

Abstract
We have recently identified a mechanistic link between disruption of the microtubule cytoskeleton and inhibition of tumor angiogenesis via the hypoxia-inducible factor-1 (HIF-1) pathway. Based on this model, we hypothesized that other microtubule-targeting drugs may have a similar effect on HIF-1α. To test that hypothesis, we studied the effects of different clinically relevant microtubule-disrupting agents, including taxotere, epothilone B, discodermolide, vincristine, 2-methox-yestradiol, and colchicine. In all cases, HIF-1α protein, but not mRNA, was down-regulated in a drug dose–dependent manner. In addition, HIF-1α transcriptional activity was also inhibited by all drugs tested. To further examine whether these effects were dependent on microtubule network disruption, we tested the ability of epothilone B to inhibit HIF-1α protein in the human ovarian cancer cell line 1A9 and its (β-tubulin mutant epothilone-resistant subclone 1A9/A8. Our data showed that epothilone B treatment down-regulated HIF-1α protein in the parental 1A9 cells but had no effect in the resistant 1A9/A8 cells. These observations were confirmed by confocal microscopy, which showed impaired nuclear accumulation of HIF-1α in parental 1A9 cells at epothilone B concentrations that induced extensive microtubule stabilization. In contrast, epothilone B treatment had no effect on either microtubules or HIF-1α nuclear accumulation in the resistant 1A9/A8 cells. Furthermore, epothilone B inhibited HIF-1 transcriptional activity in 1A9 cells, as evidenced by a hypoxia response element-luciferase reporter assay, but had no effect on HIF-1 activity in the resistant 1A9/A8 cells. These data directly link β-tubulin drug binding with HIF-1α protein inhibition. Our results further provide a strong rationale for testing taxanes and epothilones in clinical trials targeting HIF-1 in cancer patients.
Introduction
Hypoxia-inducible factor-1 (HIF-1) is a proangiogenic transcription factor critically involved in tumor progression, metastasis, and overall tumor survival (1–3). In the presence of oxygen (normoxia), HIF-1α is rapidly degraded by the proteasome. This occurs through HIF-1α protein hydroxylation on proline residues 402 and 564 (4–6) by specific HIF prolyl hydroxylases in the presence of iron and oxygen (7,8). The hydroxylated protein is then recognized by the von Hippel-Lindau tumor suppressor protein (pVHL), which functions as an E3 ubiquitin ligase. In the absence of oxygen (hypoxia), HIF-1α is not hydroxylated, which prevents its interaction with pVHL and its subsequent ubiquitination and degradation. Following hypoxic stabilization, HIF-1α is translocated to the nucleus where it heterodimerizes with HIF-1β and binds to hypoxia response elements (HRE), thereby activating the transcription of numerous genes important for adaptation and survival under hypoxia (9–11). HIF-1α activity is also controlled in an O2-independent fashion through the regulation of two main signaling pathways—the phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways (11). Tumor hypoxia confers resistance to both chemotherapy and radiation therapy while in many cases is associated with increased metastasis and poor survival (12–17). Furthermore, HIF-1α overexpression has been reported to occur in ~ 70% of all human tumors and their metastases compared with their adjacent normal tissue (18), thus making HIF an attractive yet unexplored target for cancer chemotherapy. The striking up-regulation of HIF-1α in so many different tumors and by so many different mechanisms highlights the critical role HIF-1 plays in tumor biology and makes HIF-1α a prime target for anticancer therapies (3).
Microtubules are major dynamic and structural cellular components important in several cell functions, including division, signaling, and intracellular trafficking. Thus, tubulin and microtubules are compelling cellular targets for chemotherapy, as these functions are often dysregulated in many types of cancer. In fact, among anticancer agents, those that target microtubules constitute one of the most effective classes of chemotherapeutics for survival prolongation in advanced disease (19, 20). The list of compounds, which bind to tubulin or microtubules, is large and consists of chemically unique compounds that bind to the tubulin dimers and destabilize microtubules (Vinca alkaloid– and colchicine-binding site drugs) and those that bind to the microtubule polymer and stabilize microtubules, such as the taxanes (Taxol and Taxotere) and the epothilones among others. Currently, there are no Food and Drug Administration–approved colchicine-binding site drugs used for the treatment of cancer; however, Vinca alkaloids are frequently used in the treatment of non-small cell lung cancer (NSCLC), breast cancer, and lymphomas (21, 22). Drugs that bind at the taxane site of tubulin are used extensively for the treatment of a wide variety of human cancers, including ovarian, prostate, NSCLC, and breast cancers (23–27).
We have recently identified a mechanism linking pharmacologic disruption of the microtubule cytoskeleton and downstream inhibition of tumor angiogenesis growth factors [e.g., vascular endothelial growth factor (VEGF)] that are under the control of HIF-1α (28). We have shown that disruption of interphase microtubules by the microtubule-destabilizing agent 2-methoxyestradiol impaired tumor angiogenesis and growth by inhibiting HIF-1 levels and transcriptional activity (28). Importantly, we showed in a mouse orthotopic breast cancer model that 2-methoxyestradiol inhibited tumor angiogenesis and HIF-1α at doses that disrupted tumor and endothelial microtubules (28). The question remains as to whether this inhibition of HIF-1 is 2-methoxyestradiol specific, if it is a property of only a subset of microtubule-disrupting drugs (i.e., microtubule-depolymerizing agents versus microtubule-stabilizing agents), or if it is a characteristic shared by all microtubule-disrupting drugs. In this report, we show that all microtubule-targeting drugs down-regulate HIF-1α regardless of chemical structure, binding site, or net effect on the microtubule polymer mass. We further show that HIF down-regulation occurs downstream of microtubule disruption as evidenced by the use of a β-tubulin mutant epothilone-resistant cell line, in which drug binding and hence microtubule disruption is severely impaired. Our results provide a thorough characterization of the anti-HIF effects of several microtubule-targeting drugs that bind at the taxane-, Vinca alkaloid–, and colchicine-binding sites and suggest that HIF inhibition likely contributes to the overall anticancer activity of this class of drugs.
Materials and Methods
Chemicals and antibodies.
Taxotere was a gift from Aventis (Bridge-water, NJ). Vincristine, vinblastine, epothilone B, colchicine, and MG-132 were purchased from Calbiochem (La Jolla, CA). Adriamycin was purchased from Sigma (St. Louis, MO). Stock solutions of 0.1 mmol/L were made for all compounds in DMSO and stored in aliquots at −80°C. The compounds were diluted in incubation medium immediately before each experiment. The following primary antibodies were used: rat anti-α-tubulin (Chemicon International, Temecula, CA), mouse anti-HIF-1α (BD Biosciences, San Diego, CA), rabbit polyclonal antibody to human actin (Sigma), sheep polyclonal anti-p53 (Ab7; Oncogene Science, Cambridge, MA), and mouse anti-mdm2 (Calbiochem). Secondary antibodies were horseradish peroxidase conjugated (Amersham Pharmacia Biotech, Piscataway, NJ), Alexa Fluor 488 goat anti-mouse (Molecular Probes, Eugene, OR), and Alexa Fluor 568 goat anti-rat (Molecular Probes).
Cell lines.
LN229 glioblastoma cell line, PC3 prostate cell line, A549 lung adenocarcinoma cancer cell line, MCF-7 and MDA-MB-231 breast cancer cell lines, 1A9 human ovarian carcinoma cells, and its epothilone-resistant subclone 1A9/A8 (Thrβ274Ile) were cultured in RPMI 1640 (Cellgro). All media were supplemented with 10% fetal bovine serum (Cellgro) and antibiotics (Cellgro). Cells were cultured at 37°C in a humidified atmosphere and 5% CO2. For hypoxic exposure, cells were placed in a sealed modular incubator chamber (Billups-Rothenberg, Del Mar, CA) flushed with 1% O2, 5% CO2, and 94% N2.
Western blot.
Cells were seeded in culture dishes and grown until 70% confluence. The medium was then replaced with new medium containing either vehicle (0.1% DMSO) or the different microtubule-targeting drugs at the concentrations indicated in the figures overnight at 37°C. The following day, cells were exposed to hypoxia or left under normoxia for 4 hours. Whole-cell extracts (WCE) were prepared and proteins (70 μg/lane) were resolved by 7.5% SDS-PAGE, electrotransferred to polyvinylidene difluoride membrane, and incubated with the indicated primary antibodies followed by horseradish peroxidase–conjugated secondary antiserum. For sequential blotting with additional antibodies, the membranes were stripped using a restore Western blot stripping buffer (Pierce, Rockford, IL) and reprobed with the indicated antibodies.
Transient transfections and reporter gene assay.
The cells were transfected with 1 μg/well of the reporter plasmids pNF-κB-Luc (Clontech, Palo Alto, CA) or pBI-GL-V6L as described previously (7, 28).
Isolation and analysis of RNA.
Total RNA was isolated using Qiagen RNeasy kit (Qiagen, Inc., Valencia, CA), and Northern blotting was done with probes specific for human HIF-1α, glucose transporter-1 (Glut-1) glucose transporter (gift from Dr. Erwin Van Meir, Department of Neurosurgery and Hematology-Oncology, Winship Cancer Institute, Emory University School of Medicine, Atlanta, GA), and 18S RNA (Ambion, Inc., Austin, TX) using the NorthernMax kit (Ambion).
Immunofluorescence and confocal microscopy.
Exponentially growing cells were plated on 12-mm glass coverslips (Fisher Scientific, Pittsburgh, PA) into 24-well plates and cells were allowed to attach overnight. The following day, cells were treated with the indicated drugs for 16 hours and subjected to hypoxia for an additional 4 hours. Cells were fixed and processed for double-labeling immunofluorescence as described previously (28).
Data analysis.
Experiments presented in the figures are representative of three or more different repetitions. Statistical analysis was done using a single-factor ANOVA and Tukey follow-up tests (P < 0.05 was considered statistically significant).
Results
We have shown previously that 2-methoxyestradiol, a microtubule-depolymerizing agent with antitumor and antiangiogenic properties currently in clinical development, down-regulates HIF-1α levels and its transcriptional activity. To test whether those effects on HIF-1α are a specific property of 2-methoxyestradiol or if they are also shared by other microtubule-targeting drugs that bind at any of the three well-characterized drug-binding sites on tubulin, we have done the experiments described in Fig. 1. Human ovarian 1A9 (Fig. 1A) and breast MDA-MB-231 (Fig. 1B) cancer cell lines were treated with increasing concentrations of either microtubule-stabilizing agents (top) or microtubule-destabilizing agents (bottom), and total levels of HIF-1α were assessed by Western blotting. As our group has shown the anti-HIF effects of 2-methoxyestradiol previously, this drug was included in subsequent experiments as a positive control. In all cases, HIF-1α was inhibited in a dose-dependent manner regardless of drug-binding site or drug-stabilizing/drug-destabilizing effects. Down-regulation by these six agents was specific for the a-subunit and had no effect on the protein levels of HIF-1β or other transcription factors (data not shown) in agreement with our previously reported data (28). To further investigate whether the effects on HIF-1α are solely specific for microtubule-targeting drugs, we treated 1A9 cells with the DNA-damaging agent Adriamycin. Overnight treatment with Adriamycin at concentrations able to induce p53 and its target gene mdm2 had no effect on HIF-1α levels (Fig. 1C).
Figure 1.
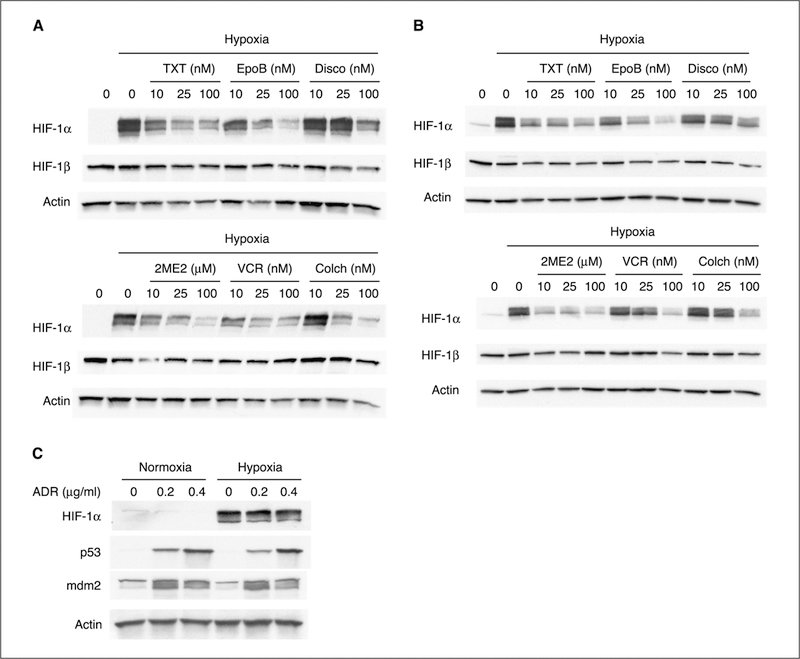
Microtubule-disrupting agents down-regulate HIF-1α. Human ovarian 1A9 (A) and human breast MDA-MB-231 (B) cancer cell lines were plated and treated with the indicated concentrations of taxotere (TXT), epothilone B (EpoB), discodermolide (Disco), 2-methoxyestradiol (2ME2), vincristine (VCR), and colchicine (Colch) for 16 hours. The following day, cells were incubated under normoxia or hypoxia (1% O2) for an additional 4 hours. WCEs were prepared and protein (75 μg) for each sample was resolved in a 7.5% SDS-PAGE gel and blotted for HIF-1α protein. The same gels were stripped and reblotted for HIF-1β and actin. The first lane of each blot represents control cells incubated under normoxic conditions. Top, include microtubule-stabilizing drugs; bottom, microtubule-destabilizing ones. C, 1A9 cells were treated overnight with the indicated concentrations of Adriamycin (ADR). WCEs were prepared and protein (75 μg) for each sample was resolved in a 7.5% SDS-PAGE gel and blotted for HIF-1α protein. The same gel was stripped and reblotted for p53, mdm2, and actin.
Our results show that treatment with both classes of microtubule-targeting drugs inhibits HIF-1α levels in a dose-dependent manner. This result differs from previously reported data (29) showing that microtubule-destabilizing drugs transiently stabilized HIF-1α protein (in <6 hours) under normoxic conditions by activating the nuclear factor-κB (NF-κB) pathway. This discrepancy prompted us to investigate effects of additional drugs and in additional cell lines. We sought to investigate whether the reported concentrations of vinblastine or colchicine were able to induce HIF-1α levels after 4 hours of incubation in different cancer cell lineages, including ovarian (1A9), lung (A549), breast (MCF-7), brain (LN229), and prostate (PC3). As shown in Fig. 2A, HIF-1α levels were not increased whatsoever following treatment with the aforementioned microtubule-depolymerizing agents. To further investigate the involvement of NF-κB in HIF-1α regulation following microtubule disruption, we did a luciferase reporter assay using a construct containing the luciferase gene under the control of four tandem repeats of the NF-κB consensus sequence from the herpes simplex virus thymidine kinase promoter (Fig. 2B ). As shown in Fig. 2B, treatment with either 100 nmol/L vincristine or epothilone B for 4 hours was unable to stimulate NF-κB-driven luciferase activity. Thus, results reported previously (29) could not be reproduced.
Figure 2.
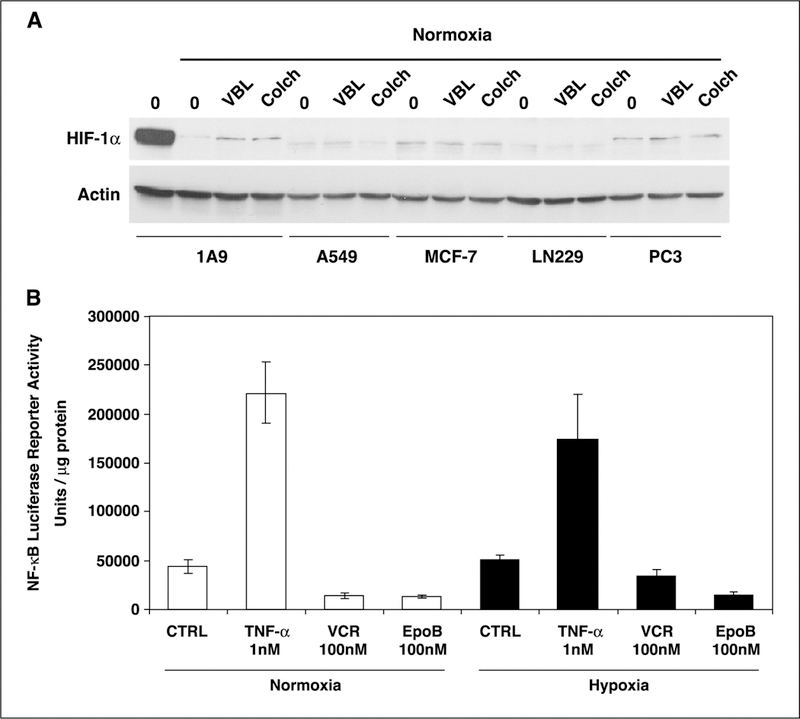
Microtubule-depolymerizing agents do not transiently increase HIF-1α levels through a NF-κB-dependent pathway. A, human ovarian (1A9), lung adenocarcinoma (A459), breast cancer (MCF-7), glioblastoma (LN229), and prostate (PC3) cancer cell lines were treated for 4 hours with 100 nmol/L vinblastine (VBL) or 1 μmol/L colchicine. WCEs were prepared and protein (75 Ag) was resolved in a 7.5% SDS-PAGE gel and blotted for HIF-1α protein and actin. A representative cell extract from 1A9 cells incubated under hypoxia was included in the first lane of the blot for comparison purposes to show the degree of HIF-1α induction under hypoxic conditions. B, 1A9 cells transiently transfected with pNF-κB-Luc vector (containing four tandem copies of the NF-κB consensus sequence from the herpes simplex virus thymidine kinase) were treated with vehicle or 100 nmol/L vincristine and epothilone B under normoxic and hypoxic conditions. Tumor necrosis factor-α (TNF-α; 1 nmol/L) was used as a positive control for NF-κB activity induction. Luciferase reporter activity was measured in the cellular extract 16 hours later. Luciferase activity represents arbitrary units per microgram of protein in each assay point.
We have shown previously that down-regulation of HIF-1α by 2-methoxyestradiol occurred at the post-transcriptional level by affecting HIF-1α protein translation. However, it is possible that other microtubule-targeting drugs down-regulate HIF-1α by a different mechanism. To determine whether additional microtubule-targeting drugs affected HIF-1α transcription, we did a Northern blot analysis of HIF mRNA (Fig. 3A). In all cases, HIF-1α mRNA levels were not significantly changed by the different treatments even at the higher doses used. On the other hand, there was an inhibition of the HIF-responsive Glut-1 mRNA transcription in the drug-treated cells compared with the hypoxic untreated control cells, in keeping with the drugs’ ability to down-regulate HIF-1α protein levels. To investigate whether both classes of microtubule-targeting drugs modulate HIF-1α protein at the post-translational level, we used the proteasome inhibitor MG-132 to prevent HIF-1α degradation. This allowed us to observe HIF-1α accumulation over time following de novo protein synthesis. Because rapid proteosomal destruction of HIF-1α occurs in normoxic cells, the accumulation rate of HIF-1α via proteosomal inhibition indirectly reflects the synthesis rate of the protein (30). As shown in Fig. 3B, HIF-1α maximally accumulated after 1 hour in the presence of MG-132 under normoxia. In contrast, treatment with either 25 nmol/L vincristine or epothilone B in the presence of MG-132 resulted in a much slower rate of HIF-1α accumulation, reflecting inhibition of HIF-1α protein synthesis. Together with the Northern blot results, these data suggest that the different microtubule-targeting drugs all down-regulate HIF-1α by inhibiting its protein synthesis. Additional studies are warranted to elucidate the exact mechanism by which microtubule-disrupting drugs disrupt protein synthesis.
Figure 3.
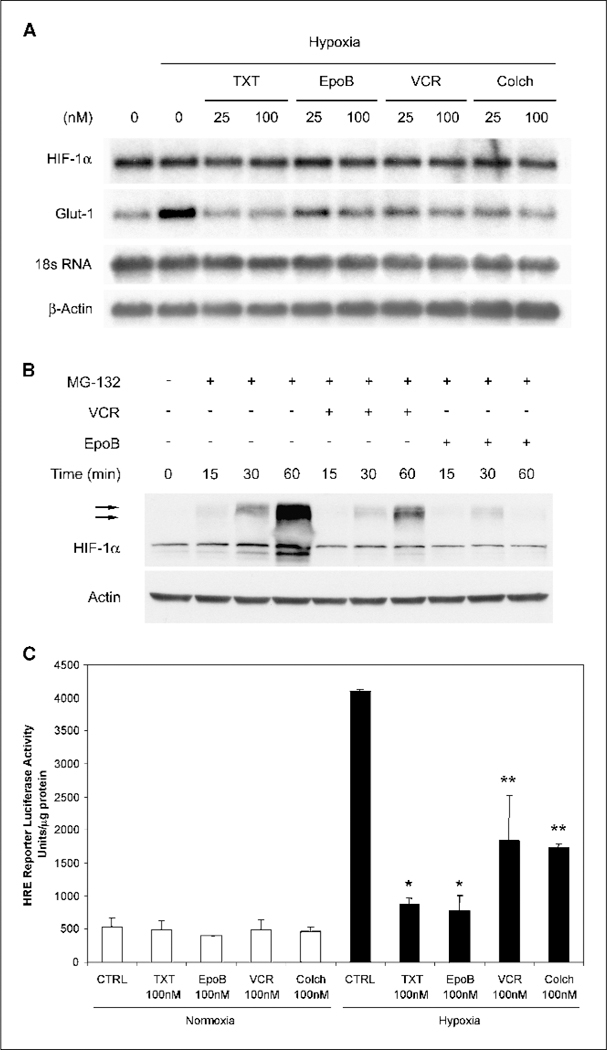
Microtubule-targeting drugs inhibit HIF-1α at the post-transcriptional level. A, total RNA was prepared from treated cells under normoxic and hypoxic conditions. Northern blotting was done with 32P-labeled probes for HIF-1α and Glut-1. 18S RNA and β-actin are shown as loading controls. B, 1A9 cells were pretreated overnight with either 25 mol/L vincristine or 25 mol/L epothilone B. The cells were then cotreated with MG-132 (10 Amol/L) in the presence or absence of the drugs for the indicated time (minutes). Equal amount of proteins from each lysate was resolved by SDS-PAGE and blotted for HIF-1α protein. The same gels were stripped and reblotted for actin. Arrows, ubiquinated HIF-1α protein species. C, 1A9 cells transiently transfected with pBI-GL V6L (6xHREs of VEGF promoter) were treated with vehicle or 100 nmol/L of each indicated drug under normoxic and hypoxic conditions. Luciferase reporter activity was measured in the cellular extract 16 hours later. Luciferase activity represents arbitrary units per microgram protein in each assay point. *, P < 0.01;**, P < 0.05.
To further investigate if down-regulation of HIF-1α protein correlated with an inhibition of HIF-1 transcriptional activation of target genes, we used a luciferase reporter gene assay with a construct containing the luciferase gene under the control of the HREs from the VEGF promoter (ref. 7; Fig. 3C). Transiently transfected 1A9 cells were subjected to hypoxia in the presence of different drugs for 16 hours and luciferase luminescence was used as a readout for HIF activation. As expected, hypoxia greatly induced the luciferase activity in control untreated cells, whereas treatment with all four drugs resulted in a significant reduction of HIF-1 transcriptional activity, consistent with the reduction of HIF-1α protein levels seen in Fig. 1A.
The microtubule dependence of HIF-1α regulation has been shown for 2-methoxyestradiol, but it has not been thoroughly shown for other microtubule-targeting drugs, including microtubule stabilizers. To further investigate this, we focused on epothilone B, which is a novel microtubule-stabilizing agent currently in phase II/III clinical development that, similarly to taxanes, stabilizes cellular microtubules leading to mitotic arrest and cell death (31,32). However, epothilone B has an important advantage over the taxanes in that it is not a substrate of P-glycoprotein—a drug efflux pump that can confer drug resistance to several chemotherapy drugs. To address the microtubule dependence of epothilone B–induced HIF down-regulation, we examined the drug’s ability to inhibit HIF-1α in the epothilone-resistant 1A9/A8 cells harboring a β-tubulin mutation at the drug’s binding site (20). As a result of this acquired β-tubulin mutation (Thrβ274Ile), these cells are >25-fold resistant to epothilone B compared with their parental 1A9 wild-type counterparts and exhibit impaired drug-induced tubulin polymerization. Using this pair of cell lines to assess the anti-HIF activity of epothilone B, we first treated them with various concentrations of epothilone B for 16 hours and then placed the cells in hypoxia for an additional 4 hours. Western blot analysis (Fig. 4A) revealed that epothilone B treatment of the 1A9 parental cells led to a dose-dependent down-regulation of HIF-1α starting at 10 nmol/L, whereas complete HIF inhibition was obtained at 25 nmol/L epothilone B. In contrast, epothilone B was unable to down-regulate HIF-1α in the epothilone B-resistant 1A9/A8 cells even when used at 50 nmol/L. Consistent with our results shown in Fig. 3A, Northern blot analysis (Fig. 4B) also confirmed that epothilone B did not have any effect on HIF-1α mRNA expression levels in the 1A9/A8 cells.
Figure 4.
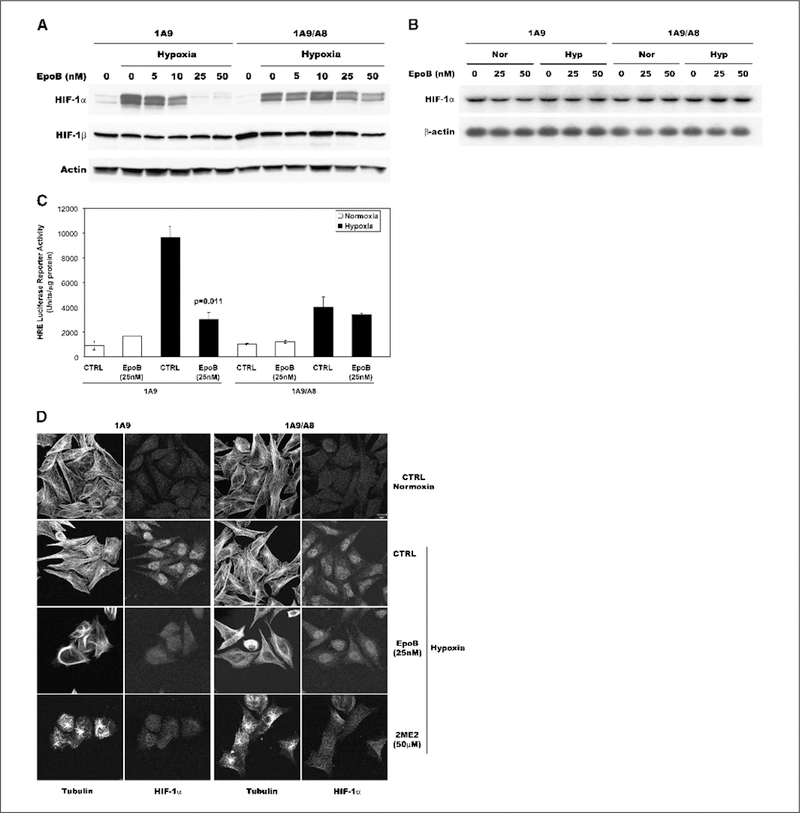
Epothilone B inhibits HIF-1α levels and activity downstream of microtubule disruption. A, parental 1A9 cells and its β-tubulin mutant epothilone-resistant subclone 1A9/A8 were treated with various concentrations of epothilone B for 16 hours and then subjected to an additional 4 hours of hypoxia as indicated. WCEs were prepared and protein (75 μg) for each sample was resolved in a 7.5% SDS-PAGE gel and blotted for HIF-1α protein. The blots were stripped and reprobed with antibodies against HIF-1β and actin. B, total RNA was prepared from 1A9 and 1A9/A8 cells treated with the indicated concentrations of epothilone B under normoxic (Nor) and hypoxic (Hyp) conditions. Northern blotting was done with 32P-labeled HIF-1α probe. β-actin RNA is shown as loading control. C, parental 1A9 and 1A9/A8 resistant cells transiently transfected with pBI-GL V6L (6xHREs of VEGF promoter) were treated with vehicle or 50 nmol/L epothilone B under normoxic and hypoxic conditions. Luciferase reporter activity was measured in the cellular extract 16 hours later. Luciferase activity represents arbitrary units per microgram protein in each assay point. D, human ovarian 1A9 and its epothilone-resistant subclone 1A9/A8 cells were treated overnight with the indicated concentrations of epothilone B and 2-methoxyestradiol and then subjected to normoxia or hypoxia for 4 hours. Cells were fixed and processed for double immunofluorescence labeling with antibodies against α-tubulin (left) and HIF-1α (right). Staining was analyzed by laser scanning confocal microscopy. Bar, 10 μm.
To further assess the effects of epothilone B on HIF transcriptional activity, we used the HRE-luciferase reporter assay as described above. These studies showed that, on hypoxic induction, epothilone B significantly inhibited HIF-driven luciferase activity in the drug-sensitive 1A9 cells in contrast to the lack of luciferase inhibition in the 1A9/A8 cells (Fig. 4C). This result was consistent with the inability of epothilone B to negatively regulate HIF-1α in the 1A9/A8 cells as shown in Fig. 4A. Laser scanning confocal microscopy in the parental and resistant cells further extended our observation and confirmed that HIF-1α inhibition requires prior microtubule disruption by epothilone B (Fig. 4D). As can be seen in the figure, both cell lines displayed similar degrees of HIF-1α induction and nuclear accumulation after hypoxic exposure, and the microtubule networks were similarly well-organized and intact. As expected, 25 nmol/L epothilone B caused microtubule hyperstabilization and bundling in the 1A9 parental cells resulting in decreased HIF-1α levels and impaired HIF-1α nuclear accumulation. However, HIF-1α staining and localization remained unchanged following epothilone B treatment of the mutant 1A9/A8 cells, in agreement with the lack of drug-induced microtubule disruption in these resistant cells. This was not the case when cells were treated with 2-methoxyestradiol, which binds to tubulin at the colchicine-binding site and retains activity in both 1A9 and 1A9/A8 cells (data not shown). 2-Methoxyestradiol caused extensive microtubule depolymerization and HIF down-regulation in both 1A9 and 1A9/A8 cells, supporting the notion that drug-tubulin interaction leading to effective microtubule disruption is required for HIF-1α inhibition.
Discussion
The data presented here indicate that all the microtubule-targeting agents tested displayed anti-HIF properties regardless of drug structure, tubulin-binding site, or microtubule-stabilizing/microtubule-destabilizing effects. In all cases, each drug down-regulated HIF-1α protein levels and activity in a dose-dependent manner with no observed effect on HIF-1α mRNA expression. Importantly, HIF down-regulation occurs downstream of microtubule disruption, as shown in Fig. 4 with the use of an epothilone-resistant β-tubulin mutant cell line. This result, together with our previous data using a taxol-resistant cell line harboring a different β-tubulin mutation (28), highlights the microtubule-dependent regulation of HIF-1α. In addition, experiments using the protea-some inhibitor MG-132 with vincristine and epothilone B (Fig. 3B) further expanded our previous findings on 2-methoxyestradiol (28), suggesting that all microtubule-disrupting agents share the ability of 2-methoxyestradiol to inhibit de novo HIF-1α protein synthesis. Given that such a wide variety of different microtubule-targeting agents all down-regulated HIF-1α in a similar manner and in multiple cell lines, this work highlights the involvement of the microtubule cytoskeleton in the physiologic regulation of the HIF pathway (Fig. 5). The detailed mechanism by which this large class of drugs exerts its anti-HIF effect is not yet clear, but it likely involves inhibition of HIF-1α translation following disruption of a functional microtubule network. One interesting hypothesis is that HIF-1α mRNA may associate with the microtubule cytoskeleton to allow for more efficient and targeted translation—a phenomenon that has been proven for other mRNA species (33). Fifteen percent to 30% of cellular mRNAs and polyribosomes are thought to associate with the cytoskeleton, and immunohistologic and biochemical approaches suggest that translation initiation and elongation factors in certain cell types follow a microtubule pattern (33). Examples of proteins that regulate translation and that associate with microtubules include eIF-2, eIF3, eIF4A, eIF-5 (34–36), the cap-binding protein eIF4E (37), eEF1a, and eEF1–2 (35). Using molecular beacons specific for HIF-1α mRNA, we observed microtubule association of HIF mRNA, which was disrupted following treatment with microtubule-depolymerizing drugs, such as 2-methoxyestradiol. In addition, linear sucrose gradient fractionation of cells treated with 2-methoxyestradiol revealed a shift of the polyribosome association profile for HIF-1α toward translation inactive state, further suggesting a microtubule-dependent control on HIF-1α translation.3 These results are preliminary in nature and we are currently investigating the molecular mechanism coupling the microtubule cytoskeleton to the regulation of HIF-1α translation.
Figure 5.
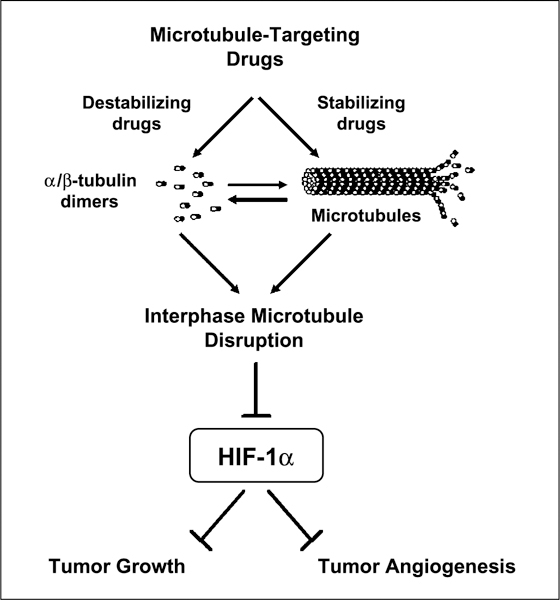
Working model for HIF-1α inhibition by microtubule-targeting drugs.
The regulation of a nuclear transcription factor by the microtubule cytoskeleton is novel and provocative given the not well-defined mechanism for such a regulation. A previous report claimed that cytoskeletal alterations by a variety of microtubule-depolymerizing agents resulted in elevated protein levels of transcriptionally active HIF-1α through a NF-κB-dependent pathway (29). However, these effects on HIF-1α and NF-κB seemed to be transient and were restored to baseline values after 5 to 7 hours of drug treatment, in agreement with our results. Our data using similar drug concentrations and time points (Fig. 2) clearly showed that treatment with vinblastine and colchicine did not increase HIF-1α protein levels nor NF-κB activity. Importantly, these observations were obtained in a variety of cancer cell lines originating from ovarian, lung, breast, brain, and prostate cancer lineages. Although the reasons for these discrepancies between our groups are not clear, one could argue that the slight differences in experimental techniques or reporter plasmids used in these studies may possibly account for the observed differences. Finally, it is important to note that the longer periods of drug treatment used primarily in our study would reflect the return to basal levels seen in the conflicting report anyway and are probably a more accurate representation of the cellular response to microtubule-targeting agents after long-term drug exposure. Furthermore, our study focused on drug treatment in cancer cells exposed to hypoxia as we reasoned that decreased oxygen tension represents the main physiologic context in which HIF-1α is up-regulated.
Our previous work has shown that HIF-1α inhibition by 2-methoxyestradiol is a result of interphase microtubule disruption and not a result of drug-induced mitotic arrest (28). Because all microtubule-disrupting agents first affect interphase microtubules ultimately resulting in mitotic arrest and cell death, it seems that HIF-1α inhibition occurs early on preceding mitotic arrest and potentially contributes to the overall antitumor activity of microtubule-targeting drugs. Most importantly, our results provide a mechanistic explanation for reports in the literature showing potent antiangiogenic effects for Taxotere and other emerging microtubule-stabilizing agents (38, 39). Taxotere in vitro is clearly an anti-HIF-1α agent, in addition to its anti-tubulin activity. It has not escaped our notice that Taxotere has been approved for the treatment of advanced osseous hormone-refractory prostate cancer (26, 27), which is known to overexpress HIF-1α as well as HIF-target genes (3, 40). As the mitotic index (cells in G2-M) of hormone-refractory prostate cancer is on average <2%, it is possible that much of the survival improvement seen with Taxotere in prostate cancer is mediated through inhibition of HIF-1 in the bone metastases rather than antimitotic effects seen in tumor systems.
The importance of HIF targeting for cancer therapy is highlighted by the emergence of novel small-molecule HIF-1α inhibitors (2, 41, 42); however, their clinical efficacy remains to be established. One of those agents is 103D5R, a novel HIF-1α inhibitor identified through a biological screen on a 10,000-membered natural product-like combinatorial library (43). However, when we tested 103D5 for potential anti-microtubule properties, no effects were observed (data not shown). On the other hand, taxanes and Vinca alkaloids are clinically established agents with proven activity in a wide variety of tumors. Our results reveal that HIF inhibition represents a novel target for these drugs downstream of interphase microtubule disruption (Fig. 5), which may contribute to their overall anticancer activity. These results further highlight the importance of a functional microtubule network for signaling and intracellular trafficking (44), revealing at the same time novel targets for microtubule-disrupting drugs. Greater understanding of this mechanism will not only provide better insight into the antitumor effects of promising new microtubule-targeting agents like epothilone B but also may result in the discovery of potentially new molecular targets for the development of cancer chemotherapeutics.
Acknowledgments
Grant support: National Cancer Institute grant 1RO1 CA100202–01, National Cancer Institute Supplement to R01 CA86335, an APRC Supplement to RO1 grant CA86335 from the division of Cancer Biology of the NCI, Entremed, Inc., Aventis, and Novartis Pharmaceuticals awards (P. Giannakakou).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Dr. Jonathan W. Simons (Winship Cancer Institute, Emory University) for critical discussions and Dr. Erwin Van Meir for kindly providing 103D5 compound.
Footnotes
Unpublished observations
References
- 1.Kaufman B, Scharf O, Arbeit J, et al. Proceedings of the Oxygen Homeostasis/Hypoxia Meeting. Cancer Res 2004;64:3350–6. [DOI] [PubMed] [Google Scholar]
- 2.Escuin D, Simons JW, Giannakakou P. Exploitation of the HIF axis for cancer therapy. Cancer Biol Ther 2004;3: 608–11. [DOI] [PubMed] [Google Scholar]
- 3.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003;3:721–32. [DOI] [PubMed] [Google Scholar]
- 4.Ivan M, Kondo K, Yang H, et al. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 2001;292:464–8. [DOI] [PubMed] [Google Scholar]
- 5.Jaakkola P, Mole DR, Tian YM, et al. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001;292:468–72. [DOI] [PubMed] [Google Scholar]
- 6.Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J 2001;20:5197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001;294:1337–40. [DOI] [PubMed] [Google Scholar]
- 8.Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001;107:43–54. [DOI] [PubMed] [Google Scholar]
- 9.Brahimi-Horn C, Berra E, Pouyssegur J. Hypoxia: the tumor’s gateway to progression along the angiogenic pathway. Trends Cell Biol 2001;11:S32–6. [DOI] [PubMed] [Google Scholar]
- 10.Maxwell PH, Ratcliffe PJ. Oxygen sensors and angiogenesis. Semin Cell Dev Biol 2002;13:29–37. [DOI] [PubMed] [Google Scholar]
- 11.Semenza G Signal transduction to hypoxia-inducible factor 1. Biochem Pharmacol 2002;64:993–8. [DOI] [PubMed] [Google Scholar]
- 12.Brizel DM, Dodge RK, Clough RW, Dewhirst MW. Oxygenation of head and neck cancer: changes during radiotherapy and impact on treatment outcome. Radio-ther Oncol 1999;53:113–7. [DOI] [PubMed] [Google Scholar]
- 13.Brizel DM, Sibley GS, Prosnitz LR, Scher RL, Dewhirst MW. Tumor hypoxia adversely affects the prognosis of carcinoma of the head and neck. Int J Radiat Oncol Biol Phys 1997;38:285–9. [DOI] [PubMed] [Google Scholar]
- 14.Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res 1998;58:1408–16. [PubMed] [Google Scholar]
- 15.Harrison LB, Chadha M, Hill RJ, Hu K, Shasha D. Impact of tumor hypoxia and anemia on radiation therapy outcomes. Oncologist 2002;7:492–508. [DOI] [PubMed] [Google Scholar]
- 16.Wouters BG, Brown JM. Cells at intermediate oxygen levels can be more important than the “hypoxic fraction” in determining tumor response to fractionated radiotherapy. Radiat Res 1997;147:541–50. [PubMed] [Google Scholar]
- 17.Wouters BG, Weppler SA, Koritzinsky M, et al. Hypoxia as a target for combined modality treatments. Eur J Cancer 2002;38:240–57. [DOI] [PubMed] [Google Scholar]
- 18.Zhong H, De Marzo AM, Laughner E, et al. Over-expression of hypoxia-inducible factor 1a in common human cancers and their metastases. Cancer Res 1999; 59:5830–5. [PubMed] [Google Scholar]
- 19.Jordan MA, Wilson L. Microtubules and actin filaments: dynamic targets for cancer chemotherapy. Curr Opin Cell Biol 1998;10:123–30. [DOI] [PubMed] [Google Scholar]
- 20.Giannakakou P, Gussio R, Nogales E, et al. A common pharmacophore for epothilone and taxanes: molecular basis for drug resistance conferred by tubulin mutations in human cancer cells. Proc Natl Acad Sci USA 2000;97: 2904–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beck W, Cass C, Houghton P. Anticancer drugs from plants: Vinca alkaloids and taxanes In: Holland J, Frei E III, Bast R, Kufe D, Morton D, Weichselbaum R, editors. Cancer medicine. 4th ed. Vol. 1 Baltimore: Williams & Wilkins; 1997. p. 1005–14. [Google Scholar]
- 22.Gidding CE, Kellie SJ, Kamps WA, de Graaf SS. Vincristine revisited. Crit Rev Oncol Hematol 1999;29: 267–87. [DOI] [PubMed] [Google Scholar]
- 23.Guastalla JP III, Dieras V. The taxanes: toxicity and quality of life considerations in advanced ovarian cancer. Br J Cancer 2003;89:S16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowak AK, Wilcken NR, Stockler MR, Hamilton A, Ghersi D. Systematic review of taxane-containing versus non-taxane-containing regimens for adjuvant and neoadjuvant treatment of early breast cancer. Lancet Oncol 2004;5:372–80. [DOI] [PubMed] [Google Scholar]
- 25.Rigas JR. Taxane-platinum combinations in advanced non-small cell lung cancer: a review. Oncologist 2004;9: 16–23. [DOI] [PubMed] [Google Scholar]
- 26.Tannock IF, de Wit R, Berry WR, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351:1502–12. [DOI] [PubMed] [Google Scholar]
- 27.Petrylak DP, Tangen CM, Hussain MH, et al. Docetaxel and estramustine compared with mitoxan-trone and prednisone for advanced refractory prostate cancer. N Engl J Med 2004;351:1513–20. [DOI] [PubMed] [Google Scholar]
- 28.Mabjeesh NJ, Escuin D, LaVallee TM, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell 2003;3: 363–75. [DOI] [PubMed] [Google Scholar]
- 29.Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. Microtubule disruption utilizes an NFκB-dependent pathway to stabilize HIF-1α protein. J Biol Chem 2003; 278:7445–52. [DOI] [PubMed] [Google Scholar]
- 30.Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF 1α. Science 2003;302:1975–8. [DOI] [PubMed] [Google Scholar]
- 31.Goodin S, Kane MP, Rubin EH. Epothilones: mechanism of action and biologic activity. J Clin Oncol 2004;22:2015–25. [DOI] [PubMed] [Google Scholar]
- 32.Mani S, McDaid H, Hamilton A, et al. Phase I clinical and pharmacokinetic study of BMS-247550, a novel derivative of epothilone B, in solid tumors. Clin Cancer Res 2004;10:1289–98. [DOI] [PubMed] [Google Scholar]
- 33.Jansen RP. mRNA localization: message on the move. Nat Rev Mol Cell Biol 2001;2:247–56. [DOI] [PubMed] [Google Scholar]
- 34.Howe JG, HersheyJW. Translational initiation factor and ribosome association with the cytoskeletal frame-work fraction from HeLa cells. Cell 1984;37:85–93. [DOI] [PubMed] [Google Scholar]
- 35.Gavrilova LP, Rutkevitch NM, Gelfand VI, et al. Immunofluorescent localization of protein synthesis components in mouse embryo fibroblasts. Cell Biol Int Rep 1987;11:745–53. [DOI] [PubMed] [Google Scholar]
- 36.Heuijerjans JH, Pieper FR, Ramaekers FC, et al. Association of mRNA and eIF-2α with the cytoskeleton in cells lacking vimentin. Exp Cell Res 1989;181:317–30. [DOI] [PubMed] [Google Scholar]
- 37.Zumbe A, Stahli C, Trachsel H. Association of a Mr 50,000 cap-binding protein with the cytoskeleton in baby hamster kidney cells. Proc Natl Acad Sci USA 1982;79:2927–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sweeney CJ, Miller KD, Sissons SE, et al. The antiangiogenic property of docetaxel is synergistic with a recombinant humanized monoclonal antibody against vascular endothelial growth factor or 2-methoxyestradiol but antagonized by endothelial growth factors. Cancer Res 2001;61:3369–72. [PubMed] [Google Scholar]
- 39.Bocci G, Nicolaou KC, Kerbel RS. Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res 2002;62:6938–43. [PubMed] [Google Scholar]
- 40.Zhong H, Agani F, Baccala AA, et al. Increased expression of hypoxia inducible factor-1α in rat and human prostate cancer. Cancer Res 1998;58:5280–4. [PubMed] [Google Scholar]
- 41.Rapisarda A, Shoemaker RH, Melillo G. Targeting topoisomerase I to inhibit hypoxia inducible factor 1. Cell Cycle 2004;3:172–5. [PubMed] [Google Scholar]
- 42.Kung AL, Zabludoff SD, France DS, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 2004;6:33–43. [DOI] [PubMed] [Google Scholar]
- 43.Tan C, de Noronha RG, Roecker AJ, et al. Identification of a novel small-molecule inhibitor of the hypoxia-inducible factor 1 pathway. Cancer Res 2005;65:605–12. [PubMed] [Google Scholar]
- 44.Gundersen GG, Cook TA. Microtubules and signal transduction. Curr Opin Cell Biol 1999;11:81–94. [DOI] [PubMed] [Google Scholar]