

Abstract
Objective:
The first live and large-scale newborn screening program of multiple mucopolysaccharidoses (MPS) was developed in Taiwan. The initial cutoff values, rates of screen positives, and genotypes were evaluated.
Study Design:
More than 100,000 dried blood spots (DBSs) were collected consecutively as part of the national Taiwan newborn screening programs. Enzyme activities were measured by tandem mass spectrometry (MS/MS) from DBS punches. Genotypes were obtained when a second newborn screening specimen again had a reduced enzyme activity. Additional clinical evaluation was then initiated based on enzyme activity and/or genotype.
Results:
Molecular genetic analysis for cases with low enzyme activity revealed 5 newborns with pathogenic IDUA mutations, 3 newborns with pathogenic IDS mutations, and one newborn was a carrier of an ARSB mutation. Several variants of unknown pathogenic significance were also identified, most likely causing pseudodeficiency.
Conclusions:
The highly robust MS/MS-based enzyme assays for MPS I, MPS II and MPS VI allow for high throughput newborn screening for these lysosomal storage disorders (LSDs). Optimized cutoff values combined with second tier testing could largely eliminate false positive results. Accordingly, newborn screening for these LSDs is possible.
Keywords: Mucopolysaccharidosis, lysosomal storage disease, newborn screening, tandem mass spectrometry, inborn errors of metabolism
Introduction
Mucopolysaccharidoses (MPSs) are a group of rare inherited metabolic disorders that result from deficiency of specific enzymes responsible for the degradation of glycosaminoglycans (GAG) present in lysosomes (1). Accumulation of GAGs causes progressive damage, which affects patient’s appearance, physical abilities, organ function and mental development (2). Seven different clinical types and numerous subtypes of the MPSs have been identified. MPS types I, II and VI are associated with deficiencies in alpha-L-iduronidase (IDUA), iduronate-2-sulfatase (IDS) and N-acetylgalactosamine-4-sulfatase (or arylsulfatase B, ARSB), respectively (3, 4). Early initiation of enzyme replacement therapy (ERT) has shown clinical benefit in patients with these MPSs. The US Food and Drug Administration approved ERT products are available for several MPS including MPS I, MPS II, MPS IVA, MPS VI, and MPS-VII diseases (5). ERT can stabilize the condition and prevent disease progression (6). Other therapeutic modalities such as hematopoietic stem cell transplantation and gene therapy have been reported to be beneficial (7). Clinical trials and recent reports have emphasized that early intervention may prevent irreversible pathology, avoid or significantly minimize disease manifestations, and improve long-term outcome (8, 9). For all of these reasons, MPSs have been incorporated into newborn screening panels. In 2016, MPS I was added to the Recommended Uniform Screening Panel for newborn screening in the USA (10). In addition to the current pilot newborn screening for lysosomal storage diseases, a retrospective epidemiological survey in Taiwan revealed that MPS II had the highest calculated birth incidence (52% of all MPS cases diagnosed), followed by MPS III (19%), MPS IV (16%), and MPS VI (7%) (11). In other Asian countries, there is also a relatively higher incidence of MPS II compared to other types of MPSs (12).
Different methods have been considered for newborn screening of MPSs including measurement of lysosomal enzymatic activity (13, 14), lysosomal enzyme abundance, and MPS biomarker quantification (15, 16). Tandem mass spectrometry (MS/MS) and fluorometry techniques for direct assay of lysosomal enzymatic activity in dried blood spots (DBS) have emerged as the most studied approaches (17). The fluorometry method using DBS for enzyme assays has been successfully developed to diagnose MPSs by using a fluorescent artificial substrate (18–20). Using the MS/MS technology, several lysosomal enzymatic activities can be determined when multiple substrates and internal standards are combined into a single buffer (21). First generation multiplexed assays were developed for six LSDs, including Pompe, Fabry, Gaucher, Niemann–Pick type A/B, Krabbe disease, and MPS-I (22). Furthermore, the second generation multiplexed assays for MPS II, MPS IIIB, MPS IV, MPS VI, MPS VII, and type 2 neuronal ceroid lipofuscinosis (NCL2) have been recently developed (23). Our group have used MS/MS multiplex technology for large scale screening for LSDs since 2010 (24, 25). Here we report the population-based screening by the MS/MS method for MPS I, MPS II, and MPS VI diseases.
Methods
Study population
For the large-scale newborn screening program in Taiwan, informed consent was obtained from subjects’ parents whose children were enrolled in the MPS study. Live screening of MPS-I, MPS-II, and MPS-VI was started on August, 2015. Until August, 2017, 130237 newborns were enrolled in the MPS-I study. For MPS-II and MPS-VI, the reporting timeframe was divided into two periods due to different cutoffs being used. From August to December, 2015, 28799 newborns were enrolled, while 101376 were enrolled from January, 2016 to August, 2017.
Enzyme activity test
Newborn DBS were collected and shipped within 3 days of birth at ambient temperature. DBS were submitted to MPS analysis on the day of arrival in the newborn screening laboratory. MS/MS assay for MPS-I (IDUA) activity was carried out by using the previously reported flow injection-MS/MS method, which was combined with Fabry, Pompe, and Gaucher screening (26). The enzyme activities of MPS-II (IDS) and MPS-VI (ARSB) in DBS were carried out by LC-MS/MS method as described (23). We used the blank (filter paper without blood), low (enzyme activity below cutoff value), medium, and high (enzyme activity above cutoff value) controls from the CDC for each run for validation (30, 31).
Establishment of reference ranges
For MPS-I, the cutoff value was adopted form our previous work, which was 3μmol/L/h (~25% of the mean activity) (20). Six clinical confirmed MPS-I patients’ enzyme activity fell under the cutoff value; five of them (0.17–0.73μmol/L/h) could be clearly distinguished, while the remaining one was a mild-form patient and had an enzyme activity around 20% of mean activity (2.7 μmol/L/h, the outlier dot in figure 1A).
figure 1.
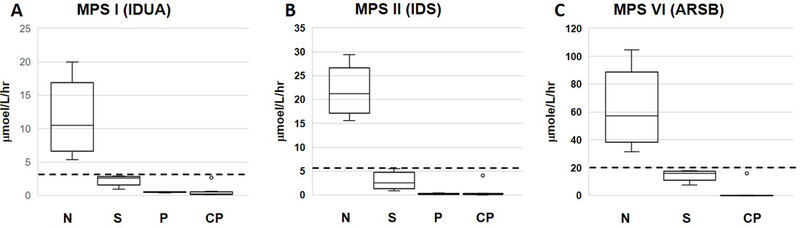
Overview of the enzyme activities from the first DBS in MPS-I, -II, and -VI screening. A, MPS-I (IDUA). B, MPS-II (IDS), C, MPS-VI (ARSB). CP, Patients found from clinics with or without ERT served as positive controls; N, normal population; P, patients found from newborn screening and confirmed with extremely low enzyme activity and/or elevated GAG disaccharide; S, range of enzymatic activities for all cases with a below cutoff value for the first DBS. Error bars are 5th and 95th percentiles. The open circles are for confirmed affected patients with or without ERT. Top and bottom of the box represent the 75th and 25th percentiles. The line through the box is the median. The dotted lines are the screening cutoff values.
For MPS-II, enzyme activities from DBS of more than 3000 anonymous newborns and 14 clinical confirmed MPS-II patients were measured. Thirteen MPS-II patients have enzymatic activities ranging from 0.02 to 0.38 μmol/L/h (<2% of mean activity; mean activity: 21.75μmol/L/h). One patient, who was under ERT treatment, had an enzyme activity around 20% of the mean activity (4.1 μmol/L/h, the outlier dot in figure 1B). Since the contribution of ERT to total enzyme activity is not known, we employed a conservative cutoff value (30% of mean activity, 6.5 μmol/L/h) for both the first and second DBS in the beginning of our study. Later, we adjusted the cutoff value from 30% to 10% of mean activity for the second DBS (Table 1).
Table1:
Cutoff values for MPS I, II, and VI and the number of screen positive and confirmed cases.
| MPS I | MPS II | MPS VI | |||
|---|---|---|---|---|---|
| Period | 2015.08–2017.08 | 2015.08–2015.12 | 2016.01–2017.08 | 2015.08–2015.12 | 2016.01–2017.08 |
| Number of newborns | 130,237 | 28,799 | 101,376 | 28,799 | 101,376 |
| cutoff for first DBS (% of mean)1 | 25 | 30 | 30 | 30 | 30 |
| cutoff for second DBS (% of mean)2 | 25 | 30 | 10 | 30 | 20 |
| Number of recalled cases (%)1 | 120 (0.09) | 56 (0.19) | 184 (0.18) | 35 (0.12) | 141 (0.14) |
| Number of referred cases (‰)2 | 4 (0.03) | 35 (1.22) | 46 (0.45) | 1 (0.03) | 1 (0.02) |
| Number of confirmed cases (‰) | 4 (0.03) | 0 | 3 (0.03) | 0 | 0 |
All the cases with enzyme activity below the cutoff value from the first DBS would be recalled for the second DBS
All the cases with enzyme activity below the cutoff value from the second DBS and/or positive results with genetic analysis were referred to the hospital for further confirmation
For MPS-VI, enzyme activities from DBS of more than 3000 anonymous newborns and 4 clinical confirmed MPS-VI patients were measured. The enzyme activities of 3 MPS VI patients (0.01–4.3 μmol/L/h) could be clearly distinguished from the normal newborns (average 61 μmol/L/h); however, one MPS VI patient receiving ERT had an enzymatic activity around 25% of mean activity (15.9 μmol/L/h, the outlier dot in figure 1C). For the same reason as in the case of MPS-II, a conservative cutoff value of 21 μmol/L/h (30% of mean activity) was then used for the first and the second DBS in the beginning of the study; and was also adjusted for the second DBS later, from 30% to 20%.
Screening algorithm
After screening, DBS with enzyme activity under the pre-set cutoff value were retested using two additional punches from the same DBS. Newborns with an average enzyme activity lower than the screening cutoff value were recalled, and the second DBS was generated. For newborns with consistent below-cutoff results, genotyping was performed. DNA was isolated from DBS using the alkaline analysis method (27). Genetic mutation assays were performed by direct exon and exon/intron boundary sequencing. Each amplicon was generated by using appropriate primers for the polymerase chain reactions (28–30). Suspicious cases with genetic variants or with consistently low enzyme activities were referred to Mackay Memorial Hospital. The percentage of referred cases is defined as the referred rate. Leukocyte enzyme activity (31, 32), urine GAG DMB examination, 2-dimensional electrophoresis (33, 34), urine GAG-disaccharide MS/MS analysis (35), physical examination (36, 37), and abdominal ultrasonography were performed.
Results
MPS-I
In the MPS-I (IDUA) live screening, 130,237 newborns joined this program, and 120 (0.09%) newborns had enzyme activities lower than the cutoff value and were recalled for a second DBS (Table 1). Most of the cases were ruled out based on the normal enzyme activities from their second DBS. This left only 5 (0.004%) newborns with decreased enzyme activity from both DBSs, DNA analysis was then carried out for these newborns. All five cases were found to have compound heterozygous mutations. They were then advised by certified genetic counselors and referred to the hospital. While the parents of case 4 refused further analysis, the other four suspected newborns (case 2 and 3 are twins, and case 1 and 5 are siblings) underwent further examinations.
In the IDUA mutation analysis, 6 variants were detected, including one intronic splicing mutation c.300–3C>G, four missense variants (c.1037T>G, c.1079T>G, c.1091C>T, c.1874A>C), and one deletion (c.1359_1384del) (Table 2). Mutations c.300–3C>G, c.1037T>G, c.1091C>T were reported as pathogenic mutations from previous databases (38, 39). Variants c.1079T>G, c.1359_1384del, and c.1874A>C have not been previously reported. Based on the very low enzyme activities (<5% of mean activity), mutation analysis, and/or the significantly high GAG-disaccharide level (Table 2), we believed all of these five cases are affected MPS-I patients; however, none of them being followed up have had any obvious clinical symptoms to date (up to two years of age). Thus, these attenuated MPS I cases will require long-term follow up.
Table 2.
Referred and/or confirmed MPS I, II, and VI cases
| DBS enzyme activity1 | WBC enzyme activity | Urine DMB GAG ratio | Urine disaccharide level2 | Clinical decision | Ref. of variants | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DS | HS | KS | ||||||||||
| Variants | Gender | Number | (umole/L/Hr) | (% of mean) | (nmol/Hr/mg protein) | (mg/mmol creatinine) | (ug/ml) | (ug/ml) | (ug/ml) | |||
| MPS I screening | ||||||||||||
| MPS I_Case 01 | c.300–3C>G | M | 1 | 0.42 | 3.50 | 0.4 | 107 | 10.9 | 2.67 | 7.75 | confirmed | Pathogenic mutation |
| c.1874A>C, p.Y625S | Novel variant | |||||||||||
| MPS I_Case 02 | c.1037T>G, p.L346R; | F | 1 | 0.59 | 4.92 | 0.2 | 204.7 | 99.60 | 13.31 | 0.83 | confirmed | Pathogenic mutation |
| c.1091C>T, p.T364M | Pathogenic mutation | |||||||||||
| MPS I_Case 03 | c.1037T>G, p.L346R; | F | 1 | 0.51 | 4.25 | 0.2 | 239.3 | 41.04 | 2.93 | 0.00 | confirmed | Pathogenic mutation |
| c.1091C>T, p.T364M | Pathogenic mutation | |||||||||||
| MPS I_Case 04 | c.1079T>G, p.F360C | M | 1 | 0.54 | 4.50 | Refused F/U | -- | -- | -- | -- | -- | Novel variant |
| c.1359_1384del, p.S453Rfs*47 | Novel variant | |||||||||||
| MPS I_Case 05 | c.300–3C>G | F | 1 | 0.56 | 4.50 | 0.1 | 65.7 | 4.76 | 3.53 | 0.17 | confirmed | Pathogenic mutation |
| c.1874A>C, p.Y625S | Novel variant | |||||||||||
| Cut-off value | >4 | >25 | >1.7 | <68.3 | <0.80 | <0.41 | <17.8 | |||||
| MPS II screening | ||||||||||||
| Non-MPS II cases 01~54 | c.103+34_56dup | 53M1F | 54 | 0.58–1.94 | 2.61–8.92 | 0.3–2.0 | 21.4–74.1 | 0–0.64 | 0–0.54 | 0–7.51 | Normal, keep F/U | likely benign polymorphism |
| c.851C>T, p.P284L | likely benign polymorphism | |||||||||||
| Non-MPS II cases 55~72 | c.1499C>T, p.T500I | M | 18 | 1.53–2.92 | 7.03–13.90 | 3.3–8.6 | 39.4–62.4 | 0–0.38 | 0–0.04 | 0–5.80 | Normal | Benign polymorphism |
| Non-MPS II cases 73~76 | c.1478G>A, p.R493H | M | 4 | 0.85–1.52 | 3.91–6.99 | 3.2–9.9 | 22.5–68.4 | 0–0.38 | 0–0.1 | 0–2.7 | Normal, keep F/U | Novel variant |
| Non-MPS II case 77 | c.890G>A, p.R297H | M | 1 | 1.14 | 5.24 | 2.3 | 69.7 | 0.08 | 0.04 | 3.47 | Normal, keep F/U | Novel variant |
| Non-MPS II case 78 | c.589C>T, p.P197S | M | 1 | 2.05 | 9.43 | 2.0 | 63.8 | 0.38 | 1.46 | 6.35 | Normal, keep F/U | Novel variant |
| MPS II_Case 01 | c.817C>T, p.R273W | M | 1 | 0.11 | 0.51 | 0.1 | 77.3 | 7.40 | 1.83 | 6.13 | confirmed | Novel variant |
| MPS II_Case 02 | c.1025A>G, p.H342R | M | 1 | 0.44 | 2.02 | 0.1 | 70.9 | 21.20 | 12.10 | 6.47 | confirmed | Novel variant |
| MPS II_Case 03 | c.311A>T, p.D104V | M | 1 | 0.20 | 0.92 | 0.1 | 114.0 | 15.60 | 103.40 | 1.40 | confirmed | Novel variant |
| Cut-off value | >6.5 | >30 | >3.2 | <68.3 | <0.8 | <0.41 | <17.8 | |||||
| MPS VI screening | ||||||||||||
| Non-MPS VI case 01 | Not found | M | 1 | 17.79 | 28.81 | 4.1 | 44.4 | 0.20 | 0.10 | 2.50 | Normal | -- |
| Non-MPS VI case 02 | c.716A>G, p.Q293R | F | 1 | 18.49 | 29.90 | 4.9 | 35.8 | 0.09 | 0.02 | 3.50 | Carrier, normal | Novel variant |
| Cut-off value | >21 | >30 | >3.5 | <68.3 | <0.80 | <0.41 | <17.8 | |||||
enzyme activities from the first DBS
DS: Dermatan sulfate; HS: Heparan sulfate; KS: Keratan sulfate
MPS-II
In the period from August to December 2015, fifty-six newborns were recalled out of 28,799 participants, and genetic sequencing was performed on 53 newborns with low enzyme activities from their recalled DBS. Among these newborns, 16 cases carry the c.301C>T variant, 18 cases carry the c.1499C>T variant, 16 cases have the linked variants of intronic variant c.103+34_56 dup and missense variant c.851C>T, and one case is with c.890G>A. The range of DBS enzyme activities from newborns with c.301 C>T was 11–25% of mean activity. Since variant c.301C>T was previously reported as a benign polymorphism (40), we did not refer these cases. Variants of c.1499C>T, c.103+34_56 dup, and c.851C>T have unknown pathogenic significance. A total of 34 patients with these variants were referred to the hospital for further examinations, including leukocyte enzyme activity, urine GAG-disaccharide analysis, and physical examination. For c.1499C>T, leukocyte enzyme activity and GAG-disaccharides from all suspicious cases were in the normal range; thus, this variant is suggested to be non-pathogenic (Table 2), and newborns harbor this variant will not be referred hereafter. For all of the cases with the linked variants (c.103+34_56dup and c.851C>T), urinary GAG-disaccharides were in the normal range, but the leukocyte enzyme activities were lower than the reference range (3.2 nmol/Hr/mg protein, Table 2). (For variant c.890G>A see below)
From January 2016 to August 2017, 184 newborns, out of 101,376, were recalled for a second DBS; and genotyping was performed on 96 of them with enzyme activities lower than 10% of mean. Among these, 50 cases were found to have the c.1499C>T variant, and 38 cases have the linked variants (c.103+34_56 dup and c.851C>T). All these 38 cases with the linked variants were referred to hospitals for follow-up. Although leukocyte enzyme activity were below the cutoff, the urinary-GAG and physical examination were normal. These results suggest that the identified variants cause pseudodeficient IDS activity (see discussion).
Six additional variants of uncertain significance (VOUS) were detected from nine cases and first reported in our study. They are c.311A>T, c.589 C>T, c.817C>T, c.890G>A, c.1025A>G, and c.1478G>A (Table 2). One case with c.890G>A and four cases with c.1478G>A had borderline leukocyte IDS enzyme activities in the range from 2.3 to 9.9 nmol/Hr/mg protein, and they all had urinary GAG-disaccharides in the normal range. One case with c.589 C>T had a decreased leukocyte enzyme activity of 2.0 nmol/Hr/mg protein, and the level of heparan sulfate was slightly elevated (1.46 ug/ml) compared to the cutoff value (<0.41 ug/ml). All six of these newborns were diagnosed as normal at the moment. However, harboring variants of unknown significance (VOUS), they will continue to be followed-up every three to six months for the next several years.
Newborns with c.311A>T, c.817C>T, or c.1025A>G had extremely low enzymatic activities of <3% of mean in both DBS and leukocytes. Urinary heparan sulfate (7.4–21.2 ug/ml) and dermatan sulfate (1.8–103.4 ug/ml) were significantly elevated in these cases compared to reference cutoff (<0.8 ug/ml for heparan sulfate and <0.41 ug/ml for dermatan sulfate). These three patients have received a diagnosis of MPS-II, but are so far asymptomatic.
MPS-VI
A total of 131,075 newborns joined this program, and 176 (0.13%) were recalled for a second DBS. From August to December 2015, one case was detected with an enzyme activity lower than 30% of mean activity from the first and the second DBSs (17.8 and 11.8 μmol/L/h). Genotype analysis revealed no variations. During the second period, another newborn was identified with low enzyme activity for both the first and second DBS (18.5 and 12.0 μmol/L/h). Genotyping revealed one novel variant c.716A>G. These two cases were referred to the hospital; leukocyte enzyme activity and urinary GAG-disaccharides were all in the normal range. Thus, no confirmed MPS-VI patient were identified in our study.
Discussion
In our initial pilot studies described here, we collected several confirmed MPS cases with or without ERT as our positive controls. Since the collection time was unavailable, the effect of residual enzyme from ERT on the DBS enzyme activity test could not be estimated. We used relatively conservative cutoffs to minimize any possibility of false negatives.
In the case of MPS-II, the vast majority of newborns with enzymatic activities below the cutoff were false positives, and thus we plan to decrease our current cutoff values to lower ones in the future. This is the typical order of events in newborn screening pilot studies. In Table 3, we summarize the recall rates and the number of the second DBSs which would have been requested as a function of the cutoff values for the first DBSs.
Table 3.
Optimum cut-off value for first DBS and the corresponding recall rate (%)
| MPS I screening number : 130,237 | ||
| Cut off (% of mean activity) | recall number | recall rate (‰) |
| 25 | 120 | 0.92 |
| 15 | 10 | 0.08 |
| 10 | 7 | 0.05 |
| MPS II screening number : 130,175 | ||
| Cut off (% of mean activity) | recall number | recall rate (‰) |
| 30 | 240 | 1.84 |
| 20 | 203 | 1.56 |
| 10 | 102 | 0.78 |
| 10 + hot spot variants sequence | 9 | 0.07 |
| MPS VI screening number : 130,175 | ||
| Cut off (% of mean activity) | recall number | recall rate (‰) |
| 30 | 176 | 1.35 |
| 20 | 36 | 0.28 |
| 10 | 3 | 0.02 |
For MPS-I (IDUA), the referred rate was found to be 5 out of 130,237 (3.8 per 100,000 live birth), including a pair of twins and a pair of siblings. All 5 newborns have mutations and/or urinary GAG levels consistent with the strong possibility to develop MPS-I disease; however, the patients are asymptomatic so far. Newborn screening labs in the USA are reporting much higher screen positive rates for MPS-I. This markedly higher screening positive rate is in part due to the prevalence of a relatively high pseudodeficiency in the US population and false positive findings (decreased enzyme activity in DBS but normal leukocyte enzyme activity). Illinois NBS laboratory later modified their method and lowered their cutoff value. In our study, the screening cutoff value could be optimized from 25% of mean to 10%. The recall rate would then be lowered from 0.92‰ to 0.05‰ (Table 3).
For MPS-II (IDS) screening we started our study with a very conservative cutoff value of 30% of mean activity. Later, we adjusted the cutoff to 10% of mean activity for the second DBS as all confirmed MPS-II patients had activity below this value except the one who was under ERT. With an optimum cutoff of 10% of mean activity for the first and second DBS, the recall rate was at most 102 per 100,000 (Table 3), and the positive rate (number need to be referred) was 49 per 100,000. However, it is noticeable that even with the optimum cutoff value, this is still a relatively large number compared to the results recently obtained in the MPS-II pilot NBS study in the Washington NBS laboratory, showing a screening positive rate of 7.5 per 100,000 (41). As full exon sequencing of the IDS gene was carried out on all below-cutoff DBS, we realized the relatively large rate of the screening positives for MPS-II was mainly due to common variants of uncertain significance in the Taiwanese population.
Variants c.301 C>T, c.1499C>T, and the linked variants were widely found in IDS mutation analysis in our population. The range of DBS enzyme activity of the c.301 C>T variant is from 11.1% to 25.3% of mean. According to a previous report, in vitro functional assay showed that the c.301 C>T containing construct expressed high activity (97 ± 9% of the wild-type activity). Thus, the c.301 C>T variant is likely to be a rare polymorphism (40). The range of DBS enzyme activities of the c.1499C>T variants is from 7.0% to 13.4% of mean. The allele count of the c.1499C>T from the East Asia population is 8/6638 (0.12%) (data obtained from the ExAC browser). All cases carrying c.301 C>T or c.1499C>T variant have normal leukocyte enzyme activities, urine GAGs and urine disaccharide levels. Hence, these two variants are now defined as non-pathogenic variants.
Fifty-four cases expressing both low DBS and leukocyte IDS enzyme activities were found with the linked variants. The linked variants include one intronic variant c.103+34_56dup and one missense variant c.851C>T. All of these 54 cases also carry one silent variant c.684A>G and one polymorphism variant c.1180+184T>C. The minor allele frequencies of c.103+34_56dup, c.684A>G, c.851C>T and c.1180+184T>C from the East Asia population are 5/1008 (0.5%), 10/6638 (0.15%), 10/6637 (0.12%), and 207/1008 (28%), respectively (from the ExAC browser and dbSNP). Since variants c.103+34_56 dup and c.1180+184T>C are located in introns, and c.684A>G is a silent variant, we believe that the low enzyme activity results from the variant c.851C>T.
Variant c.851C>T has also been reported in Japan (44). The structural modeling of IDS with p.P284L (c.851C>T variant) suggests a minor structural change occurring on the molecular surface without a predicted structural modification in the active site of IDS. In the Japanese study it was suggested that the mutated enzyme is deficient, low but finite residual activity of this enzyme may be sufficient to prevent GAG buildup in vivo. The latter suggestion is supported by the fact that all of the newborns with these linked variants had normal physical examination results, normal urinary-GAGs and disaccharide levels after more than two years of following-up. Furthermore, pedigree studies from the newborns and their relatives were performed. One of the probands’ great-grandfather (in his nineties) with the linked variants was found; and he had normal physical examination results, normal urinary-GAGs and disaccharide levels. Together, the high incidence (54 per 130,174 or one per 2,400) and family study imply that the linked variants are non-pathogenic. However, further comprehensive family studies are needed to fill the gap of knowledge.
For MPS-VI (ARSB) screening, we also adopted a very conservative cutoff of 30% of mean activity based on the level of activity seen in patients with confirmed MPS-VI disease. All of the patients had < 10% mean activity, except the patient already on ERT. In the future, we plan to lower the cutoff to 10% of mean activity as we did for MPS-II, and this would give a recall rate of 2 per 100,000 (Table 3). The data is comparable with the screening positive rate of 6 per 100,000 from the pilot study for MPS-VI in the Washington state NBS laboratory (41).
In addition to specific enzyme analyses, biomarker assay has also been regarded as one of the methods for newborn screening and diagnosis (17, 43). GAG and GAG-disaccharides are putative markers for MPS diseases progression and can be measured in DBS and urine samples (44, 45). The finding of low MPS enzyme activity combined with significantly elevated disaccharides suggests early onset type of MPS diseases and helps evaluation for treatment. Assays using MS/MS have also been recently established to measure GAG-disaccharides in DBS of patients with MPS and mucolipidoses (46). Disaccharide determination as a second tier test performed on samples with low enzyme activity could be accomplished in a newborn screening laboratory using existing equipment (47). Presumably, when enzymatic activity is low in the first-tier MS/MS test in DBS, an elevated GAG-disaccharides result as a second-tier test in DBS would mark an affected patient rather than a false positive. The feasibility of measuring DBS disaccharides as a first-tier newborn screening method is still controversial due to the higher false positive rate (0.03–0.9%) found in a recent pilot study for MPS-I, -II, and -III by measuring GAG-disaccharides by LC-MS/MS (16).
The studies reported here show that newborn screening for MPS-I, -II, and -VI in Taiwan by MS/MS is feasible. The rate of false positives is very low for MPS-I and MPS-VI, but much higher for MPS-II. In Asian population, the high frequency of psuedodeficient-like alleles in MPS-II (IDS) gene gives rise to a high false positive rate, which highlights a need of second tier test in the screening system. In this study, if GAG-disaccharides was used as the second tier test, false positive rate would remarkably decrease, as those with the linked variants would not be referred. However, cases with variants of uncertain significance (c.1478G>A. c. 890G>A, and c. 589C>T, Table 2) would therefore not be picked. In the contrary, if genotype was in use, all the false positive case would be identified and not be referred, yet cases with VOUS would be kept. The optimum cutoff value coupled with specific genotype analysis for pseudodeficiency-like variant can be used to identify real MPS cases and rule out the false positive cases due to the low-enzyme-activity-causing variants.
Abbreviations:
- MPS
Mucopolysaccharidosis
- GAG
glycosaminoglycans
- DBS
dried blood spot
- IDUA
alpha-L-iduronidase
- IDS
iduronate-2-sulfatase
- ARSB
arylsulfatase B
- MS/MS
tandem mass spectrometry
References
- 1.Dorfman A, Matalon R. The mucopolysaccharidoses (a review). Proc Natl Acad Sci U S A. 1976;73(2):630–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byers S, Rozaklis T, Brumfield LK, Ranieri E, Hopwood JJ. Glycosaminoglycan accumulation and excretion in the mucopolysaccharidoses: characterization and basis of a diagnostic test for MPS. Mol Genet Metab. 1998;65(4):282–90. [DOI] [PubMed] [Google Scholar]
- 3.Parini R, Deodato F, Di Rocco M, Lanino E, Locatelli F, Messina C, et al. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet journal of rare diseases. 2017;12(1):112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Al-Sannaa NA, Al-Abdulwahed HY, Al-Majed SI, Bouholaigah IH. The clinical and genetic Spectrum of Maroteaux-Lamy syndrome (Mucopolysaccharidosis VI) in the Eastern Province of Saudi Arabia. J Community Genet. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Whiteman DA, Kimura A. Development of idursulfase therapy for mucopolysaccharidosis type II (Hunter syndrome): the past, the present and the future. Drug Des Devel Ther. 2017;11:2467–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muenzer J, Jones SA, Tylki-Szymanska A, Harmatz P, Mendelsohn NJ, Guffon N, et al. Ten years of the Hunter Outcome Survey (HOS): insights, achievements, and lessons learned from a global patient registry. Orphanet journal of rare diseases. 2017;12(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Penati R, Fumagalli F, Calbi V, Bernardo ME, Aiuti A. Gene therapy for lysosomal storage disorders: recent advances for metachromatic leukodystrophy and mucopolysaccaridosis I. Journal of inherited metabolic disease. 2017;40(4):543–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jameson E, Jones S, Wraith JE. Enzyme replacement therapy with laronidase (Aldurazyme) for treating mucopolysaccharidosis type I. Cochrane Database Syst Rev. 2013(9):CD009354. [DOI] [PubMed] [Google Scholar]
- 9.Muenzer J, Giugliani R, Scarpa M, Tylki-Szymanska A, Jego V, Beck M. Clinical outcomes in idursulfase-treated patients with mucopolysaccharidosis type II: 3-year data from the hunter outcome survey (HOS). Orphanet journal of rare diseases. 2017;12(1):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisengart JB, Jarnes J, Ahmed A, Nestrasil I, Ziegler R, Delaney K, et al. Long-term cognitive and somatic outcomes of enzyme replacement therapy in untransplanted Hurler syndrome. Mol Genet Metab Rep. 2017;13:64–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin HY, Lin SP, Chuang CK, Niu DM, Chen MR, Tsai FJ, et al. Incidence of the mucopolysaccharidoses in Taiwan, 1984–2004. American journal of medical genetics Part A. 2009;149A(5):960–4. [DOI] [PubMed] [Google Scholar]
- 12.Lin HY, Chuang CK, Huang YH, Tu RY, Lin FJ, Lin SJ, et al. Causes of death and clinical characteristics of 34 patients with Mucopolysaccharidosis II in Taiwan from 1995–2012. Orphanet J Rare Dis. 2016;11(1):85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolfe BJ, Blanchard S, Sadilek M, Scott CR, Turecek F, Gelb MH. Tandem mass spectrometry for the direct assay of lysosomal enzymes in dried blood spots: application to screening newborns for mucopolysaccharidosis II (Hunter Syndrome). Analytical chemistry. 2011;83(3):1152–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanchard S, Sadilek M, Scott CR, Turecek F, Gelb MH. Tandem mass spectrometry for the direct assay of lysosomal enzymes in dried blood spots: application to screening newborns for mucopolysaccharidosis I. Clinical chemistry. 2008;54(12):2067–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Ruijter J, de Ru MH, Wagemans T, Ijlst L, Lund AM, Orchard PJ, et al. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Molecular genetics and metabolism. 2012;107(4):705–10. [DOI] [PubMed] [Google Scholar]
- 16.Kubaski F, Mason RW, Nakatomi A, Shintaku H, Xie L, van Vlies NN, et al. Newborn screening for mucopolysaccharidoses: a pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. Journal of inherited metabolic disease. 2017;40(1):151–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gelb MH, Scott CR, Turecek F. Newborn screening for lysosomal storage diseases. Clinical chemistry. 2015;61(2):335–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chamoles NA, Blanco M, Gaggioli D. Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clinica chimica acta; international journal of clinical chemistry. 2001;308(1–2):195–6. [DOI] [PubMed] [Google Scholar]
- 19.Chamoles NA, Blanco M, Gaggioli D, Casentini C. Gaucher and Niemann-Pick diseases--enzymatic diagnosis in dried blood spots on filter paper: retrospective diagnoses in newborn-screening cards. Clinica chimica acta; international journal of clinical chemistry. 2002;317(1–2):191–7. [DOI] [PubMed] [Google Scholar]
- 20.Chamoles NA, Niizawa G, Blanco M, Gaggioli D, Casentini C. Glycogen storage disease type II: enzymatic screening in dried blood spots on filter paper. Clinica chimica acta; international journal of clinical chemistry. 2004;347(1–2):97–102. [DOI] [PubMed] [Google Scholar]
- 21.Spacil Z, Tatipaka H, Barcenas M, Scott CR, Turecek F, Gelb MH. High-throughput assay of 9 lysosomal enzymes for newborn screening. Clinical chemistry. 2013;59(3):502–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Y, Scott CR, Chamoles NA, Ghavami A, Pinto BM, Turecek F, et al. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clinical chemistry. 2004;50(10):1785–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Yi F, Kumar AB, Kumar Chennamaneni N, Hong X, Scott CR, et al. Multiplex Tandem Mass Spectrometry Enzymatic Activity Assay for Newborn Screening of the Mucopolysaccharidoses and Type 2 Neuronal Ceroid Lipofuscinosis. Clin Chem. 2017;63(6):1118–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao HC, Chiang CC, Niu DM, Wang CH, Kao SM, Tsai FJ, et al. Detecting multiple lysosomal storage diseases by tandem mass spectrometry--a national newborn screening program in Taiwan. Clinica chimica acta; international journal of clinical chemistry. 2014;431:80–6. [DOI] [PubMed] [Google Scholar]
- 25.Chuang CK, Lin HY, Wang TJ, Huang YH, Chan MJ, Liao HC, et al. Status of newborn screening and follow up investigations for Mucopolysaccharidoses I and II in Taiwan. Orphanet journal of rare diseases. 2018;13(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang XK, Elbin CS, Chuang WL, Cooper SK, Marashio CA, Beauregard C, et al. Multiplex enzyme assay screening of dried blood spots for lysosomal storage disorders by using tandem mass spectrometry. Clinical chemistry. 2008;54(10):1725–8. [DOI] [PubMed] [Google Scholar]
- 27.Saavedra-Matiz CA, Isabelle JT, Biski CK, Duva SJ, Sweeney ML, Parker AL, et al. Cost-effective and scalable DNA extraction method from dried blood spots. Clinical chemistry. 2013;59(7):1045–51. [DOI] [PubMed] [Google Scholar]
- 28.Chou YY, Chao SC, Kuo PL, Lin SJ. A 38.8 kb deletion mutation of the iduronate-2-sulfatase gene in a patient with Hunter syndrome. J Formos Med Assoc. 2005;104(4):273–5. [PubMed] [Google Scholar]
- 29.Lam CW, Chan AO, Lai CK, Chan WH, Chan YW, Shek CC, et al. A novel mutation, Y255X, of the ARSB gene in a Chinese family with mucopolysaccharidosis type VI. Chin Med J (Engl). 2004;117(12):1850–2. [PubMed] [Google Scholar]
- 30.Venturi N, Rovelli A, Parini R, Menni F, Brambillasca F, Bertagnolio F, et al. Molecular analysis of 30 mucopolysaccharidosis type I patients: evaluation of the mutational spectrum in Italian population and identification of 13 novel mutations. Human mutation. 2002;20(3):231. [DOI] [PubMed] [Google Scholar]
- 31.Hopwood JJ, Muller V, Smithson A, Baggett N. A fluorometric assay using 4-methylumbelliferyl alpha-L-iduronide for the estimation of alpha-L-iduronidase activity and the detection of Hurler and Scheie syndromes. Clin Chim Acta. 1979;92(2):257–65. [DOI] [PubMed] [Google Scholar]
- 32.Voznyi YV, Keulemans JL, van Diggelen OP. A fluorimetric enzyme assay for the diagnosis of MPS II (Hunter disease). J Inherit Metab Dis. 2001;24(6):675–80. [DOI] [PubMed] [Google Scholar]
- 33.Chuang CK, Lin SP, Chung SF. Diagnostic screening for mucopolysaccharidoses by the dimethylmethylene blue method and two dimensional electrophoresis. Zhonghua Yi Xue Za Zhi (Taipei). 2001;64(1):15–22. [PubMed] [Google Scholar]
- 34.Chih-Kuang C, Shuan-Pei L, Shyue-Jye L, Tuen-Jen W. MPS screening methods, the Berry spot and acid turbidity tests, cause a high incidence of false-negative results in sanfilippo and morquio syndromes. J Clin Lab Anal. 2002;16(5):253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chuang CK, Lin HY, Wang TJ, Tsai CC, Liu HL, Lin SP. A modified liquid chromatography/tandem mass spectrometry method for predominant disaccharide units of urinary glycosaminoglycans in patients with mucopolysaccharidoses. Orphanet journal of rare diseases. 2014;9:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin SM, Lin HY, Chuang CK, Lin SP, Chen MR. Cardiovascular abnormalities in Taiwanese patients with mucopolysaccharidosis. Molecular genetics and metabolism. 2014;111(4):493–8. [DOI] [PubMed] [Google Scholar]
- 37.Lin HY, Shih SC, Chuang CK, Chen MR, Niu DM, Lin SP. Assessment of bone mineral density by dual energy x-ray absorptiometry in patients with mucopolysaccharidoses. Orphanet journal of rare diseases. 2013;8:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Teng YN, Wang TR, Hwu WL, Lin SP, Lee-Chen GJ. Identification and characterization of −3c-g acceptor splice site mutation in human alpha-L-iduronidase associated with mucopolysaccharidosis type IH/S. Clinical genetics. 2000;57(2):131–6. [DOI] [PubMed] [Google Scholar]
- 39.Lee-Chen GJ, Wang TR. Mucopolysaccharidosis type I: identification of novel mutations that cause Hurler/Scheie syndrome in Chinese families. Journal of medical genetics. 1997;34(11):939–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keeratichamroen S, Cairns JR, Wattanasirichaigoon D, Wasant P, Ngiwsara L, Suwannarat P, et al. Molecular analysis of the iduronate-2-sulfatase gene in Thai patients with Hunter syndrome. J Inherit Metab Dis. 2008;31 Suppl 2:S303–11. [DOI] [PubMed] [Google Scholar]
- 41.Scott CR. A high performance assay for the detection of MPS disorders, MLD, and CTX, from newborn blood spots. 14TH Annual WORLD Symposium 2018 Program; February; San Diego2018. [Google Scholar]
- 42.Tomo Sawada AT, Ken Suzuki, Hitoshi Sakuraba, Seiji Saitou, Tomoko Sakaguchi, Teruo Kitagawa. Pseudodeficiency Alleles of Iduronate 2-sulfatase Gene and the Structural Modeling of the Enzyme Protein. Journal of The Korean Society of Inherited Metabolic Disease 2012. p. 82. [Google Scholar]
- 43.Tomatsu S, Kubaski F, Sawamoto K, Mason RW, Yasuda E, Shimada T, et al. Newborn screening and diagnosis of mucopolysaccharidoses: application of tandem mass spectrometry. Nihon Masu Sukuriningu Gakkai Shi. 2014;24:19–37. [PMC free article] [PubMed] [Google Scholar]
- 44.Auray-Blais C, Lavoie P, Tomatsu S, Valayannopoulos V, Mitchell JJ, Raiman J, et al. UPLC-MS/MS detection of disaccharides derived from glycosaminoglycans as biomarkers of mucopolysaccharidoses. Anal Chim Acta. 2016;936:139–48. [DOI] [PubMed] [Google Scholar]
- 45.Minter Baerg MM, Stoway SD, Hart J, Mott L, Peck DS, Nett SL, et al. Precision newborn screening for lysosomal disorders. Genetics in medicine : official journal of the American College of Medical Genetics. 2017. [DOI] [PubMed] [Google Scholar]
- 46.Kubaski F, Suzuki Y, Orii K, Giugliani R, Church HJ, Mason RW, et al. Glycosaminoglycan levels in dried blood spots of patients with mucopolysaccharidoses and mucolipidoses. Molecular genetics and metabolism. 2017;120(3):247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matern D, Tortorelli S, Oglesbee D, Gavrilov D, Rinaldo P. Reduction of the false-positive rate in newborn screening by implementation of MS/MS-based second-tier tests: the Mayo Clinic experience (2004–2007). Journal of inherited metabolic disease. 2007;30(4):585–92. [DOI] [PubMed] [Google Scholar]