

Abstract
Topoisomerase II (TOP2) poisons as anticancer drugs work by trapping TOP2 cleavage complexes (TOP2cc) to generate DNA damage. Repair of such damage by tyrosyl DNA phosphodiesterase 2 (TDP2) could render cancer cells resistant to TOP2 poisons. Inhibiting TDP2 thus represents an attractive mechanism-based chemosensitization approach. Currently known TDP2 inhibitors lack cellular potency and / or permeability. We report herein two novel subtypes of the deazaflavin TDP2 inhibitor core. By introducing an additional phenyl ring to the N-10 phenyl ring (subtype 11), or to the N-3 site of the deazaflavin scaffold (subtype 12) we have generated novel analogues with considerably improved biochemical potency and / or permeability. Importantly, many analogues of both subtypes, particularly compounds 11a, 11e, 12a, 12b and 12h, exhibited much stronger cancer cell sensitizing effect than the best reported previous analogue 4a toward the treatment with etoposide (ETP), suggesting that these analogues could serve as effective cellular probes.
Graphical Abstract
Introduction
Topoisomerase II (TOP2) manages DNA topology during essential DNA transactions such as transcription and replication.1–2 Mechanistically, TOP2 cleaves DNA using its tyrosine residue to generate a transient TOP2 cleavage complexes (TOP2cc) in which TOP2 is covalently linked to the 5′ terminus of the DNA break via a tyrosyl phosphodiester bond. The TOP2cc is typically re-ligated at the end of each catalytic cycle, creating a dynamic DNA equilibrium between the resealed form and the TOP2cc. However, when the normally transient TOP2cc is trapped it becomes abortive and the DNA is damaged. Clinically important TOP2 poisons work by this exact mechanism as they bind to and stabilize the TOP2cc to prevent DNA re-ligation, resulting in the accumulation of abortive TOP2cc.3–4 Multiple lines of recent evidence demonstrated that tyrosyl DNA phosphodiesterase 2 (TDP2) repairs TOP2-mediated DNA damages, including the abortive TOP2cc trapped by TOP2 poisons, and causes cellular resistance to TOP2 poisons: 1) in cultured cells and animal models the lack of TDP2 led to enhanced cellular sensitivity to DNA breaks induced by TOP2 poisons;5–9 2) up-regulation of TDP2 transcription through a gain-of-function p53 mutation was linked to TOP2 poison resistance in human lung cancer.10 TOP2 poisons, such as etoposide (ETP), teniposide and doxorubicin, are widely used for treating a wide range of cancers, including lung cancer, testicular cancer, breast cancer and as a second line treatment option for platinum-resistant ovarian cancers.11 Inhibiting TDP2 represents a mechanism-based sensitizing approach which could allow these poisons to be used at lower and safer doses and against cancers that are resistant to TOP2 poisons.
In addition, TDP2 could be involved in the genome repair of certain DNA and RNA viruses, such as hepatitis B virus (HBV)12 and picornaviruses.13–14 The genome replication of these viruses is protein-primed via a tyrosine residue, which results in protein tyrosine-nucleic acid adducts similar to the TOP2cc. The repair to remove the viral proteins at the 5′ end of the viral genomes is believed to be carried out by host DNA repair machineries, possibly TDP2. Therefore, inhibiting TDP2 could also represent a novel antiviral approach.
A few compounds have recently been reported as TDP2 inhibitors (Figure 1), including benzopteridine-2,4-dione (1, Ro 08–2750),15 diaminoquinoline-2,8-dione (2, NSC111041),16 isoquinoline-1,3-diones (3),17 deazaflavins (4),15, 18–19 benzylidenepyrazolone (5, NSC375976),20 phenylacetylcystine (6, n = 0, NSC114532; n =1, NSC3198),20 quinazolinylaminopyrimidinone (7),21 triazolopyridine (8),22 indenoisoquinolines (9),23–24 and furoquinolinediones (10)25 (Figure 1). Most of these inhibitors were identified through random screening of compound libraries using biochemical assays.26 A few of them, such as 1 (reactive group), 2 (redox cycler), 5 (Michael acceptor), and 6 (redox cycler), fit the profiles of pan-assay interference structure (PAINS)27 and may not be appropriate for further development as TDP2 inhibitors. Compound 9 was reported as triple inhibitors of TOP1/TDP1/TDP2.24 More importantly, these compounds generally inhibit TDP2 only moderately with IC50 values in micromolar range. The exception is the deazaflavin chemotype (4) which inhibited TDP2 in nanomolar range with a genuine structure-activity relationship (SAR)15 and a defined binding mode,18 and sensitized cancer cells to TOP2 poison ETP,19 representing by far the most potent and the best characterized TDP2 inhibitor type. The challenge, however, is that the SAR revealed a strong dependence of TDP2 inhibition on the H-bond donating ability of the substituent on N-10 phenyl, rendering potent compounds poorly permeable.15 This is consistent with the observation that the sensitizing effect by the most potent analogue 4a (R = meta-tetrazole, chemotype 4) on DT40, chicken lymphoma cells, toward ETP treatment was not as pronounced as predicted based on its nanomolar potency in biochemical assays.19 Therefore, as potent as some of the reported 4 analogues are in biochemical assays, their use as cellular probes to study cellular functions of TDP2 is limited.
Figure 1.

Reported TDP2 inhibitors. Deazaflavin 4 is the most potent and best characterized TDP2 inhibitor type.
Ideally, molecular probes enabling cellular studies should have balanced profiles of potent inhibitory activity, good permeability and low cytotoxicity within the concentration range of studies. Interestingly, Raoof et al. also reported that an inactive compound of the same deazaflavin core exhibited good permeability and reduced efflux,15 suggesting that it is possible to develop the deazaflavin core into useful probes. We report herein our medicinal chemistry efforts toward this end. By chemically modifying the N-10 and N-3 sites (Figure 2), we have generated novel deazaflavin subtypes with enhanced potency, both biochemically and in cancer cells, and improved permeability.
Figure 2.

Novel deazaflavin subtypes 11-12 designed to improve lipophilicity by adding a second phenyl ring (B) directly off ring A (for subtype 11) or at N-3 site (for subtype 12).
Results and Discussion
Chemistry
Deazaflavin scaffold 4 was prepared according to reported procedures (Scheme 1A).15 The synthesis involved reaction of 6-chlorouracil (13) with substituted amines (14) to provide 6-amino intermediate 15 which upon reacting with 4-cyano-2-fluorobenzaldehyde (16) yielded the deazaflavin core (4). The key to the synthesis of subtype 11 was the preparation of amine intermediates 20-21 (Scheme 1B). Intermediates 20 were synthesized from an N-Boc protected 3 or 4-aminophenol (17) which was treated with substituted benzyl bromides (18) to yield corresponding O-benzyl derivatives (19). The subsequent deprotection of the N-Boc with TFA gave the aniline derivatives (20). On the other hand, amine intermediates 21 was synthesized via an SNAr reaction of phenolic alcohol 22 with 4-nitrophenyl fluoride 23 followed by the reduction of the nitro group. To advance the synthesis, amines 20 or 21 were reacted with 6-chlorouracil (13) to give intermediate 25 which upon condensation with 4-cyano-2-fluorobenzaldehyde (16) furnished subtype 11 (Scheme 1B).
Scheme 1.

Synthesis of deazaflavin scaffold 4 and new subtypes 11-12
aReagents and conditions: a) EtOH, 150 °C, 30 min, MW, 60–85%; b) DMF, 110 °C, MW, 30 min, 40–60%; c) K2CO3, DMF, r. t., 12 h; d) TFA, DCM, r. t., 6h, 60–75% over two steps; e) K2CO3, DMF, 110°C, 12h, 59%; f) 10% Pd/C, EtOAc, rt, o. n. 87%; g) EtOH, NaOH 1N, reflux, 3h, 85% h) Cu(OAc)2, DMF, air, r. t., 24–48 h, 40–52%.
N3-substituted deazaflavin subtype 12 was efficiently synthesized in one step from scaffold 4 via a modified Chan-Lam coupling28–30 with substituted boronic acids 26 under the catalysis of Cu(OAc)2 (Scheme 1C).
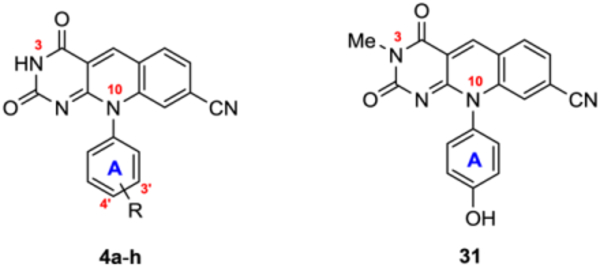
It is important to note that most of our analogues of subtype 12 bear a phenolic alcohol off phenyl ring A, which could complicate their synthesis from the corresponding 4 via the Chan-Lam coupling as both the NH at 3 position and the phenolic alcohol can be arylated under the Chan-Lam condition (Figure 3). However, in all our synthesis the N-arylated compounds of subtype 12 were obtained as the sole products, and the formation of the O-arylated subtype 11 was not observed (Figure 3). This complete chemoselectivity of N-arylation over O-arylation31 under Chan-Lam condition was confirmed by synthesizing analogue 12b, 12j-k via an alternative linear synthesis (Scheme 2). In this synthesis, phenyl ring B was introduced at the very beginning of the synthesis as part of the starting urea 27. Cyclocondensation of 27 with diethylmalonate produced the 1-phenylpyrimidine-trione (28) in excellent yield under reflux which upon treatment with POCl3 in the presence of BnEt3NCl yielded the key intermediate 3-phenylpyrimidine-2,4-dione (29).32–33 Reaction of 6-chloro intermediate (29) with anilines afforded 6-amination products in moderate to excellent yields. The desired deazaflavin subtype 12 was constructed in moderate yields by the subsequent amination and condensation reaction sequence.15 12b synthesized from this linear route was found identical to that obtained via the Chan-Lam coupling. It is also noteworthy that analogues 12j-k and 31 were synthesized via the linear route only.
Figure 3.

Chemoselectivity of the Chan-Lam coupling reaction. Under reaction conditions, the arylation occurred exclusively at the N-3 site to yield subtype 12 with O-arylated products 11 not observed.
Scheme 2.

Alternative linear synthesis for subtype 12
aReagents and conditions: a) Diethylmalonate, NaOEt, EtOH, reflux, o. n., 86%; b) POCl3, BnEt3NCl, 50 °C, 6 h, 70%; c) aniline derivative, EtOH, 150 °C, 30 min, MW, 55–80%; d) DMF, 110 °C, 30 min, MW, 50–69%.
Biology
To assess TDP2 inhibitory activity all final compounds were tested in dose-response fashion using our recently developed fluorescence-based biochemical assay.21 Most analogues were also evaluated for both the toxicity and sensitizing effect on chicken lymphoma DT40 cells toward treatment with ETP. Permeability was measured for all final compounds in Parallel Artificial Membrane Permeation Assay (PAMPA).34 Additional cytotoxicity studies were carried out in HepG2 cells for all final compounds, and in HeLa cells for most analogues. These cells are widely used for the infection of HBV and picornavirus, respectively.
The first series of compounds (Table 1) served as a control series centered around analogue 4c. IC50 values from our own biochemical assay largely agreed with reported values15 (Table 1 footnote) and followed similar SAR trends in that 1) potent TDP2 inhibition by deazaflavin subtype 4 generally required a polar substituent with H-bonding ability on phenyl ring A (4a-c); removal (4d) or etherification (4e-h) of the OH group led to activity drop by 2–10 fold; 2) analogues with a meta polar group on phenyl ring A demonstrated better potency than those with a para polar group (4b vs 4c). Unfortunately, the polar group (OH or tetrazole) critical for target binding also conferred extremely low PAMPA permeability (4a-c) with the effective permeability coefficient (Pe) in the range of 0.003–0.01 (a Pe of ≥ 1.5 is considered indicative of high permeability). PAMPA permeability was substantially improved upon removal (4d) or etherification (4e-h) of the polar OH group (Table 1). Notably analogue 4f was designed to achieve balanced potency and permeability based on the concept of lipophilic H-bond donor.35 Because the H of the difluoromethoxy group (–OCHF2) is rendered acidic by strong electron-withdrawing O and F atoms, presumably this functionality could provide the key H-bonding for potency while improving permeability. However, significantly improved TDP2 inhibition was not observed with 4f when compared with other ether analogues, particularly 4e. On the other hand, methylation of the 3-NH group (31 vs 4c) led to significantly improved permeability (13 fold) with only a moderate loss of inhibitory activity (2 fold).
Table 1.
TDP2 inhibitory activity and PAMPA permeability for the control series.
 | |||
|---|---|---|---|
| Compd | R | TDP2 IC50 (μM)a | PAMPA Pe (10−6 cm/s)b |
| 4ac | 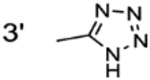 |
0.041 ± 0.003 | 0.003 |
| 4bc | 3′ OH | 0.042 ± 0.004 | 0.01 |
| 4cc | 4′ OH | 0.14 ± 0.01 | 0.008 |
| 4dc | H | 0.64 ± 0.02 | 0.4 |
| 4e | 4′ OCF3 | 1.0 ± 0.1 | 1.1 |
| 4f | 4′ OCHF2 | 0.88 ± 0.06 | 0.3 |
| 4g |  |
0.96 ± 0.02 | 0.5 |
| 4h | 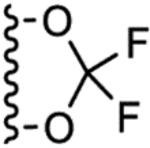 |
1.5 ± 0.1 | 1.1 |
| 31 | -- | 0.32 ± 0.02 | 0.1 |
IC50: concentration of a compound producing 50% inhibition, expressed as mean ± standard deviation from three independent experiments performed in triplicate.
Pe: effective permeability coefficients determined using the PAMPA, measured in five replicates.
Previously reported analogues. Reported IC50 values are 0.04 μM (4a), 0.05 μM (4b), 0.09 μM (4c) and 0.50 μM (4d).15
These observations prompted us to explore more drastic structural modifications over the original deazaflavin inhibitor type 4. Specifically, we decided to derivatize the polar group on phenyl ring A and / or the NH group at 3 position of the deazaflavin core with a second phenyl ring (ring B, Figure 2). We expected that such derivatization would improve permeability and hypothesized that the added phenyl ring (B) could provide additional interactions with TDP2 to possibly compensate for the loss of the polar interactions. As for the polar group on phenyl A, we chose the OH group (4b-c) mainly due to its ability to confer high potency and easy synthetic accessibility.
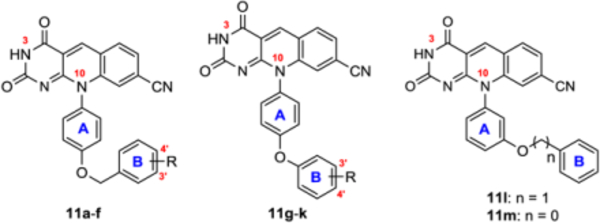
We first explored the derivatization of the OH group on phenyl ring A. Toward this end, we synthesized a panel of benzyl ethers and phenyl ethers (subtype 11, Table 2). The most prominent SAR with this series was that benzyl ethers exhibited considerably higher inhibitory potency than the corresponding phenyl ethers (11a, IC50 = 0.196 μM vs 11g, IC50 = 1.13 μM; 11b, IC50 = 1.60 μM vs 11h, IC50 > 10 μM; 11e, IC50 = 0.153 μM vs 11j, IC50 = 0.934 μM; 11f, IC50 = 0.131 μM vs 11k, IC50 = 0.186 μM). Particularly intriguing are 11a and 11e which inhibited TDP2 with essentially the same potency as 4c but exhibited drastically improved permeability over 4c (75 and 150 fold, respectively). Direct comparison between compounds with an ester functionality and a carboxylic acid functionality (11e vs 11f; 11j vs 11k) further confirmed that a polar H-bond donor benefits target binding but compromises permeability. These results validate our approach of derivatizing the polar group, particularly the phenolic OH, for balanced potency and permeability. In addition, the two analogues (11l-m) synthesized based on the meta phenolic analogue 4b also showed strong TDP2 inhibition and markedly improved permeability, though the IC50 values were much higher than 4b (8–10 fold).
Table 2.
TDP2 inhibitory activity, PAMPA permeability and cytotoxicity for analogues from phenolic OH modifications (11a-m).
 | |||
|---|---|---|---|
| Compd | R | TDP2 IC50 (μM)a | PAMPA Pe (10−6 cm/s)b |
| 4c | -- | 0.14 ± 0.01 | 0.008 |
| 11a | H | 0.20 ± 0.02 | 1.2 |
| 11b | 4′ Cl | 1.6 ± 0.2 | 0.4 |
| 11c | 4′ Br | 1.3 ± 0.0 | 0.2 |
| 11d | 3′ Cl | 0.99 ± 0.09 | 0.4 |
| 11e | 3′ CO2Me | 0.15 ± 0.01 | 0.6 |
| 11f | 3′ CO2H | 0.13 ± 0.01 | 0.01 |
| 11g | H | 1.1 ± 0.1 | 2.8 |
| 11h | 4′ Cl | >10 | 0.8 |
| 11i | 4′ F | 3.8 ± 0.3 | 2.7 |
| 11j | 3′ CO2Me | 0.93 ± 0.05 | 0.7 |
| 11k | 3′ CO2H | 0.19 ± 0.01 | 0.003 |
| 4b | -- | 0.041 ± 0.004 | 0.01 |
| 11l | -- | 0.38 ± 0.02 | 1.7 |
| 11m | -- | 0.30 ± 0.00 | 2.0 |
IC50: concentration of a compound producing 50% inhibition, expressed as mean ± standard deviation from three independent experiments performed in triplicate.
Pe: effective permeability coefficients determined using the PAMPA, measured in five replicates.
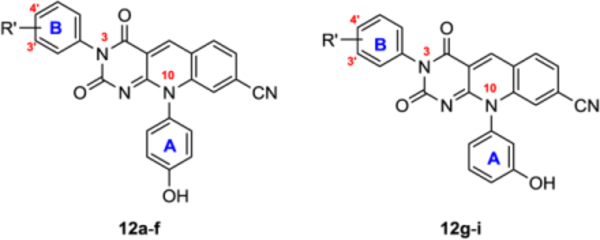
Next, we probed the impacts of introducing phenyl ring B to the N-3 site while retaining the OH group on phenyl A. For this purpose, Chan-Lam products from both 4c (para phenolic OH, 12a-f) and 4b (meta phenolic OH, 12g-i) were obtained. Comparing with subtype 11, analogues of this series all showed considerably better TDP2 inhibition. As far as SAR is concerned, for analogues (12a-f) derived from para phenolic alcohol 4c, the R′ group on phenyl ring B or its position of substitution did not significantly impact the inhibitory potency, with all analogues showing 2–4 fold of improved potency over 4c. Similar level of potency improvement (2–6 fold) was observed with analogues 12g-i over the meta phenolic alcohol 4b, though the site of Cl substitution (3′ vs 4′) seems to have a more significant impact on potency. Moderate improvement on the permeability (3–13 fold) over 4b-c was also observed with all analogues of subtype 12, with the single exception of 12f. Nevertheless, the N-3 arylation generated a novel deazaflavin subtype with improved TDP2 inhibition and PAMPA permeability.
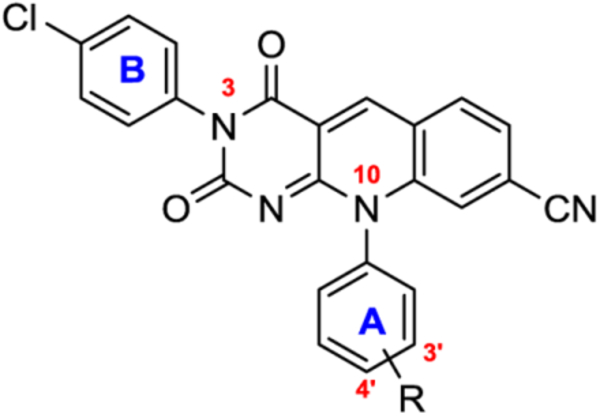
Finally, with the N-3 arylated with a para-chlorophenyl group, we examined the impact of different phenyl ring A substituents for subtype 12 (Table 4). First of all, removing the polar phenolic OH group of ring A resulted in a compound (12l) with much reduced inhibitory activity (12l vs 12b /12g, Table 4), though the permeability of 12l was among the best of all compounds tested. Secondly, replacement of the phenolic alcohol by another polar and H-bond enabling NHSO2Me group generated analogues (12j-k) showing the highest potency of all compounds tested so far, with IC50 values in single digit nM range for both para and meta substitution (Table 4). Although permeability remains poor for both, the values still reflect a moderate improvement over 4b-c (3–6 fold). Overall, our data suggest that the N-3 arylation can be a highly productive and desirable modification, and that the resulting subtype 12 represents the most active inhibitor type of TDP2 to date.
Table 4.
TDP2 inhibitory activity and PAMPA permeability for additional analogues of subtype 12 with N-10 phenyl A modifications.
 | |||
|---|---|---|---|
| Compd | R | TDP2 IC50 (μM)a | PAMPA Pe (10−6 cm/s)b |
| 12b | 4′ OH | 0.033 ± 0.002 | 0.08 |
| 12j | 4′ NHSO2Me | 0.0065 ± 0.0004 | 0.02 |
| 12g | 3′ OH | 0.0073 ± 0.0003 | 0.04 |
| 12k | 3′ NHSO2Me | 0.0061 ± 0.0006 | 0.06 |
| 12l | H | 0.27 ± 0.02 | 4.4 |
IC50: concentration of a compound producing 50% inhibition, expressed as mean ± standard deviation from three independent experiments performed in triplicate.
Pe: effective permeability coefficients determined using the PAMPA, measured in five replicates.
High permeability conferred cytotoxicity.
To gauge their potential as probes for cell-based studies, all final compounds were evaluated in two cell lines for cytotoxicity: the HepG2 cells commonly used for HBV infection36 and the HeLa cells permissible to picornavirus infection13–14 (Table 5). Overall, a strong correlation between cytotoxicity and PAMPA permeability was observed. For subtypes 4 and 11 no significant cytotoxicity was observed at concentrations up to 100 μM for compounds with a PAMP Pe ≤ 0.5, with 4f being the only exception; whereas compounds with a PAMP Pe > 0.5 all showed cytotoxicity (Table 5). For subtype 12 only analogue 12l exhibited high permeability, and hence, cytotoxicity. A few other analogues (12b-c, 12d, 12i) also demonstrated cytotoxicity despite their low permeability, although their extraordinary TDP2 inhibitory potency indicates that cellular testing of these compounds most likely will not require a concentration range close to the CC50. Therefore, their potential as molecular probes for cellular studies should not be overlooked.
Table 5.
Cytotoxicity evaluation in two cell lines.
| Compd | CC50 (μM)a | PAMPA Pe (10−6 cm/s)b | Compd | CC50 (μM)a | PAMPA Pe (10−6 cm/s)b | ||
|---|---|---|---|---|---|---|---|
| HepG2 | HeLa | HepG2 | HeLa | ||||
| 4ad | >100 | >100 | 0.003 | 11i | 14 | 12 | 2.7 |
| 4bd | >100 | -- | 0.01 | 11j | 8.7 | 9.4 | 0.7 |
| 4cd | >100 | -- | 0.008 | 11k | >100 | >100 | 0.003 |
| 4dd | >100 | -- | 0.4 | 11l | 25 | 30 | 1.7 |
| 4e | 40 | 28 | 1.1 | 11m | 9.0 | 21 | 2.0 |
| 4f | 85 | 79 | 0.3 | 12a | 50 | >100 | 0.1 |
| 4g | >100 | >100 | 0.5 | 12b | 41 | 45 | 0.08 |
| 4h | 42 | 39 | 1.1 | 12c | 46 | 32 | 0.06 |
| 31 | >100 | >100 | 0.1 | 12d | 49 | 67 | 0.05 |
| 11a | 14 | 12 | 1.2 | 12e | >100 | -- | 0.03 |
| 11b | >100 | >100 | 0.4 | 12f | >100 | -- | 0.01 |
| 11c | 17 | -- | 0.2 | 12g | 85 | 65 | 0.04 |
| 11d | 14 | 12 | 0.4 | 12h | 82 | 90 | 0.07 |
| 11e | 29 | 29 | 0.6 | 12i | 42 | 62 | 0.03 |
| 11f | >100 | >100 | 0.01 | 12j | >100 | >100 | 0.02 |
| 11g | 29 | -- | 2.8 | 12k | >100 | >100 | 0.06 |
| 11h | 5.3 | 7.4 | 0.8 | 12l | 33 | 25 | 4.4 |
CC50: concentration of a compound causing 50% cell death. ETP was used as control: CC50 = 15 μM (HepG2) and 17 μM (HeLa).
Pe: effective permeability coefficients determined using the PAMPA, measured in five replicates.
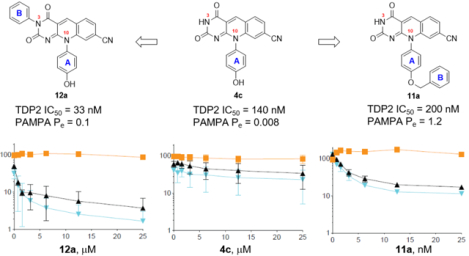
Novel inhibitors strongly sensitized DT40 cells toward ETP treatment.
The efficacy of TDP2 inhibitors in potentiating the cytotoxic action of TOP2 poison ETP was evaluated in DT40 chicken lymphoma cells.37 Cell viability was assayed using the ATPlite luminescence assay (PerkinElmer). DT40 cells were treated with combinations of ETP and TDP2 inhibitors.19 Treatment with only the TDP2 inhibitor was used as control. It is expected that TDP2 inhibition alone should not elicit cytotoxic effect. On the other hand, supplementation with a TDP2 inhibitor should amplify the cytotoxic effect associated with TOP2 poison ETP. Therefore, the decrease of cell viability with increasing concentrations of the TDP2 inhibitor at constant ETP dose would reflects the cooperative interaction of ETP and the TDP2 inhibitor in inducing toxicity. As shown in Figure 3, previously reported deazaflavin analogue 4a and our compounds 12a and 12b were non-cytotoxic up to 25μM. Although showing toxicity at 25 μM, compound 12h reached plateau in enhancing ETP action at concentrations below 10 μM. As far as subtype 11 is concerned, compounds 11a and 11e both showed general cytotoxicity at μM concentrations (see supporting information) which corroborated their cytotoxicity observed in HepG2 and HeLa cells. However, at concentrations much lower than the cytotoxic concentrations (<25 nM), 11a and 11e both sensitized DT40 to ETP treatment. This extraordinary sensitizing effect likely reflects their balanced properties between TDP2 inhibition and PAMPA permeability (Table 2). Nevertheless, all analogues of subtypes 11–12 shown in Figure 3 demonstrated stronger sensitizing effect when compared with 4a, the best analogue previously reported. The improved sensitizing effect over 4a was also observed with many other analogues (see supporting information). These results strongly validate analogues of our new subtypes 11–12 as effective cellular probes.
The overall profiles of TDP2 inhibitory activity, permeability and cytotoxicity for selected analogues of subtypes 11–12 are summarized in Table 6. These properties, along with their superior cancer cell sensitizing effects as shown in Figure 3 and supporting information, strongly validate our novel analogues as improved cellular probes over the best previous analogue 4a.
Table 6.
TDP2 inhibitory activity, permeability and cytotoxicity profiles of selected analogues.
| Compd | TDP2 IC50 (μM)a | PAMPA Pe (10−6 cm/s)b | CC50 (μM)c | |
|---|---|---|---|---|
| HepG2 | HeLa | |||
| 11a | 0.20 ± 0.02 | 1.2 | 14 | 12 |
| 11e | 0.15 ± 0.01 | 0.6 | 29 | 29 |
| 12a | 0.033 ± 0.001 | 0.1 | 50 | >100 |
| 12b | 0.033 ± 0.002 | 0.08 | 41 | 45 |
| 12g | 0.0073 ± 0.0003 | 0.04 | 85 | 65 |
| 12h | 0.020 ± 0.002 | 0.07 | 82 | 90 |
| 12j | 0.0065 ± 0.0004 | 0.02 | >100 | >100 |
| 12k | 0.0061 ± 0.0006 | 0.06 | >100 | >100 |
| 4ad | 0.041 ± 0.003 | 0.003 | >100 | >100 |
| 4bd | 0.042 ± 0.004 | 0.01 | >100 | -- |
| 4cd | 0.14 ± 0.01 | 0.008 | >100 | -- |
IC50: concentration of a compound producing 50% inhibition, expressed as mean ± standard deviation from three independent experiments.
Pe: effective permeability coefficients determined using the PAMPA, measured in five replicates.
CC50: concentration of a compound causing 50% cell death.
Previously reported15 analogues.
Molecular modeling
The co-crystal structure18 of humanized mouse TDP2 catalytic domain bound to 4c (PDB code: 5J42) elucidated the binding mode and mechanism of inhibition of deazaflavin inhibitors. Key to inhibitor binding is an extensive H-bonding network involving i) the pyrimido-dione ring of deazaflavin and the guanidine group of R266 and R268 ii) the phenolic hydroxyl group and the backbone carbonyl of N264 via water molecule (Figure 4A).18 The structure also revealed empty spaces within the binding pocket to accommodate an additional phenyl ring in pointed directions (Figure 4B) to generate two novel subtypes: subtype 11 via the O-substitution (left arrow, Figure 4B) and subtype 12 via the N-substitution (right arrow, Figure 4B). Importantly, representative analogues 11k and 12b are both docked nicely into the crystal structure and are predicted to interact extensively with the side chains of R266 and R268 using its nitrogen and carbonyl oxygen atoms within the pyrimido-dione core (Figure 4C–D), a binding feature also observed with compound 4c (Figure 4A). In addition, the ether linker of 11k allows the molecule to extend the added phenyl group into the active site and engage with active site residues H226, N264 and H349 of TDP2 using its phenyl substituent, the carboxylate group (Figure 4C). Additional docking with analogues 11a and 11e generated similar poses (SI, Figure S1), except that the additional methylene group extends phenyl ring B further to be in closer contact with H349 (pi-pi interaction) and D262 (pi-anion interaction) than phenyl ring B of 11k. The methyl carboxylate group in 11e also forms an H-bond with R206. On the other hand, docking of 12b reveals a water mediated H-bond interaction between the phenolic hydroxyl group and the side chains of N264 and R266 as observed for 4c. Nevertheless, unique to subtype 12 is that the phenyl ring at N-3 position of pyrimido-dione is oriented parallel to the R268, which could enable cation-π interactions (Figure 4D). Cation-π interactions38 are considered vital in several physiological processes which could influence biological structures and molecular recognition.39–40 These interactions are widely observed in protein-protein41 or protein-ligand40, 42 and protein-DNA43 complexes. In the biochemical assay, all analogues with an N-3 aryl group (12a-i) showed much improved potency (2–6 fold) over 4c or 4b, whereas compound 31 (N-Me analog) lacking the N-3 aryl ring was found to be 2-fold less active over 4c. These SAR observations strongly reflect the critical binding benefits gained via the predicted cation-π interaction between the N-3 aryl of subtype 12 and the R268 residue of TDP2.
Conclusion
We have generated two novel deazaflavin subtypes (11 and 12) as inhibitor types of TDP2. The synthesis of the N-3 aryl subtype featured a chemoselective Chan-Lam coupling. Most analogues of these chemotypes showed improved biochemical potency and / or PAMPA permeability. Many analogues of our new subtypes 11–12, particularly 11a, 11e, 12a, 12b and 12h, demonstrated stronger cancer cell sensitizing effect than the best previous analogue 4a toward ETP treatment. Molecular modeling revealed critical binding interactions provided by the added aryl group, particularly the cation-π interaction between the N-3 aryl and the guanidinium head of R268. Further characterization of these improved deazaflavin analogues in expanded human cancer cell lines, including ETP-resistant cell lines, is currently underway and data will be published in due course.
Experimental section
Chemistry.
General procedures.
All commercial chemicals were used as supplied unless otherwise indicated. Flash chromatography was performed on a Teledyne Combiflash RF-200 with RediSep columns (silica) and indicated mobile phase. All moisture sensitive reactions were performed under an inert atmosphere of ultrapure argon with oven-dried glassware. 1H and 13C NMR spectra were recorded on a Varian 600 MHz or Bruker 400 spectrometer. Mass data were acquired on an Agilent 6230 TOF LC/MS spectrometer capable of ESI and APCI ion sources. Analysis of sample purity was performed on a Varian Prepstar SD-1 HPLC system with a Phenomenex Gemini, 5 μm C18 column (250 mm × 4.6 mm). HPLC conditions: solvent A = H2O, solvent B = MeCN; flow rate = 1.0 mL/min; compounds were eluted with a gradient of 20% MeCN/H2O to 100% MeCN for 30 min. Purity was determined by total absorbance at 254 nm. All tested compounds have a purity ≥ 97%.
Deazaflavin analogues (4a-d) were reported earlier by Raoof et al.15
General procedure for the synthesis of deazaflavin derivatives 4, 11, 12j-k, and 31.15
To a suspension of 15, 25, or 30 (0.1 g, 1.0 eq) in DMF (2 mL) was added 4-cyano-2-fluorobenzaldehyde (16, 1.2 eq) and heated by microwave irradiation at 110 °C for 30 minutes. Water (10 mL) was added to the reaction mixture, and the resulting precipitate was filtered, washed with excess water and dried under vacuum. The crude product was purified by combiflash using 0–2% methanol in DCM and the obtained compound was triturated with methanol to furnish the desired compound. Compounds 4 (Scheme 1A), 11 (Scheme 1B), 12j-k (Scheme 2), and 31 (Scheme 2) were synthesized starting with intermediate 15, 25, 30, and 30 respectively.
10-(3-Hydroxyphenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (4b).15
Yellow solid, 75% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.22 (s, 1H), 10.04 (s, 1H), 9.13 (s, 1H), 8.39 (d, J = 8.2 Hz, 1H), 7.87 (dd, J = 8.2, 1.5 Hz, 1H), 7.49 (t, J = 8.0 Hz, 1H), 7.08 (s, 1H), 7.04 (dd, J = 8.2, 1.5 Hz, 1H), 6.86 – 6.77 (m, 2H). HRMS-ESI (+) m/z calculated for C18H11N4O3, 331.0831 [M+H]+; found: 331.0832.
10-(4-Hydroxyphenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (4c).15
Yellow solid, 68% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.21 (s, 1H), 10.03 (s, 1H), 9.13 (s, 1H), 8.39 (d, J = 8.2 Hz, 1H), 7.87 (d, J = 8.2 Hz, 1H), 7.20 (d, J = 8.6 Hz, 2H), 7.12 (s, 1H), 7.03 (d, J = 8.6 Hz, 2H). HRMS-ESI (+) m/z calculated for C18H11N4O3, 331.0831 [M+H]+; found: 331.0834.
2,4-Dioxo-10-phenyl-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (4d).15
Yellow solid (70% yield). 1H NMR (600 MHz, DMSO-d6) δ 11.25 (s, 1H), 9.17 (s, 1H), 8.42 (d, J = 8.2 Hz, 1H), 7.89 (d, J = 8.1 Hz, 1H), 7.72 (t, J = 7.6 Hz, 2H), 7.67 (t, J = 7.4 Hz, 1H), 7.44 (d, J = 7.7 Hz, 2H), 7.00 (s, 1H). HRMS-ESI (+) m/z calculated for C18H11N4O2, 315.0877 [M+H]+; found: 315.0876.
2,4-Dioxo-10-(4-(trifluoromethoxy)phenyl)-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (4e).
Yellow solid, 72% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.27 (s, 1H), 9.16 (s, 1H), 8.41 (d, J = 7.8 Hz, 1H), 7.89 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 7.6 Hz, 2H), 7.60 (d, J = 7.5 Hz, 2H), 7.19 (s, 1H). 13C NMR (150 MHz, DMSO-d6) δ 161.0, 158.7, 155.8, 148.5, 140.9, 140.8, 135.2, 132.1, 130.4, 126.2, 123.3, 122.5, 120.6, 117.8, 117.4, 115.5. HRMS-ESI (+) m/z calculated for C19H10F3N4O3, 399.0705 [M+H]+; found: 399.0707.
10-(4-(Difluoromethoxy)phenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (4f).
Yellow solid, 62% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.25 (s, 1H), 9.15 (s, 1H), 8.40 (d, J = 8.2 Hz, 1H), 7.89 (dd, J = 8.1, 1.3 Hz, 1H), 7.56 (t, J = 78 Hz, 1H), 7.52 – 7.48 (m, 4H), 7.15 (s, 1H). 13C NMR (150 MHz, DMSO-d6) δ 161.4, 159.1, 156.2, 151.7, 141.4, 141.2, 133.4, 132.5, 130.3, 126.5, 123.6, 121.0, 120.2, 118.2, 118.1 (t, J = 256.5 Hz), 117.8, 115.8. HRMS-ESI (+) m/z calculated for C19H11F2N4O3, 381.0799 [M+H]+; found: 381.0800.
10-(Benzo[d][1,3]dioxol-5-yl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (4g).
Yellow solid, 66% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.24 (s, 1H), 9.14 (s, 1H), 8.39 (d, J = 8.1 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.29 (s, 1H), 7.19 (d, J = 8.2 Hz, 1H), 7.04 (d, J = 1.5 Hz, 1H), 6.88 (dd, J = 8.1, 1.6 Hz, 1H), 6.21 (d, J = 1.9 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 159.0, 156.1, 148.5, 148.0, 141.5, 140.9, 132.2, 130.0, 126.2, 123.3, 121.6, 121.1, 117.9, 117.6, 115.6, 109.0, 108.9, 102.0. HRMS-ESI (+) m/z calculated for C19H11N4O4, 359.0780 [M+H]+; found: 359.0783.
10-(2,2-Difluorobenzo[d][1,3]dioxol-5-yl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (4h).
Yellow solid, 73% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.28 (s, 1H), 9.17 (s, 1H), 8.39 (d, J = 8.2 Hz, 1H), 7.89 (dd, J = 8.1, 1.2 Hz, 1H), 7.71 (d, J = 8.5 Hz, 1H), 7.59 (d, J = 1.9 Hz, 1H), 7.49 (s, 1H), 7.29 (dd, J = 8.5, 2.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 161.2, 159.0, 156.0, 143.7, 143.3, 141.3, 141.2, 132.6, 132.1, 126.5, 124.8, 123.3, 121.3, 117.8, 117.6, 115.8, 111.3, 111.0. HRMS-ESI (+) m/z calculated for C19H9F2N4O4, 395.0592 [M+H]+; found: 395.0596.
10-(4-(Benzyloxy)phenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (11a).
Yellow solid, 75% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.23 (s, 1H), 9.14 (s, 1H), 8.39 (d, J = 8.2 Hz, 1H), 7.87 (dd, J = 8.1, 1.2 Hz, 1H), 7.53 (d, J = 7.4 Hz, 2H), 7.44 (t, J = 7.5 Hz, 2H), 7.37–7.33 (m, 3H), 7.31 (d, J = 9.0 Hz, 2H), 7.12 (s, 1H), 5.23 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 159.0, 158.8, 156.1, 141.5, 140.8, 136.5, 132.3, 129.3, 129.2, 128.3, 127.84, 127.7, 126.1, 123.4, 120.9, 118.0, 117.6, 116.1, 115.5, 69.6. HRMS-ESI (+) m/z calculated for C25H17N4O3, 421.1301 [M+H]+; found: 421.1303.
10-(4-((4-Chlorobenzyl)oxy)phenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (11b).
Yellow solid, 58% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.23 (s, 1H), 9.15 (s, 1H), 8.40 (d, J = 8.1 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.56 (d, J = 8.1 Hz, 2H), 7.51 (d, J = 8.1 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 7.11 (s, 1H), 5.24 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.1, 158.8, 158.4, 155.9, 141.3, 140.6, 135.4, 132.2, 132.1, 129.3, 129.2, 128.2, 125.9, 125.1, 123.2, 120.7, 117.8, 117.4, 115.9, 115.3, 68.5. HRMS-ESI (+) m/z calculated for C25H16ClN4O3, 455.0911 [M+H]+; found: 455.0912.
10-(4-((4-Bromobenzyl) oxy) phenyl)-2, 4-dioxo-2, 3, 4, 10-tetrahydropyrimido [4, 5-b] quinoline-8-carbonitrile (11c).
Orange solid, 69% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.23 (s, 1H), 9.15 (s, 1H), 8.40 (d, J = 7.5 Hz, 1H), 7.88 (d, J = 7.4 Hz, 1H), 7.64 (d, J = 7.3 Hz, 2H), 7.50 (d, J = 7.2 Hz, 2H), 7.36 (d, J = 7.8 Hz, 2H), 7.31 (d, J = 7.0 Hz, 2H), 7.11 (s, 1H), 5.22 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ 161.8, 159.5, 159.1, 156.5, 142.0, 141.4, 141.2, 136.5, 131.6, 130.4, 130.2, 129.8, 129.7, 123.9, 121.4, 118.5, 118.1, 116.8, 116.4, 115.9, 69.2. HRMS-ESI (+) m/z calculated for C25H16BrN4O3, 499.0406 [M+H]+; found: 499.0407.
10-(4-((3-Chlorobenzyl)oxy)phenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinolone −8-carbonitrile (11d).
Yellow solid, 52% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.24 (s, 1H), 9.15 (s, 1H), 8.40 (d, J = 8.2 Hz, 1H), 7.88 (dd, J = 8.2, 1.2 Hz, 1H), 7.61 (s, 1H), 7.52 – 7.44 (m, 3H), 7.37 (d, J = 9.0 Hz, 2H), 7.32 (d, J = 9.0 Hz, 2H), 7.12 (s, 1H), 5.26 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 162.2, 159.9, 159.4, 156.9, 142.4, 141.7, 140.0, 133.8, 133.1, 131.1, 130.2, 128.6, 128.2, 127.1, 126.9, 124.3, 121.7, 118.9, 118.4, 117.0, 116.3, 69.5. HRMS-ESI (+) m/z calculated for C25H16ClN4O3, 455.0911 [M+H]+; found: 455.0915.
Methyl-3-((4-(8-cyano-2,4-dioxo-3,4-dihydropyrimido[4,5-b]quinolin-10(2H)-yl)phenoxy) methyl)benzoate (11e).
Yellow solid. 82% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.23 (s, 1H), 9.14 (s, 1H), 8.40 (d, J = 7.4 Hz, 1H), 8.13 (s, 1H), 7.97 (d, J = 7.4 Hz, 1H), 7.88 (d, J = 7.5 Hz, 1H), 7.82 (d, J = 7.4 Hz, 1H), 7.65 – 7.58 (m, 1H), 7.38–7.34 (m, 4H), 7.11 (s, 1H), 5.33 (s, 2H), 3.88 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 166.3, 161.7, 159.4, 159.0, 156.5, 142.0, 141.3, 137.8, 132.8, 132.7, 130.1, 129.8, 129.3, 129.0, 128.6, 126.5, 123.8, 121.3, 118.4, 118.0, 116.6, 115.9, 69.3, 52.5. HRMS-ESI (+) m/z calculated for C27H19N4O5, 479.1355 [M+H]+; found: 479.1357.
3-((4-(8-cyano-2,4-dioxo-3,4-dihydropyrimido[4,5-b]quinolin-10(2H)-yl)phenoxy) methyl)benzoic acid (11f).
Yellow solid. 52% yield; 1H NMR (600 MHz, DMSO-d6) δ 13.05 (s, 1H), 11.23 (s, 1H), 9.15 (s, 1H), 8.40 (d, J = 8.2 Hz, 1H), 8.11 (s, 1H), 7.95 (d, J = 7.7 Hz, 1H), 7.88 (dd, J = 8.2, 1.1 Hz, 1H), 7.78 (d, J = 7.6 Hz, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.37 (d, J = 8.9 Hz, 2H), 7.33 (d, J = 9.0 Hz, 2H), 7.12 (s, 1H), 5.31 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 167.5, 161.9, 159.6, 159.3, 156.7, 142.2, 141.4, 137.7, 132.9, 132.6, 131.5, 130.0, 129.3, 129.0, 126.7, 124.0, 121.5, 118.6, 118.2, 116.8, 116.1, 69.6. HRMS-ESI (+) m/z calculated for C26H17N4O5, 465.1199 [M+H]+; found: 465.1202.
2,4-Dioxo-10-(4-phenoxyphenyl)-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (11g).
Yellow solid, 79% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.25 (s, 1H), 9.15 (s, 1H), 7.89 (d, J = 8.0 Hz, 1H), 7.50 (t, J = 7.8 Hz, 2H), 7.42 (d, J = 8.6 Hz, 2H), 7.26 (t, J = 7.9 Hz, 5H), 7.22 (s, 1H). 13C NMR (150 MHz, DMSO-d6) δ 161.4, 159.2, 157.9, 156.3, 155.4, 141.6, 141.1, 132.4, 131.3, 130.3, 130.1, 126.4, 124.5, 123.6, 121.1, 119.9, 119.1, 118.2, 117.8, 115.8. HRMS-ESI (+) m/z calculated for C24H15N4O3, 407.1144 [M+H]+; found: 407.1146.
10-(4-(4-chlorophenoxy)phenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (11h).
Yellow solid, 79% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.25 (s, 1H), 9.16 (s, 1H), 8.41 (d, J = 8.2 Hz, 1H), 7.90 (dd, J = 8.2, 1.3 Hz, 1H), 7.57 – 7.50 (m, 2H), 7.50 – 7.40 (m, 2H), 7.32 – 7.28 (m, 4H), 7.24 (s, 1H). 13C NMR (150 MHz, DMSO-d6) δ 161.2, 158.9, 157.2, 156.0, 154.2, 141.3, 140.8, 132.2, 131.4, 129.9, 129.8, 128.0, 126.2, 123.4, 121.3, 120.8, 119.1, 117.9, 117.5, 115.5. HRMS-ESI (+) m/z calculated for C24H14ClN4O3, 441.0754 [M+H]+; found: 441.0756.
10-(4-(4-Fluorophenoxy)phenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (11i).
Yellow solid, 81% yield; 1H NMR (400 MHz, DMSO-d6) δ 11.24 (s, 1H), 9.15 (s, 1H), 8.40 (d, J = 8.2 Hz, 1H), 7.89 (dd, J = 8.1, 1.2 Hz, 1H), 7.41 (dd, J = 9.4, 2.5 Hz, 2H), 7.35–7.31 (m, 4H), 7.25 – 7.20 (m, 3H). 13C NMR (150 MHz, DMSO-d6) δ 161.7, 159.8 (d, J = 238.5 Hz), 159.4, 158.5, 156.5, 151.6, 141.8, 141.3, 132.7, 131.5, 130.3, 126.7, 123.9, 122.3 (d, J = 7.5 Hz), 121.3, 118.9, 118.4, 118.0, 117.2 (d, J = 24 Hz), 116.0. HRMS-ESI (+) m/z calculated for C24H14FN4O3, 425.1050 [M+H]+; found: 425.1052.
Methyl-3-(4-(8-cyano-2,4-dioxo-3,4-dihydropyrimido[4,5-b]quinolin-10(2H)-yl)phenoxy)benzoate (11j).
Yellow solid, 75% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.25 (s, 1H), 9.15 (s, 1H), 8.40 (d, J = 8.1 Hz, 1H), 7.89 (dd, J = 8.1, 1.3 Hz, 1H), 7.83 (d, J = 7.9 Hz, 1H), 7.73 – 7.68 (m, 1H), 7.65 (t, J = 7.9 Hz, 1H), 7.59 – 7.55 (m, 1H), 7.47 – 7.44 (m, 2H), 7.32 (dd, J = 9.3, 2.6 Hz, 2H), 7.23 (s, 1H), 3.87 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 165.3, 161.3, 159.0, 157.1, 156.1, 155.8, 141.4, 140.9, 132.3, 131.8, 131.5, 130.7, 130.1, 126.3, 124.8, 124.4, 123.4, 120.9, 119.6, 119.5, 118.0, 117.6, 115.6, 52.2. HRMS-ESI (+) m/z calculated for C26H17N4O5, 465.1199 [M+H]+; found: 465.1202.
3-(4-(8-Cyano-2,4-dioxo-3,4-dihydropyrimido[4,5-b]quinolin-10(2H)-yl)phenoxy)benzoic acid (11k).
Yellow solid, 67% yield; 1H NMR (400 MHz, DMSO-d6) δ 11.25 (s, 1H), 9.15 (s, 1H), 8.41 (d, J = 8.2 Hz, 1H), 7.89 (dd, J = 8.2, 1.0 Hz, 1H), 7.81 (d, J = 7.6 Hz, 1H), 7.72 (s, 1H), 7.61 (t, J = 7.9 Hz, 1H), 7.51 (dd, J = 7.9, 2.0 Hz, 1H), 7.45 (d, J = 8.8 Hz, 2H), 7.31 (d, J = 8.8 Hz, 2H), 7.24 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 166.6, 161.3, 159.0, 157.3, 156.1, 155.5, 141.4, 140.9, 133.5, 132.3, 131.6, 130.4, 130.1, 126.3, 125.0, 123.7, 123.4, 120.9, 119.8, 119.4, 118.0, 117.6, 115.6. HRMS-ESI (+) m/z calculated for C25H15N4O5, 451.1042 [M+H]+; found: 451.1046.
10-(3-(Benzyloxy)phenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (11l).
Yellow solid, 81% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.25 (s, 1H), 9.16 (s, 1H), 8.40 (d, J = 8.1 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.63 (t, J = 8.0 Hz, 1H), 7.48 (d, J = 7.3 Hz, 2H), 7.40 (t, J = 7.3 Hz, 2H), 7.36 – 7.28 (m, 2H), 7.16 (s, 1H), 7.05 (s, 1H), 7.01 (d, J = 7.5 Hz, 1H), 5.15 (s, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.3, 159.6, 158.6, 156.1, 141.0, 140.9, 137.7, 136.3, 132.3, 131.1, 128.3, 127.8, 126.2, 123.4, 120.8, 120.3, 118.0, 117.6, 116.1, 115.6, 114.7, 69.5. HRMS-ESI (+) m/z calculated for C25H17N4O3, 421.1301 [M+H]+; found: 421.1305.
2,4-Dioxo-10-(3-phenoxyphenyl)-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (11m).
Yellow solid, 71% yield; 1H NMR (600 MHz, DMSO-d6) δ 11.24 (s, 1H), 9.13 (s, 1H), 8.39 (d, J = 8.1 Hz, 1H), 7.89 (d, J = 8.1 Hz, 1H), 7.70 (t, J = 8.1 Hz, 1H), 7.43 (t, J = 7.7 Hz, 2H), 7.26 (d, J = 9.1 Hz, 2H), 7.23 – 7.15 (m, 5H). 13C NMR (150 MHz, DMSO-d6) δ 161.3, 158.7, 158.2, 156.1, 155.7, 141.0, 140.9, 137.9, 132.3, 131.6, 130.1, 126.3, 124.0, 123.5, 122.9, 120.9, 119.2, 119.1, 118.2, 118.1, 117.7, 115.7. HRMS-ESI (+) m/z calculated for C24H15N4O3, 407.1144 [M+H]+; found: 407.1148.
N-(4-(3-(4-chlorophenyl)-8-cyano-2,4-dioxo-3,4-dihydropyrimido[4,5-b]quinolin-10(2H)-yl)phenyl)methanesulfonamide (12j).
Compound 12j was synthesized using the general procedure described using compound 4a with 27q as a starting material. Yellow solid, 43% yield; 1H NMR (600 MHz, DMSO-d6) δ 10.28 (s, 1H), 9.26 (s, 1H), 8.47 (d, J = 8.2 Hz, 1H), 7.92 (dd, J = 8.2, 1.2 Hz, 1H), 7.56 – 7.52 (m, 2H), 7.49 (d, J = 8.8 Hz, 2H), 7.41 (d, J = 8.8 Hz, 2H), 7.30 – 7.26 (m, 2H), 7.25 (s, 1H), 3.20 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 161.4, 158.4, 155.5, 142.6, 141.8, 140.2, 135.7, 133.0, 132.9, 131.7, 131.0, 129.8, 129.3, 127.1, 124.1, 121.7, 120.4, 118.4, 118.2, 116.5, 40.5. HRMS-ESI (+) m/z calculated for C25H17ClN5O4S, 518.0690 [M+H]+; found: 518.0694.
N-(3-(3-(4-Chlorophenyl)-8-cyano-2,4-dioxo-3,4-dihydropyrimido[4,5-b]quinolin-10(2H)-yl)phenyl)methanesulfonamide (12k).
Compound 23p was synthesized using the procedure described for compound 4a with 27p as a starting material. Yellow solid, 52% yield; 1H NMR (600 MHz, DMSO-d6) δ 10.22 (s, 1H), 9.26 (s, 1H), 8.47 (d, J = 8.2 Hz, 1H), 7.92 (dd, J = 8.1, 1.4 Hz, 1H), 7.68 (t, J = 8.1 Hz, 1H), 7.57 – 7.51 (m, 2H), 7.46 (dd, J = 8.3, 1.4 Hz, 1H), 7.31–7.29 (m, 2H), 7.28 (d, J = 2.1 Hz, 1H), 7.27 (t, J = 2.1 Hz, 1H), 7.19 – 7.16 (m, 1H), 3.12 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 161.3, 158.1, 155.5, 142.6, 141.5, 140.9, 137.7, 135.7, 133.0, 132.9, 131.8, 131.0, 129.3, 127.2, 124.1, 123.5, 121.7, 120.8, 118.7, 118.5, 118.1, 116.6, 40.5. HRMS-ESI (+) m/z calculated for C25H17ClN5O4S, 518.0690 [M+H]+; found: 518.0691.
10-(4-Hydroxyphenyl)-3-methyl-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (31).
Yellow solid, 81% yield; 1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 9.19 (s, 1H), 8.43 (d, J = 8.1 Hz, 1H), 7.88 (dd, J = 8.1, 1.2 Hz, 1H), 7.19 (d, J = 8.7 Hz, 2H), 7.15 (s, 1H), 7.03 (d, J = 8.7 Hz, 2H), 3.22 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 160.7, 158.0, 157.3, 155.5, 141.4, 141.3, 132.2, 129.0, 127.1, 125.9, 123.3, 120.8, 117.5, 117.2, 116.5, 115.4, 27.2. HRMS-ESI (+) m/z calculated for C19H13N4O3, 345.0988 [M+H]+; found: 345.0991.
General procedure for the synthesis of deazaflavin derivatives 12 a-i.
To a suspension of compound 4b or 4c (0.1 g, 1.0 eq) in DMF (2 mL) was added phenylboronic acid derivative 26 (3.0 eq), Cu(OAc)2 (0.06 g, 0.30 mmol, 1.0 eq) and stirred at room temperature under air. The reaction was continued until the disappearance of starting material (typically between 24–48 h) and water (10 mL) was added to it. The resulting solution was extracted with EtOAc (3 × 20 mL), washed with brine and evaporated in vacuo to leave the crude product. The crude product was purified using Combi flash with 0–100% EtOAc in hexane as an eluent to furnish the desired product.
10-(4-Hydroxyphenyl)-2,4-dioxo-3-phenyl-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (12a).44
Pale yellow solid, 56% yield. 1H NMR (400 MHz, DMSO-d6) δ 10.06 (s, 1H), 9.23 (s, 1H), 8.45 (d, J = 8.2 Hz, 1H), 7.90 (dd, J = 8.2, 1.1 Hz, 1H), 7.47 (t, J = 7.5 Hz, 2H), 7.40 (t, J = 7.5 Hz, 1H), 7.26–7.22 (m, 4H), 7.19 (s, 1H), 7.05 (d, J = 8.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 161.1, 158.4, 158.1, 155.4, 142.0, 141.8, 136.5, 132.6, 129.4, 128.9, 128.7, 127.9, 127.5, 126.4, 123.7, 121.3, 118.2, 117.8, 116.9, 115.9. HRMS-ESI (+) m/z calculated for C24H15N4O3, 407.1144 [M+H]+; found: 407.1146.
3-(4-Chlorophenyl)-10-(4-hydroxyphenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (12b).
Yellow solid, 80% yield; 1H NMR (600 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.25 (s, 1H), 8.46 (d, J = 8.1 Hz, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.54 (d, J = 8.5 Hz, 2H), 7.29 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 8.6 Hz, 2H), 7.20 (s, 1H), 7.05 (d, J = 8.6 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 161.0, 158.4, 158.1, 155.2, 142.0, 141.8, 135.4, 132.6, 132.5, 130.6, 129.3, 128.9, 127.4, 126.5, 123.7, 121.3, 118.1, 117.8, 116.8, 116.0. HRMS-ESI (+) m/z calculated for C24H14ClN4O3, 441.0754 [M+H]+; found: 441.0756.
3-(4-Fluorophenyl)-10-(4-hydroxyphenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (12c).
Yellow solid, 30% yield; 1H NMR (600 MHz, DMSO-d6) δ 10.09 (s, 1H), 9.24 (s, 1H), 8.46 (d, J = 8.2 Hz, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.33 – 7.27 (m, 4H), 7.24 (d, J = 8.6 Hz, 2H), 7.20 (s, 1H), 7.05 (d, J = 8.6 Hz, 2H). HRMS-ESI (+) m/z calculated for C24H14FN4O3, 425.1050 [M+H]+; found: 425.1051.
3-(4-Bromophenyl)-10-(4-hydroxyphenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (12d).
Yellow solid, 52% yield; 1H NMR (600 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.24 (s, 1H), 8.46 (d, J = 8.2 Hz, 1H), 7.91 (dd, J = 8.2, 1.2 Hz, 1H), 7.73 – 7.63 (m, 2H), 7.25 – 7.21 (m, 4H), 7.20 (s, 1H), 7.06 – 7.03 (m, 2H). 13C NMR (150 MHz, DMSO-d6) δ 161.7, 159.1, 158.8, 155.8, 142.5, 136.5, 133.4, 132.7, 131.8, 131.5, 130.2, 129.9, 128.1, 127.2, 124.4, 121.7, 118.8, 118.5, 117.7, 116.7. HRMS-ESI (+) m/z calculated for C24H14BrN4O3, 485.0249 [M+H]+; found: 485.0250.
Methyl-3-(8-cyano-10-(4-hydroxyphenyl)-2,4-dioxo-4,10-dihydropyrimido[4,5-b]quinolin-3(2H)-yl)benzoate (12e).
Yellow solid, 59% yield; 1H NMR (600 MHz, DMSO-d6) δ 10.09 (s, 1H), 9.24 (s, 1H), 8.47 (d, J = 7.8 Hz, 1H), 7.99 (d, J = 7.4 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.85 (s, 1H), 7.64 (t, J = 7.4 Hz, 1H), 7.55 (d, J = 6.8 Hz, 1H), 7.29 – 7.18 (m, 3H), 7.06 (d, J = 7.8 Hz, 2H), 3.87 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ 165.9, 161.4, 158.6, 158.4, 155.6, 142.2, 142.0, 137.2, 134.0, 132.9, 130.7, 129.8, 129.8, 129.6, 128.9, 127.7, 126.8, 123.9, 121.6, 118.4, 118.1, 117.1, 116.2, 52.6. HRMS-ESI (+) m/z calculated for C26H17N4O5, 465.1199 [M+H]+; found: 465.1199.
3-(8-cyano-10-(4-hydroxyphenyl)-2,4-dioxo-4,10-dihydropyrimido[4,5-b]quinolin-3(2H)-yl)benzamide (12f).
Yellow solid, 72% yield; 1H NMR (600 MHz, DMSO-d6) δ 10.08 (s, 1H), 9.26 (s, 1H), 8.47 (d, J = 8.2 Hz, 1H), 8.02 (s, 1H), 7.91 (d, J = 8.1 Hz, 2H), 7.74 (s, 1H), 7.56 (t, J = 7.8 Hz, 1H), 7.44 – 7.39 (m, 2H), 7.25 (d, J = 8.7 Hz, 2H), 7.20 (s, 1H), 7.05 (d, J = 8.7 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ 167.3, 161.3, 158.6, 158.3, 155.5, 142.3, 142.0, 136.7, 135.3, 132.8, 131.8, 129.6, 129.1, 128.2, 127.7, 127.1, 126.7, 123.9, 121.6, 118.3, 118.0, 117.1, 116.2. HRMS-ESI (+) m/z calculated for C25H16N5O4, 450.1202 [M+H]+; found: 450.1202.
3-(4-Chlorophenyl)-10-(3-hydroxyphenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (12g).
Yellow solid, 65% yield; 1H NMR (600 MHz, DMSO-d6) δ 10.09 (s, 1H), 9.26 (s, 1H), 8.47 (d, J = 8.2 Hz, 1H), 7.92 (dd, J = 8.2, 1.2 Hz, 1H), 7.56 – 7.50 (m, 3H), 7.29 (d, J = 8.6 Hz, 2H), 7.17 (s, 1H), 7.07 (dd, J = 8.2, 1.7 Hz, 1H), 6.86 (d, J = 7.8 Hz, 1H), 6.83 (d, J = 2.0 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 160.8, 158.9, 157.4, 155.0, 141.9, 140.9, 137.2, 135.2, 132.4, 132.3, 131.1, 130.4, 128.7, 126.3, 123.4, 121.0, 118.2, 117.9, 117.6, 116.8, 115.8, 115.0. HRMS-ESI (+) m/z calculated for C24H14ClN4O3, 441.0754 [M+H]+; found: 441.0749.
3-(3-Chlorophenyl)-10-(3-hydroxyphenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (12h).
Yellow solid, 51% yield; 1H NMR (600 MHz, DMSO-d6) δ 10.09 (s, 1H), 9.26 (s, 1H), 8.48 (d, J = 8.2 Hz, 1H), 7.92 (dd, J = 8.1, 1.3 Hz, 1H), 7.54 – 7.48 (m, 3H), 7.40 (t, J = 1.8 Hz, 1H), 7.25 (dt, J = 7.4, 1.6 Hz, 1H), 7.18 (s, 1H), 7.07 (dd, J = 8.3, 1.7 Hz, 1H), 6.87 – 6.84 (m, 1H), 6.83 (t, J = 2.1 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 160.8, 158.9, 157.4, 155.0, 141.9, 140.9, 137.2, 135.2, 132.4, 132.3, 131.1, 130.4, 128.7, 126.3, 123.4, 121.0, 118.2, 117.9, 117.6, 116.8, 115.8, 115.0. HRMS-ESI (+) m/z calculated for C24H14ClN4O3, 441.0754 [M+H]+; found: 441.0755.
3-(3,4-Dichlorophenyl)-10-(3-hydroxyphenyl)-2,4-dioxo-2,3,4,10-tetrahydropyrimido[4,5-b]quinoline-8-carbonitrile (12i).
Yellow solid, 48% yield; 1H NMR (600 MHz, DMSO-d6) δ 10.11 (s, 1H), 9.28 (s, 1H), 8.49 (d, J = 8.2 Hz, 1H), 7.94 (dd, J = 8.2, 1.5 Hz, 1H), 7.77 (d, J = 8.5 Hz, 1H), 7.65 (d, J = 2.3 Hz, 1H), 7.52 (t, J = 8.0 Hz, 1H), 7.32 (dd, J = 8.5, 2.3 Hz, 1H), 7.19 (s, 1H), 7.07 (dd, J = 8.2, 1.5 Hz, 1H), 6.91 – 6.78 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.5, 159.7, 158.2, 155.5, 142.8, 141.7, 138.0, 137.1, 133.2, 131.9, 131.7, 131.5, 131.4, 130.0, 127.3, 124.2, 121.9, 119.0, 118.6, 118.4, 117.6, 116.7, 115.8. HRMS-ESI (+) m/z calculated for C24H13Cl2N4O3, 475.0365 [M+H]+; found: 475.0367.
Biology
14M_zTDP2 fluorescence-based biochemical assay
The reaction buffer used was composed of 50 mM Tris-HCl pH 7.4, 10 mM MgCl2, 80 mM KCl, 0.05 % (v/v) Tween-20, and 1 mM DTT. To a black 384-well plate, 10 μL of compound solution (in reaction buffer, concentration 2-fold higher than the tested concentration, final DMSO concentration of 2.5 %) was added, followed by addition of 5 μL of 14M_TDP2 enzyme (25 pM, final concentration of 6.25 pM). After a pre-incubation period of 10 minutes, 5 μL of substrate 5’-(6-FAM-NHS)(5’-tyrosine)GATCT(3’-BHQ-1)-3’ (4 μM, final concentration of 1 μM) was added, and the reaction was allowed to proceed at 25 °C for 60 minutes. The fluorescence of the product was measured using a SpectraMax M5e (Molecular Devices) (λex 285 nm; λem 325 nm; λcutoff 315nm) in kinetic-mode at 25 °C for 60 minutes.21 IC50 experiments data using 12 concentrations of inhibitor (2-fold dilutions) and vehicle-alone were fitted by GraphPad Prism software. IC50 determinations represent the means of three independent experiments performed in triplicate.
Sensitizing assay in DT40 cells
Drug cellular sensitivity was measured as previously described.19 Cells were continuously exposed to various drug concentrations for 72 h. Fifteen hundred DT40 cells/well were seeded into a 96-well white plate (PerkinElmer) and treated with drugs as single agents or in combination in 150 μL of medium. Cell viability was determined at 72 h by adding 75 μL of ATPlite solution (ATPlite 1-step kit, PerkinElmer). After 15 min incubation, luminescence was measured on an EnVision Plate Reader (PerkinElmer). The ATP level in untreated cells was defined as 100%, and viability of treated cells was defined as ATP level of treated cell/ATP level of untreated cells × 100.
Parallel artificial membrane permeability assay (PAMPA)
The membrane permeability of selected compounds were evaluated using the Corning Gentest Pre-coated PAMPA Plate System (Cat. No. 353015). The pre-coated plate assembly, which was stored at −20°C, was taken to thaw for 30 min at room temperature. The permeability assay was carried out in accordance with the manufacturer’s protocol. In summary, the 96-well filter plate, pre-coated with lipids, was used as the permeation acceptor and a matching 96 well receiver plate was used as the permeation donor. Compound solutions were prepared by diluting 10 mM DMSO stock solutions in DPBS to a final concentration of 10 μM. The compound solutions were added to the wells (300 μL/well) of the receiver plate and DPBS was added to the wells (200 μL/well) of the pre-coated filter plate. The filter plate was then coupled with the receiver plate and the plate assembly was incubated at 25 °C without agitation for five hours. At the end of the incubation, the plates were separated and the final concentrations of compounds in both donor wells and acceptor wells were analyzed using LC-MS/MS. Permeability of the compounds were calculated using the following formula Pe = {-ln [1-CA(t)/Ceq]} /[A×(1/VD+1/VA)×t], where A = filter area (0.3 cm2), VD = donor well volume (0.3 mL), VA = acceptor well volume (0.2 mL), t = incubation time (seconds), CA(t) = compound concentration in acceptor well at time t, CD(t) = compound concentration in donor well at time t, and Ceq = [CD(t)×VD+CA(t)×VA]/(VD+VA). A cutoff criterion of Pe value at 1.5 × 10−6 cm/s was used to classify the compounds into high and low permeability according to the literature report of this PAMPA plate system.45
MTT cytotoxicity assay
Hep G2 cells (human liver carcinoma) were maintained in growth media: DMEM:F12 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen). HeLa cells (human cervical adenocarcinoma) were maintained in growth media: DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen). Cells were plated in 96-well plates at 1 – 2.5 × 104 cells per well in growth media. Compounds were added to the plate in 3-fold dilution made in DMSO, starting at 20 mM, and 1 μL of compound solution was added to each well. The final volume in each well was 200 μL, yielding a final DMSO concentration of 0.5%. Compounds were tested at 9, 3x dilutions from 100 uM final concentration in growth media. Control wells contained 0.5% DMSO (positive control) or 50% DMSO (negative control) and all reactions were done in triplicate. The plate was incubated for 72 h at 37 °C in a 5% CO2/ 95% air humidified atmosphere. Measurement of cell viability was carried out using a modified method of Mosmann46 based on 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT). MTT was prepared fresh at 1 mg/mL in serum-free, phenol red-free RPMI 1640 media. MTT solution (200 μL) was added to each well, and the plate was incubated as described above for 3 h. The MTT solution was removed, and the formazan crystals were solubilized with 200 μL of isopropanol. The plate was read on a SpectraMax i3 spectrophotometer (Molecular Devices) at 570 nm for formazan and 650 nm for background subtraction. CC50 values were calculated by fitting the data in GraphPad Prism software.
Molecular modeling and docking analysis
Molecular modeling was performed using the Schrodinger small molecule drug discovery suite 2015–4.47 The crystal structure of catalytic domain of humanized mouse TDP2 in complex with 4c as reported by Hornyak et.al18 was retrieved from the protein data bank (PDB code: 5J42). The above structure was analyzed using Maestro48 (Schrodinger Inc.49) and subjected to docking protocol which involves several steps including preparing protein of interest, grid generation, ligand preparation and docking. The crystal structure was refined using protein preparation wizard48 (Schrodinger Inc.49) in which missing hydrogen atoms and side chains were added and minimized using OPLS 2005 force field50 to optimize hydrogen bonding network and converge the heavy atoms to an rmsd of 0.3 Å. In the processed crystal structure, the two chains (A & B) were separated and analyzed separately. In both chains, 4c forms a key hydrogen bond interaction between the nitrogen of pyrimido-dione ring and the side chain of R276 (R266_hTDP2). The side chain of R241 (R231_hTDP2) was positioned to make a π - π interaction with the face of the tricyclic core of 4c and the phenolic hydroxyl group of 4c was observed to make water mediated interaction with the catalytic side chain of N274 (N264_hTDP2). The tricyclic core of deazaflavin extends in to a hydrophobic pocket lined by W307 (W297_hTDP2), L323 (L313_hTDP2) and F325 (F315_hTDP2). Chain B was chosen for further docking analysis. The receptor grid generation tool in Maestro (Schrodinger Inc.)51 was used to define an active site around the native ligand (4c) to cover all the residues within 12 Å. All the compounds were drawn using Maestro and subjected to Lig Prep52 to generate conformers, possible protonation at pH of 7±3 that serves as an input for docking process. All the dockings were performed using Glide XP53 (Glide54, version 6.9) with the van der Waals radii of nonpolar atoms for each of the ligands were scaled by a factor of 0.8. The solutions were further refined by post docking and minimization under implicit solvent to account for protein flexibility. The residue numbers of TDP2 used in the discussion and the figures were based on the human TDP2.
Supplementary Material
Figure 4.

Effect of TDP2 inhibitors 4a, 12a, 12b, 12h, 11a and 11e on potentiating toxic action of ETP.
Figure 5.

Molecular modeling of 11k and 12b. (A) Binding mode of 4c within the crystal structure of catalytic domain of humanized mouse TDP2 (PDB code: 5J4218). (B) Potential vectors for designing novel deazaflavin inhibitor types. (C) Predicted binding mode of 11k within the catalytic domain of humanized mouse TDP2. (D) Predicted binding mode of 12b within the catalytic domain of humanized mouse TDP2. Key residues are highlighted in green sticks. H-bond interactions are depicted as black dotted lines. Cation- π and π-π interaction are represented as double headed arrow in black. Water molecule and magnesium ion were represented as red and blue non-bonded sphere. All the residue numberings are based on the human TDP2.
Table 3.
TDP2 inhibitory activity and PAMPA permeability for analogues from N-3 modifications (12a-i).
 | |||
|---|---|---|---|
| Compd | R′ | TDP2 IC50 (μM)a | PAMPA Pe (10−6 cm/s)b |
| 4c | -- | 0.14 ± 0.01 | 0.008 |
| 12a | H | 0.033 ± 0.001 | 0.1 |
| 12b | 4′ Cl | 0.033 ± 0.002 | 0.08 |
| 12c | 4′ F | 0.070 ± 0.003 | 0.06 |
| 12d | 4′ Br | 0.065 ± 0.003 | 0.05 |
| 12e | 3′ CO2Me | 0.038 ± 0.003 | 0.03 |
| 12f | 3′ CONH2 | 0.035 ± 0.000 | 0.01 |
| 4b | -- | 0.042 ± 0.004 | 0.01 |
| 12g | 4′ Cl | 0.0073 ± 0.0003 | 0.04 |
| 12h | 3′ Cl | 0.020 ± 0.002 | 0.07 |
| 12i | 3′, 4′ di-Cl | 0.014 ± 0.000 | 0.03 |
IC50: concentration of a compound producing 50% inhibition, expressed as mean ± standard deviation from three independent experiments performed in triplicate.
Pe: effective permeability coefficients determined using the PAMPA, measured in five replicates.
ACKNOWLEDGMENTS
This research was supported by the Academic Health Center Faculty Research Development Grant Program (FRD #14.23), University of Minnesota (to HA and ZW), and in part by the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute (Z01 BC 006161–17) and NIGMS R35 GM118047 (to HA). We acknowledge Professor Bert Semler at University of California, Irvine and Professor Haitao Guo at Indiana University School of Medicine, for providing HeLa and HepG2 Cells, respectively, and the Minnesota Supercomputing Institute (MSI) for computational resources.
ABBREVIATIONS USED
- TOP2
topoisomerase II
- TOP2cc
TOP2 cleavage complex
- TDP2
tyrosyl DNA phosphodiesterase 2
- ETP
etoposide
- HBV
hepatitis B virus
- SAR
structure-activity-relationship
- PAMPA
Parallel Artificial Membrane Permeation Assay
Footnotes
Supporting Information Available. All curves from DT40 cell sensitization experiments, additional docking poses of compound 11a and 11e with docking scores, general procedures for the synthesis of intermediates 15, 19, 20, 21, 24, 25, 28, 29, and 30 and their respective 1H NMR data, as well as 1H and 13C NMR spectra for all tested compounds. Molecular formula strings (CSV). Docked 11a with 5J42 (PDB). Docked 11e with 5J42 (PDB). Docked 11k with 5J42 (PDB). Docked 12b with 5J42 (PDB). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pommier Y; Sung YL; Huang SYN; Nitiss JL, Roles of Eukaryotic Topoisomerases in Transcription, Replication and Genomic Stability. Nat. Rev. Mol. Cell. Bio 2016, 17, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nitiss JL, DNA Topoisomerase II and Its Growing Repertoire of Biological Functions. Nat. Rev. Cancer 2009, 9, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pommier Y, Drugging Topoisomerases: Lessons and Challenges. ACS. Chem. Biol 2013, 8, 82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pommier Y; Leo E; Zhang HL; Marchand C, DNA Topoisomerases and Their Poisoning by Anticancer and Antibacterial Drugs. Chem. Biol 2010, 17, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gomez-Herreros F; Romero-Granados R; Zeng ZH; Alvarez-Quilon A; Quintero C; Ju LM; Umans L; Vermeire L; Huylebroeck D; Caldecott KW; Cortes-Ledesma F, Tdp2-Dependent Non-Homologous End-Joining Protects against Topoisomerase II-Induced DNA Breaks and Genome Instability in Cells and in Vivo. PLOS Genet. 2013, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gomez-Herreros F; Schuurs-Hoeijmakers JHM; McCormack M; Greally MT; Rultenl S; Romero-Granados R; Counihan TJ; Chaila E; Conroy J; Ennis S; Delanty N; Cortes-Ledesma F; de Brouwer APM; Cavallerit GL; El-Khamisy SF; de Vries BBA; Caldecott KW, Tdp2 Protects Transcription from Abortive Topoisomerase Activity and Is Required for Normal Neural Function. Nat. Genet 2014, 46, 516–521. [DOI] [PubMed] [Google Scholar]
- 7.Ledesma FC; El Khamisy SF; Zuma MC; Osborn K; Caldecott KW, A Human 5 ‘-Tyrosyl DNA Phosphodiesterase That Repairs Topoisomerase-Mediated DNA Damage. Nature 2009, 461, 674–678. [DOI] [PubMed] [Google Scholar]
- 8.Zeng ZH; Cortes-Ledesma F; El Khamisy SF; Caldecott KW, Tdp2/Ttrap Is the Major 5 ‘-Tyrosyl DNA Phosphodiesterase Activity in Vertebrate Cells and Is Critical for Cellular Resistance to Topoisomerase II-Induced DNA Damage. J. Biol. Chem 2011, 286, 403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maede Y; Shimizu H; Fukushima T; Kogame T; Nakamura T; Miki T; Takeda S; Pommier Y; Murai J, Differential and Common DNA Repair Pathways for Topoisomerase I- and II-Targeted Drugs in a Genetic Dt40 Repair Cell Screen Panel. Mol. Cancer Ther 2014, 13, 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Do PM; Varanasi L; Fan SQ; Li CY; Kubacka I; Newman V; Chauhan K; Daniels SR; Boccetta M; Garrett MR; Li RZ; Martinez LA, Mutant P53 Cooperates with Ets2 to Promote Etoposide Resistance. Gene Dev. 2012, 26, 830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nitiss JL, Targeting DNA Topoisomerase II in Cancer Chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koniger C; Wingert I; Marsmann M; Rosler C; Beck J; Nassal M, Involvement of the Host DNA-Repair Enzyme Tdp2 in Formation of the Covalently Closed Circular DNA Persistence Reservoir of Hepatitis B Viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maciejewski S; Nguyen JH; Gomez-Herreros F; Cortes-Ledesma F; Caldecott KW; Semler BL, Divergent Requirement for a DNA Repair Enzyme During Enterovirus Infections. MBio 2015, 7, e01931–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Virgen-Slane R; Rozovics JM; Fitzgerald KD; Ngo T; Chou W; van der Heden van Noort GJ; Filippov DV; Gershon PD; Semler BL, An Rna Virus Hijacks an Incognito Function of a DNA Repair Enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 14634–14639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raoof A; Depledge P; Hamilton NM; Hamilton NS; Hitchin JR; Hopkins GV; Jordan AM; Maguire LA; McGonagle AE; Mould DP; Rushbrooke M; Small HF; Smith KM; Thomson GJ; Turlais F; Waddell ID; Waszkowycz B; Watson AJ; Ogilvie DJ, Toxoflavins and Deazaflavins as the First Reported Selective Small Molecule Inhibitors of Tyrosyl-DNA Phosphodiesterase II. J. Med. Chem 2013, 56, 6352–6370. [DOI] [PubMed] [Google Scholar]
- 16.Kont YS; Dutta A; Mallisetty A; Mathew J; Minas T; Kraus C; Dhopeshwarkar P; Kallakury B; Mitra S; Uren A; Adhikari S, Depletion of Tyrosyl DNA Phosphodiesterase 2 Activity Enhances Etoposide-Mediated Double-Strand Break Formation and Cell Killing. DNA Repair 2016, 43, 38–47. [DOI] [PubMed] [Google Scholar]
- 17.Kankanala J; Marchand C; Abdelmalak M; Aihara H; Pommier Y; Wang ZQ, Isoquinoline-1,3-Diones as Selective Inhibitors of Tyrosyl DNA Phosphodiesterase II (Tdp2). J. Med. Chem 2016, 59, 2734–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hornyak P; Askwith T; Walker S; Komulainen E; Paradowski M; Pennicott LE; Bartlett EJ; Brissett NC; Raoof A; Watson M; Jordan AM; Ogilvie DJ; Ward SE; Atack JR; Pearl LH; Caldecott KW; Oliver AW, Mode of Action of DNA-Competitive Small Molecule Inhibitors of Tyrosyl DNA Phosphodiesterase 2. Biochem. J 2016, 473, 1869–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchand C; Abdelmalak M; Kankanala J; Huang SY; Kiselev E; Fesen K; Kurahashi K; Sasanuma H; Takeda S; Aihara H; Wang ZQ; Pommier Y, Deazaflavin Inhibitors of Tyrosyl-DNA Phosphodiesterase 2 (Tdp2) Specific for the Human Enzyme and Active against Cellular Tdp2. ACS Chem. Biol 2016, 11, 1925–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kossmann BR; Abdelmalak M; Lopez S; Tender G; Yan C; Pommier Y; Marchand C; Ivanov I, Discovery of Selective Inhibitors of Tyrosyl-DNA Phosphodiesterase 2 by Targeting the Enzyme DNA-Binding Cleft. Bioorg. Med. Chem. Lett 2016, 26, 3232–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ribeiro CJA; Kankanala J; Shi K; Kurahashi K; Kiselev E; Ravji A; Pommier Y; Aihara H; Wang ZQ, New Fluorescence-Based High-Throughput Screening Assay for Small Molecule Inhibitors of Tyrosyl-DNA Phosphodiesterase 2 (Tdp2). Eur. J. Pharm. Sci 2018, 118, 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribeiro CJA; Kankanala J; Xie J; Williams J; Aihara H; Wang Z, Triazolopyrimidine and Triazolopyridine Scaffolds as Tdp2 Inhibitors. Bioorg. Med. Chem. Lett 2019, 29, 257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beck DE; Lv W; Abdelmalak M; Plescia CB; Agama K; Marchand C; Pommier Y; Cushman M, Synthesis and Biological Evaluation of New Fluorinated and Chlorinated Indenoisoquinoline Topoisomerase I Poisons. Bioorg. Med. Chem 2016, 24, 1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang P; Elsayed MSA; Plescia CB; Ravji A; Redon CE; Kiselev E; Marchand C; Zeleznik O; Agama K; Pommier Y; Cushman M, Synthesis and Biological Evaluation of the First Triple Inhibitors of Human Topoisomerase 1, Tyrosyl-DNA Phosphodiesterase 1 (Tdp1), and Tyrosyl-DNA Phosphodiesterase 2 (Tdp2). J. Med. Chem 2017, 60, 3275–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu L-M; Hu Z; Chen Y; Ravji A; Lopez S; Plescia CB; Yu Q; Yang H; Abdelmalak M; Saha S; Agama K; Kiselev E; Marchand C; Pommier Y; An L-K, Synthesis and Structure-Activity Relationship of Furoquinolinediones as Inhibitors of Tyrosyl-DNA Phosphodiesterase 2 (Tdp2). Eur. J. Med. Chem 2018, 151, 777–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laev SS; Salakhutdinov NF; Lavrik OI, Tyrosyl-DNA Phosphodiesterase Inhibitors: Progress and Potential. Bioorg. Med. Chem 2016, 24, 5017–5027. [DOI] [PubMed] [Google Scholar]
- 27.Baell JB; Holloway GA, New Substructure Filters for Removal of Pan Assay Interference Compounds (Pains) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- 28.Chan DMT; Monaco KL; Li RH; Bonne D; Clark CG; Lam PYS, Copper Promoted C-N and C-O Bond Cross-Coupling with Phenyl and Pyridylboronates. Tetrahedron Lett. 2003, 44, 3863–3865. [Google Scholar]
- 29.Chan DMT; Monaco KL; Wang RP; Winters MP, New N- and O-Arylations with Phenylboronic Acids and Cupric Acetate. Tetrahedron Lett. 1998, 39, 2933–2936. [Google Scholar]
- 30.Lam PYS; Clark CG; Saubern S; Adams J; Winters MP; Chan DMT; Combs A, New Aryl/Heteroaryl C-N Bond Cross-Coupling Reactions Via Arylboronic Acid Cupric Acetate Arylation. Tetrahedron Lett. 1998, 39, 2941–2944. [Google Scholar]
- 31.Siva Reddy A; Ranjith Reddy K; Nageswar Rao D; Jaladanki CK; Bharatam PV; Lam PY; Das P, Copper(Ii)-Catalyzed Chan-Lam Cross-Coupling: Chemoselective N-Arylation of Aminophenols. Org. Biomol. Chem 2017, 15, 801–806. [DOI] [PubMed] [Google Scholar]
- 32.Vernekar SKV; Tang J; Wu B; Huber AD; Casey MC; Myshakina N; Wilson DJ; Kankanala J; Kirby KA; Parniak MA; Sarafianos SG; Wang Z, Double-Winged 3-Hydroxypyrimidine-2,4-Diones: Potent and Selective Inhibition against HIV-1 RNase H with Significant Antiviral Activity. J. Med. Chem 2017, 60, 5045–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu B; Tang J; Wilson DJ; Huber AD; Casey MC; Ji J; Kankanala J; Xie J; Sarafianos SG; Wang Z, 3-Hydroxypyrimidine-2,4-Dione-5-N-Benzylcarboxamides Potently Inhibit HIV-1 Integrase and RNase H. J. Med. Chem 2016, 59, 6136–6148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kansy M; Senner F; Gubernator K, Physicochemical High Throughput Screening: Parallel Artificial Membrane Permeation Assay in the Description of Passive Absorption Processes. J. Med. Chem 1998, 41, 1007–1010. [DOI] [PubMed] [Google Scholar]
- 35.Zafrani Y; Yeffet D; Sod-Moriah G; Berliner A; Amir D; Marciano D; Gershonov E; Saphier S, Difluoromethyl Bioisostere: Examining the “Lipophilic Hydrogen Bond Donor” Concept. J. Med. Chem 2017, 60, 797–804. [DOI] [PubMed] [Google Scholar]
- 36.Mabit H; Dubanchet S; Capel F; Dauguet C; Petit MA, In-Vitro Infection of Human Hepatoma-Cells (HepG2) with Hepatitis-B Virus (HBV) - Spontaneous Selection of a Stable HBV Surface Antigen-Producing HepG2 Cell-Line Containing Integrated HBV DNA-Sequences. J. Gen. Virol 1994, 75, 2681–2689. [DOI] [PubMed] [Google Scholar]
- 37.Terasaki K; Hirayama H; Kasanga CJ; Maw MT; Ohya K; Yamaguchi T; Fukushi H, Chicken B Lymphoma Dt40 Cells as a Useful Tool for in Vitro Analysis of Pathogenic Infectious Bursal Disease Virus. J. Vet. Med. Sci 2008, 70, 407–410. [DOI] [PubMed] [Google Scholar]
- 38.Kumar K; Woo SM; Siu T; Cortopassi WA; Duarte F; Paton RS, Cation-Pi Interactions in Protein-Ligand Binding: Theory and Data-Mining Reveal Different Roles for Lysine and Arginine. Chem. Sci 2018, 9, 2655–2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gallivan JP; Dougherty DA, Cation-Pi Interactions in Structural Biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma JC; Dougherty DA, The Cation−II Interaction. Chem. Rev 1997, 97, 1303–1324. [DOI] [PubMed] [Google Scholar]
- 41.Minoux H; Chipot C, Cation-Pi Interactions in Proteins: Can Simple Models Provide an Accurate Description? J. Am. Chem. Soc 1999, 121, 10366–10372. [Google Scholar]
- 42.Zacharias N; Dougherty DA, Cation-Pi Interactions in Ligand Recognition and Catalysis. Trends Pharmacol. Sci 2002, 23, 281–287. [DOI] [PubMed] [Google Scholar]
- 43.Wintjens R; Lievin J; Rooman M; Buisine E, Contribution of Cation-Pi Interactions to the Stability of Protein-DNA Complexes. J. Mol. Biol 2000, 302, 395–410. [DOI] [PubMed] [Google Scholar]
- 44.Evans DA; Katz JL; West TR, Synthesis of Diaryl Ethers through the Copper-Promoted Arylation of Phenols with Arylboronic Acids. An Expedient Synthesis of Thyroxine. Tetrahedron Lett. 1998, 39, 2937–2940. [Google Scholar]
- 45.Chen XX; Murawski A; Patel K; Crespi CL; Balimane PV, A Novel Design of Artificial Membrane for Improving the Pampa Model. Pharm. Res 2008, 25, 1511–1520. [DOI] [PubMed] [Google Scholar]
- 46.Mosmann T, Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [DOI] [PubMed] [Google Scholar]
- 47.Schrödinger Small-Molecule Drug Discovery Suite 2015–4, Schrödinger, Llc, New York, Ny, 2015. [Google Scholar]
- 48.Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W, Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aid. Mol. Des 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- 49.Schrödinger Schrödinger Release 2015–4: Schrödinger Suite 2015–4 (a)Protein Preparation Wizard; Epik, Schrödinger, Llc, New York, Ny, 2015; Impact, Schrödinger, Llc, New York, Ny, 2015; Prime, Schrödinger, Llc, New York, Ny, 2015. [Google Scholar]
- 50.Jorgensen WL; Maxwell DS; TiradoRives J, Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc 1996, 118, 11225–11236. [Google Scholar]
- 51.Schrödinger Schrödinger Release 2015–4: Schrödinger Suite 2015–4 (a)Protein Preparation Wizard; Epik, Schrödinger, Llc, New York, Ny, 2015; Impact, Schrödinger, Llc, New York, Ny, 2015; Prime, Schrödinger, Llc, New York, Ny, 2015. [Google Scholar]
- 52.Schrödinger Schrödinger Release 2015–4: Ligprep, Schrödinger, Llc, New York, Ny, 2015. [Google Scholar]
- 53.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- 54.Release Schrödinger 2015–4: Glide Version 6.9, Schrödinger, Llc, New York, Ny, 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.