± 0.31 mV, n = 11, vs −8.45 ± 0.21 mV, n = 13, respectively; ***p < 0.001) (Fig. 3C, Table 1). The slope factor (kact) was not affected by CCL2 perfusion (kcontrol = 4.73 ± 0.19 vs kCCL2 = 4.47 ± 0.29 mV; Table 1). Similar to the activation curve, the steady-state inactivation parameter, hinf, was significantly influenced by the presence of CCL2 (Fig. 3D). Indeed, the half-inactivation potential (V1/2inact) was shifted to −40.61 ± 0.09 mV (n = 10) from that of control condition (−30.55 ± 0.17 mV; n = 10) after 100 nm CCL2 treatment (***p < 0.001; Table 1). Slope values (kinact) remained unchanged between CCL2-treated (4.81 ± 0.08 mV) and control (4.72 ± 0.15 mV) neurons. Moreover, lower concentration of CCL2 (10 nm) caused a left shift in the activation (V1/2act = −13.72 ± 0.22 mV; n = 4; *p < 0.05) and steady-state inactivation (V1/2inact = −37.02 ± 0.16 mV; n = 7; **p < 0.01) curves of Nav1.8 (Fig. 3C,D; Table 1).
Table 1.
Activation and steady-state inactivation characteristics of Nav1.8 currents in small-diameter sensory neurons
| V1/2act (mV) | kact | V1/2inact (mV) | kinact | |
|---|---|---|---|---|
| Control | −8.45 ± 0.21 | 4.73 ± 0.19 | −30.55 ± 0.17 | 4.72 ± 0.15 |
| (n = 13) | (n = 10) | |||
| CCL2 (10 nm) | −13.72 ± 0.22* | 3.5 ± 0.19 | −37.02 ± 0.16** | 3.66 ± 0.14 |
| (n = 4) | (n = 7) | |||
| CCL2 (100 nm) | −18.74 ± 0.31*** | 4.47 ± 0.29 | −40.61 ± 0.09*** | 4.81 ± 0.08 |
| (n = 11) | (n = 10) | |||
| CCL2 (100 nm) +INCB3344 (100 nm) | −7.47 ± 0.26## | 5.01 ± 0.23 | −31.02 ± 0.16### | 3.59 ± 0.14 |
| (n = 7) | (n = 5) | |||
| CCL2 (100 nm) +PTX (250 ng/ml) | −7.04 ± 0.22## | 4.14 ± 0.20 | ND | ND |
| (n = 9) | ||||
| CCL2 (100 nm) +gallein (20 μm) | −6.09 ± 0.22### | 4.10 ± 0.17 | ND | ND |
| (n = 6) |
V1/2act and V1/2inact are the membrane potentials for half-maximal channel activation or inactivation, respectively. kact and kinact represent the slope factors for activation and inactivation. The numbers in parentheses reflect numbers of recorded neurons. ND, Not determined.
**p < 0.01 and ***p < 0.001 indicate statistically significant differences with control group. ##p < 0.01 and ###p < 0.001 compared with CCL2 alone.
Potentiation of Nav1.8 currents by CCL2 is blocked by the selective CCR2 antagonist
We then investigated whether these changes in the biophysical properties of Nav1.8 currents induced by CCL2 application were directly mediated by CCR2. For this purpose, electrophysiological recordings were performed on small sensory neurons following 30 min preincubation with 100 nm CCL2, in the presence of increasing concentrations of the CCR2 antagonist INCB3344 (ranging from 1 to 100 nm). As shown in Figure 2, the increase in CCL2-induced Nav1.8 sodium currents was considerably reduced in the presence of 100 nm INCB3344, thus demonstrating that the effects of CCL2 were CCR2 dependent. I–V curves of Nav1.8 currents also revealed that coincubation with INCB3344 produced a concentration-dependent inhibition of the mean peak current density, with an IC50 of 21 nm (Fig. 4A,B). At the highest concentration tested (100 nm), the selective CCR2 antagonist decreased by 85% the peak amplitude of Nav1.8 current evoked by CCL2 (−99.13 ± 2.77 pA/pF, n = 11, for CCL2, vs −59.77 ± 4.65 pA/pF, n = 7, for CCL2 plus INCB3344; ###p < 0.001; Fig. 4A,B). No significant change in Nav1.8 current density or kinetics was seen when INCB3344 (100 nm) was applied alone (Fig. 4B).
Figure 4.

INCB3344 treatment blocks the changes in the biophysical properties of Nav1.8 induced by CCL2. A, I–V curves of Nav1.8 currents obtained from small rat DRG neurons. B, Histogram showing the effects of different concentrations of INCB3344 (1, 10, 100 nm) on the increased Nav1.8 peak current induced by 100 nm CCL2 (***p < 0.001, CCL2 alone vs control; ###p < 0.001, #p < 0.05, INCB3344 plus CCL2 vs CCL2 alone; Student's t test; n = 6–11; pound signs correspond to the values in the Table 1). Error bars indicate SEM. C, D, INCB3344 also significantly inhibits the leftward shift of the activation (C) (##p < 0.01) and inactivation (D) (###p < 0.001 compared with CCL2 alone) curves of Nav1.8 current observed in the presence of 100 nm CCL2. The experimental conditions corresponding to control and 100 nm CCL2 have already been presented in Figure 3. Half-activation and half-inactivation potentials and slope factors are summarized in Table 1.
The changes in the activation and steady-state inactivation curves of Nav1.8 observed under CCL2 stimulation (100 nm) were also prevented by the presence of INCB3344. Indeed, INCB3344 at 100 nm fully blocked the left shift in the activation curves of Nav1.8 induced by CCL2 (−18.74 ± 0.31 mV, n = 11, for CCL2, vs −7.47 ± 0.26 mV, n = 7, for CCL2 plus INCB3344) (##p < 0.01; Fig. 4C, Table 1). In addition, the hyperpolarizing shift in the inactivation curve of TTX-R Nav1.8 currents observed in response to CCL2 returned to baseline values following coincubation with the CCR2 antagonist. The half-inactivation potential was −40.61 ± 0.09 mV using CCL2 alone (n = 10), and −31.02 ± 0.16 mV when incubating with 100 nm INCB3344 (###p < 0.001; n = 5) (Fig. 4D, Table 1). The slope factors, kact and kinact were unaltered by the presence of INCB3344 (Table 1).
Gi/o-proteins participate in CCL2-induced Nav1.8 current activation
CCR2 activation by CCL2 initiates intracellular signaling cascades among the ones coupling to heterotrimeric G-proteins of the Gi family (Bajetto et al., 2002; Gosselin et al., 2008). Before determining the possible role of Gi/o-proteins in the CCL2 activation of Nav1.8 current, we have first examined whether CCL2 activated CCR2 through a Gi/o-coupled mode of action to decrease neuronal cAMP production and whether treatment with the irreversible inhibitor of Gi/o-proteins, PTX (250 ng/ml) inhibited the functional coupling of CCR2 to adenylyl cyclase. As shown in Figure 5A, CCL2 applied at a concentration of 100 nm to dissociated primary sensory neurons significantly decreased basal levels of cAMP up to 30% (*p < 0.05). Moreover, overnight pretreatment with PTX reversed by 70% the CCL2-induced inhibition of adenylyl cyclase activity (#p < 0.05), thus demonstrating that CCR2 activation modulates the neuronal cAMP levels in a Gi/o-protein-dependent manner.
Figure 5.

Involvement of Gi/o-proteins in CCL2-induced potentiation of Nav1.8 currents. A, Neuronal cAMP measurement following application of CCL2 in the presence or absence of PTX. CCL2-induced inhibition of adenylyl cyclase activity is blunted by PTX pretreatment (*p < 0.05, CCL2 alone vs control; #p < 0.05, PTX plus CCL2 vs CCL2 alone; one-way ANOVA followed by Dunnett's posttest, performed in triplicate). Error bars indicate SEM. B, C, I–V curves (B) and histogram (C) showing the action of CCL2 on Nav1.8 currents (***p < 0.001 compared with Control; Student's t test) and the inhibition of this effect with PTX (250 ng/ml) pretreatment (###p < 0.001 compared with CCL2 alone; Student's t test; n = 7–11). D, Preincubation with PTX also impedes the action of CCL2 on the normalized conductance (##p < 0.01; G/Gmax). Note that the experimental conditions corresponding to control and 100 nm CCL2 have been shown previously in Figure 3.
We next determined whether PTX treatment of acutely dissociated sensory neurons affected CCL2-induced Nav1.8 current activation. Our results revealed that overnight incubation with PTX significantly prevented the Nav1.8 current amplitude change induced by 100 nm CCL2, indicating that the influence of CCL2 on Nav1.8 is dependent of the trimeric Gi-protein complex (Fig. 2). The peak current densities were −99.13 ± 2.77 pA/pF (n = 11) and −52.37 ± 3.09 pA/pF (n = 9) before and after application of PTX, respectively (###p < 0.001; Fig. 5B,C). Applied alone, PTX caused no significant changes in the Nav1.8 current amplitude (Fig. 5C). In addition, the hyperpolarized shift in the midpoint of activation, observed upon CCL2 stimulation (V1/2act = −18.74 ± 0.31 mV; n = 11) was completely inhibited in the presence of PTX (V1/2act = −7.04 ± 0.22 mV; n = 9) (##p < 0.01; Fig. 5D, Table 1). The kact value was not changed by PTX treatment (Table 1).
CCL2 activates Nav1.8 currents through the release of Gβγ dimers
There is ample evidence in the literature demonstrating that G-protein βγ subunits released from some GPCR upon receptor activation regulate a variety of downstream pathways to control various physiological functions (Smrcka et al., 2008). As a classical GPCR, CCR2 has also been shown to modulate different signaling pathways through the release of Gβγ (Bajetto et al., 2002). Therefore, we next determined the potential role of Gβγ in CCL2-mediated Nav1.8 activation. Dissociated primary sensory neurons were thus incubated overnight with 20 μm gallein, a specific Gβγ dimer signaling inhibitor (Lehmann et al., 2008; Irannejad and Wedegaertner, 2010; Ukhanov et al., 2011). As shown in Figure 2, the increase in CCL2-induced Nav1.8 sodium currents was blocked by gallein pretreatment. Under CCL2 stimulation, the peak current densities were −99.13 ± 2.77 pA/pF (n = 11) and −50.63 ± 1.73 pA/pF (n = 6) before and after application of gallein, respectively (###p < 0.001; Fig. 6A,B). Inhibition of Gβγ also completely prevented the leftward shift in the activation curves of Nav1.8 induced by CCL2 (−18.74 ± 0.31 mV, n = 11, for CCL2, vs −6.09 ± 0.22 mV, n = 6, for CCL2 plus gallein) (###p < 0.001; Fig. 6C, Table 1). The Nav1.8 current amplitude was not changed following application of gallein alone (Fig. 6B).
Figure 6.
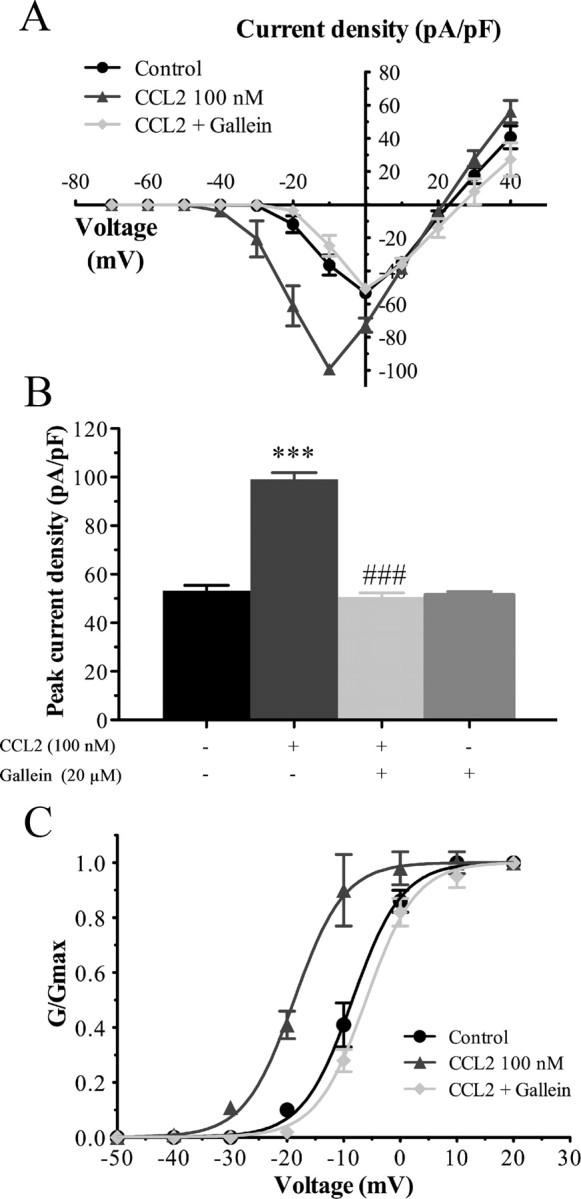
Gβγ participates in CCL2-induced Nav1.8 currents. A, B, I–V curves (A) and histogram (B) of Nav1.8 currents obtained from small rat DRG neurons showing that pretreatment with the Gβγ inhibitor gallein (20 μm) prevents the effects of CCL2 on Nav1.8 currents (***p < 0.001, CCL2 alone vs control; ###p < 0.001 gallein plus CCL2 compared with CCL2 alone; Student's t test; n = 6–11). Error bars indicate SEM. C, Gallein also completely reverses the leftward shift of activation curve of Nav1.8 observed in the presence of 100 nm CCL2 (###p < 0.001). Note that the experimental conditions corresponding to control and 100 nm CCL2 have been shown previously in previous figures. Half-activation and half-inactivation potentials and slope factors are summarized in Table 1.
Regulation of Nav1.8 currents by CCL2 in medium-sized primary sensory neurons
As shown in Figure 1, G–I, Nav1.8 channels are also expressed by medium-sized primary sensory neurons. We thus evaluated whether the biophysical properties of Nav1.8 current were differently affected in medium-diameter primary sensory neurons compared with small sensory neurons. Whole-cell patch-clamp recordings demonstrated that CCL2 significantly increased TTX-resistant Nav1.8 current densities in dissociated medium DRG neurons of naive rats (Fig. 7). The mean current amplitude at 0 mV was significantly higher after 100 nm CCL2 application (−82.60 ± 4.24 pA/pF; n = 11) compared with untreated DRG neurons (−62.19 ± 1.55 pA/pF; n = 18) (Fig. 8; ***p < 0.001). Peak current densities were also significantly reduced following exposure to INCB3344 (−64.38 ± 3.03 pA/pF; n = 7; ###p < 0.001), PTX (−62.23 ± 7.62 pA/pF; n = 5; #p < 0.05), or gallein (−62.60 ± 1.43 pA/pF; n = 11; ###p < 0.001) (Figs. 7, 8). Unlike small-sized neurons, there was, however, no change in the activation and steady-state inactivation curves of Nav1.8 following CCL2 treatment (Fig. 8C, Table 2).
Figure 7.

Representative families of Nav1.8 sodium currents in medium-sized sensory neurons. Average I–V curve family of currents recorded from medium neurons under CCL2 stimulation alone (100 nm; n = 11), and in the presence of INCB3344 (100 nm; n = 7), PTX (250 ng/ml; n = 5), or gallein (20 μm; n = 11). INCB3344, PTX, and gallein all efficiently reverse CCL2-induced enhancement of Nav1.8 currents.
Figure 8.

Density and kinetic properties of Nav1.8 currents in medium-sized sensory neurons. A, B, CCL2 increases the maximal peak current amplitude of Nav1.8 (***p < 0.001, CCL2 alone vs control). The activation of Nav1.8 currents induced by CCL2 is inhibited after treatment with INCB3344 (###p < 0.001 compared with CCL2 alone; Student's t test). C, No change in the steady-state inactivation curve of Nav1.8 was seen following CCL2 treatment (n = 6–11). D, E, Both PTX and gallein reverse CCL2-induced Nav1.8 current activation (#p < 0.05 and ###p < 0.001 compared with CCL2 alone, respectively). Half-maximal potentials (V1/2) and slope (k) values are reported in Table 2. Error bars indicate SEM.
Table 2.
Activation and steady-state inactivation characteristics of Nav1.8 currents in medium-sized sensory neurons
| V1/2act (mV) | kact | V1/2inact (mV) | kinact | |
|---|---|---|---|---|
| Control | −6.25 ± 0.44 | 5.53 ± 0.38 | −33.76 ± 0.13 | 4.19 ± 0.11 |
| (n = 18) | (n = 11) | |||
| CCL2 (100 nm) | −7.76 ± 0.12 | 4.19 ± 0.10 | −34.21 ± 0.26 | 5.12 ± 0.25 |
| (n = 11) | (n = 6) | |||
| CCL2 (100 nm) +INCB3344 (100 nm) | −9.85 ± 0.15 | 4.38 ± 0.15 | −35.63 ± 0.23 | 5.17 ± 0.23 |
| (n = 7) | (n = 7) | |||
| CCL2 (100 nm) + PTX(250 ng/ml) | −8.66 ± 0.19 | 3.62 ± 0.20 | ND | ND |
| (n = 5) | ||||
| CCL2 (100 nm) + gallein (20 μm) | −6.07 ± 0.09 | 3.72 ± 0.06 | ND | ND |
| (n = 11) |
V1/2 and k determine the half-maximal membrane potential and the slope factor for channel activation and inactivation, respectively. The numbers in parentheses reflect numbers of recorded neurons. ND, Not determined.
Discussion
Reports on the CCL2/CCR2 chemokine/receptor complex in a number of chronic pain disorders led us to investigate the peripheral mechanisms of CCL2-induced pain facilitation (Rostène et al., 2007; White et al., 2009). Using whole-cell patch-clamp recordings, we demonstrated here that CCL2 enhanced Nav1.8 current densities in both small- and medium-sized acutely dissociated sensory neurons. We also found that CCL2 increased excitability in small sensory neurons by shifting the activation and steady-state inactivation curves of Nav1.8 in the hyperpolarizing direction. This increase in Nav1.8 current was blocked by the selective CCR2 antagonist. This confirms that CCR2 is directly involved in CCL2-induced changes in Nav1.8 activity. We also found that the Gβγ released from PTX-sensitive Gi/o proteins is primarily responsible for the activation of Nav1.8 currents in response to CCL2 in primary sensory neurons.
Neuromodulation of Nav1.8 sodium channels by CCL2
Sustained pain hypersensitivity and neuronal hyperexcitability associated with nerve injury or disease-induced inflammatory responses of the peripheral nervous system are believed to result from chronic changes within DRG. In particular, modulation of voltage-gated sodium channels may underlie the increased excitability of primary afferent neurons (Dib-Hajj et al., 2009b; Docherty and Farmer, 2009; Swanwick et al., 2010). Among them, Nav1.8 has emerged as a key contributor to action potential generation in nociceptors (Cummins et al., 2007; Dib-Hajj et al., 2009a). In support, functional knockdown of Nav1.8 in rats or deletion of the Nav1.8 gene reduces hyperalgesia and allodynia in neuropathic pain models (Dib-Hajj et al., 2010; Lampert et al., 2010). Likewise, systemic and spinal administrations of Nav1.8 channel blockers partially reverse pain-related behaviors in preclinical animal models of chronic pain (Bear et al., 2009; Bhattacharya et al., 2009; Matulenko et al., 2009; Priest, 2009). Although Nav1.8 antagonists may have analgesic efficacy in patients coping with chronic pain, the high degree of structural homology within the VGSC family and side effects of existing blockers have limited their clinical use (England, 2008; Dworkin et al., 2010). Given the fact that it is actually difficult to selectively target Nav1.8, alternatives have been proposed like the indirect regulation of Nav1.8 function by acting upstream via GPCR inhibition. In that respect, inflammatory mediators, such as prostaglandin, adenosine, and serotonin have been shown to influence both activity and trafficking of TTX-R sodium channels (Gold et al., 1996; Rush and Waxman, 2004; Liu et al., 2010; Ebersberger et al., 2011).
Considerable data support the hypothesis that proinflammatory chemokines induce pain hypersensitivity, notably by causing sensory neuron hyperactivity (Wang et al., 2008; Miller et al., 2009; White et al., 2009). Accordingly, the intraganglionic release of CCL2 strongly excites CCR2-expressing nociceptive DRG neurons (Oh et al., 2001; White et al., 2005; Sun et al., 2006; Jung et al., 2008). The depolarization of these sensory neurons thus implies that a number of ion channels may be downstream targets of CCR2 activation. Here, we therefore investigated whether the chemokine CCL2 contributed to nociceptive neurons excitability through the enhancement of Nav1.8 currents. We first observed by dual immunofluorescence staining that, in both acutely dissociated cells and DRG sections, CCR2 receptors were present in most small- to medium-sized sensory neurons, previously described to express Nav1.8 (Lai et al., 2004; Devor, 2006; Cummins et al., 2007; Dib-Hajj et al., 2009a). Consistently, we demonstrated using whole-cell patch-clamp recordings that acute application of CCL2 to small DRG neurons concentration-dependently increased the magnitude of Nav1.8 currents and triggered a hyperpolarizing shift of both activation and inactivation curves. Interestingly, patch-clamp recordings from medium-sized DRG neurons revealed that CCL2 also enhanced the peak current amplitude of Nav1.8 but was, however, inefficient in shifting the conductance–voltage and the steady-state inactivation curves to more hyperpolarized potentials. Thus, these results reveal that CCL2 affects the functional properties of Nav1.8 in a cell type-specific manner within peripheral sensory ganglia.
Transactivation of Nav1.8 is not the only CCL2-mediated mechanism that is likely to be important in the excitation of sensory neurons. Indeed, TRP cation channels, TRPV1 and TRPA1, participate in CCL2-induced nociceptor sensitization (Jung et al., 2008). Similarly, inhibition of voltage-dependent K+ conductance has been hypothesized to arise from the action of CCL2 within DRG (Sun et al., 2006). In addition to sensory neuron modulation, CCL2 spinally released may also contribute to the maintenance of central sensitization by potentiating the activity of NMDA and AMPA receptors or by causing GABAergic disinhibition (Gosselin et al., 2005; Gao et al., 2009). The data reported here argue that Nav1.8 channels are targets of CCL2-induced signaling pathway and that the mechanisms by which CCL2 enhances neuronal excitability depend on the subset of DRG neurons.
Functional significance of CCR2 receptor activation to the modulation of Nav1.8 channel
To date, several lines of evidence support a critical role of CCL2/CCR2 in promoting behavioral changes associated with chronic pain (Miller et al., 2009; White and Miller, 2010). In particular, the recent progress in identifying CCR2 antagonists has revealed their effectiveness in reversing the nociceptive behaviors induced by focal nerve demyelination injury or HIV sensory neuropathy (Bhangoo et al., 2007, 2009; Jung et al., 2009). Accordingly, we recently demonstrated that the selective CCR2 antagonist INCB3344 prevented the pronociceptive action of exogenous intraspinal CCL2 and attenuated tactile allodynia in nerve-injured rats (Dansereau et al., 2008; Van Steenwinckel et al., 2011). Here, we therefore evaluated whether treatment of small- and medium-sized sensory neurons with INCB3344 abolished CCL2-induced Nav1.8 current activation. Our results demonstrated that application of INCB3344 concentration-dependently blocks CCL2-induced changes in Nav1.8 current density or kinetics. Supporting the behavioral observations, these data clearly indicate that CCL2-induced pain facilitation is mediated by direct activation of CCR2 and that CCR2 antagonists represent potential new pain-relieving drugs (Struthers and Pasternak, 2010).
Downstream CCR2 signaling in sensory neurons
Different underlying mechanisms have been proposed to explain how inflammatory mediators acting via GPCR increased Nav1.8 currents and nociceptor excitability (Priest, 2009; Lampert et al., 2010). In particular, the Nav1.8 channel was described as a final common target for several second-messenger cascades, including PKA, PKC, and p38 MAPK (Dib-Hajj et al., 2009b, 2010). As a classical GPCR, CCR2 is functionally coupled to Gα and Gβγ subunits of trimeric G-proteins and activates multiple signal transduction pathways, such as phospholipase C, ERK1/2, and p38 MAPK (Bajetto et al., 2002; Gosselin et al., 2008). Here, we thus investigated the mechanisms that governed the increase of Nav1.8 activity following CCL2 treatment. Our data strongly suggest that CCL2/CCR2 enhances Nav1.8 currents in a Gi/o-protein-dependent manner in both small- and medium-sized DRG neurons. Specifically, we showed that preventing Gi/o recruitment by PTX treatment completely reversed CCL2-induced inhibition of neuronal cAMP production, potentiation of Nav1.8 current density, as well as the shifts in activation and inactivation kinetics. The full inhibition of Nav1.8 currents by PTX further excludes the involvement of additional G-proteins in the modulation of Nav1.8 by CCL2. Since PTX inhibits the dissociation of Gβγ from Gαiβγ complex (Smrcka et al., 2008), we next examined the participation of Gβγ downstream signaling to CCL2-related Nav1.8 activation. Our results demonstrated that the specific Gβγ dimer inhibitor gallein fully blocked the activation of Nav1.8 currents in response to CCL2 in both populations of sensory neurons. Consequently, these findings suggest that the classical cAMP/PKA signaling pathway resulting from the activation of PTX-sensitive Gαi proteins is not involved in the regulation of Nav1.8 by CCL2.
Interestingly, we also found that CCL2 differently influenced the biophysical properties of Nav1.8 in DRG neuron subpopulations. In small sensory neurons, CCL2 increased Nav1.8 current amplitude, which was associated with hyperpolarizing shifts in voltage dependence of both activation and inactivation curves. In contrast, treatment of medium-sized DRG cells with CCL2 only affected the Nav1.8 current density without altering the gating properties of the channel. The different actions of CCL2 may thus reflect the implication of distinct signaling pathways. Based on previous findings (Gold et al., 1998; Vijayaragavan et al., 2004), we might speculate that, in small sensory neurons, CCL2 increases Nav1.8 current density and produces changes in voltage dependence of activation and inactivation via PKC phosphorylation of serine residues within the I–II loop of the Nav1.8 α subunit. Above all, we might propose that the stimulation of PLCβ via the release of Gβγ-dimers mediates the activation of PKCε, a PKC isoform shown to potentiate Nav1.8 current (Smrcka, 2008; Cang et al., 2009). In contrast, we might hypothesize that CCL2 regulates Nav1.8 function in medium-sized DRG neurons via a p38 MAPK-dependent pathway (Jin and Gereau, 2006; Hudmon et al., 2008), the activities of p38 MAPK being stimulated by mechanisms that involve dissociation of βγ subunits (Goldsmith and Dhanasekaran, 2007). Alternatively, as for N-type Ca2+ channels and inwardly rectifying K+ channels (Smrcka, 2008), direct binding of free Gβγ subunits to the C-terminal domain of sodium channel may induce persistent Na+ currents in neurons (Ma et al., 1997; Mantegazza et al., 2005).
Given the important role played by CCL2 in neuronal–glial interactions and in the temporal onset of pathological pain, we identified here a new mechanism by which CCL2 probably promotes peripheral sensitization and pain hypersensitivity. Since hyperexcitability and spontaneous action potential firing mediated by Nav1.8 in sensory neurons are thought to be the source of some types of chronic pain states, inhibition of CCL2/CCR2 signaling represents a promising avenue for therapeutic intervention.
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research (CIHR) (awarded to P.S. and S.M-P.). M.B. and M.-A.D. are supported by the Lavoisier and Sir Frederick Banting and Dr. Charles Best Canada Graduate Scholarships, respectively. P.S. is a CIHR new investigator. P.S. is director of the Sherbrooke's Neuroscience Center and a member of the Fonds de la Recherche en Santé du Québec-funded Centre de Recherche Clinique Étienne-Lebel.
References
- Abbadie C, Lindia JA, Cumiskey AM, Peterson LB, Mudgett JS, Bayne EK, DeMartino JA, MacIntyre DE, Forrest MJ. Impaired neuropathic pain responses in mice lacking the chemokine receptor CCR2. Proc Natl Acad Sci U S A. 2003;100:7947–7952. doi: 10.1073/pnas.1331358100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir R, Argoff CE, Bennett GJ, Cummins TR, Durieux ME, Gerner P, Gold MS, Porreca F, Strichartz GR. The role of sodium channels in chronic inflammatory and neuropathic pain. J Pain. 2006;7:S1–S29. doi: 10.1016/j.jpain.2006.01.444. [DOI] [PubMed] [Google Scholar]
- Bajetto A, Bonavia R, Barbero S, Schettini G. Characterization of chemokines and their receptors in the central nervous system: physiopathological implications. J Neurochem. 2002;82:1311–1329. doi: 10.1046/j.1471-4159.2002.01091.x. [DOI] [PubMed] [Google Scholar]
- Bear B, Asgian J, Termin A, Zimmermann N. Small molecules targeting sodium and calcium channels for neuropathic pain. Curr Opin Drug Discov Devel. 2009;12:543–561. [PubMed] [Google Scholar]
- Bhangoo S, Ren D, Miller RJ, Henry KJ, Lineswala J, Hamdouchi C, Li B, Monahan PE, Chan DM, Ripsch MS, White FA. Delayed functional expression of neuronal chemokine receptors following focal nerve demyelination in the rat: a mechanism for the development of chronic sensitization of peripheral nociceptors. Mol Pain. 2007;3:38. doi: 10.1186/1744-8069-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhangoo SK, Ripsch MS, Buchanan DJ, Miller RJ, White FA. Increased chemokine signaling in a model of HIV1-associated peripheral neuropathy. Mol Pain. 2009;5:48. doi: 10.1186/1744-8069-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya A, Wickenden AD, Chaplan SR. Sodium channel blockers for the treatment of neuropathic pain. Neurotherapeutics. 2009;6:663–678. doi: 10.1016/j.nurt.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodmerkel CM, Huber R, Covington M, Diamond S, Hall L, Collins R, Leffet L, Gallagher K, Feldman P, Collier P, Stow M, Gu X, Baribaud F, Shin N, Thomas B, Burn T, Hollis G, Yeleswaram S, Solomon K, Friedman S, et al. Discovery and pharmacological characterization of a novel rodent-active CCR2 antagonist, INCB3344. J Immunol. 2005;175:5370–5378. doi: 10.4049/jimmunol.175.8.5370. [DOI] [PubMed] [Google Scholar]
- Cang CL, Zhang H, Zhang YQ, Zhao ZQ. PKCepsilon-dependent potentiation of TTX-resistant Nav1.8 current by neurokinin-1 receptor activation in rat dorsal root ganglion neurons. Mol Pain. 2009;5:33. doi: 10.1186/1744-8069-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Waxman SG. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci. 1997;17:3503–3514. doi: 10.1523/JNEUROSCI.17-10-03503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Sheets PL, Waxman SG. The roles of sodium channels in nociception: implications for mechanisms of pain. Pain. 2007;131:243–257. doi: 10.1016/j.pain.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansereau MA, Gosselin RD, Pohl M, Pommier B, Mechighel P, Mauborgne A, Rostene W, Kitabgi P, Beaudet N, Sarret P, Melik-Parsadaniantz S. Spinal CCL2 pronociceptive action is no longer effective in CCR2 receptor antagonist-treated rats. J Neurochem. 2008;106:757–769. doi: 10.1111/j.1471-4159.2008.05429.x. [DOI] [PubMed] [Google Scholar]
- Devor M. Sodium channels and mechanisms of neuropathic pain. J Pain. 2006;7:S3–S12. doi: 10.1016/j.jpain.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Binshtok AM, Cummins TR, Jarvis MF, Samad T, Zimmermann K. Voltage-gated sodium channels in pain states: role in pathophysiology and targets for treatment. Brain Res Rev. 2009a;60:65–83. doi: 10.1016/j.brainresrev.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Black JA, Waxman SG. Voltage-gated sodium channels: therapeutic targets for pain. Pain Med. 2009b;10:1260–1269. doi: 10.1111/j.1526-4637.2009.00719.x. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- Docherty RJ, Farmer CE. The pharmacology of voltage-gated sodium channels in sensory neurones. Handb Exp Pharmacol. 2009;2009:519–561. doi: 10.1007/978-3-540-79090-7_15. [DOI] [PubMed] [Google Scholar]
- Dworkin RH, O'Connor AB, Audette J, Baron R, Gourlay GK, Haanpää ML, Kent JL, Krane EJ, Lebel AA, Levy RM, Mackey SC, Mayer J, Miaskowski C, Raja SN, Rice AS, Schmader KE, Stacey B, Stanos S, Treede RD, Turk DC, et al. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc. 2010;85:S3–S14. doi: 10.4065/mcp.2009.0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersberger A, Natura G, Eitner A, Halbhuber KJ, Rost R, Schaible HG. Effects of prostaglandin D2 on tetrodotoxin-resistant Na+ currents in DRG neurons of adult rat. Pain. 2011;152:1114–1126. doi: 10.1016/j.pain.2011.01.033. [DOI] [PubMed] [Google Scholar]
- England S. Voltage-gated sodium channels: the search for subtype-selective analgesics. Expert Opin Investig Drugs. 2008;17:1849–1864. doi: 10.1517/13543780802514559. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010;126:56–68. doi: 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29:4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Reichling DB, Shuster MJ, Levine JD. Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci U S A. 1996;93:1108–1112. doi: 10.1073/pnas.93.3.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MS, Levine JD, Correa AM. Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J Neurosci. 1998;18:10345–10355. doi: 10.1523/JNEUROSCI.18-24-10345.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith ZG, Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene. 2007;26:3122–3142. doi: 10.1038/sj.onc.1210407. [DOI] [PubMed] [Google Scholar]
- Gosselin RD, Varela C, Banisadr G, Mechighel P, Rostene W, Kitabgi P, Melik-Parsadaniantz S. Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurones. J Neurochem. 2005;95:1023–1034. doi: 10.1111/j.1471-4159.2005.03431.x. [DOI] [PubMed] [Google Scholar]
- Gosselin RD, Dansereau MA, Pohl M, Kitabgi P, Beaudet N, Sarret P, Mélik Parsadaniantz S. Chemokine network in the nervous system: a new target for pain relief. Curr Med Chem. 2008;15:2866–2875. doi: 10.2174/092986708786242822. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Choi JS, Tyrrell L, Black JA, Rush AM, Waxman SG, Dib-Hajj SD. Phosphorylation of sodium channel Nav1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J Neurosci. 2008;28:3190–3201. doi: 10.1523/JNEUROSCI.4403-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irannejad R, Wedegaertner PB. Regulation of constitutive cargo transport from the trans-Golgi network to plasma membrane by Golgi-localized G protein betagamma subunits. J Biol Chem. 2010;285:32393–32404. doi: 10.1074/jbc.M110.154963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Gereau RW., 4th Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-α. J Neurosci. 2006;26:246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Toth PT, White FA, Miller RJ. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J Neurochem. 2008;104:254–263. doi: 10.1111/j.1471-4159.2007.04969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Bhangoo S, Banisadr G, Freitag C, Ren D, White FA, Miller RJ. Visualization of chemokine receptor activation in transgenic mice reveals peripheral activation of CCR2 receptors in states of neuropathic pain. J Neurosci. 2009;29:8051–8062. doi: 10.1523/JNEUROSCI.0485-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai J, Porreca F, Hunter JC, Gold MS. Voltage-gated sodium channels and hyperalgesia. Annu Rev Pharmacol Toxicol. 2004;44:371–397. doi: 10.1146/annurev.pharmtox.44.101802.121627. [DOI] [PubMed] [Google Scholar]
- Lampert A, O'Reilly AO, Reeh P, Leffler A. Sodium channelopathies and pain. Pflugers Arch. 2010;460:249–263. doi: 10.1007/s00424-009-0779-3. [DOI] [PubMed] [Google Scholar]
- Lehmann DM, Seneviratne AM, Smrcka AV. Small molecule disruption of G protein beta gamma subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol Pharmacol. 2008;73:410–418. doi: 10.1124/mol.107.041780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Q, Su Y, Bao L. Prostaglandin E2 promotes Na1.8 trafficking via its intracellular RRR motif through the protein kinase A pathway. Traffic. 2010;11:405–417. doi: 10.1111/j.1600-0854.2009.01027.x. [DOI] [PubMed] [Google Scholar]
- Ma JY, Catterall WA, Scheuer T. Persistent sodium currents through brain sodium channels induced by G protein betagamma subunits. Neuron. 1997;19:443–452. doi: 10.1016/s0896-6273(00)80952-6. [DOI] [PubMed] [Google Scholar]
- Mantegazza M, Yu FH, Powell AJ, Clare JJ, Catterall WA, Scheuer T. Molecular determinants for modulation of persistent sodium current by G-protein βγ subunits. J Neurosci. 2005;25:3341–3349. doi: 10.1523/JNEUROSCI.0104-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulenko MA, Scanio MJ, Kort ME. Voltage-gated sodium channel blockers for the treatment of chronic pain. Curr Top Med Chem. 2009;9:362–376. doi: 10.2174/156802609788317883. [DOI] [PubMed] [Google Scholar]
- MeLean MJ, Bennett PB, Thomas RM. Subtypes of dorsal root ganglion neurons based on different inward currents as measured by whole-cell voltage clamp. Mol Cell Biochem. 1988;80:95–107. doi: 10.1007/BF00231008. [DOI] [PubMed] [Google Scholar]
- Menetski J, Mistry S, Lu M, Mudgett JS, Ransohoff RM, Demartino JA, Macintyre DE, Abbadie C. Mice overexpressing chemokine ligand 2 (CCL2) in astrocytes display enhanced nociceptive responses. Neuroscience. 2007;149:706–714. doi: 10.1016/j.neuroscience.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Miller RJ, Jung H, Bhangoo SK, White FA. Cytokine and chemokine regulation of sensory neuron function. Handb Exp Pharmacol. 2009;2009:417–449. doi: 10.1007/978-3-540-79090-7_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SB, Tran PB, Gillard SE, Hurley RW, Hammond DL, Miller RJ. Chemokines and glycoprotein120 produce pain hypersensitivity by directly exciting primary nociceptive neurons. J Neurosci. 2001;21:5027–5035. doi: 10.1523/JNEUROSCI.21-14-05027.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priest BT. Future potential and status of selective sodium channel blockers for the treatment of pain. Curr Opin Drug Discov Devel. 2009;12:682–692. [PubMed] [Google Scholar]
- Rittner HL, Brack A. Chemokines and pain. Curr Opin Investig Drugs. 2006;7:643–646. [PubMed] [Google Scholar]
- Rostène W, Kitabgi P, Parsadaniantz SM. Chemokines: a new class of neuromodulator? Nat Rev Neurosci. 2007;8:895–903. doi: 10.1038/nrn2255. [DOI] [PubMed] [Google Scholar]
- Rostène W, Dansereau MA, Godefroy D, Van Steenwinckel J, Reaux-Le Goazigo A, Mélik-Parsadaniantz S, Apartis E, Hunot S, Beaudet N, Sarret P. Neurochemokines: a menage a trois providing new insights on the functions of chemokines in the central nervous system. J Neurochem. 2011;118:680–694. doi: 10.1111/j.1471-4159.2011.07371.x. [DOI] [PubMed] [Google Scholar]
- Roy ML, Narahashi T. Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J Neurosci. 1992;12:2104–2111. doi: 10.1523/JNEUROSCI.12-06-02104.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush AM, Waxman SG. PGE2 increases the tetrodotoxin-resistant Nav1.9 sodium current in mouse DRG neurons via G-proteins. Brain Res. 2004;1023:264–271. doi: 10.1016/j.brainres.2004.07.042. [DOI] [PubMed] [Google Scholar]
- Rush AM, Craner MJ, Kageyama T, Dib-Hajj SD, Waxman SG, Ranscht B. Contactin regulates the current density and axonal expression of tetrodotoxin-resistant but not tetrodotoxin-sensitive sodium channels in DRG neurons. Eur J Neurosci. 2005;22:39–49. doi: 10.1111/j.1460-9568.2005.04186.x. [DOI] [PubMed] [Google Scholar]
- Serrano A, Paré M, McIntosh F, Elmes SJ, Martino G, Jomphe C, Lessard E, Lembo PM, Vaillancourt F, Perkins MN, Cao CQ. Blocking spinal CCR2 with AZ889 reversed hyperalgesia in a model of neuropathic pain. Mol Pain. 2010;6:90. doi: 10.1186/1744-8069-6-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV. G protein betagamma subunits: central mediators of G protein-coupled receptor signaling. Cell Mol Life Sci. 2008;65:2191–2214. doi: 10.1007/s00018-008-8006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smrcka AV, Lehmann DM, Dessal AL. G protein betagamma subunits as targets for small molecule therapeutic development. Comb Chem High Throughput Screen. 2008;11:382–395. doi: 10.2174/138620708784534761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struthers M, Pasternak A. CCR2 antagonists. Curr Top Med Chem. 2010;10:1278–1298. doi: 10.2174/156802610791561255. [DOI] [PubMed] [Google Scholar]
- Sun JH, Yang B, Donnelly DF, Ma C, LaMotte RH. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J Neurophysiol. 2006;96:2189–2199. doi: 10.1152/jn.00222.2006. [DOI] [PubMed] [Google Scholar]
- Swanwick RS, Pristerá A, Okuse K. The trafficking of NaV1.8. Neurosci Lett. 2010;486:78–83. doi: 10.1016/j.neulet.2010.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Minami M, Nakagawa T, Satoh M. Enhanced production of monocyte chemoattractant protein-1 in the dorsal root ganglia in a rat model of neuropathic pain: possible involvement in the development of neuropathic pain. Neurosci Res. 2004;48:463–469. doi: 10.1016/j.neures.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Ukhanov K, Brunert D, Corey EA, Ache BW. Phosphoinositide 3-kinase-dependent antagonism in mammalian olfactory receptor neurons. J Neurosci. 2011;31:273–280. doi: 10.1523/JNEUROSCI.3698-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Steenwinckel J, Reaux-Le Goazigo A, Pommier B, Mauborgne A, Dansereau MA, Kitabgi P, Sarret P, Pohl M, Mélik Parsadaniantz S. CCL2 released from neuronal synaptic vesicles in the spinal cord is a major mediator of local inflammation and pain after peripheral nerve injury. J Neurosci. 2011;31:5865–5875. doi: 10.1523/JNEUROSCI.5986-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaragavan K, Boutjdir M, Chahine M. Modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by protein kinase A and protein kinase C. J Neurophysiol. 2004;91:1556–1569. doi: 10.1152/jn.00676.2003. [DOI] [PubMed] [Google Scholar]
- Wang JG, Strong JA, Xie W, Yang RH, Coyle DE, Wick DM, Dorsey ED, Zhang JM. The chemokine CXCL1/growth related oncogene increases sodium currents and neuronal excitability in small diameter sensory neurons. Mol Pain. 2008;4:38. doi: 10.1186/1744-8069-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Miller RJ. Insights into the regulation of chemokine receptors by molecular signaling pathways: functional roles in neuropathic pain. Brain Behav Immun. 2010;24:859–865. doi: 10.1016/j.bbi.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Sun J, Waters SM, Ma C, Ren D, Ripsch M, Steflik J, Cortright DN, Lamotte RH, Miller RJ. Excitatory monocyte chemoattractant protein-1 signaling is up-regulated in sensory neurons after chronic compression of the dorsal root ganglion. Proc Natl Acad Sci U S A. 2005;102:14092–14097. doi: 10.1073/pnas.0503496102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Feldman P, Miller RJ. Chemokine signaling and the management of neuropathic pain. Mol Interv. 2009;9:188–195. doi: 10.1124/mi.9.4.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27:12396–12406. doi: 10.1523/JNEUROSCI.3016-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Li GD, Zhao ZQ. State-dependent phosphorylation of epsilon-isozyme of protein kinase C in adult rat dorsal root ganglia after inflammation and nerve injury. J Neurochem. 2003;85:571–580. doi: 10.1046/j.1471-4159.2003.01675.x. [DOI] [PubMed] [Google Scholar]