

Abstract
The glyoxalase system is the most important pathway for the detoxification of methylglyoxal (MG), a highly reactive dicarbonyl compound mainly formed as a by-product of glycolysis. MG is a major precursor of advanced glycation end products (AGEs), which are associated with several neurodegenerative disorders. Although the neurotoxic effects of MG and AGEs are well characterized, little is known about the glyoxalase system in the brain, in particular with regards to its activity in different neural cell types. Results of the present study reveal that both enzymes composing the glyoxalase system [glyoxalase-1 (Glo-1) and Glo-2] were highly expressed in primary mouse astrocytes compared with neurons, which translated into higher enzymatic activity rates in astrocytes (9.9- and 2.5-fold, respectively). The presence of a highly efficient glyoxalase system in astrocytes was associated with lower accumulation of AGEs compared with neurons (as assessed by Western blotting), a sixfold greater resistance to MG toxicity, and the capacity to protect neurons against MG in a coculture system. In addition, Glo-1 downregulation using RNA interference strategies resulted in a loss of viability in neurons, but not in astrocytes. Finally, stimulation of neuronal glycolysis via lentiviral-mediated overexpression of 6-phosphofructose-2-kinase/fructose-2,6-bisphosphatase-3 resulted in increased MG levels and MG-modified proteins. Since MG is largely produced through glycolysis, this suggests that the poor capacity of neurons to upregulate their glycolytic flux as compared with astrocytes may be related to weaker defense mechanisms against MG toxicity. Accordingly, the neuroenergetic specialization taking place between these two cell types may serve as a protective mechanism against MG-induced neurotoxicity.
Introduction
The glyoxalase system is a ubiquitous enzymatic pathway that catalyzes the glutathione (GSH)-dependent detoxification of methylglyoxal (MG) and other reactive dicarbonyl compounds, thereby playing a major role in the cellular defense against glycation and oxidative stress (Thornalley, 1993; Kalapos, 2008b). It comprises two enzymes: Glo-1 and Glo-2. Glo-1 catalyzes the conversion of the hemithioacetal formed by the nonenzymatic reaction of GSH with MG, to S-d-lactoylglutathione. S-d-Lactoylglutathione is then converted to d-lactate by Glo-2, which recycles GSH in the process (Fig. 1) (Thornalley, 1993).
Figure 1.

Schematic representation of the main metabolic pathways involved in MG production and elimination. A, B, MG is formed mainly by the fragmentation of the glycolytic intermediates glyceraldehyde-3P and DHAP (A), but also from the metabolism of lipids and proteins (B). PFK is the rate-limiting step of glycolysis and thus constitutes an important regulatory site, and one of its most potent allosteric activators is fructose-2,6-P2. Fructose-2,6-P2 levels are controlled by the Pfkfb, which is most abundantly expressed in astrocytes compared with neurons (Herrero-Mendez et al., 2009). C, MG is detoxified principally via the glyoxalase system, which consists of the enzymes Glo-1 and Glo-2. The first step of MG detoxification requires its spontaneous reaction with GSH to form a hemithioacetal, which is used as a substrate by Glo-1 to form S-lactoylglutathione. Glo-2 then catalyzes the transformation of S-lactoylglutathione into d-lactate, recycling GSH in the process. D, The pentose phosphate pathway is linked to MG detoxification via the formation of NADPH, which is required for the recycling of GSH from its oxidized form (GSSG) via the action of glutathione reductase (GR).
The accumulation of MG is highly deleterious, as this metabolite is one of the most potent glycating agents present in cells. It readily reacts with lipids and nucleic acids and with lysine and arginine residues of proteins to form advanced glycation end products (AGEs) such as the hydroimidazolone MG-H1, argpyrimidine, Nϵ-(1-carboxyethyl)lysine (CEL), and MG-derived lysine dimer (Rabbani and Thornalley, 2010). AGEs are implicated in various pathophysiological mechanisms, including those associated with diabetic complications (cataracts, retinopathy, nephropathy, angiopathy), aging, and neurodegenerative disorders (Wautier and Guillausseau, 2001; Münch et al., 2010).
MG is inevitably produced (either spontaneously or enzymatically) as a by-product of fatty acid, protein, and glucose metabolism (Thornalley, 1996; Kalapos, 1999). The glycolytic pathway is the most important endogenous source of MG via the fragmentation of the triosephosphates glyceraldehyde-3-phosphate (glyceraldehyde-3P) and dihydroxyacetone phosphate (DHAP) (Fig. 1). It is estimated that MG production accounts for 0.1–0.4% of the glycolytic flux (Kalapos, 2008a).
The brain has very high energy requirements: although it represents only 2% of the body weight, it is responsible for 25% of the overall glucose consumption (Magistretti, 2008). As the cellular production of MG is linked to the glycolytic pathway, this suggests that the glyoxalase system is of primary importance to protect the brain against the accumulation of AGEs. A large body of evidence demonstrates that cerebral glucose consumption is not homogenous and that cell-type-specific differences exist in terms of energy metabolism. For instance, according to the astrocyte–neuron lactate shuttle (ANLS), glutamate uptake into astrocytes during synaptic activation causes a stimulation of anaerobic glycolysis and glucose uptake in astrocytes. Lactate produced in astrocytes is shuttled to neurons, where it may be used as an energy substrate in the Krebs cycle following its conversion to pyruvate, thus bypassing glycolysis (Magistretti, 2009). Interestingly, it has been demonstrated that in contrast to astrocytes, neurons have a poor capacity to upregulate glycolysis (Almeida et al., 2001; Porras et al., 2004; Herrero-Mendez et al., 2009). Recent evidence shows that one mechanism explaining this characteristic might be the constitutive proteasomal degradation of 6-phosphofructose-2-kinase/fructose-2,6-bisphosphatase-3 (Pfkfb3), which results in low levels of fructose-2,6-bisphosphate (fructose-2,6-P2), a potent activator of phosphofructokinase (Fig. 1) (Herrero-Mendez et al., 2009).
Despite the fact that the brain is highly dependent upon glucose consumption, little is known about its glyoxalase system, especially with regards to potential differences and complementarities between neural cell types, which may arise from their specific energy metabolic profiles. The aim of this study was thus to characterize the glyoxalase system in astrocytes and neurons in relation to the formation of AGEs.
Materials and Methods
Cell cultures
Experiments were conducted in accordance with the Swiss Federal Guidelines for Animal Experimentation and were approved by the Cantonal Veterinary Office for Animal Experimentation (Vaud, Switzerland).
Astrocytic cultures.
Primary cultures of cerebral cortical astrocytes were prepared from newborn (1- to 2-d-old) OF1 mice (Charles River Laboratories) as previously described (Gavillet et al., 2008). Cells were plated in 35 mm dishes and maintained in DMEM (D7777, Sigma-Aldrich) containing 25 mm glucose and supplemented with 44 mm NaHCO3, 10 ml/L antibiotic/antimycotic solution (Invitrogen), and 10% FCS (Bioconcept), and incubated at 37°C in an atmosphere containing 5% CO2 and 95% air. Experiments were performed on confluent 21-d-old cultures.
Neuronal cultures.
Primary cultures of cortical neurons were prepared from E17 OF1 mice embryos (Charles River Laboratories) as previously described (Allaman et al., 2010). Neurons were plated in 35 mm dishes and maintained in Neurobasal medium (which contains 25 mm glucose) supplemented with B27, GlutaMAX, penicillin (50 U/ml), and streptomycin (50 μg/ml) (Invitrogen) at 37°C in a humidified atmosphere containing 5% CO2 and 95% air and were used at DIV 14. These culture conditions typically produced 93% pure neuronal cultures, as assessed by MAP2 and GFAP coimmunostaining.
CHO.
CHO cells were obtained from LGC Standards (Molsheim). Stock cultures were kept in 150 ml flasks in DMEM supplemented with 10% FCS, penicillin (50 U/ml), and streptomycin (50 μg/ml), and were passaged once a week. To perform experiments, cells were plated in 35 mm dishes until they reached confluence, at which point they were treated as described in the following section. Cells under passage 15 were used.
Cell treatment with MG
If not otherwise specified, culture medium was renewed 2 h before treatment with MG. To avoid interference with subsequent biochemical assays, serum- and phenol red-free culture medium was used. For astrocytes, this medium consisted of DMEM (D2902; Sigma-Aldrich) supplemented with 44 mm NaHCO3 and 10 ml/L antibiotic/antimycotic solution (Invitrogen) and complemented to 25 mm glucose to give rise to a medium that is strictly equivalent to the normal culture medium except for phenol red and serum. This medium was also used for the treatment of CHO cells with MG.
For neurons, Neurobasal medium without phenol red was obtained from Invitrogen (catalog no. 12348-017) and complemented to give rise to a medium otherwise identical to that in which cells were grown, with the other exception that B27 supplement without antioxidants was used. Regular B27 supplement contains GSH and other antioxidants, which may interfere or mask some effects of MG, in particular those related to the changes of cellular redox status. GSH is also used by the glyoxalase system to detoxify MG, and thus had to be removed to get an accurate evaluation of the intrinsic response of neurons to MG. Treatment with MG was induced by the addition of a 100× stock solution in the culture medium.
Coculture experiments
Bilaminar cocultures were used to evaluate the effects of MG treatment on neuronal survival in the presence or absence of astrocytes. Astrocytes were rinsed and medium was replaced with 2 ml of fresh Supplemented Neurobasal medium (see above, Cell treatment with MG). Cells were incubated at 37°C in an atmosphere containing 5% CO2 and 95% air for 2 h, after which cocultures were initiated by transferring neurons grown on coverslips on top of the astrocyte layer (neurons facing up). Neurons cocultured with astrocytes were compared with primary neuronal cultures prepared in parallel and treated exactly the same way, i.e., they were transferred in new dishes containing identical Neurobasal medium preincubated for 2 h but in the absence of astrocytes. Two hours after transferring the coverslip, primary neurons and cocultures were exposed to MG as described above. Neuronal viability was assessed 24 h following treatment with MG using the MTT reduction assay.
Glyoxalase activity
Cell culture medium was replaced (see above, Cell treatment with MG) 24 h before the assay. Cells were rinsed with ice-cold PBS and lysed with 300 μl of buffer containing 10 mm HEPES, pH 7.0, 0.02% Triton X-100, and 100 μg/ml BSA. Samples were harvested and sonicated briefly on ice and centrifuged at 16,000 × g for 30 min at 4°C. Glo-1 and -2 activities were measured using a modification from a previously published method (Thornalley and Tisdale, 1988). To measure Glo-1 activity, 50 μl of sample was loaded onto a UV microplate (Microtiter; ThermoFisher Scientific) and 200 μl of reaction mix was added. Reaction mix consisted of 60 mm sodium phosphate buffer, pH 6.6, containing 4 mm GSH and 4 mm MG, and was preincubated for 10 min at 37°C. S-Lactoylglutathione synthesis was followed by measuring the absorbance at 240 nm every 15 s for 5 min at 25°C (Safire 2; Tecan). To measure Glo-2 activity, 100 μl of sample was loaded onto a UV microplate and 150 μl of reaction mix was added. Reaction mix consisted of 65 mm Tris-HCl, pH 7.4, containing 350 μm S-lactoylglutathione (Wako Pure Chemicals). S-Lactoylglutathione consumption was followed by measuring the absorbance at 240 nm every 30 s for 10 min at 25°C (Safire 2; Tecan). The protein content was measured using the remaining cell lysate using the BCA protein assay reagent kit (Thermo Scientific) according to the manufacturer's instructions. Glo-1 and -2 activities were expressed as milliunits per milligram of protein, where 1 unit is the amount of enzyme required to catalyze the formation (Glo-1) or utilization (Glo-2) of 1 μmol of S-lactoylglutathione per minute under assay conditions.
Western blot analysis
Cells were harvested in 80 μl of lysis buffer (62 mm Tris-HCl, pH 6.8, 50 mm DTT, 0.3% SDS) containing protease (Complete; Roche) and phosphatase (Sigma-Aldrich) inhibitor cocktails. Loading buffer (giving rise to final concentrations of 62 mm Tris-HCl, pH 6.8, 10% glycerol, 1% SDS, 0.1 mm DTT, and 0.005% bromophenol blue) was added to protein samples (10–20 μg) before denaturing at 100°C for 5 min. Proteins were resolved on NuPAGE 10% Bis-Tris minigels using MES SDS running buffer (Invitrogen) and transferred to Immobilon-P PVDF membranes (Millipore). The membranes were blocked for 1 h in PBS containing 0.1% Tween 20 (PBST) and 5% dry milk and then incubated overnight at 4°C with antibodies directed against Glo-1 (rabbit polyclonal, 1:75,000 dilution; Santa Cruz Biotechnology), Glo-2 (goat polyclonal, 1:10,000 dilution; Santa Cruz Biotechnology), AGEs [mouse monoclonal; clone 6D12, 1:2000 (see Fig. 7C) or 1:5000 (see Fig. 8B) dilution; TransGenic] or argpyrimidine (mouse monoclonal, 1:500 dilution; JaICA). After washing with PBST, the blots were probed with the appropriate HRP-conjugated secondary antibody (GE Healthcare) diluted 1:15,000 in PBST plus 5% dry milk. After extensive washing with PBST, the peroxidase activity was detected by chemiluminescence using the ECL detection system (GE Healthcare) and Kodak BioMax MR film. β-Actin expression was assessed to ensure equal loading. The PVDF membranes were stripped for 30 min at 50°C in buffer containing 62 mm Tris-HCl, pH 6.8, 100 mm β-mercaptoethanol, and 2% SDS. Membranes were washed several times with PBST and probed as described above using a mouse monoclonal antibody against β-actin diluted 1:500,000 (Sigma-Aldrich). Film was digitized using a CoolSNAP cf CCD camera (Photometrics).
Figure 7.
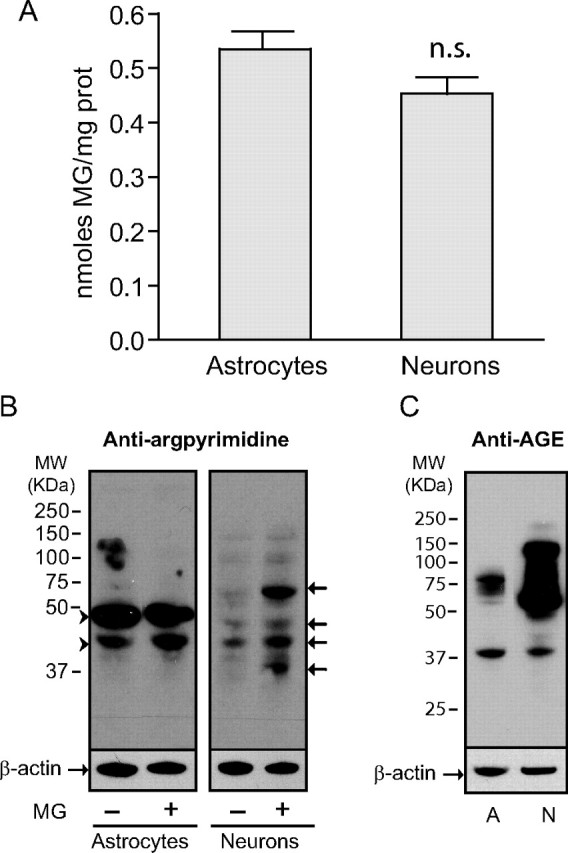
Neurons are more susceptible to MG-induced protein modifications. A, Similar levels of MG were measured in astrocytes and neurons by HPLC. Results are means ± SEM of at least 13 determinations from at least four independent experiments. Data were statistically analyzed using a Student's t test (n.s., not significantly different). B, Argpyrimidine levels were assessed by Western blotting in astrocytes and neurons. Some cultures were exposed to 0.5 mm MG for 24 h before protein extraction. Arrowheads mark proteins highly modified by argpyrimidine in astrocytes. Arrows mark neuronal proteins showing increased argpyrimidine immunoreactivity following exposure to MG. C, AGE levels were assessed by Western blotting in control cultures of astrocytes (A) and neurons (N). B, C, Representative bands are shown. Similar results were obtained in three independent experiments (n = 6).
Figure 8.
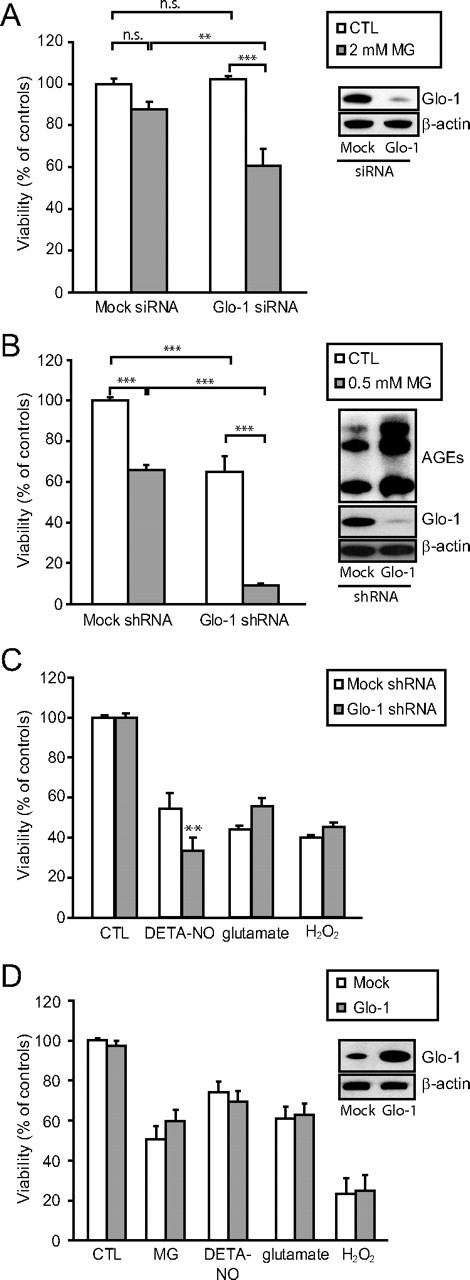
Effects of RNA interference against Glo-1 in astrocytes and neurons. A, Astrocytes were transfected with siRNA against Glo-1 or negative control siRNA (mock siRNA). Representative Western blot bands demonstrating the resulting decrease in Glo-1 expression are shown. Cells were subsequently exposed to vehicle (white bars) or 2 mm MG (gray bars), and cellular viability was assessed 24 h later using the MTT viability assay. Results are expressed as a percentage of controls (CTL) and are means ± SEM of at least six determinations from at least two independent experiments. Data were statistically analyzed with two-way ANOVA followed by Tukey's multiple-comparisons test (n.s., not significantly different; **p ≤ 0.01; ***p ≤ 0.001). B, Glo-1-targeted shRNAs (or mock shRNAs) were delivered in neurons using lentiviral vectors. Representative Western blot bands demonstrating the resulting decrease in Glo-1 expression and increase in AGE levels are shown. Cells were subsequently exposed to vehicle (white bars) or 0.5 mm MG (gray bars) and cellular viability was assessed 24 h later. Results are expressed as a percentage of controls and are means ± SEM of six determinations from two independent experiments. Data were statistically analyzed with two-way ANOVA followed by Tukey's multiple-comparison test (***p ≤ 0.001). C, Glo-1-targeted shRNAs (gray bars) or mock shRNAs (white bars) were delivered in neurons using lentiviral vectors. Neurons were subsequently exposed to various cellular stresses (DETA-NO, 100 μm for 24 h; glutamate, 10 μm for 4 h; and H2O2, 25 μm for 4 h) before assessing cellular viability using the MTT assay. Results are expressed as a percentage of controls and are means ± SEM of at least six determinations from at least two independent experiments. Data were statistically analyzed with two-way ANOVA followed by Bonferroni's test (**p ≤ 0.01). D, Glo-1 (gray bars) or a mock construction (white bars) were delivered in neurons using lentiviral vectors. Neurons were subsequently exposed to various cellular stresses (MG, 500 μm for 24 h; DETA-NO, 100 μm for 24 h; glutamate, 10 μm for 4 h; and H2O2, 25 μm for 4 h) before assessing cellular viability using the MTT assay. Results are expressed as percentage of controls and are means ± SEM of at least nine determinations from at least three independent experiments. Data were statistically analyzed with two-way ANOVA followed by Bonferroni's test and no statistical differences could be found between mock and Glo-1 groups in any of the conditions.
Quantitative PCR
Total RNA was isolated from cultured cells using a NucleoSpin RNA II kit (Macherey-Nagel) according to the manufacturer's instructions. RNA concentration was determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies). The first strand of cDNA was synthesized from 100 ng of total RNA (60 min at 37°C, followed by 5 min at 95°C) using the High Capacity RNA-to-cDNA Reverse Transcription system (Applied Biosystems). One twenty-fifth of the resulting cDNA was amplified by quantitative PCR (qPCR) with the ABI PRISM 7900 system (Applied Biosystems). PCR mix was composed of 6 ng of cDNA and 200 nm forward and reverse primers in 10 μl of 1× SYBR Green PCR Master Mix (Applied Biosystems). Primer sequences were designed using primer Express 3.0 software (Applied Biosystems) and oligonucleotides were synthesized by Microsynth. Forward and reverse primer sequences were as follows: 5′-CCT GAT GAC GGG AAA ATG AAA G-3′ and 5′-GCC GTC AGG GTC TTG AAT GA-3′ (Glo-1; GenBank NM_025374) and 5′-AGG CGC TGC TTG AAG TCT TG-3′ and 5′-AAG GTT GTT TAC GGT GTA TTC ATG AC-3′ (Glo-2; GenBank accession number NM_024284.2). The specificity of PCR amplification for each set of primers was checked by the presence of a single sharp peak in the melting curve analysis. Data were computed using the sequence detector software SDS 2.3 (Applied Biosystems) and analyzed using a macro developed by the genomic platform of Geneva University (Frontiers in Genetics, UNIGE). Delta-CT relative quantification (ΔΔCt) was used and data were expressed as percentage of astrocytic expression.
Immunofluorescence and image acquisition
Female OF1 mice were perfused-fixed with 4% paraformaldehyde and brains were cryoprotected in a 30% sucrose solution and frozen at −80°C until use. Free-floating coronal sections (30 μm) were obtained by cryostat sectioning and kept in PBS containing 0.05% sodium azide at 4°C until use. For double immunofluorescence staining, sections were rinsed in PBS and permeabilized for 30 min at room temperature with PBS containing 0.25% Triton X-100. Sections were then incubated in PBS containing 5% BSA and 0.05% Triton X-100 (PBS-BT) for 1 h at room temperature to block nonspecific binding before incubation with a rabbit polyclonal antibody against Glo-1 (1/250 dilution; catalog no. sc-67351; Santa-Cruz Biotechnology) in PBS-BT overnight at 4°C. Sections were rinsed thoroughly and incubated for 1 h at room temperature in PBS-BT containing 1/250 donkey anti-rabbit secondary antibody labeled with the green fluorescent Alexa Fluor 488 dye (Invitrogen). After extensive washing, sections were sequentially stained (using the procedure described above for Glo-1) with mouse monoclonal antibodies directed against either GFAP (1/600 dilution; catalog no. G3893; Sigma-Aldrich) or NeuN (1/600 dilution; catalog no. MAB377; Millipore) followed by incubation with donkey anti-mouse secondary antibody labeled with the red fluorescent Alexa Fluor 568 dye (Invitrogen). Sections were finally incubated with 0.2 μg/ml Hoechst to stain nuclei, rinsed in phosphate buffer (120 mm, pH 7.4), and mounted with an anti-bleaching medium (DABCO; Sigma-Aldrich). Single staining with each primary antibody (Glo-1, GFAP, or NeuN) and its respective secondary antibody was also performed as a control experiment according to the same protocol. For each individual target, the staining patterns observed in single and double immunofluorescence experiments were similar.
Image acquisitions were performed as the maximum projection of a stack of 12 images with 1 μm step size (except for Fig. 3C, which consisted of 30 images with 300 nm step size) on a confocal microscope (LSM 710; Zeiss) with a Plan Apochromat 40× 1.3 oil-immersion objective. Image processing was performed using the Imaris software v6.4.2, in surpass mode. To avoid any cross talk, the images were acquired in sequential mode using laser lines at 405 nm (30 mW), 488 nm (25 mW), and 561 nm (15 mW) with 1–4.5% transmission; 8 average line method with a pixel dwell of 0.79 μs; and bandpass of 410–480 nm for DAPI, 490–550 nm for Alexa 488, and 565–700 nm for Alexa 568. To obtain an appropriate resolution, a format of 1024 × 1024 with a zoom at 1.2 was used, corresponding to 148 nm/pixel (except for Fig. 3C; format = 354 × 222, zoom = 1.4, and pixel size = 148 nm). The pinhole aperture was 1 airy unit.
Figure 3.

High expression of Glo-1 in astrocytes of the mouse cerebral cortex. Coronal sections of mouse brain were immunostained with Glo-1 and with the astrocytic marker GFAP (A, C) or the neuronal marker NeuN (B). Glo-1 immunostaining was strongest in astrocytes (A, arrows), but was also present in neurons at lower levels (B, arrowheads). C, Scaled-up image of a single astrocyte in the mouse cerebral cortex shows that Glo-1 immunoreactivity is located in the cell body and processes along the GFAP+ filaments. Representative images from one of three animals are shown. Nuclei are stained using Hoechst. Scale bars: 15 μm (A, B) and 7 μm (C).
Cell dissociation and FACS analysis
Young C57BL/6-Tg (hGFAP-eGFP) mice (23–27 d old) were deeply anesthetized with isoflurane (Attane; Minrad) and killed by decapitation. The brain was rapidly removed and cerebral cortices were dissected out and placed in ice-cold calcium and magnesium-free modified HBSS (mHBSS) containing 25 mm d-glucose and 10 mm HEPES (Invitrogen). Cortices were cut into 1 mm3 pieces and enzymatically digested with 20 U/ml papain (Worthington) in mHBSS containing 0.12 mg/ml l-cysteine (Sigma-Aldrich) and 1 U/ml DNaseI (Worthington) for 45 min at 35°C under gentle agitation. Papain activity was quenched for 5 min using a 1% solution of BSA diluted in FCS and tissue was centrifuged 2 min at 200 × g to remove the enzyme solution. The tissue was then dissociated by gentle trituration in ice-cold mHBSS and the cell suspension was collected in a tube containing DNaseI. This step was repeated three times. Cells were then centrifuged for 8 min at 500 × g, the pellet was resuspended in 2 ml of mHBSS, and cell clusters and debris were removed by filtration through a 40 μm cell strainer (Falcon; BD Biosciences). Finally, 2 μl of propidium iodide (Fluka), a fluorescent cell death indicator, was added to the cell suspension.
Cell suspensions were individually sorted by FACS using a flow cytometer with Diva option (FACS Vantage SE; Becton Dickinson) equipped with a Coherent Enterprise laser simultaneously emitting 360 and 488 nm in position 1 (5 fluorescence), and a Coherent Helium-Neon emitting 630 nm in the third position (2 fluorescence). Data were collected and analyzed using FACSDiva software (Becton Dickinson). One hundred thousand to 130,000 viable GFP+ cells were typically obtained using this procedure. Sorted cells were harvested in PBS, pelleted, and resuspended in lysis buffer, following which Glo-1 enzyme activity was assessed as described above. The enrichment of the cell fractions was assessed using qPCR analysis. Astrocytic markers were 10- to 50-fold higher in GFP+ cell fraction, while the oligodendrocytes and the neuronal and the microglial markers were 5- to 20-fold enriched in the GFP− cell fraction.
MTT viability assay
For MTT determination in coculture and primary culture experiments, a 20× stock solution of MTT was added directly to the culture medium to give rise to a final concentration of 0.2 mg/ml (cocultures and neurons) or 0.1 mg/ml (astrocytes), and cells were incubated for 20 min at 37°C in an atmosphere containing 5% CO2 and 95% air. The medium was then removed by aspiration, and the reaction was stopped by the addition of 1 ml of DMSO. For coculture experiments, the glass coverslips bearing neurons were placed in a new dish before the addition of DMSO. The amount of reduced MTT (formazan) solubilized in DMSO was then determined spectrophotometrically using absorbance at 560 nm (Safire 2; Tecan). Results are expressed as percentage of control values.
GSH assay
Total intracellular GSH levels ([GSx]i, where GSx = reduced form GSH + oxidized form GSSG) were determined spectrophotometrically as previously described (Allaman et al., 2010). Absolute values were calculated from a standard curve of known amounts of GSSG, and intracellular GSH levels were expressed as nanomoles of GSx equivalent per milligram of protein.
NADP/NADPH assay
Cycling assays for nicotinamide adenine dinucleotides were adapted from previously published methods (Matsumura and Miyachi, 1980; Zerez et al., 1987). Cells were rinsed three times with ice-cold PBS, harvested in 300 μl of ice-cold carbonate-bicarbonate buffer (100 mm Na2CO3, 20 mm NaHCO3 containing 10 mm nicotinamide to inhibit NADase), and quickly frozen on dry ice. Samples were thawed in a room temperature water bath and immediately chilled on ice. Extracts were centrifuged at 12,000 × g for 30 min at 4°C and half of the supernatant was heated at 60°C for 30 min to denature NADP. Fifty microliters of both the heated extract (containing NADPH only) and the unheated extract (containing NADP and NADPH) were loaded onto a 96-well microplate along with blanks (carbonate-bicarbonate buffer) and standards of known NADPH (Roche) concentrations dissolved in carbonate-bicarbonate buffer. Then 150 μl of a reaction mixture was added into each well and the absorbance was followed spectrophotometrically at 570 nm every 15 s over a 5 min period (Safire 2; Tecan). Reaction mixture was prepared right before use and contained 130 mm bicine, 5.3 mm EDTA, 0.6 mm MTT, 2.2 mm phenazine ethosulfate, 3.3 mm glucose-6-phosphate, and 2.5 U/ml glucose-6-phosphate dehydrogenase (G6378; Sigma-Aldrich). Blanks values were subtracted from all samples and NADP amounts were calculated by subtracting NADPH values from total NADP + NADPH values. The protein content was measured using the remaining cell lysate using the BCA protein assay reagent kit according to the manufacturer's instructions and used for normalization. Values are expressed as nanomoles per milligram protein.
HPLC
HPLC determinations were conducted using a method previously described for the measurement of free and reversibly bound MG (Chaplen et al., 1998). Briefly, cells were rinsed twice with PBS and harvested in 0.45N perchloric acid (PCA) containing 10 μm 5-methylquinoxaline (5-MQ) (Sigma-Aldrich) as an internal standard. For each sample, two 35 mm dishes were pooled in a total volume of 150 μl and derivatized with 1 mm o-phenylenediamine (Sigma-Aldrich) for 15 h at room temperature and centrifuged at 12,000 × g for 30 min to remove PCA-precipitated materials. The resulting 2-methylquinoxaline (2-MQ) was measured by HPLC using a Waters Alliance 2795 HPLC system (Waters) with a Waters 996 photodiode array detector (scanning wavelength, 315 nm) as previously described (Chaplen et al., 1996) with modifications. A 150 μl sample was injected onto the reverse-phase column (BDS Hypersil C18 100 × 4.6 mm; 3 μm particle size) fitted with a BDS C18 5 μm Guard precolumn (ThermoFisher Scientific). Isocratic elution was performed with buffer A consisting of 80% 10 mm KH2PO4, pH 2.5, and 20% HPLC grade acetonitrile (Fisher Scientific) at a constant flow rate of 1 ml/min. A 2 min wash step with buffer B (20% 10 mm KH2PO4, pH 2.5, and 80% acetonitrile) was included between each sample. Peak area was determined using an automated integrator (MassLynx v 4.0, Waters), and 2-MQ concentrations were calculated in relation to the internal standard (5-MQ) using a standard curve of MG (ranging from 0 to 500 pmol) derivatized and processed in parallel with the samples. Average retention times for 2-MQ and 5-MQ were 5.1 and 9.9 min, respectively. Average protein concentrations for each treatment group were determined in a separate set of dishes using the BCA assay and used for normalization.
siRNA transfection
Specific Glo-1 downregulation in astrocytes was achieved using Stealth Select RNAi siRNAs (ref. no. Glo1/MSS27278; Invitrogen). For each 35 mm dish, 100 pmol of siRNA and 5.5 μl of HiPerFect Transfection Reagent (Qiagen) were diluted in 90 μl of DMEM (without serum and antibiotics) and vortexed at low speed for 2 s. After a 10 min incubation period at room temperature, the transfection complex was added dropwise to 80% confluent astrocytic cultures. Control groups (mock siRNA) were transfected with Stealth siRNA negative controls (Lo GC complex #2; Invitrogen). siRNA efficacy was assessed using the glyoxalase enzyme activity assay and by Western blotting. Glo-1 siRNA treatment typically resulted in a 75% decrease in Glo-1 enzyme activity specifically, beginning 24 h following treatment and lasting for at least the following 96 h (data not shown). Experiments were conducted within this time frame.
Lentiviral vector production
The cDNA coding for Glo-1 (GenBank accession number NM_025374) or for Pfkfb3 (GenBank accession number NM_133232.2) was cloned downstream of the phosphoglycerate kinase (PGK) promoter in a self-inactivating lentiviral transfer vector (SIN-W-PGK) (Déglon et al., 2000). A Kozak consensus sequence was inserted upstream of the ATG codon. To produce a negative control virus (mock virus), a short (42 bp) noncoding sequence was also cloned into the SIN-W-PGK vector and processed in parallel with the target genes. For gene silencing in neuronal cultures, small hairpin RNA (shRNA) constructs specifically targeted against mouse Glo-1 and cloned into the pLKO1 lentiviral vector (ref. no. TRCN0000114844) were purchased from Open Biosystems. The empty pLKO1 vector was also obtained and used as a negative control for experiments using shRNA (mock shRNA).
Lentiviral vectors were generated using a four-plasmid system [i.e., SIN-W-PGK (or pLKO1 for shRNA delivery), pCMVΔR8.92, pRSV-Rev, and pMD2.G] by transient calcium phosphate transfection of 293T cells as previously described (Towne and Aebischer, 2009; Barde et al., 2010). pCMVΔR8.92, pRSV-Rev, and pMD2.G plasmids were purchased from Fasteris. Supernatants were collected 72 h later and filtered, and viral particles were concentrated by ultracentrifugation. Pellets were resuspended in PBS containing 0.5% BSA and the p24 antigen content was determined by ELISA (Gentaur) according to the manufacturer's instructions. Viral stocks were stored at −80°C until use. Neurons were infected on DIV 3 and experiments were conducted 1 week later. The amount of virus added was 25 ng for Glo-1 gene silencing and overexpression and 200 ng per 35 mm dish for Pfkfb3 overexpression experiments. All experiments using lentiviral vectors were performed in a biosafety level 2 laboratory.
[3H]-2-Deoxyglucose uptake
Cells were treated with viruses as specified and [3H]-2-deoxyglucose ([3H]-2-DG) uptake experiments were conducted as previously described (Allaman et al., 2004). Incubation medium consisted of glucose-free Neurobasal medium supplemented with 5 mm glucose, GlutaMAX, B27 minus antioxidants, and 1 μCi/ml [3H]-2-DG (specific activity, 30–60 Ci/mmol; ANAWA). Results, which represent transporter-mediated glucose uptake and subsequent phosphorylation, were calculated by subtracting from total counts the portion that was not inhibited by the glucose transporter inhibitor cytochalasin B (50 μm) added 15 min before and during [3H]-2-DG incubation. [3H]-2-DG uptake is expressed as percentage of control values.
Lactate release
l-Lactate release into the medium was determined as previously described (Allaman et al., 2010). Measurements were made on aliquots of the culture medium used for [3H]-2-DG, which were collected at the end of the incubation period. Briefly, aliquots (200 μl) were mixed with 1 ml of glycine buffer (0.2 m glycine, 0.2 m semicarbazide, pH 10) containing 2 mg/ml NAD and 7 U/ml l-lactate dehydrogenase (Roche) and incubated at 40°C for 1 h. After 20 min at room temperature, the amount of NADH produced was measured in 200 μl aliquots loaded on a 96-well black plate with a spectrophotometer (increase in fluorescence; 340 nm excitation/450 nm emission) (Safire 2; Tecan) and absolute values were determined from a standard curve of l-lactate.
d-Lactate released into the extracellular space following treatment with MG was measured using the abovementioned protocol with some modifications. Seventy microliter aliquots of culture medium were directly loaded on a 96-well black plate and 180 μl of glycine buffer (0.2 m glycine, 0.2 m semicarbazide, pH 10) containing 2 mg/ml NAD and 40 U/ml d-lactate dehydrogenase (CPC Biotech) was added. Samples were incubated for 2 h at room temperature, after which NADH was measured as described above. Absolute values were determined from a standard curve of known d-lactate concentrations. l-Lactate and MG up to 5 mm were undetectable using this assay. In some cases, samples were diluted with fresh culture medium in order to obtain values within the linear part of the standard curve.
Statistical analysis
Data were analyzed for statistical significance by Student's t test, or by one-way or two-way ANOVA. Statistically significant one-way ANOVAs were followed by a post hoc Dunnett's multiple-comparison test when all groups were compared with the control group, or by a Bonferroni's multiple-comparison test when comparing all pairs of groups (Prism 4.0; GraphPad). Two-way ANOVAs were followed by a post hoc Bonferroni's multiple-comparison test, except when requiring comparison between all pairs of groups, in which case post hoc analysis was made using Tukey's multiple-comparison test (R 2.12.0). Significance was accepted at p ≤ 0.05 for all statistical tests.
Results
The glyoxalase system in astrocytes and neurons
To evaluate the intrinsic capacity of astrocytes and neurons to detoxify MG, Glo-1 and -2 activity rates were determined in primary cortical cultures. Both enzymes displayed significantly higher activity in astrocytes compared with neurons. Glo-1 and -2 activity were, respectively, 9.8 times higher (943.4 ± 69.5 vs 95.4 ± 9.0 mU/mg protein) and 2.5 higher (18.7 ± 0.5 vs 7.4 ± 0.2 mU/mg protein) in astrocytes as compared with neurons (Fig. 2A,B). It is also worth noting that Glo-1 activity rates were markedly higher compared with Glo-2 in both cell types (Fig. 2A vs 2B), supporting the notion that the rapid conversion of GSH and MG to S-lactoylglutathione plays a critical role in the cellular defense against MG toxicity (Thornalley, 2008).
Figure 2.

Characterization of the glyoxalase system in astrocytes and neurons. A, B, Glo-1 (A) and Glo-2 (B) activity in primary cultured astrocytes and neurons prepared from mouse cortex. Results are means ± SEM of at least 12 determinations from three independent experiments. Data were statistically analyzed using a Student's t test (***p ≤ 0.001 vs astrocytes). C, D, Glo-1 (C) and Glo-2 (D) protein expression in cultured astrocytes and neurons (designated by A and N, respectively) was assessed by Western blotting. Representative bands are shown. Similar results were obtained in three independent experiments (n = 6). E, F, Glo-1 (E) and Glo-2 (F) mRNA expression was assessed in cultured astrocytes and neurons by qPCR. Results are expressed as a percentage of astrocytic expression and are means ± SEM of six determinations from two independent experiments. Data were statistically analyzed using a Student's t test (***p ≤ 0.001 vs astrocytes; n.s., not significantly different). G, Time course analysis of d-lactate release into the extracellular space following exposure of astrocytes and neurons to MG (500 μm). Results are means ± SEM of at least nine determinations from at least three independent experiments. Data were statistically analyzed with two-way ANOVA followed by Bonferroni's post hoc test (**p ≤ 0.01 and ***p ≤ 0.001 vs neurons). H, Dose–response analysis of d-lactate release into the extracellular space 24 h following exposure to MG in astrocytes and neurons. Results are means ± SEM of at least nine determinations from at least three independent experiments. Data were statistically analyzed with two-way ANOVA followed by Bonferroni's post hoc test (**p ≤ 0.01 and ***p ≤ 0.001 vs neurons).
In accordance with the enzymatic activity measurements, Western blot analysis revealed that Glo-1 and -2 protein expression is higher in astrocytes compared with neurons, and this difference is more pronounced for Glo-1 than for Glo-2 (Fig. 2C,D). At the mRNA level, while Glo-1 expression is 3.3 times higher in astrocytes than in neurons (Fig. 2E), Glo-2 expression is not significantly different between both cell types (Fig. 2F), suggesting that posttranscriptional mechanisms differentially regulate Glo-2 expression in astrocytes and neurons.
The end product of MG detoxification via the glyoxalase system is d-lactate. In contrast to the l-lactate enantiomer, d-lactate is poorly metabolized in mammalian neural cells (Brandt et al., 1984). We thus measured d-lactate release into the extracellular space as an index to monitor the detoxification of MG by the glyoxalase pathway in astrocytes and neurons. Importantly, neither l-lactate nor MG interfered with the d-lactate assay at concentrations up to 5 mm (data not shown). For the time course analysis, astrocytes and neurons were exposed to 500 μm MG and aliquots of the culture medium were taken over the next 5 h (Fig. 2G). The production of d-lactate occurred significantly more rapidly in astrocytes than in neurons, in agreement with the higher Glo-1 and -2 enzyme activity rates reported above. Furthermore, astrocytes completely metabolized the MG bolus within 5 h, as the addition of 500 μm MG resulted in the production of 513.3 ± 25.4 μm d-lactate, which is consistent with the predicted stoichiometry of the glyoxalase system reaction. In contrast, neurons were unable to metabolize the entire bolus of MG added within the same period ([d-lactate] = 310.0 ± 18.7 μm after 5 h; Fig. 2G). A similar pattern (i.e., faster and more complete conversion of MG to d-lactate in astrocytes vs neurons within 5 h) was obtained following exposure to a lower dose (250 μm) of MG (data not shown).
We next exposed astrocytes and neurons to increasing concentrations of MG and measured the amount of d-lactate produced after 24 h. As shown in Figure 2H, astrocytes at concentrations higher than 250 μm had a capacity to metabolize significantly higher amounts of MG as compared with neurons. Exposure of astrocytes to MG up to 2 mm resulted in the nearly equimolar production of d-lactate, while at higher concentrations MG was not entirely converted into d-lactate. In neurons, MG was entirely metabolized to d-lactate only up to a concentration of 250 μm (209.2 ± 10.7 μm d-lactate produced), after which MG detoxification was incomplete—d-lactate production reaching a plateau at 535.4 ± 4.6 μm despite the presence of MG in the millimolar range (Fig. 2H). The incomplete detoxification of MG at high concentrations is likely accompanied by a toxic effect of MG, since it significantly decreases astrocytic and neuronal viability at high concentrations (see below).
Results showing an equimolar production of d-lactate from subtoxic concentrations of MG suggest that the vast majority of MG is detoxified via the glyoxalase pathway in astrocytes and neurons, which is consistent with reports demonstrating that the glyoxalase system is by far the primary MG detoxification route in most cell types (Thornalley, 1996). In addition to the glyoxalase system, other enzymes such as aldose reductase, aldehyde dehydrogenase, and carbonyl reductase can also metabolize MG (Thornalley, 1993; Kalapos, 1999). The potential compensatory upregulation of these alternative detoxification enzymes following exposure to MG (2 mm for 24 h) was assessed in astrocytes by qPCR; however, no such effect could be observed (data not shown).
Glo-1 enrichment in astrocytes in vivo
The enrichment of Glo-1 in astrocytes compared with neurons was confirmed by immunofluorescence in the cerebral cortex of mice. Glo-1 immunofluorescence showed diffuse staining throughout the parenchyma with some cells showing more intense immunoreactivity (Fig. 3A,B). Consistent with previous reports (Chen et al., 2004; Kuhla et al., 2006), Glo-1 immunoreactivity could be observed in both astrocytes (GFAP-positive cells, arrows) and neurons (NeuN-positive cells, arrowheads); however, the strongest levels of Glo-1 immunostaining colocalized with the astrocytic marker GFAP (Fig. 3A). In astrocytes, Glo-1 immunostaining was present in the cell body and in the main astrocytic processes along the GFAP-positive filaments (Fig. 3C).
To ensure that the higher Glo-1 immunostaining observed in astrocytes in the mouse brain has functional implications, we assessed Glo-1 enzyme activity in astrocytes acutely isolated from the brain of transgenic mice expressing eGFP under the control of the GFAP promoter. To do so, cerebral cortices were gently dissociated and FACS was used to isolate astrocytes (GFP+ cells) from other neural cell types (GFP− cells). Our results show that in acutely isolated brain cells, Glo-1 activity is 2.0 ± 0.3-fold higher in the astrocyte-enriched cell fraction compared with other brain cells (n = 6 from three independent experiments; p ≤ 0.001). It is worth noting that these results are consistent with those of Cahoy et al. (2008), who have shown, using a similar technique, a 2.5-fold enrichment of Glo-1 mRNA expression in acutely isolated mouse astrocytes.
Together, these results indicate that our in vitro observation that the glyoxalase system is highly efficient in cortical astrocytes compared with neurons reflects the in vivo situation and support the notion that this cell-specific distribution has physiological significance in the intact brain.
MG toxicity in astrocytes and neurons
To determine whether the striking differences observed in the glyoxalase system capacity of astrocytes and neurons could result in different resistance to MG toxicity, cultured astrocytes and neurons were exposed to increasing concentrations of MG and cell viability was assessed 24 h later using the MTT assay. Dose–response analysis shows that astrocytes were six times more resistant to MG toxicity as compared with neurons (LD50 = 3.39 ± 0.16 mm in astrocytes vs 0.55 ± 0.05 mm in neurons, p ≤ 0.001; Fig. 4A). MG toxicity was also assessed using an alternative cell viability assay (calcein-AM uptake). LD50 values were not significantly different from those obtained with the MTT assay for both cell types (data not shown).
Figure 4.
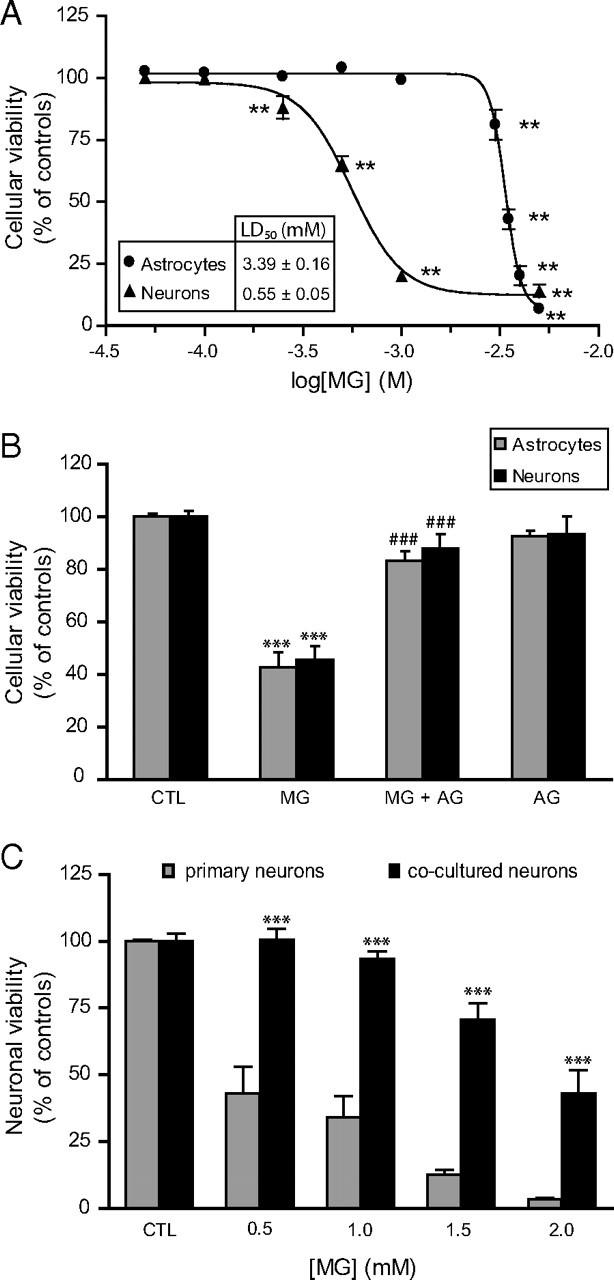
MG toxicity in astrocytes and neurons. A, Dose-dependent decrease in astrocytic and neuronal viability following exposure to MG. Cells were treated with MG and cellular viability was assessed 24 h later using the MTT assay. Results are expressed as a percentage of control values and are means ± SEM of at least nine determinations from at least three independent experiments. Data were statistically analyzed with one-way ANOVA followed by Dunnett's test (**p ≤ 0.01 vs controls). B, Pretreatment with the carbonyl scavenger AG protects astrocytes and neurons against MG-induced toxicity as assessed using the MTT assay. Different concentrations of MG were chosen to produce robust toxicity in both cell types (3.5 and 1 mm MG for astrocytes and neurons, respectively). Where indicated, an equimolar amount of AG (3.5 and 1 mm for astrocytes and neurons, respectively) was added 30 min before the addition of MG and maintained throughout the incubation period (24 h). Results are expressed as a percentage of control (CTL) values and are means ± SEM of at least nine determinations from at least three independent experiments. Data were statistically analyzed with ANOVA followed by Bonferroni's test (***p ≤ 0.001 vs controls; ###p ≤ 0.001 vs MG). C, Astrocytes protect neurons against MG toxicity in astrocyte–neuron cocultures. Primary neurons and cocultures were exposed to the indicated doses of MG, and cellular viability was assessed in the neuronal compartment using the MTT assay. Results are expressed as a percentage of control values and are means ± SEM of at least eight determinations from at least three independent experiments. Data were statistically analyzed with two-way ANOVA followed by Bonferroni's test (***p ≤ 0.001 vs primary neurons).
MG-induced cell death could be prevented in astrocytes and neurons by the addition of an equimolar amount of the carbonyl scavenger aminoguanidine (AG) 30 min before MG (Fig. 4B). Furthermore, AG reduced by 88 and 86% the amount of d-lactate produced from MG in astrocytes and neurons, respectively (data not shown), which is consistent with the direct scavenging of MG by AG, thereby neutralizing MG and making it unavailable for the glyoxalase system.
Previous characterization of the glyoxalase system in CHO cells—which is a rapidly dividing and thus high energy-consuming cell line—has shown that they display very high metabolic fluxes to MG and d-lactate compared with other cell types (Kingkeohoi and Chaplen, 2005; Paoli et al., 2010). We thus measured Glo-1 and -2 activities as well as MG toxicity in this cell line to obtain a comparison point for the data obtained in astrocytes and neurons. Glo-1 and -2 activity rates in CHO cells were comprised between those observed in neurons and astrocytes (Glo-1, 189.6 ± 4.2 mU/mg protein; Glo-2, 10.7 ± 1.6 mU/mg protein). When comparing CHO cells to astrocytes, our results show that Glo-1 and -2 activity rates were, respectively, 5.0-fold and 1.7-fold higher in astrocytes, suggesting that astrocytes possess a particularly efficient glyoxalase system. Consistent with the enzymatic activity data, CHO cells were more resistant than neurons, but less than astrocyte, to MG toxicity as assessed by the MTT reduction assay (LD50, 0.91 ± 0.17 mm).
Considering that astrocytes possess a more efficient glyoxalase system for the detoxification of MG and are significantly more resistant to MG toxicity than neurons, we next sought to determine whether astrocytes could provide significant neuroprotection against MG toxicity. Coculture experiments demonstrate that the rapid detoxification of MG by astrocytes can indeed be protective for surrounding neurons, since neurons cultured in the presence of astrocytes were significantly more resistant to MG toxicity than primary neurons (Fig. 4C). Indeed, a concentration of 2 mm MG was necessary to reduce neuronal viability by half in our coculture system, which is close to four times the concentration required to achieve a similar effect in primary neurons (Fig. 4A,C).
Differential effects of MG on the redox status of astrocytes and neurons
The first step for the detoxification of MG via the glyoxalase system is its reaction with GSH to form a hemithioacetal, which is subsequently used as a substrate by Glo-1 to form S-lactoylglutathione. Although GSH is recycled in the following step by the action of Glo-2, exposure to MG is likely to cause a transient GSH depletion, especially since Glo-2 displays much slower activity rates than Glo-1 in both astrocytes and neurons (Fig. 2). According to the extent and duration of this depletion, it may have important consequences for the cell's oxidative stress status, since GSH represents one of the most important antioxidants in mammalian cells (Dringen, 2000). Because it has previously been reported that GSH concentrations are higher in astrocytes than in neurons (Dringen, 2000), this suggests that differences in GSH metabolism may explain, together with higher Glo-1 and -2 activities, the greater resistance of astrocytes against MG toxicity. To assess this, we measured total intracellular GSH levels ([GSx]i) as well as NADP and NADPH (the latter is essential for GSH recycling by GSH reductase; Fig. 1) in astrocytes and neurons, following exposure to MG.
In control conditions, intracellular GSx concentrations were on average ninefold higher in astrocytes compared with neurons (118.7 ± 7.6 and 13.7 ± 0.5 nmol/mg protein in astrocytes and neurons, respectively), and NADPH levels were threefold higher in astrocytes compared with neurons (0.605 ± 0.020 and 0.205 ± 0.011 nmol/mg protein in astrocytes and neurons, respectively). For dose–response analysis, cells were exposed to increasing concentrations of MG for 24 h. Subtoxic doses of MG (1 and 2 mm) induced a large increase of intracellular GSH levels in astrocytes, whereas higher doses produced a gradual decrease in GSH levels (Fig. 5A), which correlated with MG toxicity in astrocytes (Fig. 4A). In contrast, no significant increase in intracellular GSH could be observed following exposure to subtoxic doses of MG in neurons (Fig. 5B). Instead, GSH levels gradually decreased starting at 500 μm and became undetectable following exposure to 2 mm MG (Fig. 5B). NADPH concentration was significantly increased in astrocytes following exposure to 2.5 mm MG but decreased gradually, together with NADP, in response to higher concentrations of MG (Fig. 5C). Similar to the intracellular GSH pattern, neurons did not display any compensatory increase in NADPH; instead both NADP and NADPH levels were significantly decreased starting at 0.5 and 1 mm MG, respectively (Fig. 5D).
Figure 5.
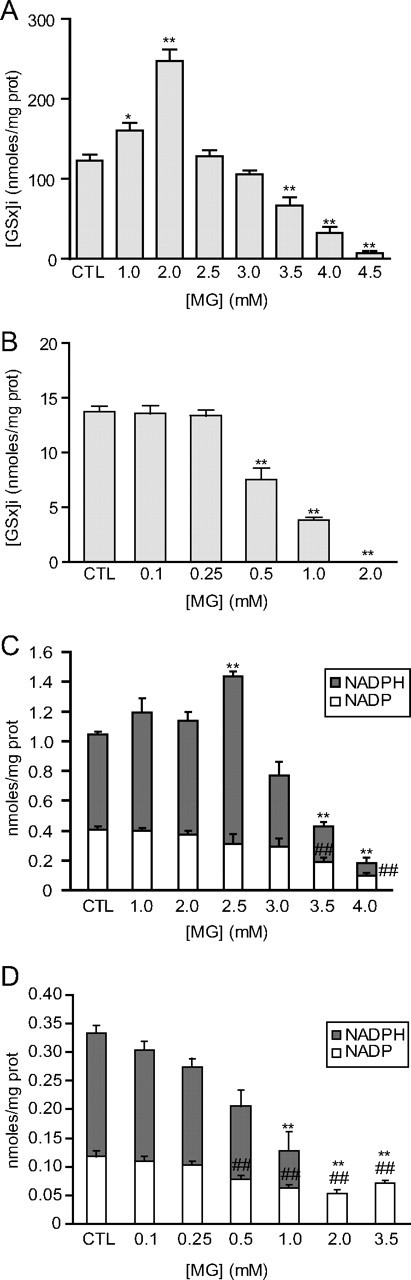
Dose–response effect of MG on the redox state of astrocytes and neurons. A, B, Astrocytes (A) and neurons (B) were exposed to the indicated concentrations of MG, and total intracellular GSH ([GSx]i) was measured 24 h later. Results are expressed as a percentage of control (CTL) values and are means ± SEM of at least eight determinations from at least three independent experiments. Data were statistically analyzed with ANOVA followed by Dunnett's test (*p ≤ 0.05; **p ≤ 0.01). C, D, Dose–response analysis of NADP and NADPH levels in astrocytes (C) and neurons (D) following exposure to the indicated concentrations of MG for 24 h. Results are means ± SEM of at least nine determinations from at least three independent experiments. Data were statistically analyzed with ANOVA followed by Dunnett's test (**p ≤ 0.01 vs CTL NADPH levels; ##p ≤ 0.01 vs CTL NADP levels).
For time course analyses, MG doses in the lower toxicity range were chosen (2.5 mm and 500 μm for astrocytes and neurons, respectively) to create a significant cellular stress. Exposure to MG lowered intracellular GSH levels in both cell types, with a maximum decrease occurring 1 h following exposure to MG and reaching 37 and 48% of control levels in astrocytes and neurons, respectively (Fig. 6A,B). In astrocytes, GSH levels returned to control values within 24 h (Fig. 6A), while in neurons GSH levels did not recover within 24 h (Fig. 6B), despite the fact that they were exposed to five times less MG than astrocytes. In astrocytes, NADPH levels gradually increased over the first 3 h following exposure to MG and remained significantly elevated for up to 6 h (Fig. 6C). NADP levels remained unchanged following exposure to MG, suggesting that compensatory mechanisms are activated in astrocytes, leading to a net increase in NADPH synthesis. In contrast, no significant change in NADP and NADPH levels could be observed in neurons for up to 6 h following exposure to MG (Fig. 6D).
Figure 6.
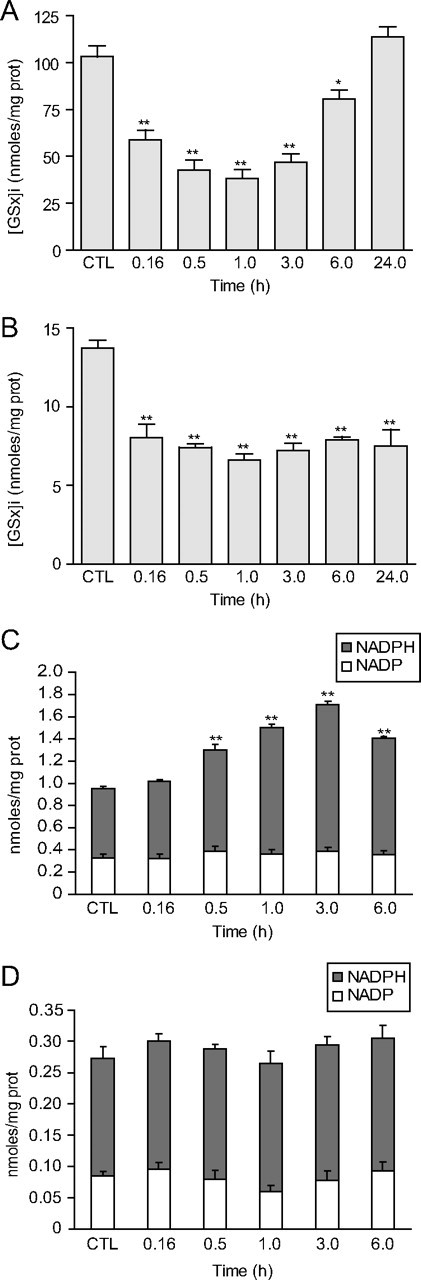
Time course analysis of the effects of MG on the redox state of astrocytes and neurons. Astrocytes and neurons were, respectively, exposed to 2.5 and 0.5 mm MG. A, B, Time course analysis of total intracellular GSH ([GSx]i) in astrocytes (A) and neurons (B) following exposure to MG. Results are expressed as percentage of control (CTL) values and are means ± SEM of at least eight determinations from at least three independent experiments. Data were statistically analyzed with ANOVA followed by Dunnett's test (*p ≤ 0.05; **p ≤ 0.01). C, D, Time course analysis of NADP and NADPH levels in astrocytes (C) and neurons (D) following exposure to MG. Results are means ± SEM of at least nine determinations from at least nine independent experiments. Data were statistically analyzed with ANOVA followed by Dunnett's test (**p ≤ 0.01 vs CTL NADPH levels).
The observed increases in the astrocytic GSH levels following exposure to subtoxic doses of MG suggest that nuclear factor (erythroid-derived 2)-like 2 (Nrf2)-dependent mechanisms might be activated in this cell type. Indeed, this transcription factor (which is primarily expressed in astrocytes in the CNS) can be activated by decreases in the intracellular concentration of GSH and plays a central role in inducible astrocytic defense against oxidative stress, especially with regards to GSH synthesis and release (Thimmulappa et al., 2002; Vargas and Johnson, 2009). Interestingly, we observed that exposure to MG induced the expression of several antioxidant genes known to be under the control of Nrf2, such as thioredoxin reductase, GSH synthetase, GSH reductase, and the glutamate-cysteine ligase modifier subunit (data not shown). This suggests that Nrf2-dependent oxidative stress defense mechanisms might participate in the compensatory increase in intracellular GSH levels observed in astrocytes in response to subtoxic doses of MG (Fig. 5A) and contribute to their high resistance to MG toxicity.
Differential MG-related protein modification in astrocytes and neurons
We next conducted HPLC analyses to determine whether the differences observed in the MG detoxification capacity of astrocytes and neurons have an impact on the cellular levels of MG present in these two cell types. Results show that MG levels are not significantly different between astrocytes and neurons (Fig. 7A). In addition, Western blot analysis using an antibody against argpyrimidine (an MG-derived AGE) shows that control cultures of astrocytes and neurons display similar levels of argpyrimidine, with the notable exception of two bands that were highly stained for argpyrimidine in astrocytes (Fig. 7B, arrowheads). Interestingly, several proteins bearing arginine hotspot sites for MG modifications have previously been identified (Rabbani and Thornalley, 2010). Further studies are warranted to determine the identity of these argpyrimidine-modified proteins in astrocytes. This may provide useful information about the physiological consequences of MG toxicity in this cell type.
When cultures were exposed to 500 μm MG, no increase in argpyrimidine levels could be observed in astrocytes, while in contrast, several proteins appeared to be modified by MG in neurons (Fig. 7B, arrows), consistent with the fact that neurons detoxify MG less efficiently than astrocytes. Another indication of this was provided by Western blot analysis using the 6D12 anti-AGE antibody, which has been shown to recognize several AGE epitopes, including the glyoxal-derived Nϵ-(carboxymethyl)lysine and the MG-derived CEL (Koito et al., 2004; Nagai et al., 2008). Results revealed that cultured neurons display very high levels of AGEs compared with astrocytes in basal conditions (Fig. 7C).
Effects of modulations in the expression levels of Glo-1
RNA interference strategies against Glo-1 were used to determine whether the Glo-1 system is necessary for astrocytic and neuronal survival. Transfection of astrocytes with siRNA targeted against Glo-1 resulted in a 70% decrease of its activity as measured by the glyoxalase enzymatic assay (30.4 ± 3.1% of control values; Student's t test, p ≤ 0.001; n = 7), while Glo-2 activity was not significantly modified (95. 3 ± 2.3% of control values; n = 7). Glo-1 downregulation was confirmed by Western blot analysis (Fig. 8A). Glo-1 silencing did not itself induce any significant decrease in cell viability in astrocytes (Fig. 8A); however, astrocytes in which Glo-1 expression was downregulated were significantly more vulnerable to MG toxicity (Fig. 8A).
Robust transfection of neuronal cultures with siRNA could not be achieved in our hands. We thus opted for a lentivirus-mediated shRNA knockdown strategy to achieve Glo-1 gene silencing in neurons. Expression of Glo-1-specific shRNA in neurons decreased Glo-1 activity (as assessed with the glyoxalase enzymatic assay) in a similar range to that observed with siRNA in astrocytes (21.1 ± 4.0% of control values; Student's t test, p ≤ 0.001; n = 6), while Glo-2 activity was not significantly decreased (95. 5 ± 6.7% of control values; n = 6). Glo-1 downregulation was confirmed by Western blot analysis (Fig. 8B). In contrast to astrocytes, Glo-1 silencing alone induced a significant decrease in neuronal viability (64.8 ± 3.1% of negative control shRNA; p ≤ 0.001) (Fig. 8B). This was accompanied by a marked increase in AGE levels (Fig. 8B), suggesting that an increase in dicarbonyl compounds leading to the accumulation of toxic AGEs contributes to the loss of neuronal viability following Glo-1 downregulation. Furthermore, neurons in which Glo-1 was silenced were much more vulnerable to MG toxicity than neurons expressing normal levels of Glo-1 (Fig. 8B).
To determine whether Glo-1-deficient neuronal cultures were more sensitive to various substances known to induce neurodegeneration, Glo-1 shRNA-treated neuronal cultures were exposed to 100 μm diethylenetriamine-nitric oxide (DETA-NO) for 24 h, 10 μm glutamate for 4 h, or 25 μm H2O2 for 4 h. Glo-1 silencing resulted in a significantly increased neuronal vulnerability to NO, but did not significantly modify the susceptibility to glutamate or H2O2 (Fig. 8C). Since NO has been shown to decrease Glo-1 expression and activity (Mitsumoto et al., 1999; Miller et al., 2006), one can postulate that NO-induced stress may impair the residual Glo-1 activity in Glo-1-deficient neurons, leading to further neuronal death.
Considering the high susceptibility of neurons to Glo-1 silencing, we next sought to determine whether Glo-1 upregulation could conversely protect neurons against various cellular stresses. To this end, Glo-1 was overexpressed in neurons using lentiviral-mediated gene delivery. The efficacy of Glo-1 overexpression using this approach was confirmed by Western blotting (Fig. 8D) and by the glyoxalase enzymatic assay. Glo-1 overexpression resulted in a 6.8-fold increase in Glo-1 activity (678.4 ± 49.1% of control values; n ≥ 10 from three independent experiments; p ≤ 0.001). This effect was specific to this enzyme, as Glo-2 activity was not significantly changed (104.0 ± 6.5% of control values; n ≥ 10 from three independent experiments) in neurons overexpressing Glo-1. Despite this large increase in enzymatic activity, Glo-1 overexpression did not significantly protect neurons against MG (500 μm for 24 h) or other neurotoxic substances, including the NO donor DETA-NO (100 μm for 24 h), glutamate (10 μm for 4 h), or H2O2 (25 μm for 4 h) (Fig. 8D). These results suggest that low expression levels of Glo-1 may not be the only factor accounting for the high neuronal vulnerability to MG toxicity.
Enhanced neuronal glycolysis leads to increase production of MG
Recent reports have demonstrated that the poor capacity of neurons to upregulate glycolysis (Almeida et al., 2001) is explained by the constitutive downregulation of Pfkfb3 (also known as PFK-2, isoform 3). This enzyme is responsible for the generation of fructose-2,6-P2, which is a key regulator of glycolysis (Herrero-Mendez et al., 2009) (Fig. 1). Since MG production is tightly linked to the glycolytic rate, the low protein expression levels of Pfkfb3 may represent a neuronal regulatory mechanism preventing MG toxicity, especially considering that, as observed here, neurons have low MG detoxification capacity. To test this hypothesis, we took advantage of the observation made by Herrero-Mendez et al. (2009) that overexpression of Pfkfb3 results in an increase of the glycolytic rate in cultured rat neurons to manipulate glucose utilization in our neuronal cultures. Long-term Pfkfb3 overexpression was achieved using lentiviral-mediated gene delivery and confirmed by Western blot analysis (Fig. 9A, top panel). As expected, increased Pfkfb3 expression in neurons resulted in a significant stimulation of glucose utilization (119.8 ± 3.1% of controls; p ≤ 0.001; Fig. 9A) together with a concomitant increase in lactate release (265.3 ± 38.6% of controls; p ≤ 0.001; Fig. 9B). Remarkably, this stimulation of the neuronal glycolytic rate was accompanied by a significant increase in intracellular MG levels as determined by HPLC (Fig. 9C). In addition, Western blotting analysis showed enhanced levels of argpyrimidine-modified proteins in neurons overexpressing Pfkfb3 (Fig. 9D), consistent with the observed increase in MG levels. As a whole, these results suggest that a moderate increase in neuronal glycolytic rate can readily overwhelm the glyoxalase system, leading to the accumulation of MG and in turn resulting in protein damage in this cell type.
Figure 9.
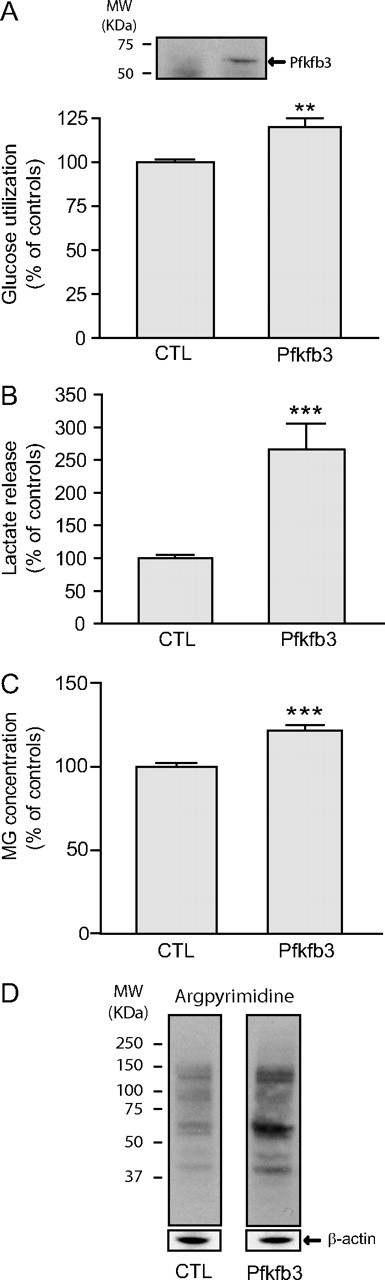
Enhanced neuronal glycolysis via Pfkfb3 overexpression results in elevated MG levels. Pfkfb3 was overexpressed in neurons using lentiviral vectors and the following measurements were made 1 week later. A, Top panel, Pfkfb3 overexpression was confirmed by Western blotting; bottom panel, glucose utilization as measured by [3H]-2-DG uptake (data are expressed as a percentage of control values and are means ± SEM of at least 9 determinations from at least 3 independent experiments; control values were 211.8 ± 29.5 fmol/mg protein/20 min). B, Lactate release into the extracellular medium (data are expressed as percentage of control values and are means ± SEM of at least 9 determinations from at least 3 independent experiments; control values were 282.9 ± 50.1 nmol/mg protein/20 min). C, MG levels by HPLC (values are means ± SEM of at least 10 determinations from at least 4 independent experiments). A–C, Data were statistically analyzed using a Student's t test (**p ≤ 0.01; ***p ≤ 0.001). D, Western blot analysis shows increased neuronal argpyrimidine levels following Pfkfb3 overexpression. Representative bands are shown. Similar results were obtained in three independent experiments (n ≥ 8).
Discussion
For the first time, results of the present study demonstrate major differences in the glyoxalase system of astrocytes and neurons, which, together with marked disparities in GSH metabolism, impact their respective capacity for cellular defense against glycation. Astrocytes displayed a much more efficient glyoxalase system than neurons, as demonstrated by higher Glo-1/-2 protein expression and enzymatic activity in vitro, as well as higher Glo-1 immunoreactivity and activity in the mouse brain. Astrocytic Glo-1 activity rates observed here were also severalfold higher compared with the high MG-producing CHO cells and with those reported for various other cell types (Ranganathan et al., 1995; Shinohara et al., 1998; Wu et al., 2001; Ahmed et al., 2003; Kumagai et al., 2009), supporting the notion that astrocytes possess a particularly efficient glyoxalase system.
The detoxification of MG via the glyoxalase pathway requires GSH as a cofactor. It has previously been reported that GSH levels are significantly higher in astrocytes than in neurons (Dringen, 2000). Consistent with these reports, our results show that intracellular GSH and NADPH levels were, respectively, ninefold and threefold higher in astrocytes. Furthermore, astrocytes displayed greater metabolic plasticity with regards to GSH and NADPH, as demonstrated by a compensatory increase of both metabolites in response to subtoxic doses of MG, by the activation of Nrf2-dependent antioxidant defense mechanisms, and by the capacity of astrocytes to recover normal GSH levels following a transient MG-induced decrease. These differences in GSH metabolism, together with a higher glyoxalase activity, confer to astrocytes greater resistance to exogenous MG. Consistently, previous studies have reported a high neuronal susceptibility to MG toxicity, which was associated with oxidative stress leading to apoptosis (Kikuchi et al., 1999; Di Loreto et al., 2008; Chen et al., 2010).
Considering that CSF levels are between 10 and 20 μm (Kuhla et al., 2005), and cellular levels of free MG are typically in the low micromolar range (Rabbani and Thornalley, 2010), the LD50 values observed here for MG in astrocytes and neurons may appear quite high. However, MG is highly reactive and is expected to have a short half-life in the cellular environment; therefore, local concentration at the time and site of production may be significantly higher (Kalapos, 2008b). Interestingly, it is estimated that 90–99% of cellular MG is bound to macromolecules, and assessments of total (bound + free) MG showed that cellular concentrations up to 300 μm can be measured (Thornalley, 1996; Chaplen et al., 1998).
Despite the fact that cultured astrocytes display >3 times higher glycolytic rate than neurons in basal conditions (Magistretti and Pellerin, 1996), we did not see any differences in endogenous MG levels between these two cell types, which may be explained by the more efficient glyoxalase system present in astrocytes. Consistent with this, Western blot analysis showed higher levels of MG and glyoxal-derived AGEs in neurons compared with astrocytes using the 6D12 anti-AGE antibody.
Previous reports have demonstrated that Glo-1 overexpression protects against increased AGE formation and/or oxidative stress associated with aging, hyperglycemia, and ischemia-reperfusion injury (Shinohara et al., 1998; Morcos et al., 2008; Kumagai et al., 2009; Schlotterer et al., 2009; Brouwers et al., 2011). However, in our hands Glo-1 overexpression did not protect cortical neurons against MG toxicity. One possible explanation for this is that, as discussed above, cultured cortical neurons have particularly low levels of GSH and a poor capacity to replenish the GSH pool following exposure to MG. GSH levels may therefore be a limiting factor for MG detoxification in neurons, in which case Glo-1 overexpression would not be sufficient to afford neuroprotection. Further studies are warranted to clarify this issue. In contrast with the lack of effect of Glo-1 overexpression, its downregulation induced a significant loss of viability in neurons but not in astrocytes, indicating that (1) Glo-1 is essential for neuronal viability and that (2) in contrast with neurons, astrocytes express Glo-1 in excess of the levels required for their survival in the absence of a major cellular stress.
Astrocytes are key players in the maintenance of brain homeostasis, and several cooperative metabolic processes and interdependencies between astrocytes and neurons have been identified (Bélanger and Magistretti, 2009; Allaman et al., 2011). For example, it is well established that astrocytes protect neurons against oxidative stress and excitotoxicity (Rothstein et al., 1996; Johnson et al., 2008; Vargas and Johnson, 2009). Here, we show that astrocytes protect neurons against MG toxicity in a coculture model. This conceivably represents another level of astrocyte–neuron cooperativity whereby a highly efficient glyoxalase system in astrocytes may contribute to neuronal protection against MG potentially leaking from the periphery, the CSF, or surrounding cells.
The differential Glo-1 efficiency observed in astrocytes and neurons may relate to their dissimilar metabolic profile. Numerous reports have shown that astrocytes display high metabolic plasticity in terms of glucose utilization, which is not the case for neurons (Pellerin and Magistretti, 1994; Almeida et al., 2001; Prapong et al., 2002; Porras et al., 2004; Gavillet et al., 2008; Allaman et al., 2010; Bélanger et al., 2011). One striking example is the differential response of astrocytes and neurons following the inhibition of mitochondrial respiration by NO. Unlike neurons in which NO leads to a massive ATP depletion and apoptosis, astrocytes respond by increasing glycolysis, which limits the fall in ATP levels and prevents apoptosis (Almeida et al., 2001). This astrocytic response is dependent upon the synthesis of fructose-2,6-P2—a potent activator of the glycolytic enzyme phosphofructokinase-1—by the enzyme Pfkfb (Almeida et al., 2004). While this enzyme is highly expressed in astrocytes, it is virtually absent in neurons due to its constant proteasomal degradation (Herrero-Mendez et al., 2009). Furthermore, the activation of neuronal glycolysis via Pfkfb3 overexpression leads to oxidative stress and apoptosis (Herrero-Mendez et al., 2009). Interestingly, similar to Pfkfb3, the enzymatic machinery for glycogen synthesis is tightly suppressed in neurons—hence the virtual absence of glycogen in this cell type—and failure to do so also results in neuronal apoptosis (Vilchez et al., 2007). Thus, two pathways directly linked to triosephosphate metabolisms are tightly repressed in neurons and lead to apoptosis if activated.
Our results suggest that high neuronal susceptibility to MG toxicity may in part be involved in the deleterious effects of increasing the glycolytic fluxes in these cells. While Pfkfb3 overexpression did not induce significant neuronal death in our hands—possibly because the increases in glucose utilization achieved here using a different species and another gene transduction system were modest compared with those previously reported (Herrero-Mendez et al., 2009)—even a moderate increase in neuronal glycolysis was sufficient to significantly increase MG and AGE levels. This observation suggests that a limited capacity to face deleterious dicarbonyl stress may in part explain why neurons constitutively suppress pathways potentially leading to MG formation.
These observations may also provide insight into the reason why astrocytes can sustain a higher basal glycolytic rate than neurons, and why neurons depend on astrocytes to meet their energy needs—as proposed by the ANLS model and substantiated by several in vitro, ex vivo, and in vivo data (Kasischke et al., 2004; Porras et al., 2004; Rouach et al., 2008; Magistretti, 2009; Chuquet et al., 2010). Considering that glucose metabolism inevitably leads to MG formation, lactate utilization may provide a means for neurons to alleviate MG toxicity by decreasing their glucose utilization, yet meet their high energetic requirements by using astrocyte-derived lactate, which can be oxidized in the Krebs cycle following its conversion to pyruvate. Most of the MG burden associated with a high glycolytic flux would thus be left to the astrocytes, which according to our data are better equipped to handle it.
Interestingly, modulations of the glyoxalase system have been observed in aging and various neuropathological conditions. For instance, Glo-1 expression was reported to decrease progressively in the human brain starting from the fifth decade (Kuhla et al., 2006), in correlation with increased AGE levels in neurons and astrocytes (Lüth et al., 2005). Impaired Glo-1 activity may thus contribute to the pathogenesis of neurodegenerative disorders, considering that MG induces oxidative stress and AGE formation, which are major factors involved in many of these age-dependent pathologies. For example, in Alzheimer's disease (AD), Glo-1 expression is increased in early stages, but then decreases below normal levels at later stages, correlating with a marked increase in AGE accumulation (Lüth et al., 2005; Kuhla et al., 2007). AGEs colocalize with amyloid-β plaques and neurons containing hyperphosphorylated tau protein in AD brains, but astrocytes are also affected as they show increased AGE and inducible nitric oxide synthase immunoreactivity (Wong et al., 2001; Lüth et al., 2005). While further studies are warranted to clarify the specific effects on different neural cell types, strong evidence suggests that the glyoxalase system is impaired in AD, which may participate in the vicious circle of amyloid-β deposition, AGE formation, and oxidative stress associated with this disease.
In conclusion, results of the present study show that the glyoxalase pathway differs significantly between astrocytes and neurons in a way that renders neurons more vulnerable to carbonyl stress and AGE accumulation. These results support the notion that the metabolic specialization taking place in brain cells—astrocytes are more glycolytic than neurons—may help protect neurons from MG toxicity.
Footnotes
This work was supported by grants from the Swiss National Science Foundation to P.J.M. (no. 3100AO-108336/1). M.B. was supported by the Fonds de la Recherche en Santé du Québec. P.J.M. is the recipient of the Asterion Foundation Chair. We thank Sylvain Lengacher for his assistance with virus production, Diego Chiappe from Ecole Polytechnique Fédérale de Lausanne's Proteomics Core Facility, as well as Cendrine Barrière Borgioni, Elsy Dunand, Joël Gyger, and Evelyne Ruchti, for expert technical assistance.
The authors declare no competing financial interests.
References
- Ahmed N, Battah S, Karachalias N, Babaei-Jadidi R, Horányi M, Baróti K, Hollan S, Thornalley PJ. Increased formation of methylglyoxal and protein glycation, oxidation and nitrosation in triosephosphate isomerase deficiency. Biochim Biophys Acta. 2003;1639:121–132. doi: 10.1016/j.bbadis.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Allaman I, Pellerin L, Magistretti PJ. Glucocorticoids modulate neurotransmitter-induced glycogen metabolism in cultured cortical astrocytes. J Neurochem. 2004;88:900–908. doi: 10.1046/j.1471-4159.2003.02235.x. [DOI] [PubMed] [Google Scholar]
- Allaman I, Gavillet M, Bélanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ. Amyloid-β aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci. 2010;30:3326–3338. doi: 10.1523/JNEUROSCI.5098-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaman I, Bélanger M, Magistretti PJ. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 2011;34:76–87. doi: 10.1016/j.tins.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Almeida A, Almeida J, Bolaños JP, Moncada S. Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci U S A. 2001;98:15294–15299. doi: 10.1073/pnas.261560998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida A, Moncada S, Bolaños JP. Nitric oxide switches on glycolysis through the AMP protein kinase and 6-phosphofructo-2-kinase pathway. Nat Cell Biol. 2004;6:45–51. doi: 10.1038/ncb1080. [DOI] [PubMed] [Google Scholar]
- Barde I, Salmon P, Trono D. Production and titration of lentiviral vectors. Curr Protoc Neurosci. 2010;53:4.21.1–4.21.23. doi: 10.1002/0471142301.ns0421s53. [DOI] [PubMed] [Google Scholar]
- Bélanger M, Magistretti PJ. The role of astroglia in neuroprotection. Dialogues Clin Neurosci. 2009;11:281–295. doi: 10.31887/DCNS.2009.11.3/mbelanger. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bélanger M, Allaman I, Magistretti PJ. Differential effects of pro- and anti-inflammatory cytokines alone or in combinations on the metabolic profile of astrocytes. J Neurochem. 2011;116:564–576. doi: 10.1111/j.1471-4159.2010.07135.x. [DOI] [PubMed] [Google Scholar]
- Brandt RB, Waters MG, Rispler MJ, Kline ES. d- and l-lactate catabolism to CO2 in rat tissues. Proc Soc Exp Biol Med. 1984;175:328–335. doi: 10.3181/00379727-175-41803. [DOI] [PubMed] [Google Scholar]
- Brouwers O, Niessen PM, Ferreira I, Miyata T, Scheffer PG, Teerlink T, Schrauwen P, Brownlee M, Stehouwer CD, Schalkwijk CG. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation endproducts and oxidative stress in diabetic rats. J Biol Chem. 2011;286:1374–1380. doi: 10.1074/jbc.M110.144097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplen FW, Fahl WE, Cameron DC. Method for determination of free intracellular and extracellular methylglyoxal in animal cells grown in culture. Anal Biochem. 1996;238:171–178. doi: 10.1006/abio.1996.0271. [DOI] [PubMed] [Google Scholar]
- Chaplen FW, Fahl WE, Cameron DC. Evidence of high levels of methylglyoxal in cultured Chinese hamster ovary cells. Proc Natl Acad Sci U S A. 1998;95:5533–5538. doi: 10.1073/pnas.95.10.5533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Wollmer MA, Hoerndli F, Münch G, Kuhla B, Rogaev EI, Tsolaki M, Papassotiropoulos A, Götz J. Role for glyoxalase I in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:7687–7692. doi: 10.1073/pnas.0402338101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YJ, Huang XB, Li ZX, Yin LL, Chen WQ, Li L. Tenuigenin protects cultured hippocampal neurons against methylglyoxal-induced neurotoxicity. Eur J Pharmacol. 2010;645:1–8. doi: 10.1016/j.ejphar.2010.06.034. [DOI] [PubMed] [Google Scholar]
- Chuquet J, Quilichini P, Nimchinsky EA, Buzsáki G. Predominant enhancement of glucose uptake in astrocytes versus neurons during activation of the somatosensory cortex. J Neurosci. 2010;30:15298–15303. doi: 10.1523/JNEUROSCI.0762-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Déglon N, Tseng JL, Bensadoun JC, Zurn AD, Arsenijevic Y, Pereira de Almeida L, Zufferey R, Trono D, Aebischer P. Self-inactivating lentiviral vectors with enhanced transgene expression as potential gene transfer system in Parkinson's disease. Hum Gene Ther. 2000;11:179–190. doi: 10.1089/10430340050016256. [DOI] [PubMed] [Google Scholar]
- Di Loreto S, Zimmitti V, Sebastiani P, Cervelli C, Falone S, Amicarelli F. Methylglyoxal causes strong weakening of detoxifying capacity and apoptotic cell death in rat hippocampal neurons. Int J Biochem Cell Biol. 2008;40:245–257. doi: 10.1016/j.biocel.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Gavillet M, Allaman I, Magistretti PJ. Modulation of astrocytic metabolic phenotype by proinflammatory cytokines. Glia. 2008;56:975–989. doi: 10.1002/glia.20671. [DOI] [PubMed] [Google Scholar]
- Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. 2009;11:747–752. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Johnson DA, Kraft AD, Calkins MJ, Jakel RJ, Vargas MR, Chen PC. The Nrf2-ARE pathway: an indicator and modulator of oxidative stress in neurodegeneration. Ann N Y Acad Sci. 2008;1147:61–69. doi: 10.1196/annals.1427.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalapos MP. Methylglyoxal in living organisms: chemistry, biochemistry, toxicology and biological implications. Toxicol Lett. 1999;110:145–175. doi: 10.1016/s0378-4274(99)00160-5. [DOI] [PubMed] [Google Scholar]
- Kalapos MP. Methylglyoxal and glucose metabolism: a historical perspective and future avenues for research. Drug Metabol Drug Interact. 2008a;23:69–91. doi: 10.1515/dmdi.2008.23.1-2.69. [DOI] [PubMed] [Google Scholar]
- Kalapos MP. The tandem of free radicals and methylglyoxal. Chem Biol Interact. 2008b;171:251–271. doi: 10.1016/j.cbi.2007.11.009. [DOI] [PubMed] [Google Scholar]
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science. 2004;305:99–103. doi: 10.1126/science.1096485. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, Shinpo K, Moriwaka F, Makita Z, Miyata T, Tashiro K. Neurotoxicity of methylglyoxal and 3-deoxyglucosone on cultured cortical neurons: synergism between glycation and oxidative stress, possibly involved in neurodegenerative diseases. J Neurosci Res. 1999;57:280–289. doi: 10.1002/(SICI)1097-4547(19990715)57:2<280::AID-JNR14>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Kingkeohoi S, Chaplen FW. Analysis of methylglyoxal metabolism in CHO cells grown in culture. Cytotechnology. 2005;48:1–13. doi: 10.1007/s10616-005-1920-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koito W, Araki T, Horiuchi S, Nagai R. Conventional antibody against Nepsilon-(carboxymethyl)lysine (CML) shows cross-reaction to Nepsilon-(carboxyethyl)lysine (CEL): immunochemical quantification of CML with a specific antibody. J Biochem. 2004;136:831–837. doi: 10.1093/jb/mvh193. [DOI] [PubMed] [Google Scholar]
- Kuhla B, Lüth HJ, Haferburg D, Boeck K, Arendt T, Münch G. Methylglyoxal, glyoxal, and their detoxification in Alzheimer's disease. Ann N Y Acad Sci. 2005;1043:211–216. doi: 10.1196/annals.1333.026. [DOI] [PubMed] [Google Scholar]
- Kuhla B, Boeck K, Lüth HJ, Schmidt A, Weigle B, Schmitz M, Ogunlade V, Münch G, Arendt T. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol Aging. 2006;27:815–822. doi: 10.1016/j.neurobiolaging.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Kuhla B, Boeck K, Schmidt A, Ogunlade V, Arendt T, Münch G, Lüth HJ. Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer's disease brains. Neurobiol Aging. 2007;28:29–41. doi: 10.1016/j.neurobiolaging.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Kumagai T, Nangaku M, Kojima I, Nagai R, Ingelfinger JR, Miyata T, Fujita T, Inagi R. Glyoxalase I overexpression ameliorates renal ischemia-reperfusion injury in rats. Am J Physiol Renal Physiol. 2009;296:F912–F921. doi: 10.1152/ajprenal.90575.2008. [DOI] [PubMed] [Google Scholar]
- Lüth HJ, Ogunlade V, Kuhla B, Kientsch-Engel R, Stahl P, Webster J, Arendt T, Münch G. Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer's disease brains. Cereb Cortex. 2005;15:211–220. doi: 10.1093/cercor/bhh123. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ. Brain energy metabolism. In: Squire L, Bloom FE, Spitzer NC, du Lac S, Ghosh A, Berg D, editors. Fundamental neuroscience. San Diego: Academic; 2008. pp. 271–292. [Google Scholar]
- Magistretti PJ. Role of glutamate in neuron-glia metabolic coupling. Am J Clin Nutr. 2009;90:875S–880S. doi: 10.3945/ajcn.2009.27462CC. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L. Cellular mechanisms of brain energy metabolism. Relevance to functional brain imaging and to neurodegenerative disorders. Ann N Y Acad Sci. 1996;777:380–387. doi: 10.1111/j.1749-6632.1996.tb34449.x. [DOI] [PubMed] [Google Scholar]
- Matsumura H, Miyachi S. Cycling assay for nicotinamide adenine dinucleotides. Methods Enzymol. 1980;69:465–470. [Google Scholar]
- Miller AG, Smith DG, Bhat M, Nagaraj RH. Glyoxalase I is critical for human retinal capillary pericyte survival under hyperglycemic conditions. J Biol Chem. 2006;281:11864–11871. doi: 10.1074/jbc.M513813200. [DOI] [PubMed] [Google Scholar]
- Mitsumoto A, Kim KR, Oshima G, Kunimoto M, Okawa K, Iwamatsu A, Nakagawa Y. Glyoxalase I is a novel nitric-oxide-responsive protein. Biochem J. 1999;344:837–844. [PMC free article] [PubMed] [Google Scholar]
- Morcos M, Du X, Pfisterer F, Hutter H, Sayed AA, Thornalley P, Ahmed N, Baynes J, Thorpe S, Kukudov G, Schlotterer A, Bozorgmehr F, El Baki RA, Stern D, Moehrlen F, Ibrahim Y, Oikonomou D, Hamann A, Becker C, Zeier M, et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell. 2008;7:260–269. doi: 10.1111/j.1474-9726.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- Münch G, Westcott B, Menini T, Gugliucci A. Advanced glycation endproducts and their pathogenic roles in neurological disorders. Amino Acids. 2010 doi: 10.1007/s00726-010-0777-y. [DOI] [PubMed] [Google Scholar]
- Nagai R, Fujiwara Y, Mera K, Motomura K, Iwao Y, Tsurushima K, Nagai M, Takeo K, Yoshitomi M, Otagiri M, Ikeda T. Usefulness of antibodies for evaluating the biological significance of AGEs. Ann N Y Acad Sci. 2008;1126:38–41. doi: 10.1196/annals.1433.001. [DOI] [PubMed] [Google Scholar]
- Paoli T, Faulkner J, O'kennedy R, Keshavarz-Moore E. A study of d-lactate and extracellular methylglyoxal production in lactate re-utilizing CHO cultures. Biotechnol Bioeng. 2010;107:182–189. doi: 10.1002/bit.22757. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porras OH, Loaiza A, Barros LF. Glutamate mediates acute glucose transport inhibition in hippocampal neurons. J Neurosci. 2004;24:9669–9673. doi: 10.1523/JNEUROSCI.1882-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prapong T, Buss J, Hsu WH, Heine P, West Greenlee H, Uemura E. Amyloid beta-peptide decreases neuronal glucose uptake despite causing increase in GLUT3 mRNA transcription and GLUT3 translocation to the plasma membrane. Exp Neurol. 2002;174:253–258. doi: 10.1006/exnr.2001.7861. [DOI] [PubMed] [Google Scholar]
- Rabbani N, Thornalley PJ. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids. 2010 doi: 10.1007/s00726-010-0783-0. [DOI] [PubMed] [Google Scholar]
- Ranganathan S, Walsh ES, Tew KD. Glyoxalase I in detoxification: studies using a glyoxalase I transfectant cell line. Biochem J. 1995;309:127–131. doi: 10.1042/bj3090127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Rouach N, Koulakoff A, Abudara V, Willecke K, Giaume C. Astroglial metabolic networks sustain hippocampal synaptic transmission. Science. 2008;322:1551–1555. doi: 10.1126/science.1164022. [DOI] [PubMed] [Google Scholar]
- Schlotterer A, Kukudov G, Bozorgmehr F, Hutter H, Du X, Oikonomou D, Ibrahim Y, Pfisterer F, Rabbani N, Thornalley P, Sayed A, Fleming T, Humpert P, Schwenger V, Zeier M, Hamann A, Stern D, Brownlee M, Bierhaus A, Nawroth P, et al. C. elegans as model for the study of high glucose- mediated life span reduction. Diabetes. 2009;58:2450–2456. doi: 10.2337/db09-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, Thornalley PJ, Giardino I, Beisswenger P, Thorpe SR, Onorato J, Brownlee M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J Clin Invest. 1998;101:1142–1147. doi: 10.1172/JCI119885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- Thornalley PJ. The glyoxalase system in health and disease. Mol Aspects Med. 1993;14:287–371. doi: 10.1016/0098-2997(93)90002-u. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification—a role in pathogenesis and antiproliferative chemotherapy. Gen Pharmacol. 1996;27:565–573. doi: 10.1016/0306-3623(95)02054-3. [DOI] [PubMed] [Google Scholar]
- Thornalley PJ. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems—role in ageing and disease. Drug Metabol Drug Interact. 2008;23:125–150. doi: 10.1515/dmdi.2008.23.1-2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornalley PJ, Tisdale MJ. Inhibition of proliferation of human promyelocytic leukaemia HL60 cells by S-d-lactoylglutathione in vitro. Leuk Res. 1988;12:897–904. doi: 10.1016/0145-2126(88)90016-1. [DOI] [PubMed] [Google Scholar]
- Towne C, Aebischer P. Lentiviral and adeno-associated vector-based therapy for motor neuron disease through RNAi. Methods Mol Biol. 2009;555:87–108. doi: 10.1007/978-1-60327-295-7_7. [DOI] [PubMed] [Google Scholar]
- Vargas MR, Johnson JA. The Nrf2-ARE cytoprotective pathway in astrocytes. Expert Rev Mol Med. 2009;11:e17. doi: 10.1017/S1462399409001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D, Ros S, Cifuentes D, Pujadas L, Vallès J, García-Fojeda B, Criado-García O, Fernández-Sánchez E, Medraño-Fernández I, Domínguez J, García-Rocha M, Soriano E, Rodríguez de Córdoba S, Guinovart JJ. Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci. 2007;10:1407–1413. doi: 10.1038/nn1998. [DOI] [PubMed] [Google Scholar]
- Wautier JL, Guillausseau PJ. Advanced glycation end products, their receptors and diabetic angiopathy. Diabetes Metab. 2001;27:535–542. [PubMed] [Google Scholar]
- Wong A, Lüth HJ, Deuther-Conrad W, Dukic-Stefanovic S, Gasic-Milenkovic J, Arendt T, Münch G. Advanced glycation endproducts co-localize with inducible nitric oxide synthase in Alzheimer's disease. Brain Res. 2001;920:32–40. doi: 10.1016/s0006-8993(01)02872-4. [DOI] [PubMed] [Google Scholar]
- Wu L, Eftekharpour E, Davies GF, Roesler WJ, Juurlink BH. Troglitazone selectively inhibits glyoxalase I gene expression. Diabetologia. 2001;44:2004–2012. doi: 10.1007/s001250100004. [DOI] [PubMed] [Google Scholar]
- Zerez CR, Lee SJ, Tanaka KR. Spectrophotometric determination of oxidized and reduced pyridine nucleotides in erythrocytes using a single extraction procedure. Anal Biochem. 1987;164:367–373. doi: 10.1016/0003-2697(87)90506-9. [DOI] [PubMed] [Google Scholar]