

Abstract
Objective
To investigate the long-term safety and efficacy of weekly subcutaneous IgPro20 (Hizentra, CSL Behring) in chronic inflammatory demyelinating polyneuropathy (CIDP).
Methods
In a 48-week open-label prospective extension study to the PATH study, patients were initially started on 0.2 g/kg or on 0.4 g/kg weekly and—if clinically stable—switched to 0.2 g/kg weekly after 24 weeks. Upon CIDP relapse on the 0.2 g/kg dose, 0.4 g/kg was (re)initiated. CIDP relapse was defined as a deterioration by at least 1 point in the total adjusted Inflammatory Neuropathy Cause and Treatment score.
Results
Eighty-two patients were enrolled. Sixty-two patients initially received 0.4 g/kg, 20 patients 0.2 g/kg weekly. Seventy-two received both doses during the study. Sixty-six patients (81%) completed the 48-week study duration. Overall relapse rates were 10% in 0.4 g/kg–treated patients and 48% in 0.2 g/kg–treated patients. After dose reduction from 0.4 to 0.2 g/kg, 51% (27/53) of patients relapsed, of whom 92% (24 of 26) improved after reinitiation of the 0.4 g/kg dose. Two-thirds of patients (19/28) who completed the PATH study without relapse remained relapse-free on the 0.2 g/kg dose after dose reduction in the extension study. Sixty-two patients had adverse events (AEs) (76%), of which most were mild or moderate with no related serious AEs.
Conclusions
Subcutaneous treatment with IgPro20 provided long-term benefit at both 0.4 and 0.2 g/kg weekly doses with lower relapse rates on the higher dose. Long-term dosing should be individualized to find the most appropriate dose in a given patient.
Classification of evidence
This study provides Class IV evidence that for patients with CIDP, long-term treatment with SCIG beyond 24 weeks is safe and efficacious.
Recently, subcutaneous immunoglobin (SCIG) IgPro20 (Hizentra, CSL Behring, Marburg, Germany/King of Prussia, PA) was shown to be efficacious and well tolerated as maintenance treatment for chronic inflammatory demyelinating polyneuropathy (CIDP) in a randomized, placebo-controlled trial: the PATH study.1 This study showed that 2 doses of IgPro20 were efficacious in preventing disease relapse over 24 weeks with a good safety profile in patients who were previously shown to be dependent on intravenous immunoglobulin (IVIG) treatment. Systemic side effects were reduced using SCIG compared with IVIG, which might be explained by differences in pharmacokinetics.2–6 SCIG increases patient autonomy and quality of life and may lead to cost savings (including cost for premedication and complication management, as well as time of various health care professionals) depending on local country-specific rules and pricing.7–10 Increased patient satisfaction has also been suggested in patients with CIDP treated with SCIG.11
However, the long-term effect of weekly SCIG in CIDP beyond 24 weeks has not been studied adequately with appropriate disability outcome measures.12 We conducted a multicenter, open-label extension study to the PATH study, designed to provide long-term safety and efficacy data of the 2 doses of IgPro20 (0.2 and 0.4 g/kg weekly) in patients with CIDP and to confirm that SCIG is an alternative long-term maintenance treatment option for patients with CIDP who were previously treated with IVIG.
Methods
Design
This open-label prospective extension study was conducted by the PATH study group. The primary study objective was to determine the long-term safety of SCIG IgPro20 in patients with CIDP, whereas the secondary study objective was to determine the long-term efficacy. The study was designed to provide Class IV evidence that for patients with CIDP, long-term treatment with SCIG beyond 24 weeks is safe and efficacious and provides an alternative to IVIG.
The study design in the original extension study protocol and the study design after a protocol amendment differed in the requirements for patient entry from the PATH study and in the SCIG dosing regimens (see below). The change in protocol arose from the interest to observe maintenance of patients who were started on the 0.2 g/kg dose.
Standard protocol approvals, registrations, and patient consents
All participants gave written informed consent before any study-specific procedures were performed. The study protocol was approved by the ethics committees of all participating centers. The study was overseen by a steering committee, which provided overall supervision and scientific support for the study, and a safety review committee (SRC), which periodically reviewed and evaluated safety data to provide recommendations regarding the patient safety. The SRC had the competence to continue as planned, to stop, to temporarily suspend, or to amend the study. The PATH study is registered with Clinicaltrials.gov, number NCT01545076.
Participants
Patients who had completed the PATH study or who had successfully been rescued from a relapse during the PATH study were eligible for participation in the extension study. Patients eligible for the PATH study were aged at least 18 years and had been diagnosed with definite or probable CIDP according to the European Federation of Neurological Societies/Peripheral Nerve Society criteria13 and had responded to IVIG. Inclusion criteria of the original extension protocol: written informed consent was obtained; randomized and received SCIG or placebo in the PATH study; and gap between studies was not longer than 8 weeks (exceptions applied if the extension study was not yet open for enrollment at the site). Inclusion criteria after protocol amendment: the patient had completed the PATH study (SC week 25) or was successfully rescued from a relapse during SCIG or placebo treatment.
Exclusion criteria were similar to those used in the original PATH study.14 The following additional exclusion criteria were applied after the protocol amendment: gap between studies of more than 1 day; new medical condition and/or social behavior (i.e., alcohol, drug, or medication abuse) during the PATH study that in the judgment of the investigator could increase risk to the patient, interfere with outcome evaluation and/or the conduct of the study.
Enrollment into the extension study started in 2014, more than 2 years after the start of the PATH study. As a consequence, most patients had a time gap between completion of PATH and entry into the extension study during which they were treated with IVIG.
Treatment and follow-up
In the PATH study, patients were treated with SCIG (0.2 or 0.4 g/kg weekly) or with placebo. The PATH study was blinded until after all extension study patients were enrolled. In the original extension study protocol, 0.4 g/kg was used as the initial weekly dose. Patients who did not relapse for 24 weeks were then switched to (open-label) 0.2 g/kg weekly (low dose) for another 24 weeks. After a protocol amendment, the low dose was chosen as the initial dose. Patients remained on low dose for 48 weeks unless relapse occurred. Patients who relapsed on low-dose SCIG were given the option to switch to high-dose SCIG. Patients who relapsed on high-dose SCIG were given the option to remain on high dose or be discontinued depending on the patient and investigator's judgment. Patients who remained on high dose had to successfully recover from relapse within 4 weeks (±2 days) and remain on this dose until week 48 or were otherwise discontinued from the study.
Patients were previously trained on the SC technique during the PATH study. All patients enrolled in this study received weekly SCIG. SCIG volume was administered in 1 infusion session for the low dose, and in 2 approximately equal-volume infusion sessions for the high dose, for 1 or 2 consecutive days. The total volume infused could be subdivided over multiple SC infusion sites. Moreover, 2 infusion pumps, if needed, could be used in parallel. The maximum infusion rate was 35 mL/h, as tolerated. For patients enrolled under the original extension study protocol, the maximum infusion volume was 40 mL/per site, but for patients enrolled after amendment 1, the maximum volume was 50 mL/per site, as tolerated.
It was recommended to change the infusion site(s) with each administration. Appropriate infusion sites were, e.g., the abdomen, thighs, and lateral hip. The number of infusion sites depended on the total volume administered. If needed, additional infusion sites were used consecutively. The goal was to increase the patient's comfort while decreasing pain and infusion time. A patient who did not tolerate a specific volume per infusion site could reduce the volume per site as needed. Any change of volume or rate per infusion site was approved by the investigator.
The following concomitant CIDP treatments were not permitted during the study: other nonstudy IgGs, rituximab, alemtuzumab, plasma exchange, interferon, tumor necrosis factor alpha inhibitors, fingolimod, cyclophosphamide, and any other systemic immunosuppressive medications, except those medications permitted during the PATH study. Concomitant CIDP treatments other than those listed above were permitted, provided that their dose and frequency were kept stable during the whole study: e.g., methotrexate, azathioprine, mycophenolate, or corticosteroids (maintenance dose ≤20 mg/d).
Treatment compliance was monitored. Patients were required to fill a “drug accountability form,” which was checked by the local investigator and sponsor. Patients also had to return empty and not fully used vials.
Definition of relapse, successful recovery, and outcome measures
CIDP relapse was defined as a deterioration (i.e., increase) by at least 1 point in the total adjusted Inflammatory Neuropathy Cause and Treatment (INCAT) score (range 0 [healthy] to 10 [unable to make any purposeful movements with arms or legs])15 compared with baseline. Successful recovery after a relapse was defined as the return of the adjusted INCAT score back to (or better than) the baseline score. Baseline scores were defined as the scores assessed at the enrollment visit; for patients enrolled after protocol amendment, baseline assessments were performed at the same time as the completion visit of the PATH study. Other efficacy measures were mean grip strength assessed for both hands separately using the hand-held Martin Vigorimeter,16 Medical Research Council (MRC) sum score (range 0–80; including shoulder abduction, elbow flexion, wrist extension, index finger abduction, hip flexion, knee extension, foot dorsiflexion, and great toe dorsiflexion),17 and Inflammatory Neuropathy-Rasch-Built Overall Disability Scale (I-RODS) (range 0 [most severe activity and social participation limitations] to 100 [no activity and social participation limitations]).18 INCAT scores, grip strength, MRC sum score, and I-RODS were assessed at baseline; week 2, 9, 25, 33, and at the completion visit (week 49); at any unscheduled visit; and at a 4-week follow-up visit after a relapse. To assess the safety and tolerability of SCIG, adverse events (AEs) per infusion and the number and percentage of patients with AEs were determined. We assessed the patient's preference with a questionnaire.14 Furthermore, we measured serum IgG trough concentrations (before administration of the study drug).
Safety analysis
As most patients received both doses during the study, AEs were allocated to either the high or low dose, depending on the dose when the AE started.
Statistical analysis
This study included patients from the preceding PATH study. Descriptive statistics were used to analyze the results. The Statistical Analysis System (SAS) software package (SAS Institute, Cary, NY) version 9.2 or higher was used for statistical analyses.
Data availability
CSL will only consider requests to share individual patient data that are received from systematic review groups or bona-fide researchers (see table e-1, links.lww.com/NXI/A124 for more information).
Results
A total of 82 patients of 172 PATH study patients were enrolled in the extension study between 2014 and 2016 from 33 sites. Baseline characteristics of these patients are shown in table 1, and the patient's disposition by treatment and relapse status in the PATH trial is presented in table 2. Sixty-three patients (77%) were enrolled under the original extension study protocol (i.e., with 0.4 g/kg planned as the initial weekly high dose), and 19 (23%) were enrolled after approval of a protocol amendment (i.e., with weekly low dose [0.2 g/kg] planned as the initial dose) (figure). Two patients who were planned to start with the high dose actually were started on the low dose, and 1 patient planned to start with the low dose was started on the high dose. Thus, 62 patients started on the high dose and 20 on the low dose. Fifty-two patients who started on the high dose were switched to the low dose after 24 weeks of treatment, which was a planned procedure according to the protocol. In addition, patients who relapsed on the low dose were switched to the high dose (again). As a consequence, a total of 73 patients received low-dose treatment, and 72 patients received high-dose treatment at some time during the study. Ten patients on low dose (of which, 8 completed the study) and 9 patients on high dose (of which, 1 completed the study) never had any change in dose during the study.
Table 1.
Baseline characteristics (N = 82)
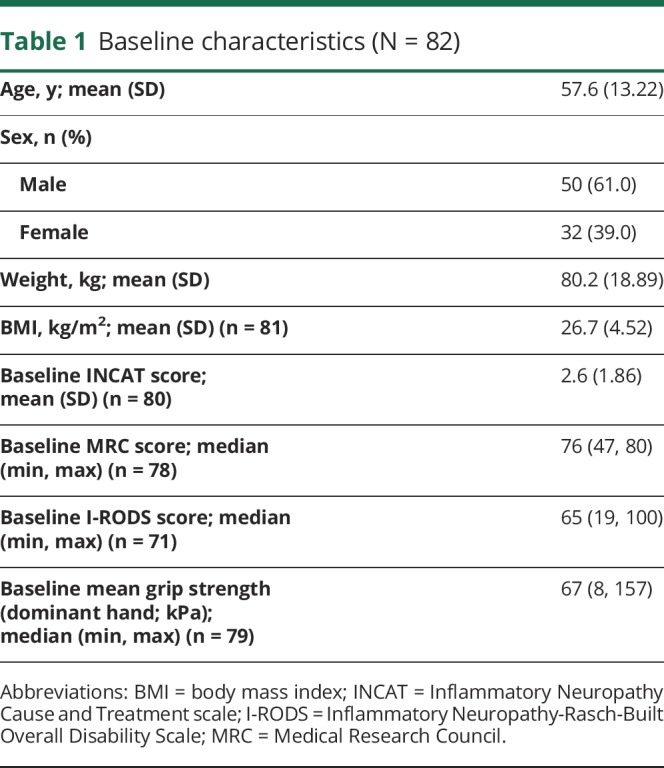
Table 2.
Subject disposition by treatment and relapse status in the PATH study
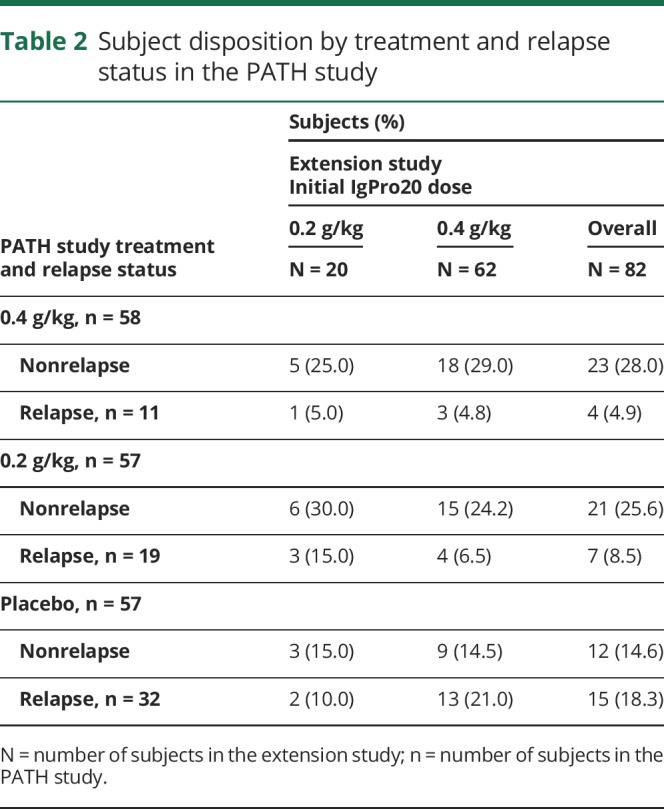
Figure. Study profile.
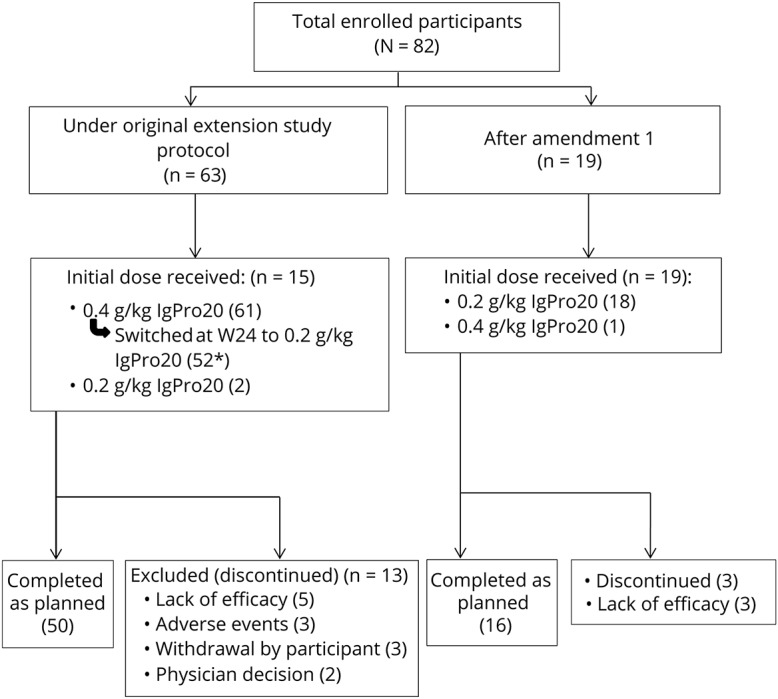
*Including one subject who relapsed twice on HD but switched to LD at W24 and discontinued due to lack of efficacy.
A total of 66 patients (80%) completed the study, and 16 patients (20%) discontinued because of lack of efficacy (relapse according to the investigator; n = 8), physician decision (n = 2), AEs (n = 3), or withdrawal by patient (n = 3).
Relapse rates of patients treated with high dose (0.4 g/kg weekly)
Overall, relapse on high dose occurred in 7 of 72 patients (10%) (tables 3 and 4). Six of these relapses occurred in patients who were started on high dose and 1 in a patient who was switched from low dose to high dose. Three of the 7 relapses occurring on the high dose were self-limiting, meaning that they improved spontaneously without further intervention.
Table 3.
Relapse rates and time to first relapse by treatment and status at end of the PATH study

Table 4.
Subject preference for treatment responses (last postdose observation)

Extension study patients who previously relapsed in PATH had a relapse rate on high dose of 8% (2 of 24), and those who did not relapse in PATH had a relapse rate of 10% (5 of 48). Similarly, relapse rates on high dose in the extension study were 8% (2 of 24) and 9% (2 of 23) for patients treated with high or low dose in PATH irrespective if they were relapsers or nonrelapsers. Only 1 of the 15 patients on high dose in the extension study (7%) had a relapse who previously relapsed on placebo in PATH. The lowest relapse rate (1 of 21, 5%) was observed in patients who started high-dose treatment in PATH and continued high-dose treatment in the extension study.
Of the 62 patients who started on the high dose, 52 were switched to the low dose at W24, as was foreseen by the original protocol (including 1 patient started on high dose erroneously, when amendment was active and 1 patient switching incorrectly after experiencing relapses on high dose). Of those who relapsed under low dose (see below), 92% (24 of 26) recovered after reinitiating high dose. Thirty-three patients completed the study on high-dose treatment.
Relapse rates of patients treated with low dose (0.2 g/kg weekly)
Overall, relapses occurred in 35 of 73 patients (48%) treated with low dose (tables 3 and 4). Nine relapses occurred in 20 patients (45%) who started treatment on the low dose, and 26 relapses occurred in 52 patients (50%) who had a planned dose decrease from high to low dose.
Extension study patients who previously relapsed in PATH had a relapse rate on low dose of 67% (16 of 24), and those who did not relapse in PATH had a relapse rate of 39% (19 of 49). Similarly, relapse rates on low dose in the extension were 46% (11 of 24) and 38% (9 of 24) for patients treated with high or low dose in PATH irrespective if they were relapsers or nonrelapsers. In addition, 67% (10 of 15) of patients relapsed in this study who also had a relapse on placebo in PATH. However, patients who did not relapse on placebo in the PATH study had a 50% chance to relapse on low dose (5 of 10) and 20% on high dose (2 of 10).
Of the 62 patients who started on high dose, 52 were switched to low dose after 24 weeks of whom 26 relapsed (50%). From the 28 patients who were switched from high to low dose and completed the PATH study without relapse on both IgPro20 doses, 9 (32%) experienced a relapse. Thirty-three patients completed the study on low-dose treatment.
MRC, I-RODS, and grip strength
Patients presented with similar baseline median scores for MRC, I-RODS, and grip strength (table e-2, links.lww.com/NXI/A124). At relapse, all clinical scores showed significant deterioration. MRC sum scores deteriorated by 3 points in low-dose patients and 8 points in high-dose patients. I-RODS scores deteriorated by 8 and 13 centile points and grip strength deteriorated by 6 and 12 kPa.
Safety
Most patients were treated with both high- and low-dose SCIG. Seventy-three patients received the low dose (0.2 g/kg), and 72 patients received the high dose (0.4 g/kg) weekly during the study. Sixty-two patients (76%) had 180 AEs (table 5 and table e-3, links.lww.com/NXI/A124). More patients had AEs during high-dose treatment than during low-dose treatment. However, the rate of AEs per infusion in the high-dose group was one half that of the low-dose group.
Table 5.
All AEs reported in ≥2% of subjects

There was an overall low frequency of systemic side effects; headaches were not reported by any patient during low-dose treatment and in 4 patients (5.6%) under the 0.4 g/kg dose. Nausea occurred in 2 patients (2.8%). No case of hypertension was reported. A total of 18 patients (22%) had 40 local reactions. The majority of AEs were either mild (62%) or moderate (29%). Twelve severe AEs were reported in 8 patients (10%). None of the severe AEs were causally related, except 3 local reactions experienced by 1 patient. All severe AEs resolved, except for a preexisting vitamin D deficiency in 1 patient in the low-dose group, and 1 AE of infusion site swelling and 1 AE of infusion site erythema, occurring in the same patient in the high-dose group. Seven patients (9%) had 8 serious AEs (SAEs); none of these events were causally related to IgPro20. All patients recovered from these SAEs. Two patients discontinued the study because of AEs while on low dose and 1 patient while on high dose.
IgG serum levels
Serum trough IgG levels were similar at baseline for low- and high-dose-treated patients (median 21.1 and 21.2 g/L). After 24 weeks of treatment, trough levels declined in low-dose patients but were stable in high-dose patients (median change −1.0 and −0.4 g/L, respectively.). This was more pronounced when patients had a relapse where trough levels had decreased by −5.3 g/L in low-dose patients and −0.2 g/L in high-dose patients.
Patient preference
At study exit, overall 82.4% of patients preferred their current SC treatment (table 4). The most frequent reason was that treatment was believed to offer more independence (71.6%), followed by “spend less time with current therapy” (40.5%), “preferred frequency of administration” (37.8%), and “seem to feel fewer side effects” (31.1%). In comparison, overall 12.2% of patients preferred IV treatment, and 5.4% had no preference (table 4).
Discussion
This study was designed to investigate the long-term safety and efficacy of IgPro20 in the maintenance treatment of CIDP. Overall, the safety findings in the extension study were consistent with those from the PATH study and the already known safety profile for IgPro20. There was a low frequency of systemic side effects such as headache, nausea, or hypertension. In IVIG marketing approval studies, headaches are frequently reported in approximately 30% of patients, hypertension in 9%–14%, and nausea in 6%–11%.15,19 One in 5 patients had a local reaction (e.g., infusion site swelling or erythema) with a lower rate in the 0.2 g/kg dosed patients.
No new safety findings were identified. Most AEs were mild or moderate, and no SAEs were assessed as related to therapy.
Patients who were treated with the 0.4 g/kg dose showed a low relapse rate (overall approximately 10%) compared with patients treated with the 0.2 g/kg dose (44%). When considering the treatment assignment in the preceding PATH study, it appeared that those patients who were treated with the high dose (in PATH) and continued high-dose treatment in the extension study had a very low relapse rate in the extension study (5%). In addition, some patients relapsing on the high dose recovered without further intervention, indicating that these relapses were most likely disease fluctuations. On the other hand, patients on low dose showed consistently higher relapse rates, regardless whether initially started on this dose or after dose switch from high- to low-dose SCIG. Consistent with clinical relapse were the findings of IgG level decreases in the low-dose group, particularly in those who relapsed.
An important finding in this study was that patients who relapsed on low dose could successfully be switched back to high dose with 92% (24 of 26) of patients fully recovering back to previous clinical levels. This has practical implications, as physicians might want to reduce a patient's dose after a period of clinical stability. Reinitiating the high dose can be seen as an effective “rescue strategy” for relapsed patients on lower doses.
Despite these differences in relapse rates, the study also showed that approximately 68% of patients who completed PATH without a relapse remained stable after switching from high to low dose in the extension study. We conclude from this observation that for a considerable number of patients, the low dose was an appropriate treatment, albeit that more than two-thirds of these patients were treated with high dose for at least 24 weeks first.
When exiting the study, more than 80% of patients preferred their SC treatment over IV treatment, which they had received before entry into the PATH and/or extension study. The most common named advantages were gain of independence, less time spent for SC therapy, preferred frequency of administration, and less felt side effects. However, important to realize is that this may be a selected group of patients preferring SCIG (as they volunteered for the PATH trial and then again for the extension study).
The PATH study was enriched with IVIG responders, as the main goal was to investigate maintenance treatment. Leaving out patients who had successfully been rescued from a relapse in the PATH study would have biased this study even more toward treatment responders.
Only a few studies with long-term SCIG treatment in CIDP have been published. Markvardsen et al. followed up on 17 patients from their placebo-controlled study for 12 months, of which 16 completed the study.20,21 Four patients experienced a decline in muscle strength, 10 experienced an increase, and 2 remained unchanged. None of the patients experienced an increase in disability. The SCIG dosage used was 0.33–0.37 g/kg/wk, which is close to the 0.4 g/kg/wk used in our study. Cocito et al.22 reported a 2-year study of 6 Italian neurological centers, which included 45 patients with CIDP. The SCIG dose administered was equivalent to a 1:1 conversion from the previous IVIG dose (usually corresponding to a 0.33 g/kg weekly SCIG dose). In only about 13% of patients were dose adjustments required.
In summary, efficacy findings confirmed that high-dose IgPro20, 0.4 g/kg weekly, offered a higher likelihood for maintaining patients' health status than low dose, 0.2 g/kg weekly. Yet, a substantial proportion of patients could be down-titrated to low dose without further worsening. This emphasizes that in clinical practice, dose reductions are recommended, tailored to the individual needs of patients, and that by increasing the dose, patients who relapse can be rescued.
Glossary
- AE
adverse event
- CIDP
chronic inflammatory demyelinating polyneuropathy
- INCAT
Inflammatory Neuropathy Cause and Treatment
- I-RODS
Inflammatory Neuropathy-Rasch-Built Overall Disability Scale
- MRC
Medical Research Council
- SAE
serious AE
- SRC
Safety Review Committee
Appendix 1. Authors

Appendix 2. PATH study group: coinvestigators
Footnotes
Class of Evidence: NPub.org/coe
Study funding
This study was supported by CSL Behring. The funder of the study together with a steering committee was responsible for the design of the study and the data analysis and contributed to the data interpretation and writing of the manuscript. A statistician was a member of the steering committee and critically reviewed all results. The funder had no role in the data collection. The authors had full access to all data in the study, and the corresponding author had final responsibility for the decision to submit for publication.
Disclosure
I.N. van Schaik chairs a steering committee for CSL Behring and received departmental honoraria for serving on scientific advisory boards for CSL Behring and Baxter. He received departmental research support from The Netherlands Organization for Scientific Research and from the Dutch Prinses Beatrix Fonds. All lecturing and consulting fees for INS were donated to the Stichting Klinische Neurologie, a local foundation that supports research in the field of neurologic disorders. He served on the editorial board of the Cochrane Neuromuscular Disease Group and is a member of the scientific board of the Kreuth III meeting on the optimal use of plasma-derived medicinal products, especially coagulation factors and normal immunoglobulins organized under the auspices of the European Directorate for the Quality of Medicines & HealthCare (EDQM). V. Bril is consultant to CSL Behring, Grifols, UCB, Bionevia, ArgenX, Shire, Alnylam, and Alexion. She serves on international scientific advisory boards of MGFA and the CIDP/GBS International Foundation and has received research support from CSL Behring, Grifols, Bionevia, Baxter, UCB, and ArgenX. N. van Geloven received departmental honoraria for serving at a scientific advisory board of CSL Behring. H.P. Hartung received fees for consulting and serving on steering committees or ad boards from Baxter, Bayer Healthcare, Biogen, CSL Behring, GeNeuro, Kedrion, LFB, MedImmune, Merck, Novartis, Octapharma, Receptos Celgene, Roche, Sanofi Genzyme, and Teva with approval by the Rector of Heinrich Heine University Düsseldorf. R. A. Lewis is chair of the Inflammatory Neuropathy Consortium (INC), a standing committee of the Peripheral Nerve Society, a member of the board of Directors of the Peripheral Nerve Society, and a member of the medical advisory board of the GBS-CIDP Foundation, MGF of America, and MGF of California. He is a paid consultant for CSL Behring, Novartis, Pharnext, Axelacare, Biotest, and NuFactor. G. Sobue served on the scientific advisory boards of the Kanae Science Foundation for the Promotion of Medical Science and the Takeda Foundation. He serves on a steering committee for CSL Behring. He received funding for travel and speaker honoraria from Mitsubishi Tanabe Pharma Co, Shionogi Co Ltd, Bristol-Myers Squibb, Sumitomo Dainippon Pharma Co Ltd, Novartis Pharma KK, Bayer Yakuhin Ltd, Pfizer Japan Inc, Boehringer Ingelheim Japan, Inc, Kissei Pharmaceutical Co Ltd, Janssen Pharmaceutical KK, Teijin Pharma Ltd, FP Pharmaceutical Co, Nihon Pharmaceutical Co Ltd, Japan Blood Products Organization, Kowa Pharmaceutical Co Ltd, Ono Pharmaceutical Co Ltd, and Eisai Co Ltd. He also received grants from the Ministry of Health, Labour and Welfare Japan, Japanese Ministry of Education, Culture, Sports, Science and Technology, and Japan Society for the Promotion of Science. J.P. Lawo and M. Praus are CSL employees and biostatisticians for the PATH study. O. Mielke is a CSL employee and program director for the PATH study. B.L. Durn is a CSL employee and clinical scientist for the PATH study. D.R. Cornblath is consultant for Annexon Biosciences, argenx BVBA, Biotest Pharmaceuticals, Inc., Cigna Health Management, Inc., CSL Behring, DP Clinical, Inc., Grifols S.A., Hansa Medical Inc., Merrimack Pharmaceuticals, Inc., Neurocrine Biosciences, Novartis Corp., Octapharma AG, Pharnext SAS, Seattle Genetics, Inc., Sun Pharmaceuticals, and Syntimmune. He is a member of Data Safety Monitoring Boards for Pfizer Inc., Axovant Sciences LTD., Ampio Pharmaceuticals, PledPharma, Momenta Pharma, and Sanofi; Technology Licensing for the Total Neuropathy Score to AstraZeneca Pharmaceuticals, LP, Calithera Biosciences, Genentech Inc, Neurocrine Biosciences, Merrimack Pharmaceuticals, Inc., and Seattle Genetics, Inc. He is a nonvoting member of the Board of Directors of The Peripheral Nerve Society. He is on the Global Medical Advisory Board of the GBS-CIDP Foundation International. He is Editor-in-Chief of the Journal of the Peripheral Nervous System. I.S.J. Merkies received funding for research from the Talecris Talents program, the GSB CIDP Foundation International, Princes Beatrix foundation, and from the European Union 7th Framework Programme (grant n°602273). Furthermore, a research foundation at the University of Maastricht received honoraria on behalf of him for participation in steering committees of the Talecris ICE Study, LFB, CSL Behring, Novartis, Grifols, and Octapharma. He serves on the editorial board of the Journal of Peripheral Nervous system and is a member of the Inflammatory Neuropathy Consortium (INC) and the Peripheral Nerve Society. Go to Neurology.org/NN for full disclosures.
References
- 1.van Schaik IN, Bril V, van Geloven N, et al. Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy (PATH): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol 2018;17:35–46. [DOI] [PubMed] [Google Scholar]
- 2.Markvardsen LH, Christiansen I, Andersen H, Jakobsen J. Headache and nausea after treatment with high-dose subcutaneous versus intravenous immunoglobulin. Basic Clin Pharmacol Toxicol 2015;117:409–412. [DOI] [PubMed] [Google Scholar]
- 3.Markvardsen LH, Christiansen I, Harbo T, Jakobsen J. Hemolytic anemia following high dose intravenous immunoglobulin in patients with chronic neurological disorders. Eur J Neurol 2014;21:147–152. [DOI] [PubMed] [Google Scholar]
- 4.Berger M, Jolles S, Orange JS, Sleasman JW. Bioavailability of IgG administered by the subcutaneous route. J Clin Immunol 2013;33:984–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berger M, Rojavin MA, Kiessling P, Zenker O. Pharmacokinetics of subcutaneous immunoglobulin and their use in dosing of replacement therapy in patients with primary immunodeficiencies. Clin Immunol 2011;139:133–141. [DOI] [PubMed] [Google Scholar]
- 6.Berger M. Adverse effects of IgG therapy. J Allergy Clin Immunol 2013;1:558–566. [DOI] [PubMed] [Google Scholar]
- 7.Gardulf A, Andersen V, Bjorkander J, et al. Subcutaneous immunoglobulin replacement in patients with primary antibody deficiencies: safety and costs. Lancet 1995;345:365–369. [DOI] [PubMed] [Google Scholar]
- 8.Kittner JM, Grimbacher B, Wulff W, JÄGer B, Schmidt RE. Patients' attitude to subcutaneous immunoglobulin substitution as home therapy. J Clin Immunol 2006;26:400–405. [DOI] [PubMed] [Google Scholar]
- 9.Lazzaro C, Lopiano L, Cocito D. Subcutaneous vs intravenous administration of immunoglobulin in chronic inflammatory demyelinating polyneuropathy: an Italian cost-minimization analysis. Neurol Sci 2014;35:1023–1034. [DOI] [PubMed] [Google Scholar]
- 10.Cocito D, Paolasso I, Peci E, Spagone E, Lopiano L. Improvement of quality of life in patients with chronic inflammatory demyelinating polyneuropathy shifting from 16 to 20% subcutaneous immunoglobulins. Neurol Sci 2013;34:2061–2062. [DOI] [PubMed] [Google Scholar]
- 11.Hadden RDM, Marreno F. Switch from intravenous to subcutaneous immunoglobulin in CIDP and MMN: improved tolerability and patient satisfaction. Ther Adv Neurol Disord 2015;8:14–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Racosta JM, Sposato LA, Kimpinski K. Subcutaneous versus intravenous immunoglobulin for chronic autoimmune neuropathies: a meta-analysis. Muscle Nerve 2017;55:802–809. [DOI] [PubMed] [Google Scholar]
- 13.Van den Bergh PYK, Hadden RDM, Bouche P, et al. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society—First Revision. Eur J Neurol 2010;17:356–363. [DOI] [PubMed] [Google Scholar]
- 14.van Schaik IN, van Geloven N, Bril V, et al. Subcutaneous immunoglobulin for maintenance treatment in chronic inflammatory demyelinating polyneuropathy (The PATH Study): study protocol for a randomized controlled trial. Trials 2016;17:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes RA, Donofrio P, Bril V, et al. Intravenous immune globulin (10% caprylate-chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol 2008;7:136–144. [DOI] [PubMed] [Google Scholar]
- 16.Vanhoutte EK, Latov N, Deng C, et al. Vigorimeter grip strength in CIDP: a responsive tool that rapidly measures the effect of IVIG – the ICE study. Eur J Neurol 2013;20:748–755. [DOI] [PubMed] [Google Scholar]
- 17.Kleyweg RP, Van der Meché FGA, Schmitz PI. Interobserver agreement in the assessment of muscle strength and functional abilities in Guillain-Barré syndrome. Muscle Nerve 1991;14:1103–1109. [DOI] [PubMed] [Google Scholar]
- 18.Van Nes SI, Vanhoutte EK, Van Doorn PA, et al. Rasch-built overall disability scale (R-ODS) for immune-mediated peripheral neuropathies. Neurology 2011;76:337–345. [DOI] [PubMed] [Google Scholar]
- 19.Leger JM, De Bleecker JL, Sommer C, et al. Efficacy and safety of Privigen® in patients with chronic inflammatory demyelinating polyneuropathy: results of a prospective, single-arm, open-label Phase III study (the PRIMA study). J Peripher Nerv Syst 2013;18:130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Markvardsen LH, Debost JC, Harbo T, et al. Subcutaneous immunoglobulin in responders to intravenous therapy with chronic inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol 2013;20:836–842. [DOI] [PubMed] [Google Scholar]
- 21.Markvardsen LH, Harbo T, Sindrup SH, et al. Subcutaneous immunoglobulin preserves muscle strength in chronic inflammatory demyelinating polyneuropathy. Eur J Neurol 2014;21:1465–1470. [DOI] [PubMed] [Google Scholar]
- 22.Cocito D, Merola A, Romagnolo A, et al. Subcutaneous immunoglobulin in CIDP and MMN: a different long-term clinical response? J Neurol Neurosurg Psychiatry 2016;87:791–793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
CSL will only consider requests to share individual patient data that are received from systematic review groups or bona-fide researchers (see table e-1, links.lww.com/NXI/A124 for more information).