

Abstract
Brain-derived neurotrophic factor (BDNF) is an important mediator of activity-dependent functions of the nervous system and its expression is dysregulated in several neuropsychiatric disorders. Regulation of rodent BDNF neuronal activity-dependent transcription has been relatively well characterized. Here, we have studied regulation of human BDNF (hBDNF) transcription by membrane depolarization of cultured mouse or rat primary cortical neurons expressing hBDNF gene or transfected with hBDNF promoter constructs, respectively. We identified an asymmetric E-box-like element, PasRE [basic helix-loop-helix (bHLH)-PAS transcription factor response element], in hBDNF promoter I and demonstrate that binding of this element by bHLH-PAS transcription factors ARNT2 (aryl hydrocarbon receptor nuclear translocator 2) and NPAS4 (neuronal PAS domain protein 4) is crucial for neuronal activity-dependent transcription from promoter I. We show that binding of CREB (cAMP response element-binding protein) to the cAMP/Ca2+-response element (CRE) in hBDNF promoter IV is critical for activity-dependent transcription from this promoter and that upstream stimulatory factor (USF) transcription factors also contribute to the activation by binding to the upstream stimulatory factor binding element (UBE) in hBDNF promoter IV. However, we report that full induction of hBDNF exon IV mRNA transcription is dependent on ARNT2 and NPAS4 binding to a PasRE in promoter IV. Finally, we demonstrate that CRE and PasRE elements in hBDNF promoter IX are required for the induction of this promoter by neuronal activity. Together, the results of this study have identified the cis-elements and transcription factors regulating neuronal activity-dependent transcription of human BDNF gene.
Introduction
Neuronal activity-responsive transcription is an important component in development and in adaptive functions of the nervous system. Neuronal activity leads to expression of ∼300 genes (Lin et al., 2008), including brain-derived neurotrophic factor (BDNF), one of the major mediators of activity-dependent functions (Poo, 2001). BDNF was first described to support survival of embryonic sensory neurons (Barde et al., 1982). Currently, BDNF is implicated in survival and differentiation of several neuronal populations and in regulation of synaptic plasticity, long-term potentiation, learning, memory, and neurogenesis (Ernfors et al., 1994; Jones et al., 1994; Patterson et al., 1996; Lyons et al., 1999; Hall et al., 2000; Binder and Scharfman, 2004). Moreover, neuronal activity-dependent transcription of BDNF has been shown to be essential for development of cortical inhibition (Hong et al., 2008).
Regulation of BDNF has been thoroughly studied in rodents. Rat BDNF (rBDNF) gene was first described to have four promoters driving expression of transcripts containing different 5′ exons spliced to a single coding exon (Timmusk et al., 1993). To date, it has been found that rodent BDNF (rodBDNF) contains nine exons (Aid et al., 2007), and several regulators of calcium-dependent upregulation have been identified. Foremost, rBDNF promoter IV (pIV) is upregulated via cAMP response element-binding protein (CREB) (alias CREB1) binding to a cAMP/Ca2+-response element (CRE) (Shieh et al., 1998; Tao et al., 1998). This is modulated by upstream stimulatory factors (USFs) and calcium-responsive transcription factor (CaRF) binding to an E-box element and a CaRE1 (Ca2+-response element 1) element, respectively (Tao et al., 2002; Chen et al., 2003a). Additionally, MeCP2 (methyl-CpG-binding protein 2), BHLHB2 (basic helix-loop-helix B2) and NFκB (nuclear factor κB) have been shown to regulate rodent pIV (Chen et al., 2003b; Jiang et al., 2008). However, CRE plays a central role because knock-in mutation of CRE blocks mouse BDNF (mBDNF) pIV activity-responsiveness in vivo (Hong et al., 2008). Exon I of rBDNF is also induced by neuronal activity (Timmusk et al., 1993). CREB, USFs, MEF2D (myocyte enhancer factor 2D), and NFκB have been implicated in mediating the induction (Tabuchi et al., 2002; Lubin et al., 2007; Flavell et al., 2008). Additionally, NPAS4 (neuronal PAS domain protein 4) has been shown to bind pI and pIV of mBDNF (Lin et al., 2008), but the binding sites were not specified. Finally, BDNF exon IXa transcripts are also upregulated, whereas levels of mRNAs with other 5′ exons are elevated to a lesser extent or not influenced by neuronal activity (Koppel et al., 2009).
Decreased BDNF expression has been documented in neurodegenerative diseases and in cognitive, mood, and anxiety disorders (Binder and Scharfman, 2004). To enable studying human BDNF (hBDNF) transcription, hBDNF gene structure was described. Similarly to rodBDNF, multiple 5′ exons are spliced to a single coding exon in human, and altogether, the hBDNF gene contains 11 exons and nine promoters (Pruunsild et al., 2007). Importantly, there are two human-specific exons in hBDNF and, additionally, the locus comprises a non-coding antisense gene that is absent in rodents (Pruunsild et al., 2007). Although much is known about regulation of rodBDNF, regulation of hBDNF by neuronal activity has been poorly studied. Here, we identify cis-elements and transcription factors (TFs) involved in hBDNF transcriptional activation by membrane depolarization of cultured cortical neurons.
Materials and Methods
Primary neuron cultures.
All animal procedures were performed in compliance with the local ethics committee. Primary neuron cultures from human BDNF-BAC (bacterial artificial chromosome) mice, line C3 (Koppel et al., 2009), were generated from postnatal day 0 mouse (male and female) brains, and the rat primary neuron cultures for luciferase assays were generated from embryonic day 21 rat embryo (male and female) brains (Sprague Dawley). Briefly, the cortices, together with hippocampi, were dissected and cells were dissociated with 0.25% trypsin (Invitrogen), treated with 0.05% DNase I (Roche), and the cell suspension was plated on poly-l-lysine-coated dishes in Neurobasal A medium (Invitrogen) with B27 supplement (Invitrogen), penicillin (PAA Laboratories, 100 U/ml), streptomycin (PAA Laboratories, 0,1 mg/ml), and 1 mm l-glutamine (PAA Laboratories). Mitotic inhibitor 5-fluoro-2′-deoxyuridine (Sigma) was added to the medium (10 μm) at 2 days in vitro (DIV). For modeling neuronal activity by activation of voltage-sensitive calcium channels, final concentration of 25 mm KCl was added to the medium at 7 DIV (mouse primary neurons) and BDNF gene expression was analyzed by reverse transcription-quantitative PCR (RT-qPCR) using RNA isolated at indicated time points. Transfection and KCl treatment of rat primary neurons are specified in DNA transfection and luciferase assay (Materials and Methods). Amaxa nucleofection with green fluorescent protein (GFP) or dominant-negative aryl hydrocarbon receptor nuclear translocator 2 (delTAD-ARNT2) and NPAS4 (delTAD-NPAS4) coding constructs was performed with the Amaxa Rat Neuron Nucleofector Kit (Lonza). Briefly, 107 embryonic day 19 rat primary neurons were electroporated with 5 μg of indicated plasmid using the predefined program O-003 of the Nucleofector II (Lonza) according to the manufacturer's instructions. Nucleofected neurons were treated with KCl at 5 DIV and endogenous rat BDNF transcript levels were measured by RT-qPCR using RNA isolated at indicated time points.
DNA constructs and mutagenesis.
hBDNF promoter regions and rBDNF pI were amplified from human genomic DNA and rat genomic DNA, respectively, using the Expand High Fidelity PCR System (Roche). All BDNF promoters were cloned into the pGL4.15[luc2P/Hygro] (Promega) in front of firefly luciferase (FFluc) coding sequence. For normalization of the luciferase assays, promoter of human EF1α (elongation factor-1α) was cloned into the pGL4.83[hRlucP/Puro] in front of humanized Renilla luciferase (hRluc). Mutagenesis was performed using complementary primers against the target sequence containing the respective mutation (listed in supplemental Table S1, available at www.jneurosci.org as supplemental material) with Phusion High-Fidelity DNA Polymerase (Finnzymes) according to the manufacturer's instructions. After 25 cycles of PCR, the samples were treated with the DpnI restriction enzyme (Fermentas) to degrade the wild-type (WT) template plasmid and were transformed into TOP10 competent cells (Invitrogen). All promoter sequences were verified by sequencing and are shown in supplemental Figures 1 and 2 (available at www.jneurosci.org as supplemental material). Full-length coding regions of human CREB1, USF1, USF2, ARNT2, NPAS4, SIM2, and DREAM family members were amplified from human cDNA using Phusion High-Fidelity DNA Polymerase (Finnzymes) (primers listed in supplemental Table S1, available at www.jneurosci.org as supplemental material) and cloned into the pcDNA3.1 vector (Invitrogen). The viral transcription activator domain VP16 was obtained from the vector pACT (Promega). The delTAD-ARNT2 and delTAD-NPAS4 constructs expressing ARNT2 and NPAS4 proteins without C-terminal transcription activator domains were generated by removing the 3′ part of cDNAs. In human ARNT2, DNA sequence downstream of the ClaI (Bsu15I, Fermentas) restriction site was removed, and in human NPAS4, DNA sequence downstream of the more 5′ located XmnI (PdmI, Fermentas) restriction site of two XmnI sites was removed, creating constructs coding for ARNT2 amino acids 1–Arg402 and NPAS4 amino acids 1–Glu348.
DNA transfection and luciferase assays.
Rat primary neurons were transfected at 6–7 DIV with BDNF promoter/pGL4.15[luc2P/Hygro] plasmids using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's instructions. A DNA/Lipofectamine ratio of 1 μg/2 μl and 0.5 μg of DNA per 0.75 cm2 well of a 48-well cell culture plate were used. The effector protein pcDNA3.1 expression constructs were cotransfected in 1:1 ratio with the BDNF promoter construct where indicated. For normalization, pEF1α/pGL4.83[luc2P/Hygro] (1:100 of BDNF promoter construct quantity) was cotransfected. At 8–9 DIV, neuronal membranes were depolarized by addition of 25 mm KCl to the medium, and luciferase assays were performed with the Dual-Glo Luciferase Assay System (Promega) using cell lysates prepared at indicated time points. At least three independent experiments were performed. For presentation of data in relative luciferase units (RLUs), the background signals from untransfected neurons were subtracted from signals obtained from transfected cells, and the FFluc signal values were normalized to EF1α promoter-dependent hRluc signals. All data were log-transformed and autoscaled using the data of the longest WT promoter, in the case of cis-element mapping, data of the pRC-cotransfected WT promoter in the case of effector overexpression analyses, and using all data in the case of induction time course analyses. Means and SDs were calculated and t tests for analyses of statistical significance were performed. For graphical representation, the data were back-transformed to the original scale. Error bars represent upper and lower limits back-transformed as mean + SD and mean − SD, respectively.
RNA isolation and RT-qPCR.
Total RNA from primary neurons or HEK293 cells was purified using an RNeasy Micro Kit (Qiagen) as recommended by the manufacturer. First-strand cDNAs were synthesized from 500 to 2000 ng of total RNA with Superscript III reverse transcriptase (Invitrogen) according to the manufacturer's recommendations using oligo(dT) primers. The cDNA was used for quantitative PCR with the qPCR Core kit for SYBR Green I No Rox (Eurogentec) and LightCycler 2.0 (Roche). At least three independent experiments were performed and each sample was measured in triplicates with primers targeting the respective transcript. Expression of the housekeeping gene HPRT1 was used for normalization. All primers used are listed in supplemental Table S1, available at www.jneurosci.org as supplemental material. The data were log-transformed, autoscaled, means and SDs were calculated, and t tests were performed. Then, the data were back-transformed into the original scale. Error bars represent upper and lower limits back-transformed as mean + SD and mean − SD, respectively. For quantitative measurement of BDNF exon I to exon IV ratio with qPCR, human exon I and exon IV PCR products were cloned together in tail-to-tail orientation in one plasmid construct and the respective mouse PCR products in another construct. Plasmid template standard curves spanning at least five orders of magnitude were generated with the same 10-fold serial dilutions with exactly a 1:1 ratio of template for hBDNF or mBDNF exon I and IV qPCR primers, respectively. These directly comparable plasmid standard curves were used to quantify the relative abundance of BDNF exon I transcripts compared with exon IV transcripts in control untreated hBDNF-BAC mice primary neurons and neurons depolarized 8 h with KCl. Three independent experiments were used for calculations. Error bars represent SD.
Electrophoretic mobility shift assay.
Neuronal lysates were prepared for electrophoretic mobility shift assay (EMSA) as follows. The cells on a 10 cm dish were washed with PBS and scraped in 200 μl of ice-cold sonication buffer (containing, in mm: 20 HEPES-KOH, pH 7.9, 25% glycerol, 500 KCl, 1.5 MgCl2, 0.4 EDTA, 5 DTT, 1 EGTA, 0.5 PMSF, and protease inhibitors) (Roche Complete Protease Inhibitor Cocktail). After sonication, the lysates were centrifuged at maximum speed for 10 min in a tabletop centrifuge at 4°C, the pellet was discarded, and the lysates were stored in −80°C. The in vitro translated proteins were produced using the TnT Quick Coupled Transcription/Translation System (Promega) according to the manufacturer's instructions, using unlabeled methionine. Oligos used in EMSAs were radioactively labeled with T4 polynucleotide kinase (Fermentas) according to the manufacturer's instructions. For oligo binding reaction, 10 μg of protein in 15 μl of reaction buffer (containing, in mm: 10 HEPES-KOH, pH 7.9, 10% glycerol, 0.1 EDTA, 8 MgC12, and 1 dithiothreitol) containing 0.2 μg of poly(dI-dC) was preincubated 10 min on ice. For obtaining the ARNT2-NPAS4 shift, the reaction buffer contained (in mm): 10 Tris-HCl, pH 7.5, 50 NaCl, 1 EDTA, 1 dithiothreitol, 5% glycerol and 0.02% β-mercaptoethanol, and 1 μg of poly(dI-dC) was added. Then, 0.15 pmol of radioactively labeled oligonucleotide was added and incubated 20 min at room temperature. For DNA competition, 10-fold excess of unlabeled double-stranded oligonucleotide was added 5 min before the probe. Where indicated, protein lysate was incubated with 0.5 μg of the respective antibody (ab) in reaction buffer before incubation with the radioactively labeled probe. The abs used were rabbit anti-CREB1 (Millipore catalog #06-863), rabbit anti-phospho-CREB1(Ser133) (Millipore catalog #06-519), rabbit anti-USF1 (CeMines catalog #HLH230), rabbit anti-USF2 (CeMines catalog #HLH240), rabbit anti-ARNT2 (Santa Cruz Biotechnology, catalog #sc-5581X) and rabbit anti-NPAS4 (Aviva Systems Biology, catalog #ARP32556_P050). The DNA–protein complexes were resolved in 5% nondenaturing polyacrylamide gel and visualized by autoradiography.
Western blot.
Proteins were separated by 8% SDS-PAGE and transferred to polyvinylidene fluoride membrane (Millipore). For blotting, membranes were blocked in 5% skim milk and 0.05% Tween 20 in PBS for 30 min at room temperature. Next, membranes were incubated with the respective primary ab for 1 h and then with secondary ab for 1 h at room temperature. Three washing steps with 0.05% Tween 20 in PBS followed both incubations. Primary abs were diluted in 5% skim milk and 0.05% Tween 20 in PBS as follows: rabbit anti-NPAS4 (Aviva Systems Biology, catalog #ARP32556_P050) 1:2000, rabbit anti-ARNT2 (Santa Cruz Biotechnology, catalog #sc-5581X) 1:5000, rabbit anti-CREB1 (Millipore catalog #06-863) 1:2000, mouse anti-V5 (Invitrogen catalog #R960-25) 1:5000, mouse anti-VP16 (clone 2GV-4) (Eurogentec) 1:5000. The HRP-conjugated goat anti-mouse or anti-rabbit IgG secondary abs (Pierce) were diluted 1:5000. Chemiluminescent signal was detected using SuperSignal West Femto Chemiluminescent Substrate (Pierce).
Chromatin immunoprecipitation.
For chromatin immunoprecipitation (ChIP) with HEK293 cells, cells grown on 10 cm dishes were transfected with plasmids coding V5-tagged or untagged proteins, as indicated, by LipoD293 DNA In Vitro Transfection Reagent (SignaGen) according to the manufacturer's instructions. A 1 μg/2 μl ratio of DNA/LipoD293 and 5 μg of DNA per 10 cm cell culture dish were used. Crosslinking with 1% formaldehyde was performed for 10 min at 24 h posttransfection. Crosslinking reaction was quenched by the addition of 1/10th volume of 1.25 m glycine to the medium, and the cells were washed twice with 1× PBS. Cells were lysed with lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8.0, with protease inhibitors). Then, the lysate was sonicated to obtain 200–1000 bp fragments of genomic DNA, and insoluble material was spun down for 5 min at maximum speed in a tabletop centrifuge. For ChIP with human (male) postmortem parietal cortex samples, frozen tissue was chopped into small pieces with a razor blade and inserted into 1× PBS. Crosslinking was performed with final concentration of 1% formaldehyde for 15 min at room temperature and quenched by adding glycine to a final concentration of 0.125 m. The samples were transferred on ice and washed twice with 1× PBS. A Dounce homogenizer was used to disaggregate the tissue. Cells were lysed with lysis buffer (0.5% NP40, 85 mm KCl, 5 mm PIPES, pH 8.0, with protease inhibitors) on ice for 15 min. Then, nuclei were pelleted and resuspended in nuclei lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8.0, with protease inhibitors) and incubated on ice for 20 min. The lysate was sonicated as described for ChIP with HEK cells. Anti-V5 agarose (Sigma) or protein A Sepharose (GE Healthcare) for HEK293 or tissue ChIP, respectively (both 40 μl of 50% slurry per reaction), was preabsorbed with 200 μg/ml BSA and 10 μg/ml sheared salmon sperm DNA by rotating overnight at 4°C in dilution buffer (1% Triton X-100, 150 mm NaCl, 2 mm EDTA, 20 mm Tris-HCl, pH 8.0, with protease inhibitors). For HEK293 ChIP, anti-V5 agarose was then added to the lysate that was diluted 1:9 with dilution buffer and the samples were rotated for 6 h at 4°C. For tissue ChIP the lysate was diluted 1:9 with dilution buffer and incubated with rabbit anti-CREB1 (Millipore catalog #06-863), rabbit anti-ARNT2 (Santa Cruz Biotechnology, catalog #sc-5581X) ab, both 1:1000, or without ab, overnight at 4°C, and then the preabsorbed protein A Sepharose was added for 2 h. The steps for both ChIP experiments were as follows. Agarose–chromatin or Sepharose–chromatin complexes were washed 3 times with wash buffer (1% Triton X-100, 0.1% SDS, 150 mm NaCl, 2 mm EDTA, 20 mm Tris-HCl, pH 8.0, with protease inhibitors) and once with final wash buffer (1% Triton X-100, 0.1% SDS, 500 mm NaCl, 2 mm EDTA, 20 mm Tris-HCl, pH 8.0, with protease inhibitors). The immune complexes were eluted 3 times by addition of 50 μl of elution buffer (1% SDS and 100 mm NaHCO3) and the eluates of the same sample were combined. Cross-links were reversed by incubating the eluates with 200 mm NaCl at 65°C overnight. DNA was purified with a QIAquick PCR Purification Kit (Qiagen) and analyzed by qPCR with the qPCR Core kit for SYBR Green I No Rox (Eurogentec) and LightCycler 2.0 (Roche). Three independent experiments were performed, and each sample was measured in triplicate with primers detecting the indicated genomic regions (supplemental Table S1, available at www.jneurosci.org as supplemental material). For HEK293 ChIP, the data were normalized to the levels of the respective target in input DNA. All data were log-transformed and autoscaled, means and SDs were calculated, and t tests for analyses of statistical significance were performed. For graphical representation, the data were back-transformed into the original scale. The error bars represent upper and lower limits back-transformed as mean + SD and mean − SD, respectively.
Results
Human BDNF exon I and IV transcripts are upregulated in primary cortical neurons by KCl depolarization
BAC transgenic mice with the human BDNF gene generated in our group beforehand (Koppel et al., 2009) were used to determine whether hBDNF expression is upregulated in response to neuronal activity in cultured primary cortical neurons. Depolarization of primary neuronal membranes with KCl treatment was used as the model of neuronal activity. Primary neurons cultured 7 DIV were treated with 25 mm KCl and the levels of the transgenic human and endogenous mouse BDNF mRNAs were measured by RT-qPCR. In addition, neurons were treated with KCl for different time periods to compare the time courses of upregulation between mBDNF and hBDNF mRNAs. First, the results showed that total hBDNF mRNA, measured with primers detecting all of the hBDNF transcripts, was upregulated in these neurons similarly to the endogenous total mBDNF (Fig. 1A). However, mBDNF mRNA levels were elevated more than the transgenic hBDNF mRNA levels (Fig. 1A). Next, we analyzed the upregulation of different BDNF exon-specific transcripts. As evidenced by Figure 1B, both hBDNF and mBDNF exon I- and exon IV-containing transcripts were the most upregulated BDNF mRNAs by neuronal depolarization. hBDNF exon I and IV transcripts reached to ∼120- and 27-fold higher levels, respectively, and mBDNF exon I and IV transcripts ∼230- and 32-fold higher levels, respectively, than the levels of these transcripts in untreated neurons (Fig. 1B). The time courses of induction of hBDNF and mBDNF exon I-containing transcripts were similar—after 2 h of KCl treatment the levels of exon I mRNAs were ∼10% of full induction that was reached by 4 and 8 h, respectively (Fig. 1B). Although not statistically significant, the induction time course of exon IV transcripts differed to some extent between mBDNF and hBDNF − mBDNF exon IV mRNA levels reached near to the maximum already after 2 h of KCl treatment whereas hBDNF exon IV levels were approximately half of maximum after 2 h and continued to rise also after 4 h of KCl treatment (Fig. 1B). With a slightly weaker rise in levels compared with mBDNF exon IV transcripts, mBDNF transcripts with exon IX as the 5′ exon (IX-5′) were also induced (Fig. 1B). Interestingly, the levels of mBDNF IX-5′ mRNA were considerably more elevated than the respective hBDNF mRNA levels (Fig. 1B). Both hBDNF and mBDNF exon II and VI transcript levels were also elevated in response to KCl treatment, but the induction was by far weaker than the induction of exon I and exon IV transcripts, respectively (Fig. 1B). Therefore, as BDNF exons are divided into clusters in the genome (Timmusk et al., 1993; Pruunsild et al., 2007), it is plausible that exon I is the major activity regulated exon in the first cluster and exon IV is the major activity-regulated exon in the second cluster. Of note, expression of transcripts containing other 5′ exons of hBDNF was undetectable in cultured neurons from hBDNF-BAC mice. Collectively, these data show that hBDNF transcripts are upregulated in response to depolarization of primary neurons and that rodent neurons can be used as a model for studying neuronal activity-dependent regulation of hBDNF promoters.
Figure 1.

Characterization of neuronal activity-dependent hBDNF mRNA expression in primary neurons and determination of activity-induced hBDNF promoters. A, Cortical neurons from BAC-transgenic mice with hBDNF inserted in the genome were used for studying hBDNF regulation in response to neuronal activity modeled by KCl depolarization. Neurons (7 DIV) were treated with 25 mm KCl for the time indicated and both endogenous mBDNF and transgenic hBDNF levels were measured by RT-qPCR using species-specific primers for the coding region of BDNF exon IX. mRNA levels are shown as fold induced levels over levels of total BDNF in untreated neurons (CNTR). B, The same neurons as in A were used to study changes in expression levels of BDNF transcripts with indicated 5′ exons upon KCl treatment by RT-qPCR using species-specific primers. mRNA levels at each time point are shown as fold induced levels over levels of respective transcripts in untreated neurons. The data in A and B were obtained from three independent experiments with triplicate measurements and normalized to HPRT1 expression. C, The same neurons as in A were used to measure the ratio of BDNF exon I to exon IV mRNA for both hBDNF and mBDNF gene in untreated neurons and in neurons treated 8 h with KCl. For quantification, plasmid serial dilutions with exactly 1:1 ratio of exon I and IV PCR templates of hBDNF or mBDNF, respectively, were used. Results of three independent experiments with triplicate measurements are shown. D, Rat primary neurons transfected with the indicated hBDNF promoter-dependent FFluc construct and hEF1α promoter-dependent hRluc construct were treated with 25 mm KCl for 8 h and luciferase activities were measured. Data are represented as fold induced promoter activities over activities measured from untreated control neurons for each hBDNF promoter. Results of at least three independent experiments with duplicate measurements normalized to hRluc activities are shown. Error bars represent SD. Statistical significance denoted by asterisks is relative to respective untreated control in A and B, and between the bars indicated with lines in C and D (***p < 0.001; **p < 0.01; *p < 0.05; NS, not significant; t test).
Provided that induction of both hBDNF and mBDNF exon I transcripts was particularly strong, we measured the relative abundance of exon I transcripts compared with exon IV transcripts in transgenic mouse primary neurons before and after KCl-depolarization. For that we cloned the human exon I and exon IV PCR products together in one plasmid construct and the respective mouse PCR products in another construct, which enabled us to use the dilution series of these plasmids as reference for calculating the ratio of exon I to exon IV mRNA. The results showed that hBDNF exon I transcripts reached to comparable levels with exon IV transcript levels upon depolarization of neurons (Fig. 1C). mBDNF exon I transcript levels were elevated to ∼60% of mouse exon IV transcript levels (Fig. 1C). We detected a significant difference in the ratio of exon I to exon IV mRNAs transcribed from hBDNF compared with mBDNF in uninduced cells: while the levels of mouse exon I transcripts were ∼20-fold lower compared with the levels of mouse exon IV transcripts, the human exon I mRNAs were only 4-fold less abundant than human exon IV mRNAs (Fig. 1C). Considering this and that the rBDNF exon I mRNAs have been reported to constitute <5% of total BDNF mRNAs in the adult rat cortex (Timmusk et al., 1994), whereas exon I transcripts in adult human parietal cortex have been found to be half as abundant as exon IV mRNAs (Garzon and Fahnestock, 2007), we conclude that BDNF exon I transcripts could represent a relatively larger proportion of total BDNF mRNAs in human brain than in rodent brain.
Next, we cloned the promoter regions of hBDNF major exons and the human-specific 5′ exon Vh in front of luciferase reporter and analyzed their ability to induce luciferase activity in response to KCl-depolarization in rat primary neurons. We used rat neurons because in majority of previous studies that have determined the regulatory elements for rodent BDNF, rat primary neuronal cultures have been used (Shieh et al., 1998; Tao et al., 1998; Tabuchi et al., 2002; Chen et al., 2003a,b). Our results showed that hBDNF promoters I and IV, that were induced ∼7- and 6-fold, respectively, were the most upregulated BDNF promoters by KCl treatment (Fig. 1D). In addition, pIX was induced ∼1.7-fold, which was statistically significant (p < 0.05; n = 3; t test), whereas pII 1.2-fold induction was not (p > 0.05; n = 3; t test; Fig. 1D). pVh was not induced and pVI activity was reduced upon depolarization (Fig. 1D).
Human BDNF promoter I is induced by neuronal activity primarily by bHLH-PAS transcription factor ARNT2-NPAS4 heterodimer
Tabuchi et al. (2002) have reported that a CRE-like element contributes to the activity-mediated activation of rat BDNF pI. To test whether this element is also involved in hBDNF pI regulation we generated reporter constructs containing the hBDNF pI linked to luciferase reporter gene with or without mutation in the orthologous CRE-like element. We mutated the CRE correspondingly to the mutation used previously for the rat pI (Tabuchi et al., 2002). The results showed that mutating the CRE-like element in hBDNF pI did not significantly affect the induction of hBDNF pI. Both WT hBDNF pI and hBDNF pI with mutated CRE were induced nearly 6.5-fold (Fig. 2A). Next, by removing half of the promoter we generated hBDNF pI 5′ and 3′ constructs that contained the 5′ or the 3′ transcription start site (TSS), respectively. As demonstrated in Figure 2B, the 5′ region of hBDNF pI was significantly more induced (24-fold) than the 3′ region of hBDNF pI (2-fold). This indicated that the region responsible for the induction of hBDNF pI upon neuronal activity is localized to the 5′ half of the cloned promoter. The CRE-like element resides in this half, but mutation in the CRE did not alter the induction of hBDNF pI 5′ (Fig. 2B). To map the cis-element that was needed for the induction we generated several 5′-deletion constructs. We found that deleting the region from −242 to −166 bp upstream of the TSS1 almost abolished hBDNF pI upregulation (Fig. 2B). Bioinformatic analysis indicated that this region contains two potential cis-elements that could regulate calcium-dependent activation. First, there is an activator protein 1 (AP1)-like element known to bind FOS and JUN proteins (TCACTCA; Fig 2C) (Morgan and Curran, 1991), and second, an asymmetric E-box-like element (CACGAC) (Fig. 2C) that has been shown to bind bHLH-PAS factors (Ooe et al., 2004). Notably, the E-box-like element is situated in reverse orientation on the complementary DNA strand compared with previously published data on such elements (Ooe et al., 2004). We mutated these elements and analyzed the effect of the mutations on pI depolarization-induced upregulation. Mutation of the AP1-like element (m1) did not affect induction, whereas mutation of the asymmetric E-box-like element (m2) reduced the induction from 24-fold to 5-fold (Fig. 2B). Collectively, these results indicate that the proximal promoter region sufficient for induction of hBDNF exon I transcription by neuronal activity is located between −242 and +25 bp relative to the TSS1 and that the novel E-box-like element is the most important activity-responsive cis-element in this region. Also, the results argue that the NFκB binding sites described for the rBDNF pI (Lubin et al., 2007) and the distally located enhancer bound by MEF2D in mice (Flavell et al., 2008) are dispensable for induction of the hBDNF pI as the orthologous sites of hBDNF reside outside the hBDNF pI 5′SphI promoter construct. However, we cannot rule out possible induction-modulating role of these sites.
Figure 2.

hBDNF pI neuronal activity-regulated induction depends on ARNT2-NPAS4 heterodimer. A, Rat primary neurons transfected with WT or cAMP/Ca2+-response element-like element mutated (CREm) hBDNF pI-dependent FFluc construct and hEF1α promoter-dependent hRluc construct were treated with 25 mm KCl and luciferase activities were measured. B, Rat primary neurons transfected with the indicated derivative of the WT hBDNF pI-dependent FFluc construct shown in A and hEF1α promoter-dependent hRluc construct were treated with 25 mm KCl and luciferase activities were measured. Data are represented as fold induced promoter activities over activities measured from untreated control neurons both in A and B. Numbers in the schematic representations of the promoters indicate nucleotide positions relative to the most 5′ TSS (TSS1) of hBDNF exon I. TSS2, 3′ transcription start site of hBDNF exon I. 5′, 5′ part of hBDNF pI; 3′, 3′ part of hBDNF pI; SphI and E31I, SphI and Eco31I restriction sites used to generate 5′ deletion constructs of hBDNF pI; m1 and m2, mutations in the AP1-like and asymmetric E-box-like (named PasRE here) cis-regulatory DNA elements, respectively. C, Alignment of the region responsible for neuronal activity-dependent induction of hBDNF pI and the region containing the CRE-like element in hBDNF pI with respective sequences in the rat promoter. Numbers indicate nucleotide positions relative to the TSS1 of hBDNF exon I. AP1-like, binding site for activator protein 1 complex; PasRE, asymmetric E-box-like bHLH-PAS TF response element; UBE, USF binding element. Note that 4 of the 6 nucleotides defined by Tabuchi et al. (2002) as critical for USF binding in the rat pI (boxed) are not conserved in human. D, EMSA showing CREB1 and USF complexes binding to the hBDNF pI CRE oligo and bHLH-PAS TF ARNT2-NPAS4 dimer binding to the pI AP1/PasRE oligo. Addition of 10-fold excess (10×) of respective unlabeled oligonucleotide or addition of ab to the reaction is indicated above the panel. Lysate used in the reaction is indicated below the panel where CNTR designates untreated neurons, 30 min KCl and 2 h KCl designate KCl-treated neurons and in vitro designates in vitro synthesized proteins. SS, supershift generated by the addition of ab. Representative image of at least three independent experiments is shown. E, Rat primary neurons transfected with hBDNF pI 5′-dependent FFluc construct and hEF1α promoter-dependent hRluc construct together with empty vector control (pRC) or a construct coding the indicated protein, were left untreated (CNTR) or were treated for 8 h with 25 mm KCl and luciferase activities were measured. VP16, a viral transcription activator domain fused to the respective protein where indicated. delTAD represents deletion of the transcription activator domain. F, Rat primary neurons transfected with rBDNF pI-dependent FFluc construct and hEF1α promoter-dependent hRluc construct together with empty vector control (pRC) or a construct coding the indicated protein, were left untreated (CNTR) or were treated for 8 h with 25 mm KCl and luciferase activities were measured. Promoter activities in E and F are presented in RLUs (specified in Materials and Methods) relative to the promoter activity measured from untreated pRC-transfected neurons that was arbitrarily set as 1. Results of at least three independent experiments with duplicate measurements are shown in A, B, E, and F. Error bars represent SD. Statistical significance denoted by asterisks is relative to pI 5′ construct fold induction in B and relative to promoter activity in pRC-transfected untreated control or pRC-transfected KCl-treated neurons, respectively, in E and F (***p < 0.001; **p < 0.01; *p < 0.05; NS, not significant; t test).
The TFs binding the region responsible for the induction of hBDNF pI were determined by EMSAs. We used two different oligonucleotides and protein lysates of untreated primary rat neurons or neurons treated for 30 min or 2 h with KCl. First, we confirmed that the region in hBDNF pI orthologous to the rBDNF pI region that has previously been shown to bind CREB1 and USF proteins (Tabuchi et al., 2002), also bound CREB1 and USF1 (oligo pI CRE; Fig. 2D). Second, when we used the region of the hBDNF pI containing the AP1-like and the asymmetric E-box-like element we detected a strong signal of mobility shift only when lysate from neurons treated 2 h with KCl was used (Fig. 2D). In addition, we verified with mutated oligos that the protein complex that shifted this oligo was bound to the asymmetrical E-box-like element (Fig. 2D). Ooe et al. (2004) have shown that bHLH-PAS factor NPAS4-ARNT2 dimers bind such elements and that they bind with the highest affinity to the sequence identical to the one we mapped in hBDNF pI (Fig. 2C). Moreover, it has been demonstrated that NPAS4 protein is expressed in primary cortical neurons only after KCl treatment (Lin et al., 2008). Therefore, we used NPAS4 and ARNT2 antibodies and in vitro synthesized ARNT2 and NPAS4 to verify that the complex that binds to the hBDNF pI cis-element responsible for the depolarization-induced induction contains ARNT2-NPAS4. The results showed that ARNT2 ab blocked formation of the complex and NPAS4 ab weakened considerably the intensity of the shifted complex in EMSA (Fig. 2D). In vitro synthesized ARNT2 and NPAS4 proteins were able to generate a shift of the pI AP1/PasRE oligo only when added together to the reaction (Fig. 2D) indicating they bind the hBDNF promoter as a dimer. Unlabeled oligo with mutated E-box-like sequence could not out-compete the labeled oligo whereas unlabeled oligo with mutated AP1-like element abolished the shift obtained with the labeled WT pI AP1/PasRE oligo (Fig. 2D). Moreover, addition of ARNT2 ab completely lost the formation of in vitro synthesized ARNT2–NPAS4 complex (Fig. 2D). Of note, the detected complexes did not contain FOS or JUN proteins since the respective abs did not change the intensity or mobility of oligo shifts (data not shown). Combined, these data showed that the asymmetric E-box-like element responsible for the activity-dependent induction of hBDNF pI binds the ARNT2-NPAS4 dimer and therefore we named the element PasRE (bHLH-PAS transcription factor response element).
To confirm these findings we transfected the hBDNF pI 5′-luciferase construct into rat primary neurons together with constructs coding for different effector proteins. Our experiments demonstrated that excess of CREB1 in primary neurons was not sufficient to enhance either basal or KCl-induced activity of hBDNF pI (Fig. 2E). Overexpression of constitutively active CREB1 (VP16-CREB) (Barco et al., 2002) was able to increase hBDNF pI activity in uninduced cells but not in depolarized cells (Fig. 2E). These results, together with the EMSA results and our observation that the CRE-like element mutation lowers basal activity of hBDNF pI to 65% or 28% of WT promoter basal activity in pI 5′ or pI construct context, respectively (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material), indicate that CREB1 binds to hBDNF pI, but most probably does not regulate its neuronal activity-dependent induction. Tabuchi et al. (2002) have described that the rBDNF pI CRE-like element is overlapping with a USF-binding element (UBE). We found that although not conserved in human (Fig. 2C), the orthologous sequence was bound by USF1 in EMSA (Fig. 2D). To study whether USF proteins could regulate hBDNF pI, we overexpressed USF1 and -2 fused to VP16 in neurons. The viral transcription activation domain VP16-USF fusion constructs instead of WT USF constructs were used to get a strong and clear activation of the promoter upon USF binding. We saw a small but statistically significant increase in hBDNF pI activity only in uninduced neurons (p < 0.05, n = 3; t test; Fig. 2E). The KCl-induced levels were slightly reduced in these neurons (Fig. 2E), suggesting that the USF proteins are not involved in hBDNF pI activity-dependent regulation. Conversely, the strongest increase in KCl-induced pI activity was obtained when ARNT2 and NPAS4 together were overexpressed, reaching to ∼3-fold higher levels than that seen for the empty vector control (pRC) (Fig. 2E). NPAS4 overexpression could also significantly increase the activity of hBDNF pI, whereas ARNT2 overexpression decreased the activity (Fig. 2E). NPAS4 overexpression enhanced pI activity in uninduced state most likely because ARNT2, as the bHLH-PAS general factor (Kewley et al., 2004), was expressed in uninduced cells (Keith et al., 2001) and NPAS4 addition allowed heterodimer formation. In contrast, ARNT2 overexpression did not enhance KCl-induced pI activity probably because the balance in the levels of ARNT2 and NPAS4 was disrupted leading to potential formation of ARNT2 homodimers. ARNT2 homodimer formation has been demonstrated before by Ooe et al. (2004). To strengthen the evidence that bHLH-PAS factors were responsible for hBDNF pI induction we overexpressed the bHLH-PAS transcriptional repressor SIM2 that has been shown to interfere with ARNT2-NPAS4-mediated transcriptional activation (Ooe et al., 2004). As can be seen in Figure 2E, SIM2 could effectively impede the rise of pI activity, indicating that bHLH-PAS TFs were involved in hBDNF pI upregulation. Furthermore, we deleted the C-terminal transactivation domains (TAD) of ARNT2 (Hirose et al., 1996) and NPAS4 (Ooe et al., 2004) to generate constructs coding for delTAD-ARNT2 and delTAD-NPAS4 that would act as dominant-negative bHLH-PAS factors similar to the IPAS protein (Makino et al., 2001). By coexpressing delTAD-ARNT2 and delTAD-NPAS4 proteins we were able to reduce depolarization-mediated induction of hBDNF pI by 8-fold compared with pRC-transfected cells (Fig. 2E). Overexpression of delTAD-NPAS4 alone also significantly reduced depolarization-induced activity of pI (Fig. 2E). However, delTAD-ARNT2 alone did not affect pI induction (Fig. 2E). Together, these results show that KCl-mediated activation of hBDNF pI in cortical neurons is dependent on the novel cis-regulatory element PasRE and bHLH-PAS TF ARNT2-NPAS4 dimer that binds the PasRE.
To answer the question whether the predominant role of ARNT2 and NPAS4 in inducing neuronal activity-dependent upregulation of BDNF pI is human-specific, we cloned the rat BDNF pI region corresponding to hBDNF pI 5′ in front of the luciferase reporter and analyzed the effect of CREB1 and ARNT2-NPAS4 overexpression on this promoter. Our experiments demonstrated that excess of CREB1 in primary neurons was sufficient to significantly enhance only the basal activity, but not KCl-induced activity of rBDNF pI (Fig. 2F). With ARNT2 and NPAS4 overexpression, on the other hand, there was strong increase in rBDNF pI activity both in uninduced and KCl-treated neurons compared with the control pRC-transfected cells (Fig. 2F). Relying on these results we suggest that, similarly to hBDNF pI, neuronal activity-dependent upregulation of rBDNF pI is predominantly mediated by ARNT2-NPAS4 dimer and not by CREB1.
CREB, ARNT2-NPAS4, and USFs are the key factors required for activation of human BDNF promoter IV by neuronal activity
To analyze the molecular mechanism of hBDNF pIV KCl-induced activation, we generated hBDNF pIV-luciferase constructs with mutations in the corresponding cis-elements that have been shown to regulate rBDNF pIV in response to neuronal activity (for review, see Greer and Greenberg, 2008). In addition, we mutated a novel element upstream of the described CaRE, UBE, and CRE with strong resemblance to the PasRE that we found to be important for the induction of hBDNF pI by neuronal activity (TTCGTG; Fig. 3A). In hBDNF pIV, the PasRE element is located in the same orientation as has been shown before for Drebrin promoter (Ooe et al., 2004).
Figure 3.

hBDNF pIV neuronal activity-regulated induction depends on CREB, ARNT2-NPAS4, and USFs. A, Alignment of the region responsible for neuronal activity-dependent induction of hBDNF pIV and downstream regions containing cis-elements mutated in this study with respective sequences in the rat promoter. PasRE, BHLHB2-RE, NFκB-RE, and NFAT-RE designate response elements for the respective TFs. B, Rat primary neurons transfected with WT hBDNF pIV or PasRE, CaRE, UBE, CRE, BHLHB2-RE, NFκB-RE or NFAT-RE mutated hBDNF pIV-dependent FFluc construct and hEF1α promoter-dependent hRluc construct, were treated with 25 mm KCl and luciferase activities were measured. Data are represented as fold induced promoter activities over activities measured from untreated control neurons. Numbers in A and in the schematic representation of the promoter in B indicate nucleotide positions relative to the most 5′ TSS (TSS1) of hBDNF exon IV. C, EMSA showing USF and CREB1 complexes binding to the pIV UBE/CRE oligo and bHLH-PAS TF ARNT2-NPAS4 dimer binding to the pIV PasRE oligo. Addition of unlabeled oligo or ab to the reaction and the lysates used are designated as in Figure 2D. Representative image of at least three independent experiments is shown. D, Rat primary neurons transfected with WT or UBEm hBDNF pIV-dependent FFluc construct and hEF1α promoter-dependent hRluc construct together with empty vector control (pRC) or constructs coding USF1 and USF2 fused to VP16 domain, were left untreated (CNTR) or were treated for 8 h with 25 mm KCl and luciferase activities were measured. E, Rat primary neurons transfected with WT or CREm hBDNF pIV-dependent FFluc construct and hEF1α promoter-dependent hRluc construct together with pRC or a construct coding CREB1 or CREB1 fused to VP16, were left untreated or were treated for 8 h with 25 mm KCl and luciferase activities were measured. F, Rat primary neurons transfected with WT or PasREm hBDNF pIV-dependent FFluc construct and hEF1α promoter-dependent hRluc construct together with pRC or with a construct coding the indicated protein, were left untreated or were treated for 8 h with 25 mm KCl and luciferase activities were measured. Promoter activities in D–F are presented in RLUs relative to the promoter activity measured from untreated pRC-transfected neurons that was arbitrarily set as 1. Results of at least three independent experiments with at least duplicate measurements are shown in B, D–F. Error bars represent SD. Statistical significance denoted by asterisks is relative to WT construct fold induction in B and between the bars indicated with lines in D–F (***p < 0.001; **p < 0.01; *p < 0.05; NS, not significant; t test).
Luciferase assays were performed similarly to hBDNF pI analyses. As demonstrated in Figure 3B, mutation in the CRE-element orthologous to the CRE described for the rBDNF pIV (Shieh et al., 1998; Tao et al., 1998) almost abolished the depolarization-induced upregulation of hBDNF pIV activity. In addition, mutation in the novel PasRE element decreased the induction of the promoter from ∼6-fold to 2-fold, indicating that in hBDNF pIV this element is also needed for responsiveness to neuronal activity (Fig. 3B). The UBE mutation reduced induction, but was statistically less significant than the effect of the PasRE mutation (Fig. 3B). The effect of CaRE mutation was not significant and the effect of the mutation in the binding site for BHLHB2 had an opposite effect compared with the results obtained by Jiang et al. (2008) for the rat pIV (Fig. 3B). The mutations in the binding sites for NFκB (Jiang et al., 2008) and NFAT (Vashishta et al., 2009) did not affect KCl-mediated induction of pIV (Fig. 3B). These data showed that the most important cis-regulatory elements required for the induction of hBDNF pIV are CRE, PasRE and UBE.
Next, we conducted EMSA experiments to verify that the hBDNF pIV CRE and UBE are bound by CREB1 and USF, respectively, and to identify whether the novel PasRE element in pIV is also bound by the ARNT2-NPAS4 dimer as was pI PasRE. The results obtained with the UBE and CRE cis-elements containing hBDNF pIV oligonucleotide (pIV UBE/CRE) showed that the oligo was bound by similar protein complexes when untreated or KCl-treated primary neuron lysates were used (Fig. 3C). Addition of WT or mutated oligos confirmed that the UBE and CRE elements were bound and the complexes were verified to contain USF1 or CREB1 proteins using abs against USF1, USF2, CREB1 and pSer133-CREB1 (Fig. 3C). Notably, CREB1-containing complexes were detected as two separate bands with slower and faster mobility. The anti-CREB1 ab shifted only the slower-migrating complex and the anti-pSer133-CREB1 ab shifted both of the complexes (Fig. 3C). In addition, the USF1-containing complex was detected as a partially overlapping band with the CREB1-containing faster-migrating complex (Fig. 3C). Finally, in vitro synthesized CREB1 and USF1 and -2 proteins could shift the hBDNF pIV UBE/CRE oligo, whereas USF1 binding was considerably stronger than USF2 binding (Fig. 3C). When hBDNF pIV PasRE-containing oligo was used, a mobility shift pattern highly similar to that obtained with the hBDNF pI AP1/PAS oligo was seen—a strong signal was observed only when lysates of neurons treated 2 h with KCl were used (Fig. 3C), suggesting that this PasRE element was also bound by ARNT2-NPAS4 dimer. Moreover, the complex migrated with the same speed in the gel as in vitro synthesized ARNT2–NPAS4 complex (Fig. 3C). The complex was bound to the PasRE element since it could be out-competed with the WT but not with PasRE mutated unlabeled oligo and contained ARNT2 as the shift could be abolished with the anti-ARNT2 ab (Fig. 3C). Of note, there was a faster migrating complex that we were unable to identify (Fig. 3C).
We performed transient transfection experiments with hBDNF pIV-luciferase constructs and USF, CREB1, ARNT2 and NPAS4 expression constructs to confirm the results obtained with EMSA. In addition, because several elements were involved in human pIV induction, we used mutated promoter constructs to show specificity of effects. USF-VP16 overexpression elevated uninduced as well as KCl-induced levels of hBDNF pIV driven luciferase activity in primary neurons (Fig. 3D) and this effect was lost when UBE mutated hBDNF pIV-luciferase construct was used (Fig. 3D). In contrast to hBDNF pI, on which CREB1 overexpression had no effect (Fig. 2E), KCl-induction of hBDNF pIV was significantly enhanced by overexpression of CREB1 since induction of hBDNF pIV was increased from ∼7-fold in pRC-transfected neurons to ∼15-fold in CREB1-transfected neurons (Fig. 3E). VP16-CREB1 overexpression enhanced both uninduced and depolarization-induced hBDNF pIV activity (Fig. 3E). These effects were lost when CRE mutated pIV was used (Fig. 3E). The effect of ARNT2 and NPAS4 overexpression on hBDNF pIV was similar to that seen for hBDNF pI—when ARNT2 and NPAS4 were overexpressed together, pIV activity measured from uninduced neurons was 3.5-fold higher and pIV activity measured from KCl-induced neurons was 4-fold higher than the respective activities obtained from pRC-transfected cells (Fig. 3F). NPAS4 alone also significantly enhanced pIV activity, whereas ARNT2 overexpression had no effect (Fig. 3F). Although not statistically significant, overexpression of the bHLH-PAS transcriptional repressor SIM2 reduced depolarization-dependent induction of pIV activity (Fig. 3F). Interestingly, the dominant-negative forms of ARNT2 and NPAS4 (delTAD-ARNT2 and delTAD-NPAS4, respectively) did not lower the depolarization-induced hBDNF pIV driven luciferase activity, but instead, enhanced the uninduced levels and by that lowered the fold induction of pIV (Fig. 3F), indicating that the PasRE element in hBDNF pIV might recruit a repressor in the uninduced state. The effects seen with SIM2, delTAD-ARNT2 and delTAD-NPAS4 constructs were not as clear as seen for promoter I most probably because of more complex regulation of pIV compared with pI. Last, ARNT2-NPAS4 overexpression did not enhance PasRE mutated hBDNF pIV activity, showing that the effect of ARNT2-NPAS4 coexpression on WT hBDNF pIV was specifically dependent on the novel PasRE element (Fig. 3F). Notably, one study has suggested transcriptional repressor DREAM (Carrión et al., 1999) to be involved in the regulation of CREB1-dependent mBDNF exon IV mRNA expression (Fontán-Lozano et al., 2009). However, we did not detect involvement of DREAM proteins in the regulation of hBDNF pIV in rat cultured cortical neurons with EMSA, using abs specific for the DREAM family members (supplemental Fig. 3C, available at www.jneurosci.org as supplemental material), or with luciferase reporter assays, using WT as well as Ca2+-insensitive EF-hand mutant DREAM constructs and constructs coding for other DREAM family members (Pruunsild and Timmusk, 2005) with mutated EF-hand motifs (supplemental Fig. 3D, available at www.jneurosci.org as supplemental material). Collectively, with these experiments we showed that CREB1, ARNT2-NPAS4 and USF bind to CRE, PasRE and UBE elements in the hBDNF pIV, respectively, and are the central factors required for activation of hBDNF pIV by KCl-mediated membrane depolarization.
CRE mediates initiation and PasRE enhancement of human BDNF promoter IV induction by neuronal activity
Next, we studied the role of the novel PasRE element in the activity-responsive regulation of hBDNF pIV more thoroughly. It has been shown that CREB1-dependent rBDNF pIV transcription is induced rapidly after depolarization of neuronal membranes (Shieh et al., 1998; Tao et al., 1998). The novel PasRE element, on the other hand, is bound by the ARNT2-NPAS4 complex in which the NPAS4 protein has to be synthesized by neurons after the initial depolarizing signal (Lin et al., 2008). Moreover, our EMSA results showed that ARNT2-NPAS4 DNA binding was detectable only after 2 h of KCl treatment. To control the hypothesis that CREB1, through activation by phosphorylation, is the initial target of depolarization-induced pathways on BDNF pIV and subsequently ARNT2-NPAS4 dimer enhances the transcriptional activity, we studied induction of hBDNF pIV driven luciferase activity in rat primary neurons transfected with hBDNF pIV constructs over the time course of 1–10 h after addition of 25 mm KCl. The results shown in Figure 4 revealed that the CRE element is crucial for induction of hBDNF pIV by neuronal activity. While the WT hBDNF pIV was induced 5.5-fold, mutation in the CRE element nearly abolished induction by KCl-mediated depolarization (Fig. 4). Mutating the PasRE element allowed hBDNF pIV activation with identical kinetics compared with the WT promoter (Fig. 4). However, 3 h after KCl addition mutation in the PasRE caused a significant decrease in further induction of pIV activity and 10 h after KCl addition the PasRE mutated hBDNF pIV was ∼2-fold less induced than WT hBDNF pIV (Fig. 4). These results argue that the CRE element in hBDNF pIV is needed for initiating transcription by neuronal depolarization and the PasRE element is necessary for enhancing further the transcriptional activity driven by hBDNF pIV.
Figure 4.
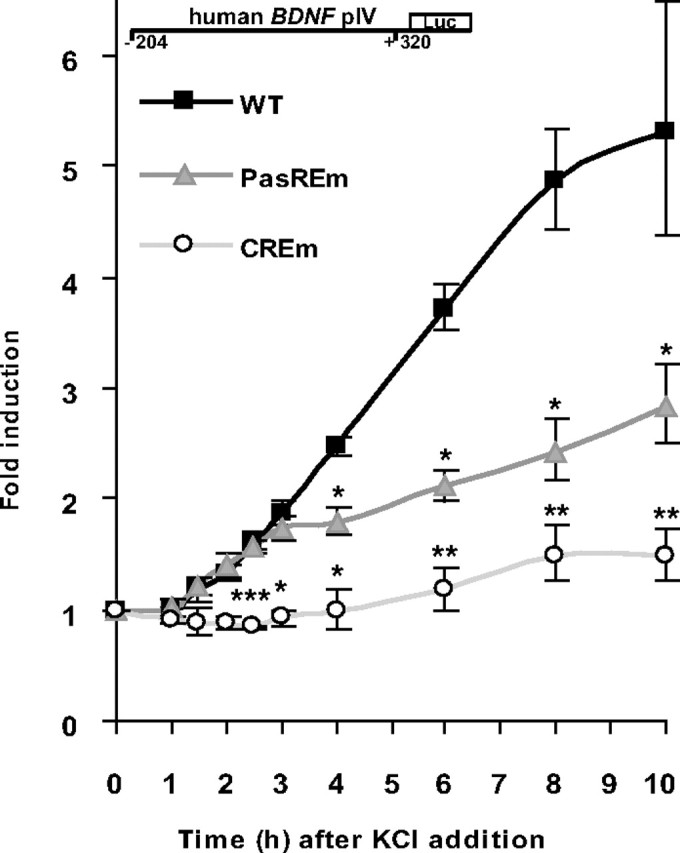
CRE is needed for initiation and PasRE for enhancement of hBDNF pIV induction by neuronal activity. Rat primary neurons transfected with WT or with CRE or PasRE mutated (CREm or PasREm) hBDNF pIV-dependent FFluc construct and hEF1α promoter-dependent hRluc construct, were treated with 25 mm KCl and luciferase activities were measured at time points from 1 to 10 h after KCl addition. Data at different time points are represented as fold induced promoter activities over activities measured from untreated control neurons. Results of three independent experiments with duplicate measurements normalized to hRluc levels are shown. Error bars represent SD. Statistical significance denoted by asterisks is relative to WT construct fold induction at respective time point (***p < 0.001; **p < 0.01; *p < 0.05; t test).
Partially overlapping PasRE and CRE-like elements regulate human BDNF promoter IX induction
In a previous study, our group has demonstrated that in addition to exon I and IV, kainic acid treatment of transgenic mice carrying the hBDNF gene induces expression of human as well as endogenous mBDNF IX-5′ mRNAs (Koppel et al., 2009). Furthermore, here we found that although hBDNF pIX promoter region of ∼1200 bp was induced considerably less than pI and pIV in response to KCl treatment, the induction was statistically significant (p < 0.05; n = 3; t test; Fig. 1D). Therefore, to map the inducible promoter region, we generated 5′ and 3′ deletion constructs of the promoter and used these constructs in luciferase assays. The results indicated that the region responsible for the neuronal activity-regulated induction locates to the 3′ part of the cloned hBDNF pIX as the 5′ part of the promoter was not induced and the 3′ region sustained inducibility (Fig. 5A). With 5′-deletions, we reduced the inducible region to a 112 bp fragment of hBDNF pIX (Fig. 5A), where we identified an asymmetric E-box-like element (CTCGTG) partially overlapping with a CRE-like element (TGACAGCA) (Fig. 5B). Mutations in these elements (mut) eliminated inducibility of the 112bp hBDNF pIX by KCl depolarization (Fig. 5A). With EMSA analyses we detected CREB1 and USF binding to the pIX DNA region responsible for KCl-induction as shown by supershift experiments with respective abs using lysates of uninduced neurons (Fig. 5C). With lysates from neurons treated for 2 h with KCl, an additional slower-migrating band was detected (Fig. 5C). Anti-ARNT2 ab eliminated formation of this complex indicating that the complex contained ARNT2. Usage of 10-fold excess of either WT or mutated unlabeled oligos showed that the protein complexes were binding the partially overlapping PasRE and CRE-like sites (Fig. 5C). Of note, the intensities of the obtained signals in EMSAs with hBDNF oligo pIX PasRE/CRE-like were much weaker than the signals acquired with hBDNF pI and pIV oligonucleotides.
Figure 5.
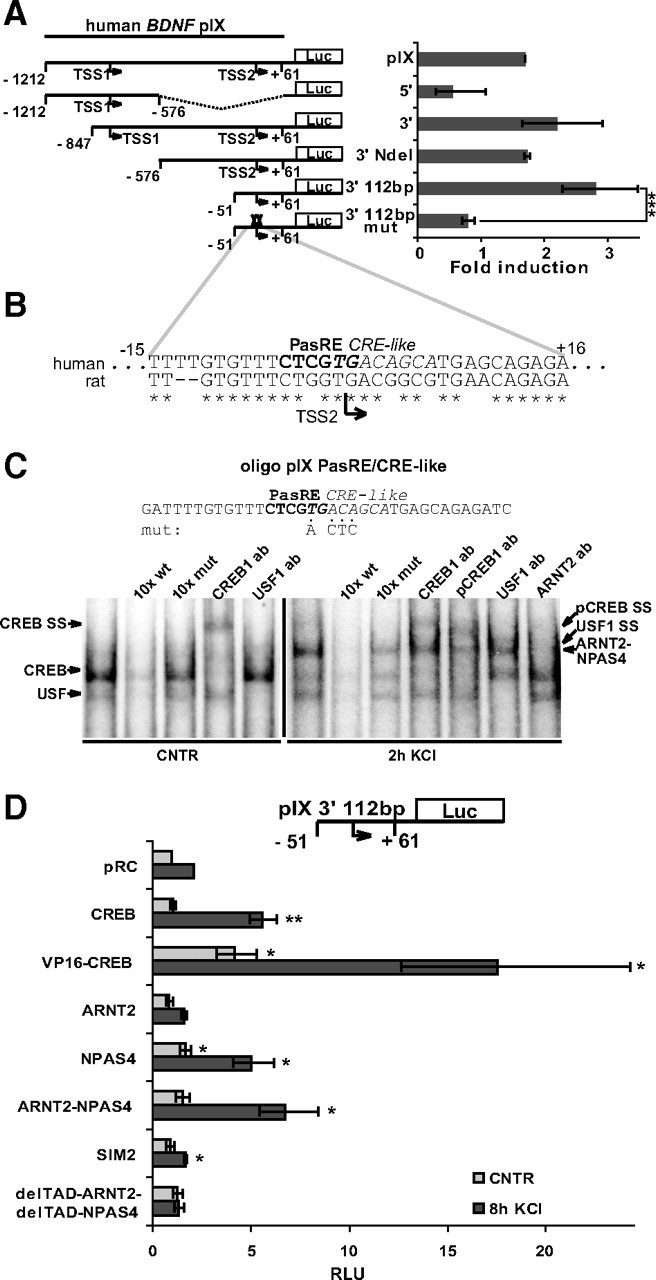
PasRE and CRE-like elements regulate hBDNF pIX induction. A, Rat primary neurons transfected with hBDNF pIX-dependent FFluc construct or indicated derivative thereof and hEF1α promoter-dependent hRluc construct, were treated with 25 mm KCl and luciferase activities were measured. Data are represented as fold induced promoter activities over activities measured from untreated control neurons. Numbers in the schematic representations of the promoters indicate nucleotide positions relative to the 3′ TSS (TSS2) of hBDNF exon IX. TSS1, 5′ transcription start site in hBDNF exon IX. 5′, 5′ part of hBDNF pIX; 3′, 3′ part of hBDNF pIX; NdeI, NdeI restriction site used to generate the pIX 3′ NdeI construct. 3′ 112bp, hBDNF pIX-luciferase construct with minimal KCl inducible sequence. mut, mutations in the PasRE and CRE-like elements. B, Alignment of the region responsible for neuronal activity-dependent induction of hBDNF pIX with the respective region in the rat promoter. Numbers indicate nucleotide positions relative to the TSS2 of hBDNF exon IX. C, EMSA showing CREB1-, USF-, and ARNT2-NPAS4-containing complexes binding to the PasRE and CRE-like element containing oligo pIX PasRE/CRE-like. Addition of unlabeled oligo or ab to the reaction and the lysates used are designated as in Figure 2D. Representative image of three independent experiments is shown. D, Rat primary neurons transfected with 3′ 112bp hBDNF pIX-dependent FFluc construct and hEF1α promoter-dependent hRluc construct together with pRC or with a construct coding the indicated protein, were left untreated or were treated for 8 h with 25 mm KCl and luciferase activities were measured. Promoter activities are presented in RLUs relative to the promoter activity measured from untreated pRC-transfected neurons that was arbitrarily set as 1. Results of at least three independent experiments with duplicate measurements are shown. Error bars represent SD. Statistical significance denoted by asterisks is between the bars indicated by lines in A and relative to promoter activity in pRC-transfected untreated control or pRC-transfected KCl-treated neurons, respectively, in D (***p < 0.001; **p < 0.01; *p < 0.05; t test).
Next, we analyzed whether the TFs detected in EMSA regulate the activity of hBDNF pIX-luciferase construct when overexpressed in neurons. USF-VP16 was found not to influence the activity of hBDNF pIX (data not shown). Overexpression of CREB1 enhanced the induction and overexpression of VP16-CREB1 enhanced both the uninduced and induced levels of hBDNF pIX driven luciferase activity (Fig. 5D). ARNT2 overexpression did not enhance, whereas overexpression of NPAS4 alone or with ARNT2 enhanced the induced luciferase levels by 2- or 3-fold, respectively (Fig. 5D). SIM2 reduced the induced levels of pIX activity, whereas overexpression of delTAD-ARNT2 together with delTAD-NPAS4 slightly increased uninduced levels and abolished induction of hBDNF pIX-dependent luciferase activity (Fig. 5D). Altogether the results show that: (1) KCl-induced activation of hBDNF pIX is weak compared with induction of pI and pIV and (2) activation of hBDNF pIX is mediated by ARNT2-NPAS4 and CREB1 binding to the PasRE and CRE-like elements.
Overexpression of dominant-negative ARNT2-NPAS4 reduces neuronal activity-dependent transcription of endogenous rat BDNF exon I, IV, and IX-5′ mRNAs
Since all cis-elements except the novel PasREs that we have described here for hBDNF promoters have been shown before to be important for rodent BDNF activity-dependent induction, we chose to validate the role of the PasRE elements also at the level of endogenous rodBDNF regulation. For that we used overexpression of the delTAD-ARNT2 and delTAD-NPAS4 that have the ability to compete with endogenous factors by binding to BDNF PasRE elements and interfering with activity-regulated BDNF transcription. We electroporated delTAD-ARNT2 and delTAD-NPAS4 or control GFP constructs into rat primary neurons using Amaxa nucleofection and treated the cells at 5 DIV with 25 mm KCl for 2 to 8 h. We analyzed the ability of delTAD-ARNT2 and delTAD-NPAS4 to hinder endogenous BDNF mRNA upregulation by RT-qPCR. The results showed that the upregulation of rBDNF exon I, IV, and IX-5′ mRNAs in the delTAD-ARNT2- and delTAD-NPAS4-expressing cells was considerably weaker than in GFP-electroporated cells (Fig. 6). In the GFP-electroporated neurons exon I mRNA levels were ∼5-fold more induced than in the delTAD-ARNT2 and delTAD-NPAS4-electroporated cells (Fig. 6). Similarly, induction of exon IV mRNA levels was 2.5-fold weaker (Fig. 6) and induction exon IX-5′ mRNA levels was 3-fold weaker (Fig. 6) at the time point of 4 h of KCl treatment in delTAD-ARNT2 and delTAD-NPAS4-electroporated neurons than in GFP-electroporated neurons. The results of this experiment suggest that the full induction of activity-responsive transcription of endogenous rBDNF exon I, IV and IX-5′ mRNAs is to a great extent dependent on bHLH-PAS TFs ARNT2 and NPAS4 binding the PasRE elements.
Figure 6.

Dominant-negative ARNT2-NPAS4 interfere with neuronal activity-dependent transcription of endogenous rat BDNF exon I, IV and IX-5′ mRNAs. Control GFP coding construct or a construct coding ARNT2 without the transactivation domain (delTAD-ARNT2) together with a construct coding NPAS4 without the transactivation domain (delTAD-NPAS4), were electroporated into rat primary neurons. At 5 DIV, neurons were treated with 25 mm KCl for the time indicated and mRNA levels of rat endogenous BDNF exon I, IV and IX-5′ transcripts were measured with RT-qPCR. mRNA levels are shown as fold induced levels over levels of the respective transcript in untreated neurons. The data are from two independent experiments measured in triplicates and normalized to HPRT1 expression levels. Error bars represent SD.
ARNT2-NPAS4 and CREB activate endogenous human BDNF transcription by binding human BDNF promoters I and IV
We performed chromatin immunoprecipitation (ChIP) experiments to demonstrate that ARNT2-NPAS4 and CREB1 bind to PasRE and CRE elements, respectively, on endogenous BDNF promoters in human cells. Moreover, to demonstrate that transcription of endogenous BDNF is enhanced in human cells by ARNT2-NPAS4 and CREB1 we analyzed hBDNF mRNA expression in human embryonic kidney HEK293 cells overexpressing ARNT2, NPAS4 or CREB1.
For ChIP, either constructs coding for ARNT2-V5 or NPAS4-V5 alone or together with NPAS4 or ARNT2 coding constructs, respectively, were transfected into HEK293 cells (Fig. 7A). ChIP was performed with V5-agarose enabling us to monitor whether ARNT2-V5 and NPAS4-V5 binding to DNA was enhanced by addition of the dimer partner. Also, CREB1-V5 was expressed in HEK293 cells (Fig. 7A). In qPCR analyses of the immunoprecipitated DNA, we focused on hBDNF pI, pIV and pIX and used primers for an unrelated region on human chromosome 11 as a negative control (Chr11URR). We observed strong binding of ARNT2-V5 and NPAS4-V5 on hBDNF pI and pIV only when the V5-tagged proteins were expressed together with NPAS4 and ARNT2, respectively (Fig. 7B), supporting the data that ARNT2 and NPAS4 bind DNA as a dimer. In contrast, we detected very weak binding of ARNT2-V5 and NPAS4-V5 to pIX and to the control Chr11URR in all transfection combinations (Fig. 7B). CREB1-V5 was also significantly enriched on hBDNF pI and IV and not bound to pIX or the unrelated control region (Fig. 7B). ARNT2-NPAS4 dimer was enriched relatively more on pI than on pIV and CREB1 was enriched relatively more on pIV (Fig. 7B). Additionally, these results suggest that ARNT2-NPAS4 and CREB1 were either not bound to pIX or the binding was under the detection limit of the experiment.
Figure 7.

ARNT2-NPAS4 and CREB bind endogenous hBDNF promoters and activate hBDNF transcription. A, Western blot showing ARNT2–V5, NPAS4–V5 or combination of ARNT2–V5 with NPAS4 and NPAS4–V5 with ARNT2, and CREB1–V5 expression in HEK293 cells transfected with the respective constructs. Protein expression was verified with anti-NPAS4, anti-ARNT2, anti-CREB1 and anti-V5 abs. B, ChIP experiment with anti-V5 ab using the same cell lysates as described in A, demonstrating ARNT2-NPAS4 heterodimer and CREB1 binding to endogenous hBDNF pI and pIV. The data are represented as fold enrichment of immunoprecipitated DNA from cells overexpressing the indicated proteins over empty vector pRC-transfected cells. Results from three independent experiments measured in triplicates and normalized to the levels of the respective target in input DNA are shown. Statistical significance denoted by asterisks is relative to enrichment values of Chr11URR obtained from each transfection. C, Western blot showing VP16-CREB1 or ARNT2–V5, NPAS4–V5 or combination of ARNT2–V5 with NPAS4–V5 expression in HEK293 cells transfected with the respective constructs. Protein expression was verified with anti-VP16 and anti-V5 abs. D, RT-qPCR analyses showing hBDNF exon I, IV or IX-5′ transcript expression in HEK293 cells described in C. The data are presented as fold change in BDNF transcript levels in indicated protein overexpressing cells compared with levels of the respective transcript in control pRC-transfected cells. Results of three independent experiments measured in triplicates and normalized to the levels of HPRT1 mRNA levels are shown. Statistical significance denoted by asterisks is relative to mRNA levels in pRC-transfected cells. E, ChIP experiment with anti-CREB1 or anti-ARNT2 ab using postmortem human parietal cortex lysates, demonstrating CREB1 binding to endogenous hBDNF pIV and ARNT2 binding to endogenous hBDNF pI and pIV. The data are represented as enrichment of ab immunoprecipitated DNA relative to −ab control precipitated DNA. Statistical significance denoted by asterisks is relative to enrichment values of Chr11URR obtained with the indicated ab. Results with three independent tissue samples measured in triplicates are shown. Immunoprecipitated DNA was analyzed by qPCR with primers amplifying fragments of indicated genomic regions in B and E. pI, hBDNF pI; pIV, hBDNF pIV; pIX, hBDNF pIX; and Chr11URR, unrelated region in human chromosome 11 serving as a negative control (***p < 0.001; **p < 0.01; *p < 0.05; t test). Error bars represent SD.
Next, we overexpressed VP16-CREB1, ARNT2, NPAS4 or ARNT2 together with NPAS4 in HEK239 cells (Fig. 7C) and measured hBDNF mRNA expression by RT-qPCR. As shown in Figure 7D, hBDNF exon I transcript levels were elevated ∼80-fold in ARNT2-NPAS4-cotransfected cells compared with control pRC-transfected cells and ∼10-fold induction was seen upon VP16-CREB1 or NPAS4 transfection. Similarly, the highest levels of exon IV transcripts were detected when ARNT2 and NPAS4 were cotransfected (Fig. 7D). VP16-CREB1 induced hBDNF exon IV transcription ∼7-fold and NPAS4 ∼2.5-fold. Notably, VP16-CREB1 induced hBDNF exon IV transcription relatively more than exon I transcription when the effect of ARNT2-NPAS4 on the respective transcripts is considered. In accordance to the results of the ChIP and EMSA experiments, only weak upregulation was detected for hBDNF exon IX-5′ transcripts. ARNT2-NPAS4 transfection elevated BDNF exon IX-5′ mRNA levels ∼2.5-fold compared with pRC-transfected cells (Fig. 7D). VP16-CREB1 overexpression did not have a significant effect on hBDNF IX-5′ transcript levels in HEK293 cells (Fig. 7D). These results imply that TFs ARNT2, NPAS4 and CREB1 bind hBDNF promoters I and IV in chromatin context and enhance transcription of endogenous hBDNF. Also, these experiments suggest that ARNT2-NPAS4 dimer binds and induces endogenous hBDNF pI relatively more than pIV and CREB1, on the other hand, binds and induces endogenous hBDNF pIV relatively more than pI.
Finally, we performed ChIP experiments with anti-ARNT2 and anti-CREB1 abs and human postmortem parietal cortex samples to analyze whether ARNT2 and CREB1 bind hBDNF promoters in the human brain in vivo. With qPCR analyses of the immunoprecipitated DNA we detected significant enrichment of ARNT2 on hBDNF pIV and pI compared with the control Chr11URR (Fig. 7E). CREB1 was significantly enriched only on hBDNF pIV and not on hBDNF pI (Fig. 7E). ARNT2 binding to hBDNF pIX was also statistically significant when compared with the control Chr11URR (Fig. 7E). However, as the enrichment of the pIX region in the precipitated DNA was low relative to the −ab control (Fig. 7E), we suggest that the detection limit of our ChIP experiment is not enough to answer whether ARNT2 binds hBDNF pIX in vivo. Detecting ARNT2 on pIX might be complicated in postmortem tissue because the ARNT2–NPAS4 complex probably binds hBDNF pIX with weaker affinity than it binds hBDNF pI and pIV as is indicated by our results with EMSA and the results of the ChIP experiments in HEK cells. Together, the results show that ARNT2 binds hBDNF pI and pIV and CREB1 binds hBDNF pIV in the human parietal cortex in vivo, which strongly supports our previous results of this study showing that ARNT2 and NPAS4 regulate neuronal activity-dependent transcription of hBDNF exon I and exon IV and CREB1 regulates neuronal activity-dependent transcription of hBDNF exon IV.
Discussion
Results presented in this study reveal several novel findings on the regulation of neuronal activity-dependent BDNF transcription. First, exon I and IV transcripts of human BDNF are the most upregulated hBDNF mRNAs by depolarization of primary cortical neurons. Second, activity-regulated induction of hBDNF pI depends primarily on a novel PasRE element. Thirdly, activity-regulated induction of hBDNF pIV depends predominantly on CRE, PasRE and UBE elements. And last, the novel PasRE elements in pI and pIV are conserved in human and rodents and are bound by bHLH-PAS transcription factor ARNT2-NPAS4 heterodimers.
In this study, we analyzed neuronal activity-dependent regulation of BDNF transcription in cultured primary cortical neurons from hBDNF-BAC transgenic mice (Koppel et al., 2009) and demonstrated that exon I and IV transcripts are the most upregulated BDNF mRNAs by membrane depolarization in both human and mouse BDNF genes. Our results showed that there was a slight difference in the time course of upregulation between hBDNF and mBDNF exon IV transcripts as human transcripts accumulated with a delay compared with respective mouse transcripts. Human exon IX-5′ transcripts were considerably less upregulated than the respective mouse transcripts and in addition, the ratio of exon I to exon IV transcripts in uninduced cells was significantly higher for human BDNF than for mouse BDNF. These results raise the possibility that dissimilarities might exist in the regulation of BDNF gene between mouse and human in vivo. However, two important aspects should be considered. First, we used rodent cells to study hBDNF regulation. Therefore, one might ask how accurately these results mirror the regulation of BDNF in human. Second, the results could be influenced by the fact that all regulatory sequences of hBDNF might not be present in the BAC construct (Koppel et al., 2009). It is recognized that differences in cis-regulatory elements rather than trans-acting factors explain many of the interspecies variations in gene regulation (Wilson and Odom, 2009). Moreover, human regulatory elements or a human chromosome have been shown to direct human-specific gene expression and TF binding in transgenic mice (Prabhakar et al., 2008; Wilson et al., 2008). Therefore, we believe it is plausible to use rodent neurons as a tool for studying regulation of a human gene. On the other hand, we admit that the hBDNF transgene could be missing regulatory elements. For example, the antisense BDNF (Pruunsild et al., 2007) or the distal 5′ region that has been found to influence hBDNF expression in vivo (Gray et al., 2006). Nevertheless, in principle, depolarization of mouse primary neurons activated hBDNF transcription and therefore, rodent primary neurons are credible for hBDNF promoter studies.
We showed that the CRE-like element that has been described for the rat pI (Tabuchi et al., 2002) was not the regulator of activity-dependent induction of hBDNF pI. Instead, we found that a novel element upstream of the CRE-like element was responsible for the upregulation of hBDNF pI. However, we propose that this effect is not human-specific. According to our results on the regulation of the rBDNF pI, CREB overexpression in primary neurons significantly enhances basal but not depolarization-induced levels of pI activity. Excess of ARNT2 and NPAS4 in cells, on the other hand, drastically increases both uninduced as well as induced levels of rBDNF pI activity. Moreover, the data of Tabuchi et al. (2002) show that deletion of the CRE-like element in rBDNF pI decreases the basal activity of the promoter, whereas deletion of a region upstream of the CRE specifically reduces fold induction of rBDNF pI upon depolarization. Thus, CREB or a related factor is regulating the basal activity of BDNF pI, but the TFs contributing most significantly to the activity-induced activation of BDNF pI are bHLH-PAS TFs ARNT2 and NPAS4 that bind the novel activity regulated cis-element named PasRE here. bHLH-PAS proteins regulate many crucial physiological and developmental processes (Kewley et al., 2004). Our result that they regulate hBDNF pI is supported by previous studies that have shown that: (1) a region upstream of the CRE-like element is involved in activity-dependent regulation of rat pI (Tabuchi et al., 2002); (2) NPAS4 binds DNA in the pI region of mBDNF (Lin et al., 2008); (3) NPAS4 forms a heterodimer preferably with ARNT2 in brain (Ooe et al., 2009); (4) ARNT2-NPAS4 dimer has the highest affinity to a DNA sequence identical to the PasRE element in hBDNF pI (Ooe et al., 2004); and (5) mBDNF levels are reduced in NPAS4 knock-out mice (Lin et al., 2008).
Our data showing that the CRE element is fundamental for the induction of hBDNF pIV is in accordance with the finding that CRE mutation in the mBDNF pIV loses its inducibility in vivo (Hong et al., 2008). Additionally, our results indicated that a novel PasRE element contributes to the depolarization-induced upregulation of pIV by significantly enhancing activity of the promoter. While initial induction of PasRE mutated pIV upon KCl depolarization was identical to the WT promoter, significant decrease in induction was detected from 3 h onward. This is in good agreement with the data presented by Lin et al. (2008) that the NPAS4 protein, shown here to bind BDNF PasREs as a heterodimer with ARNT2, is produced in primary cultured neurons after depolarization of neuronal membranes. Moreover, we found that EMSA shift by ARNT2-NPAS4 heterodimer was detectable only with lysates of depolarized neurons. Interestingly, the predominant rise of both human and mouse BDNF exon I transcript levels took place later after KCl addition than the major rise in BDNF exon IV transcript levels as was shown here using primary neurons from hBDNF-BAC mice. This supports the finding that NPAS4 accumulation is needed for induction of pI. pIV, on the other hand, is induced by CREB as an immediate-early promoter. One might argue also that in the hBDNF-BAC mouse neurons hBDNF exon IV transcript levels were elevated more slowly than the respective mouse transcripts due to relatively bigger role of the PasRE element compared with the CRE element in hBDNF pIV. Proving this suggestion needs additional experiments though.
The mutation in the UBE of hBDNF pIV decreased induction of pIV by half, suggesting involvement of USF proteins, which was confirmed by EMSA and luciferase assays. Conversely, we saw only modest effect on induction when the CaRE element in the hBDNF pIV was mutated. Recently it was reported that CaRF is dispensable for induction of mBDNF exon IV expression (McDowell et al., 2010). Considering this, we suggest that CaRF is not one of the major contributors to the depolarization-induced upregulation of hBDNF pIV activity in cortical neurons. Also, we showed here that mutations in NFκB or NFAT sites had no effect and mutation in BHLHB2 binding site had induction-decreasing effect on hBDNF pIV activation by depolarization, respectively. These results differed from the results described earlier (Jiang et al., 2008; Vashishta et al., 2009), but could be explained by use of glutamate receptor agonists instead of KCl-mediated depolarization for modeling neuronal activation in previous studies. In addition, we found that, at least in primary neurons, the DREAM family TFs do not contribute to activity-dependent regulation of hBDNF pIV as has been proposed for mBDNF pIV (Fontán-Lozano et al., 2009). Again, further experiments are needed to elucidate these issues.
Here we also showed that upregulation of hBDNF pIX was modest, being induced only ∼2-fold upon KCl treatment of primary neurons. Nevertheless, we identified partially overlapping CRE-like and PasRE elements in the region responsible for activity-dependent induction of hBDNF pIX. As can be suggested from our ChIP assay results, human pIX was induced less than pI and pIV by neuronal activity probably because of weaker binding of CREB and ARNT2-NPAS4 to pIX. Interestingly, the activity-regulated cis-elements described here for the human pIX are not conserved in rodents. However, we found that induction of endogenous rat exon IX-5′ transcription was partly dependent on bHLH-PAS factors. This is conceivable though, because only one-third of TF binding events take place on an aligned site in an orthologous promoter between human and mouse (Odom et al., 2007) suggesting another PasRE location in rodBDNF pIX.
Transcriptional regulation of rodBDNF has been used as a model for studying neuronal activity-responsive gene expression (for review, see Greer and Greenberg, 2008). Our analysis of hBDNF regulation showed that, in general, activity-dependent upregulation of BDNF promoters is similar between human and rodent. Importantly, the role of PasRE elements in hBDNF promoters was described here. Because these elements had not been studied in rat beforehand we confirmed that the usage of PasREs in BDNF is conserved also in rodents since we could interfere with the induction of endogenous rBDNF by overexpressing dominant-negative forms of ARNT2 and NPAS4 proteins. We validated the findings obtained in rodent cells by showing with ChIP experiments that endogenous hBDNF PasRE elements are bound by overexpressed ARNT2-NPAS4 in human cells. Furthermore, we demonstrated that these cis-elements are bound by endogenous ARNT2 in human brain, implying that the BDNF PasRE sites are active in human brain in vivo. It is significant that PasRE elements regulate hBDNF since ARNT2 has been found to be a key TF in the hippocampus, where it is potentially crucial for synaptic plasticity (Valen et al., 2009), and both ARNT2 as well as NPAS4 have been demonstrated to be neuroprotective TFs, which could be good therapeutic targets (Drutel et al., 1999; Hester et al., 2007; Zhang et al., 2009). Altogether, our study specifies the cis-elements and transcription factors regulating neuronal activity-dependent transcription of human BDNF and provides insight into the molecular mechanisms impairment of which might be one of the causes for diseases and disorders with altered BDNF expression.
Footnotes
This work was supported by the Wellcome Trust (Grant 067952), the Estonian Ministry of Education and Research (Grant 0140143), Estonian Science Foundation (Grants 7257 and 8844), and Estonian Enterprise (Grant EU27553). We thank Enn Jõeste from North Estonian Regional Hospital, Tallinn, for collaboration and Epp Väli for technical assistance.
The authors declare competing financial interests. The authors have filed a patent application based on data presented in this paper.
References
- Aid T, Kazantseva A, Piirsoo M, Palm K, Timmusk T. Mouse and rat BDNF gene structure and expression revisited. J Neurosci Res. 2007;85:525–535. doi: 10.1002/jnr.21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell. 2002;108:689–703. doi: 10.1016/s0092-8674(02)00657-8. [DOI] [PubMed] [Google Scholar]
- Barde YA, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982;1:549–553. doi: 10.1002/j.1460-2075.1982.tb01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. 2004;22:123–131. doi: 10.1080/08977190410001723308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrión AM, Link WA, Ledo F, Mellström B, Naranjo JR. DREAM is a Ca2+-regulated transcriptional repressor. Nature. 1999;398:80–84. doi: 10.1038/18044. [DOI] [PubMed] [Google Scholar]
- Chen WG, West AE, Tao X, Corfas G, Szentirmay MN, Sawadogo M, Vinson C, Greenberg ME. Upstream stimulatory factors are mediators of Ca2+-responsive transcription in neurons. J Neurosci. 2003a;23:2572–2581. doi: 10.1523/JNEUROSCI.23-07-02572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003b;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- Drutel G, Héron A, Kathmann M, Gros C, Macé S, Plotkine M, Schwartz JC, Arrang JM. ARNT2, a transcription factor for brain neuron survival? Eur J Neurosci. 1999;11:1545–1553. doi: 10.1046/j.1460-9568.1999.00562.x. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Lee KF, Jaenisch R. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature. 1994;368:147–150. doi: 10.1038/368147a0. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ, Markenscoff-Papadimitriou E, Bear DM, Greenberg ME. Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron. 2008;60:1022–1038. doi: 10.1016/j.neuron.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontán-Lozano A, Romero-Granados R, del-Pozo-Martín Y, Suárez-Pereira I, Delgado-García JM, Penninger JM, Carrión AM. Lack of DREAM protein enhances learning and memory and slows brain aging. Curr Biol. 2009;19:54–60. doi: 10.1016/j.cub.2008.11.056. [DOI] [PubMed] [Google Scholar]
- Garzon DJ, Fahnestock M. Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J Neurosci. 2007;27:2628–2635. doi: 10.1523/JNEUROSCI.5053-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray J, Yeo GS, Cox JJ, Morton J, Adlam AL, Keogh JM, Yanovski JA, El Gharbawy A, Han JC, Tung YC, Hodges JR, Raymond FL, O'Rahilly S, Farooqi IS. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55:3366–3371. doi: 10.2337/db06-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59:846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Hall J, Thomas KL, Everitt BJ. Rapid and selective induction of BDNF expression in the hippocampus during contextual learning. Nat Neurosci. 2000;3:533–535. doi: 10.1038/75698. [DOI] [PubMed] [Google Scholar]
- Hester I, McKee S, Pelletier P, Thompson C, Storbeck C, Mears A, Schulz JB, Hakim AA, Sabourin LA. Transient expression of Nxf, a bHLH-PAS transactivator induced by neuronal preconditioning, confers neuroprotection in cultured cells. Brain Res. 2007;1135:1–11. doi: 10.1016/j.brainres.2006.11.083. [DOI] [PubMed] [Google Scholar]
- Hirose K, Morita M, Ema M, Mimura J, Hamada H, Fujii H, Saijo Y, Gotoh O, Sogawa K, Fujii-Kuriyama Y. cDNA cloning and tissue-specific expression of a novel basic helix-loop-helix/PAS factor (Arnt2) with close sequence similarity to the aryl hydrocarbon receptor nuclear translocator (Arnt) Mol Cell Biol. 1996;16:1706–1713. doi: 10.1128/mcb.16.4.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Tian F, Du Y, Copeland NG, Jenkins NA, Tessarollo L, Wu X, Pan H, Hu XZ, Xu K, Kenney H, Egan SE, Turley H, Harris AL, Marini AM, Lipsky RH. BHLHB2 controls Bdnf promoter 4 activity and neuronal excitability. J Neurosci. 2008;28:1118–1130. doi: 10.1523/JNEUROSCI.2262-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KR, Fariñas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B, Adelman DM, Simon MC. Targeted mutation of the murine arylhydrocarbon receptor nuclear translocator 2 (Arnt2) gene reveals partial redundancy with Arnt. Proc Natl Acad Sci U S A. 2001;98:6692–6697. doi: 10.1073/pnas.121494298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kewley RJ, Whitelaw ML, Chapman-Smith A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int J Biochem Cell Biol. 2004;36:189–204. doi: 10.1016/s1357-2725(03)00211-5. [DOI] [PubMed] [Google Scholar]
- Koppel I, Aid-Pavlidis T, Jaanson K, Sepp M, Pruunsild P, Palm K, Timmusk T. Tissue-specific and neural activity-regulated expression of human BDNF gene in BAC transgenic mice. BMC Neurosci. 2009;10:68. doi: 10.1186/1471-2202-10-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim TK, Hu LS, Malik AN, Greenberg ME. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature. 2008;455:1198–1204. doi: 10.1038/nature07319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubin FD, Ren Y, Xu X, Anderson AE. Nuclear factor-kappa B regulates seizure threshold and gene transcription following convulsant stimulation. J Neurochem. 2007;103:1381–1395. doi: 10.1111/j.1471-4159.2007.04863.x. [DOI] [PubMed] [Google Scholar]
- Lyons WE, Mamounas LA, Ricaurte GA, Coppola V, Reid SW, Bora SH, Wihler C, Koliatsos VE, Tessarollo L. Brain-derived neurotrophic factor-deficient mice develop aggressiveness and hyperphagia in conjunction with brain serotonergic abnormalities. Proc Natl Acad Sci U S A. 1999;96:15239–15244. doi: 10.1073/pnas.96.26.15239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, Cao Y, Berkenstam A, Poellinger L. Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature. 2001;414:550–554. doi: 10.1038/35107085. [DOI] [PubMed] [Google Scholar]
- McDowell KA, Hutchinson AN, Wong-Goodrich SJ, Presby MM, Su D, Rodriguiz RM, Law KC, Williams CL, Wetsel WC, West AE. Reduced cortical BDNF expression and aberrant memory in Carf knock-out mice. J Neurosci. 2010;30:7453–7465. doi: 10.1523/JNEUROSCI.3997-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JI, Curran T. Stimulus-transcription coupling in the nervous system: involvement of the inducible proto-oncogenes fos and jun. Annu Rev Neurosci. 1991;14:421–451. doi: 10.1146/annurev.ne.14.030191.002225. [DOI] [PubMed] [Google Scholar]
- Odom DT, Dowell RD, Jacobsen ES, Gordon W, Danford TW, MacIsaac KD, Rolfe PA, Conboy CM, Gifford DK, Fraenkel E. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nat Genet. 2007;39:730–732. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooe N, Saito K, Mikami N, Nakatuka I, Kaneko H. Identification of a novel basic helix-loop-helix-PAS factor, NXF, reveals a Sim2 competitive, positive regulatory role in dendritic-cytoskeleton modulator drebrin gene expression. Mol Cell Biol. 2004;24:608–616. doi: 10.1128/MCB.24.2.608-616.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooe N, Saito K, Kaneko H. Characterization of functional heterodimer partners in brain for a bHLH-PAS factor NXF. Biochim Biophys Acta. 2009;1789:192–197. doi: 10.1016/j.bbagrm.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Prabhakar S, Visel A, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Morrison H, Fitzpatrick DR, Afzal V, Pennacchio LA, Rubin EM, Noonan JP. Human-specific gain of function in a developmental enhancer. Science. 2008;321:1346–1350. doi: 10.1126/science.1159974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruunsild P, Timmusk T. Structure, alternative splicing, and expression of the human and mouse KCNIP gene family. Genomics. 2005;86:581–593. doi: 10.1016/j.ygeno.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Pruunsild P, Kazantseva A, Aid T, Palm K, Timmusk T. Dissecting the human BDNF locus: bidirectional transcription, complex splicing, and multiple promoters. Genomics. 2007;90:397–406. doi: 10.1016/j.ygeno.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Tabuchi A, Sakaya H, Kisukeda T, Fushiki H, Tsuda M. Involvement of an upstream stimulatory factor as well as cAMP-responsive element-binding protein in the activation of brain-derived neurotrophic factor gene promoter I. J Biol Chem. 2002;277:35920–35931. doi: 10.1074/jbc.M204784200. [DOI] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Tao X, West AE, Chen WG, Corfas G, Greenberg ME. A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron. 2002;33:383–395. doi: 10.1016/s0896-6273(01)00561-x. [DOI] [PubMed] [Google Scholar]
- Timmusk T, Palm K, Metsis M, Reintam T, Paalme V, Saarma M, Persson H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron. 1993;10:475–489. doi: 10.1016/0896-6273(93)90335-o. [DOI] [PubMed] [Google Scholar]
- Timmusk T, Belluardo N, Persson H, Metsis M. Developmental regulation of brain-derived neurotrophic factor messenger RNAs transcribed from different promoters in the rat brain. Neuroscience. 1994;60:287–291. doi: 10.1016/0306-4522(94)90242-9. [DOI] [PubMed] [Google Scholar]
- Valen E, Pascarella G, Chalk A, Maeda N, Kojima M, Kawazu C, Murata M, Nishiyori H, Lazarevic D, Motti D, Marstrand TT, Tang MH, Zhao X, Krogh A, Winther O, Arakawa T, Kawai J, Wells C, Daub C, Harbers M, Hayashizaki Y, Gustincich S, Sandelin A, Carninci P. Genome-wide detection and analysis of hippocampus core promoters using DeepCAGE. Genome Res. 2009;19:255–265. doi: 10.1101/gr.084541.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashishta A, Habas A, Pruunsild P, Zheng JJ, Timmusk T, Hetman M. Nuclear factor of activated T-cells isoform c4 (NFATc4/NFAT3) as a mediator of antiapoptotic transcription in NMDA receptor-stimulated cortical neurons. J Neurosci. 2009;29:15331–15340. doi: 10.1523/JNEUROSCI.4873-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Odom DT. Evolution of transcriptional control in mammals. Curr Opin Genet Dev. 2009;19:579–585. doi: 10.1016/j.gde.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Wilson MD, Barbosa-Morais NL, Schmidt D, Conboy CM, Vanes L, Tybulewicz VL, Fisher EM, Tavaré S, Odom DT. Species-specific transcription in mice carrying human chromosome 21. Science. 2008;322:434–438. doi: 10.1126/science.1160930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Zou M, Lu L, Lau D, Ditzel DA, Delucinge-Vivier C, Aso Y, Descombes P, Bading H. Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 2009;5:e1000604. doi: 10.1371/journal.pgen.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]