

Case presentation
A 54-year-old woman presented with complaints of myalgias, fatigue, and progressive leg weakness of several weeks duration, followed by acute loss of vision in her left eye. Her medical history was notable for long-standing systemic lupus erythematosus (SLE) that was actively treated with prednisone, mycophenolate mofetil, and quinacrine.
Initially, it was thought she was exhibiting evidence of an exacerbation in her SLE and was treated with IV methylprednisolone in conjunction with plasma exchange. Despite a week of continuous treatment, she developed acute vision loss in her right eye, hearing changes, and difficulty swallowing. Given the progression of symptoms, she was transferred to the Hospital of the University of Pennsylvania for further assessment.
On admission, her neurologic examination demonstrated normal mental status. The cranial nerve examination was most notable for absent light perception bilaterally, nonreactive pupils, severe diffuse ophthalmoparesis, loss of facial sensation in the left V1 and V2 trigeminal distribution, and diminished hearing to finger rubbing on the right and right palate deviation. Motor deficits included mild bilateral upper extremity weakness and paraplegia. Deep tendon reflexes were pathologically exaggerated in the upper extremities, but absent in the lower extremities. Vibratory sensation was decreased throughout all 4 limbs, whereas pinprick sensation was reduced in the lower limbs in association with a sensory level at T10.
Blood studies were notable for lymphopenia (absolute lymphocyte count 200), positive double-stranded DNA antibody, and mildly reduced C3 and C4 complement levels. MRI of the brain (figure 1, A–D) demonstrated multifocal nonenhancing parenchymal lesions and patchy, noncontinuous enhancements of multiple cranial nerves (optic chiasm, left III, bilateral V, and right VIII), in conjunction with leptomeningeal enhancement. MRI of the spine (figure 1, E–H) was notable for T2 signal prolongation, spanning the level from T2 to the conus medullaris. The enhancement pattern was restricted to the caudal aspect of the spinal cord.
Figure 1. MRIs.

(A and B) Axial FLAIR images of the brain demonstrate multifocal parenchymal lesions including the right hippocampus, right midbrain, left temporal-occipital and left anterior temporal areas, and the left thalamus (arrow heads). There is also evidence of intraventricular debris with a fluid level in the bilateral occipital horns (C, arrow heads). Although there were some confluent changes to the parenchyma in a periventricular distribution, other lesions appeared more leptomeningeal (C, arrow head pointing toward a linear hyperintensity, which on closer inspection is localized to the leptomeningeal zone of a left parietal gyrus lesion). (D) T1 post-gadolinium images demonstrated enhancement of the optic chiasm, several other cranial nerves, and the tentorium (arrow heads). (E and F) Sagittal and axial T2-weighted images of the thoracic spine reveal a longitudinally extensive, centrally predominant hyperintensity (arrow heads, seen in NMO), although enhancement was not observed. We further illustrate the caudal extent of the lesion, which clearly involves the conus and the exiting nerve roots of the cauda equine, both of which demonstrate enhancement on T1 post-gadolinium images (G and H, respectively; arrow heads). FLAIR = fluid attenuation inversion recovery.
CSF showed xanthochromia, 44 white blood cell count/mm3 (61% lymphocytes, 34% neutrophils, and 5% monocytes), elevated total protein (115 mg/dL), and 6 CSF-restricted oligoclonal bands.
Ophthalmologic consultation revealed bilateral disseminated retinal nerve fiber layer lesions, most consistent with cotton wool spots, in conjunction with multifocal retinal hemorrhages. Taken together, the evidence was most consistent with severe, bilateral, outer retinal necrosis (figure 2).1
Figure 2. Fundus of a patient with progressive outer retinal necrosis from VZV infection.
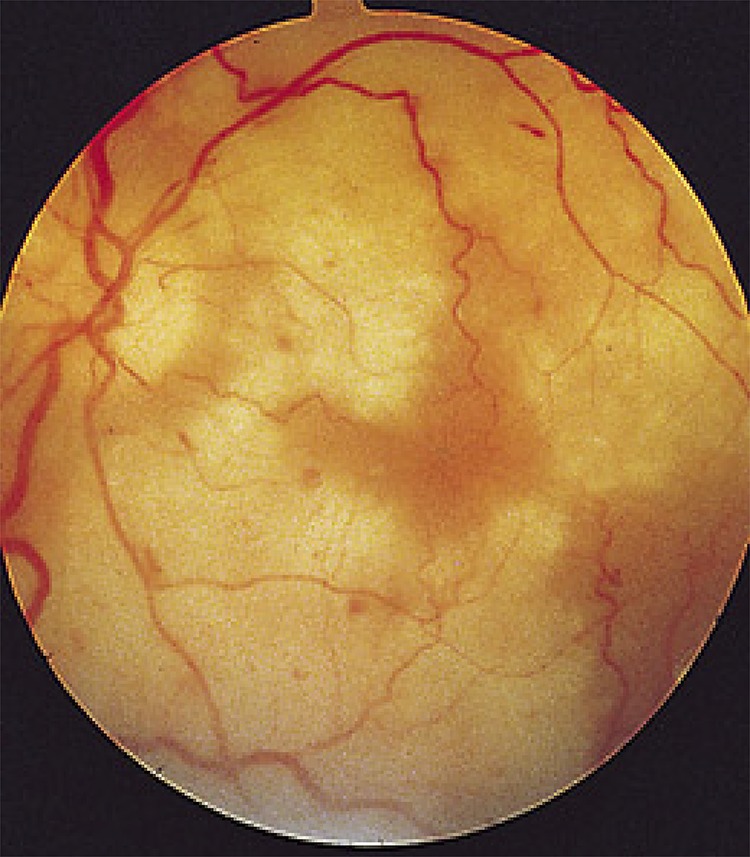
The virus replicates rapidly and spreads through the outer layer of the retina, producing necrotic (white) coalescing lesions and intraretinal hemorrhages. The vitreous typically does not reflect signs of intraocular inflammation, likely due to an immunosuppressed state. Relative preservation of the retinal vasculature also helps to differentiate progressive outer retinal necrosis from retinal artery occlusions. VZV = varicella zoster virus.
Differential diagnosis
This patient presented with a disseminated and pathologic cataclysmic syndrome that included subacute paraplegia (the derivative of a centrally predominant and longitudinally extensive, edematous, transverse myelitis [LETM] with multiple areas of patchy focal enhancement on MRI), bilateral cranial neuropathies, and bilateral optic nerve involvement with severe retinal necrosis.
The combination of these findings listed in the preceding paragraph is suggestive of a disease process capable of rapidly moving across the rostral-caudal extent of the neuraxis; with a meningitic infectious process being the most likely etiology (table). Notwithstanding this likelihood, in a patient such as ours, SLE is often posited as a chief diagnostic consideration when characteristics of a diffuse vasculitis are present. Nevertheless, an important and highly salient “pitfall principle” is that a bona-fide lupus-related vasculitis is truly exceptional.2 Alternately, SLE can certainly account for a diffuse vasculopathy, with semiologic features typically including headaches, seizures, psychiatric manifestations, encephalopathy, and stroke like symptoms; all of which were lacking in our patient.3 Furthermore, spinal cord and root involvement in SLE is extraordinarily rare.4,5
Table.
Differential diagnosis for basilar meningitis
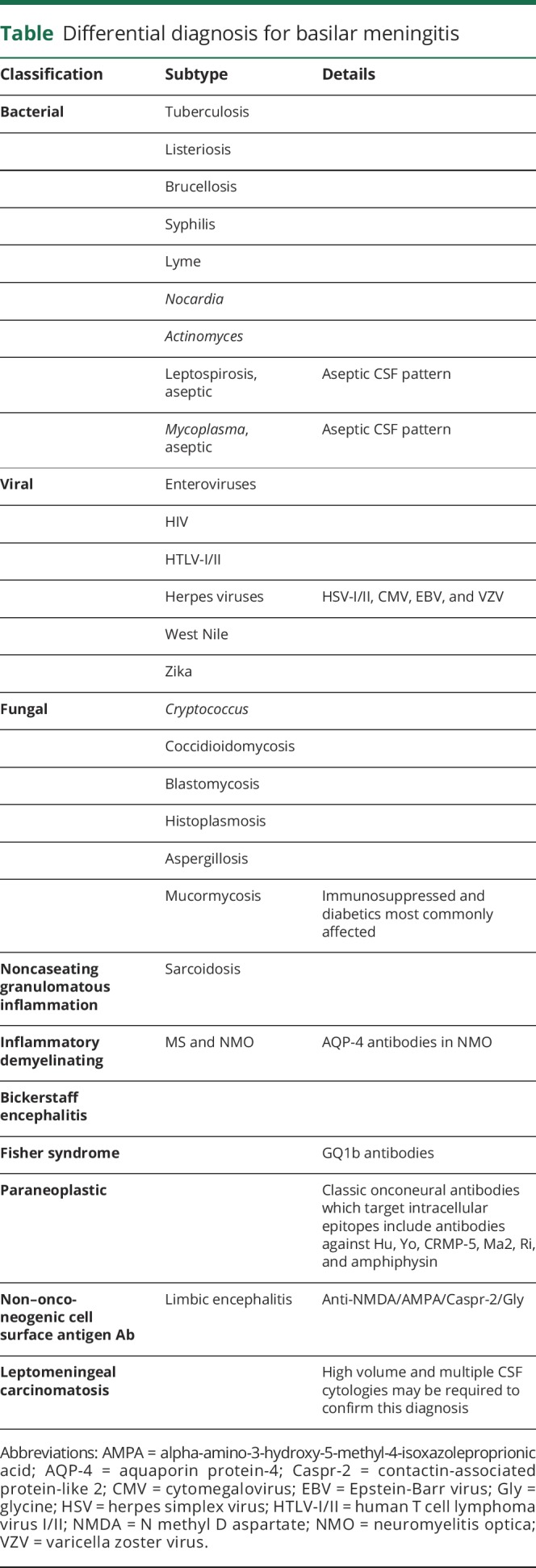
As our patient already had 1 autoimmune disorder, disorders of immune dysregulation such as neuromyelitis optica (NMO) spectrum disorder, Sjogren syndrome, neurosarcoidosis, and Susac syndrome were considered in the differential (see below).6,7
NMO can be a legitimate overlap disorder and can account for a constellation of neurologic deficits, such as optic neuropathy, retinal vascular disturbances, multifocal brainstem processes, and the LETM, which was documented in our patient. However, the extra-axial patchy enhancement of multiple cranial nerves and the multiplicity of them would be highly exceptional disease characteristics, thereby making NMO a highly untenable diagnostic consideration.
Sjogren syndrome can produce myelitis, even the longitudinal extensive type, which is characteristic of NMO. Alternately, the diffuse inflammatory constellation of targeted structures across the rostral-caudal extent would be highly unusual, and our patient had none of the features of keratoconjunctival sicca that is typical for Sjogren syndrome.
Neurosarcoidosis must be a prime consideration in a patient with evidence of CSF pleocytosis and a clinical course, which localizes to, even if only in part, a basilar meningitis.
Susac syndrome could affect both retina and hearing; however, the retinal pathology is distinctive in producing branch retinal artery occlusions with “cloud-like” abnormalities, often in conjunction with intravascular Gass plaques, whereas the pathology identified in our patient was instead most consistent with a severe retinal necrotic process, most likely secondary to an infectious etiology.7,8 Furthermore, the presence of a concomitant and longitudinally extensive myelitis and the absence of callosal lesions make this diagnosis unlikely.7
Although the diffuse nature of the disease process raises consideration of malignant disorders, the rapid evolution of symptoms could be considered atypical.9 The rapid and broadly disseminated involvement of the rostral to caudal extent of the neuroaxis should arouse suspicion for an infectious disorder, which includes a basilar meningitis, particularly given the multifocal and bilateral involvement of cranial nerves.
Pathoetiologic infectious agents that can cause a basilar meningitic process with a propensity for spread via CSF and leptomeningeal surfaces include tuberculosis, listeriosis, leptospirosis, mycoplasma, enteroviruses, syphilis, HIV, human T cell lymphoma virus I/II, members of the herpes family of viruses, and a number of unusual viral agents such as West Nile and Zika.10
Given our patient's known immune suppression, infiltration of the subarachnoid space by a viral infection could produce the rapid, diffuse, and widespread involvement of multiple areas of the CNS. Several specific microbial infectious agents including Mycoplasma, enterovirus, and herpes viruses such as cytomegalovirus (CMV), varicella zoster virus (VZV), and herpes simplex virus can all cause cranial neuropathies and myelitis.11,12
Narrowing the differential further, progressive outer retinal necrosis is most commonly seen as a reactivation of a latent herpetic infection in immunocompromised patients.8 These findings made investigation for a basilar meningitis a priority.
Final diagnosis
VZV infection resulting in oculo-meningo-encephalo-myelo-radiculitis was confirmed by positive VZV PCR testing in the CSF. Treatment was initiated urgently with 14 days of IV acyclovir.
After 3 months, the patient regained the ability to swallow, recovered full strength in both upper extremities, and had partial motor improvement of both lower extremities. Brain and spinal cord imaging abnormalities nearly resolved, but she remains blind bilaterally. The presence of fulminant outer zonal retinal necrosis, accompanied by disseminated cotton wool spots and retinal nerve fiber hemorrhages at presentation, reflects the massive degree of destruction targeting the retinal ganglion cell neurons and the axonal fiber pathways of the optic nerve.
The pathology affecting the eyes in this patient could not seriously be characterized as an optic neuropathy, but rather was a fulminant and rapidly evolving infectious process eventually leading to vascular occlusions, ischemia, hemorrhage, and ultimately necrosis. Such cases require prompt recognition and treatment with antimicrobial therapy, potentially in conjunction with steroids, to mitigate the often associated inflammatory burden that can contribute to tissue damage in conjunction with the infectious process itself.8,13 Furthermore, severe consequences can follow from the acute changes in pressure dynamics within an anatomically restricted compartment such as the eye.
In the context of severe edema, optic nerve sheath decompression needs to be considered urgently with the objective of preventing or mitigating a compartment syndrome, where the rapid intensification of pressure can lead to both vascular stasis and thereby attenuation or even abolishment of retinal perfusion and interrupted axonal transport in the retinal ganglion cell fibers that become the optic nerve. By analogy, fulminant spinal cord edema can result in extensive vascular deficits across the spinal cord when edema intensifies and exceeds the space it resides in, or it interrupts the circumferentially organized vaso corona, a collateral vascular supply to the spinal cord, which can be compressed and thereby fail to provide rescue perfusion in the setting of severe swelling, especially when there is restricted spinal canal compliance.
Discussion
Estimating infection risk
VZV is a ubiquitous, neurotropic virus. Although primary infection has declined since the introduction of the live-attenuated vaccine (Zostavax), in 1995, the majority of adults (and the patient in this case) acquired the infection in childhood.14,15 Following the primary infection, VZV becomes latent in the dorsal roots, cranial nerves, and autonomic ganglia throughout the neuroaxis. The virus is primarily contained by T-cell–mediated immunity for much of adulthood.15,16
Certain risk factors are associated with virus reactivation. Advancing age (>50 years) is associated with immune senescence, which either primarily or predominantly impacts memory T cells, and results in diminished immune protection.17 While B-cell–derived antibodies to primary infection or vaccination may exist as surrogates for immunity, these do not necessarily convey protection as T-cell function declines.17 In fact, it is estimated that 50% of individuals will experience VZV reactivation by age 85 years. As a means to boost immunity in the setting of immune senescence, the CDC recommends recombinant shingles vaccine (Shingrix) for all immunocompetent patients aged 50 years and older.15 Our patient is older than 50 years and had not yet received the shingles vaccine. Moreover, immunosuppressive conditions from disease states such as HIV or from medications (evidenced by lymphopenia in this case) also increase the risk of VZV reactivation.
Manifestations of VZV reactivation
The most common result of reactivation of VZV is herpes zoster, a dermatomal vesicular eruption that is often painful. A trigeminal nerve ganglionitis (the majority affecting V1) or dorsal root ganglionitis in the cervical or thoracic regions is most common. Importantly, a rash is not always a heralding sign for viral reactivation, and there was no evidence of rash during or preceding this patient's hospitalization.14–16,18
VZV reactivation can result in a number of cranial neuropathies, producing optic neuritis, ophthalmoplegias, facial palsy, and pharyngeal and hypoglossal dysfunction. Within the peripheral nervous system, VZV reactivation can affect anterior and posterior spinal roots resulting in a radiculitis and paresis.12–16,18
Notwithstanding these observations, the majority of patients experience a meningoencephalitis, whereas VZV myelitis is a rare complication.15 Most cases of VZV myelitis occur as a postinfectious, self-limited paraparesis. However, in the setting of immunosuppression, VZV can invade the spinal cord, causing progressive myelitis and longitudinally extensive cord lesions, as observed in our patient.18 If widespread parenchymal lesions are present in the brain, an association with VZV vasculopathy must also be considered, particularly given the destructive nature of infection-related vasculitis.
Following reactivation in the trigeminal and other cranial nerve ganglia, the virus spreads transaxonally via afferent fibers to the adventitia of intracranial blood vessels (and across the spectrum from arterioles to large arteries) potentially resulting in (or causing in lieu of resulting) stroke.19
Ocular disease can be devastating in the immunosuppressed patient, with VZV representing the most common cause of severe progressive outer retinal necrosis, whereby rapid replication of the virus leads to focal areas of necrosis and intraretinal hemorrhage (figure 2).1 The retinal vasculature and vitreous are often normal. In contrast to the more common CMV retinitis, VZV-related lesions accumulate and coalesce rapidly leading to blindness; making early recognition imperative, as vision loss is often extensive and irreversible.8
Detection and treatment of VZV infection
Any concern for VZV affecting the CNS should prompt CSF analysis, the current gold standard over more invasive brain biopsy. In the first 7 days of symptoms, the best test for diagnosis is VZV PCR. After several days, the sensitivity of the PCR starts to decrease, so the measurement of intrathecal synthesis of VZV antibody (both immunoglobulin G and immunoglobulin M) in the CSF is recommended after 7 days.20
Early treatment is paramount. If clinical suspicion is high (known immunosuppression and evidence on examination of CNS involvement), empiric treatment is warranted. The treatment of choice is IV acyclovir. The duration of treatment is 7 days in immunocompetent patients and 10–14 days in immunocompromised patients.13,20 As in this case, there can be significant recovery with treatment.
Conclusion
This case underscores the importance of considering a broad differential diagnosis in patients with autoimmune conditions that present with acute, multifocal and rapidly disseminating neurologic manifestations, particularly when traversing the rostral to caudal extent of the entire neuraxis. Such patients are truly at potential risk of cataclysmic infectious complications as a consequence of prolonged immunosuppressive therapy. The rapid evolution of this patient's symptoms and several rare manifestations of VZV reactivation in the absence of rash highlight the importance of early consideration, detection, and urgent treatment intervention.
Acknowledgment
The National Multiple Sclerosis Society Case Conference Proceedings are both a live educational Webinar, in conjunction with a peer review publishing initiative from the monthly “Diagnostic and Treatment Challenges in MS and Neuroimmunology Webinars,” sponsored by the National Multiple Sclerosis Society Fellowship Training Program and Directed by Elliot M. Frohman, MD, PhD, and Teresa C. Frohman, MPAS, MSCS, PA-C, from the Dell Medical School at the University of Texas at Austin.
Author contributions
C. Perrone: concept, manuscript drafting, clinical and imaging review, literature review, and critical revision of the manuscript. R. Lisak: conception and critical revision of the manuscript for intellectual content. E. I. Meltzer, P. Sguigna, E. Tizazu, D. Jacobs, E. Melamed, A. Lucas, L. Freeman, G. Pardo, A. Goodman, E. J. Fox, K. Costello, and M. S. Parsons: critical revision of the manuscript for intellectual content. S. Zamvil and E. M. Frohman: conception and critical revision of the manuscript for intellectual content. T. C. Frohman: critical revision of the manuscript for intellectual content.
Study funding
Diagnostic and Treatment Challenges in MS and Neuroimmunology Webinars is sponsored by the National Multiple Sclerosis Fellowship Training Program.
Disclosure
C. Perrone received a clinical fellowship funded in part by the NMSS Institutional Clinical Training Award. R.P. Lisak has been funded for research support by the NIH, National Multiple Sclerosis Society (USA), Mallinckrodt Pharmaceuticals, Genentech, Teva Pharmaceuticals, Novartis, Medimmune, Ra Pharmaceuticals, Alexion, Argenx, and Chugai; served as a consultant to Novartis, Argenx, Syntimmune, Alexion, GLG Consulting, Alpha Sites, Insights Consulting, Informa Pharma Consulting, and Slingshot Consulting; and served on the speaker's bureau of Teva Pharmaceuticals (nonbranded talks only). E. I. Meltzer received speaker fees from Sanofi Genzyme. P. Sguigna and E. Tizazu report no disclosures. D.A. Jacobs received consulting fees from Biogen and Sanofi Genzyme and lecture fees from EMD Serono. E. Melamed served as consultant for EMD Serono. A. Lucas reports no disclosures. L. Freeman served as consultant for Celgene and received honorarium from McGraw-Hill Education, speaker fees from the National Multiple Sclerosis Society, research funding from Race to Erase MS and PCORI, and program sponsorship from Biogen. G. Pardo serves as a member of the National Medical Advisory Council of the National Multiple Sclerosis Society and has served as a consultant and/or been in the speaker bureau of Biogen, Celgene, Genentech, Sanofi Genzyme, EMD Serono, Novartis, and Teva Pharmaceuticals. A.D. Goodman received consulting fees during the past 2 years from Acorda, Adamas, AbbVie, EMD Serono, and Teva; his employer has received research support for clinical trials from the following sponsors: Atara, Biogen, Roche, Sanofi Genzyme, Novartis, Sun Pharma, and Teva. E. J. Fox received compensation for research, consulting, speakers' bureau, and/or advisory work from Acorda, Bayer, Biogen, Celgene, Chugai, EMD Serono, Genentech/Roche, Mallinckrodt, MedDay, Novartis, Sanofi Genzyme, Teva, and TG Therapeutics. K. Costello and M. parson report no disclosures. S.S. Zamvil is Deputy Editor of Neurology: Neuroimmunology & Neuroinflammation; is a member of the advisory board for the International Society of Neuroimmunology; has served as a consultant and received honoraria from Biogen Idec, EMD Serono, Genzyme, Novartis, Roche/Genentech, and Teva Pharmaceuticals, Inc.; has served or serves on Data Safety Monitoring Boards for Lilly, BioMS, Teva, and Opexa Therapeutics; and receives research grant support from the NIH, the NMSS, The Maisin Foundation, Biogen, and Celgene. E. M. Frohman received speaker honoraria from Sanofi Genzyme, Novartis, Alexion, and Acorda. T. C. Frohman received consulting fees from Genzyme and Acorda. Go to Neurology.org/NN for full disclosures.
References
- 1.Bryan RG, Myers FL. Progressive outer retinal necrosis in a patient with rheumatoid arthritis. Arch Ophthalmol 1998;116:1249. [PubMed] [Google Scholar]
- 2.Clancy R, Marder G, Martin V, Belmont H, Abramson SB, Buon J. Circulating activated endothelial cells in systemic lupus erythematous: further evidence for diffuse vasculopathy. Arthritis Rheum 2001;44:1203–1208. [DOI] [PubMed] [Google Scholar]
- 3.van Dam AP. Diagnosis and pathogenesis of CNS lupus. Rheumatol Int 1991;11:1–11. [DOI] [PubMed] [Google Scholar]
- 4.Johnson RT, Richardson EP. The neurological manifestations of systemic lupus erythematosus. Medicine (Baltimore) 1968;47:337–369. [DOI] [PubMed] [Google Scholar]
- 5.The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum 1999;42:599–608. [DOI] [PubMed] [Google Scholar]
- 6.Asgari N, Jarius S, Laustrup H, et al. Aquaporin-4-autoimmunity in patients with systemic lupus erythematosus: a predominantly population-based study. Mult Scler 2018;24:331–339. [DOI] [PubMed] [Google Scholar]
- 7.Kleffner I, Dörr J, Ringelstein M, et al. Diagnostic criteria for Susac syndrome. J Neurol Neurosurg Psychiatry 2016;87:1287–1295. [DOI] [PubMed] [Google Scholar]
- 8.Holland GN. The progressive outer retinal necrosis syndrome. Int Ophthalmol 1994;18:163–165. [DOI] [PubMed] [Google Scholar]
- 9.Balbuena-Merle RI, Sante-Prez M, Perez-Berenguer J, Velez-Rosario R, Correa-Rivas M, Jimenez A. Leptomeningeal carcinomatosis as the initial manifestation of metastatic disease diagnosed in post-mortem examination: a case series. PR Health Sci J 2019;38:64–67. [PubMed] [Google Scholar]
- 10.Smith JE, Aksamit AJ Jr. Outcome of chronic idiopathic meningitis. Mayo Clin Proc 1994;69:548–556. [DOI] [PubMed] [Google Scholar]
- 11.Jacob A, Weinshenker BG. An approach to the diagnosis of acute transverse myelitis. Semin Neurol 2008;28:105–120. [DOI] [PubMed] [Google Scholar]
- 12.Brizzi KT, Lyons JL. Peripheral nervous system manifestations of infectious disease. Neurohospitalist 2014;4:230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilden D. Varicella zoster virus and central nervous system syndromes. Herpes 2004;11(suppl 2):89A–94A. [PubMed] [Google Scholar]
- 14.Pergam SA, Limaye AP. Varicella zoster virus (VZV). Am J Transpl 2009;9:S108–S115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen JI. Clinical practice: herpes zoster. N Engl J Med 2013;369:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilden D, Nagel MA, Cohrs RJ, Mahalingam R. The variegate neurological manifestations of varicella zoster virus infection. Curr Neurol Neurosci Rep 2013;13:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wenjuan T, Rao S. Mechanisms underlying T cell immunosenescence: aging and cytomegalovirus infection. Front Microbiol 2016;7:2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skripuletz T, Pars K, Schulte A, et al. Varicella zoster virus infections in neurological patients: a clinical study. BMC Infect Dis 2018;18:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nagel MA, Gilden D. Update on varicella zoster vasculopathy. Curr Infect Dis Rep 2014;16:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grahn A, Studahl M. Varicella-zoster virus infections of the central nervous system: prognosis, diagnostics and treatment. J Infect 2015;71:281–293. [DOI] [PubMed] [Google Scholar]