

Abstract
Enhancers activate transcription in a distance-, orientation-, and position-independent manner, which makes them difficult to be identified. Self-transcribing active regulatory region sequencing (STARR-seq) measures the enhancer activity of millions of DNA fragments in parallel. Here we used STARR-seq to generate a quantitative global map of rice enhancers. Most enhancers were mapped within genes, especially at the 5′ untranslated regions (5′UTR) and in coding sequences. Enhancers were also frequently mapped proximal to silent and lowly-expressed genes in transposable element (TE)-rich regions. Analysis of the epigenetic features of enhancers at their endogenous loci revealed that most enhancers do not co-localize with DNase I hypersensitive sites (DHSs) and lack the enhancer mark of histone modification H3K4me1. Clustering analysis of enhancers according to their epigenetic marks revealed that about 40% of identified enhancers carried one or more epigenetic marks. Repressive H3K27me3 was frequently enriched with positive marks, H3K4me3 and/or H3K27ac, which together label enhancers. Intergenic enhancers were also predicted based on the location of DHS regions relative to genes, which overlap poorly with STARR-seq enhancers. In summary, we quantitatively identified enhancers by functional analysis in the genome of rice, an important model plant. This work provides a valuable resource for further mechanistic studies in different biological contexts.
Keywords: Plant, Enhancer, Functional analysis, Epigenetic modification, Gene expression
Introduction
Gene expression is tightly regulated, which is critical for plant development and responses to alterations in the environment and hormone levels [1]. Promoters proximal to transcription start sites (TSS) are frequently considered sufficient for the initiation and elongation of transcription, but the level of promoter-driven expression is generally low [1]. High level of gene expression requires the participation of enhancers to increase the efficiency of transcription initiation and elongation to produce more mRNAs, although the exact mechanisms remain poorly understood.
Different from promoters, whose function correlates with genomic location, enhancers regulate the expression of target genes in a distance-, orientation-, and position-independent manner [2]. Thus, defined location-to-function relationship cannot be easily established between an enhancer and its target gene, which makes the identification of enhancers challenging. In mammalian genomes, one gene can be regulated simultaneously by multiple enhancers, or by different enhancers in different tissues and at different developmental stages. Meanwhile, one enhancer can regulate multiple genes [2], [3], [4], [5].
Advancements in molecular biology and computational techniques have enabled the characterization of enhancers genome-wide based on epigenetic marks [6], [7], [8], [9], [10], [11], [12], [13], [14], [15] or by direct measurement of transcription enhancing activity of candidate sequences [16], [17], [18], [19], [20], [21], [22]. Intergenic enhancers have been predicted in Arabidopsis according to the location of DNase hypersensitive sites (DHSs). Basically a DHS is considered as an enhancer if located more than 1.5 kb away from the TSS, and not inside any gene body [23]. However, the arbitrary exclusion of DHSs close to the TSS may lead to the exclusion of substantial number of potential enhancers. To date, only a handful of enhancers have been identified in plants [24], [25], [26], [27], [28], and no genome-wide annotation of enhancers has yet been performed based on functional analysis.
Self-transcribing active regulatory region sequencing (STARR-seq) has been successfully used to measure enhancer activity of millions of fragments in Drosophila melanogaster and mammalian genomes [16], [18], [19], [20], [21], [22]. Here, we used STARR-seq and successfully mapped the locations and quantified the strengths of enhancers in the model plant rice. We analyzed the epigenetic marks of the identified enhancers and revealed that histone modifications and chromatin states for rice enhancers are quite different from those for STARR-seq enhancers identified in animal models. Our work provides a functional enhancer resource and shows that plant and animal enhancers may be different at least in epigenetic features.
Results
Global quantitative enhancer discovery using STARR-seq
To comprehensively identify sequences with enhancer activity, we constructed a reporter library from randomly fragmented genomic DNA of the rice cultivar Nipponbare (Oryza sativa L. ssp japonica). The plasmid DNA of the reporter library was transfected in replicates into protoplasts isolated from the stem of 2-week-old rice seedlings. Plasmid DNA and mRNA were isolated from transfected cells 16 h after transfection. Sequencing libraries of plasmids and mRNA were generated and subjected to paired-end sequencing on Illumina HiSeq X Ten platform (Figure S1). For the two transfection replicates, 30.6 million and 15.5 million (Table S1) independent fragments were recovered from input plasmid libraries, with a median length of ∼670 bp (Figure S2A and B). As a result, ∼90% of the rice genome was covered with at least one unique fragment for each single nucleotide (Figure S2C and D). For the cDNA libraries generated from isolated mRNA samples, 13.7 million and 6.1 million independent fragments were produced (Table S1, Figure S2E and F). The quality of the libraries were checked and the enrichment of cDNA over plasmid input was determined for 600 bp bins across the genome and potential enhancers were identified (Figures S2–S7). Two identified enhancer sites located on chromosome 9 are shown in Figure 1A as examples.
Figure 1.

Genome-wide quantitative enhancer discovery
A. STARR-seq cDNA (red) and input plasmid (gray) fragment densities at representative genomic loci. Light and dark blue boxes denote the identified enhancers in two replicates, respectively. B. Gene expression indicated by STARR-seq enrichment and real-time PCR quantification is linearly correlated (r = 0.79). STARR-seq enhancers were arbitrarily grouped into weak, medium, and strong categories based on the enrichment of cDNA over input plasmid with FC ranging below 2.0, 2.0 to 4.0, and above 4.0, respectively. Error bars indicate total four real-time qPCR quantifications, two qPCRs for each independent biological replicate; inset, the same data depicted as a box plot. Significant difference between groups was determined using Wilcoxon rank-sum test (*, P < 0.05; ***, P < 0.001). r, Pearson correlation coefficient; FC, fold change.
To validate the identified peaks, we chose 29 sites (Table S2) with weak (<2 fold enrichment), medium (2–4 fold enrichment) or strong (>4 fold enrichment) enhancer strength. These sites were cloned into luciferase reporter vectors and the reporter gene expression was quantified using real-time RT-PCR and normalized against the expression of the co-transfected Renilla luciferase reporter gene. Our data showed that STARR-seq enrichment intensity is highly correlated with the reporter gene expression levels across a wide activity range for enhancers (Pearson correlation coefficient, r = 0.79; Figure 1B). The activities of the weak, medium, and strong enhancers also showed significant differences between different groups (P < 0.05, P < 0.001, and P < 0.001 for weak vs. medium, weak vs. strong, and medium vs. strong enhancers, respectively, Wilcoxon test) (Figure 1B).
The Pearson correlation coefficient for the two replicates was 0.604 (Figure S5), demonstrating that STARR-seq is reproducible in plant system. 15,208 and 12,210 regions (Figure 2A) were significantly enriched from the replicates 1 and 2, respectively (P < 0.001). Among them, 9642 enriched peaks were identified in both biological replicates (Table S3) and used for further analysis (Figure 2A).
Figure 2.

Distribution of STARR-seq enhancers in rice genome
A. Enhancers identified in two independent STARR-seq experiments. Totally 9642 enhancers were commonly discovered in both replicates. B. and C. Distribution and relative enrichment of identified enhancers in the rice genome. D. Distribution of enhancers relative to the gene body. TSSs and TTSs of all genes are aligned at the beginning and the end of gene body, respectively. Extended regions of 2.4 kb (the median size of genes in rice) upstream of TSS and downstream of TTS are divided into 10 bins of equal size (240 bp), respectively. The horizontal gray dotted line shows the mean density of enhancers from control elements, which was calculated from 10,000 randomly selected regions of 700 bp in length and repeated for at least 10 times. TE, transposable element; TSS, transcription start site; TTS, transcription termination site.
Enhancers are enriched within gene body
STARR-seq enhancers identified in the Drosophila genome are mostly located within genes and at promoter regions, especially in introns (55.6%), whereas only 22.6% of enhancers are in intergenic regions. Meanwhile Drosophila enhancers are significantly underrepresented in repetitive sequences [16]. To reveal if the distribution pattern of enhancers in rice is different from that in Drosophila, we calculated the percentage of rice STARR-seq enhancers in different functional genomic regions. Surprisingly, more than 50% (5020) of enhancers were mapped in repetitive sequences, most of which are transposable elements (TEs) (Figure 2B). Moreover, nearly all of these enhancers (4831/5020) overlap with one type of repetitive elements such as short interspersed nuclear elements (SINEs), long interspersed nuclear elements (LINEs), long terminal repeats (LTRs), DNA transposons, satellite DNA, or simple repeats (Table S4). The enrichment of STARR-seq enhancers in TE-related sequences in rice may be consistent with the hypothesis that TEs may regulate gene expression or even give rise to new genes during evolution [29], [30]. In addition to the repeats, identified enhancers are overrepresented in the 5′ untranslated regions (5′UTR) and coding sequences, but underrepresented in introns (Figure 2C). These observations demonstrate a strikingly difference in the distribution pattern of enhancers between the Drosophila and rice genomes [16].
To reveal if enhancers show different distribution patterns in different chromatin contexts, we further divided the rice genome into repetitive sequences enriched with TEs (TE regions) and sequences without TEs (non-TE regions) and analyzed the enhancer distribution relative to proximal genes. Overall, the enhancer distribution patterns are similar in both TE and non-TE regions (Figure 2D) despite the different genetic composition in these two types of sequences. Enhancers are located mostly within genes at the 5′ end, and their abundance gradually declines to background level toward the 3′ end of genes (Figure 2D).
The majority of genes lack proximal enhancers in the rice genome
The majority of STARR-seq enhancers were mapped within or close to genes (gene body ±5 kb), which suggests that proximal regulation by enhancers may be a prevalent choice in the rice genome. Compared to the total number of annotated and predicted genes (∼56,000) in rice [31], the number of STARR-seq enhancers is relatively low (9642), i.e., less than 0.2 enhancers per gene on average, which suggests that most genes may not be directly regulated by enhancers in close proximity (gene body ±5 kb). Further analysis shows that 28.7% (15,997) of genes (Figure 3A and B) have at least one proximal enhancer, suggesting that many enhancers have to be in proximity to more than one gene.
Figure 3.

The proximal sequences of most genes are lack of identified enhancers
Number (A) and percentage (B) of total genes with or without STARR-seq enhancer in proximity in the whole genome. Enhancers located within 5 kb upstream of TSS, inside gene body, and within 5 kb downstream of TTS are considered to be proximal to genes. Number (C) and percentage (D) of genes expressed at different levels with or without enhancers in proximity in non-TE regions. Number (E) and percentage (F) of genes expressed at different levels with or without enhancers in proximity in TE regions. Genes are grouped into four categories based on their expression levels. Silent, RPKM = 0; low, 0 < RPKM ≤ 1; medium, 1 < RPKM ≤ 10; high, RPKM > 10. The horizontal blue lines show the percentage of genes with enhancers in proximity in the whole genome.
To investigate if enhancers are preferentially enriched for active genes, we separated genes into four groups according to their expression levels (silent, low, medium, and high) and calculated the percentage of genes with a proximal enhancer for each group. The percentage of genes with proximal enhancers changes little (from 22.7% to 26.1%) for genes with the different expression levels in non-TE regions (Figure 3C and D). In contrast to non-TE regions, about 40% of silent and lowly-expressed genes were found with proximal enhancers in TE regions (Figure 3E and F). These results suggest that STARR-seq enhancers are not necessarily enriched at actively transcribed genes in vivo. Our observations agree with previous reports that episomal analysis may not reflect the endogenous chromatin state of the assayed sequences [16].
Genes in repetitive sequences are enriched with enhancers
Not only silent and lowly-expressed genes in TE regions are enriched with proximal enhancers, in fact, 52.1% of identified STARR-seq enhancers are located in TE regions (Figure 2B), which account for about 42.8% of the rice genome (Figure 2B) and harbor only 28.3% (15,839) of total genes (∼56,000) (Figure 3E). Most genes inside TE regions (15,318, 96.7% of total genes in TE regions) were lowly expressed (960 and 1389 genes with or without enhancer in proximity, respectively) or silent (5181 and 7788 genes with or without enhancer in proximity, respectively) (Figure 3E). And 39.6% (6277, sum of four gene groups of different expression level; Figure 3E) of total genes located in TE regions (15,839) contained at least one proximal enhancer. For genes of higher transcription levels or genes in non-TE regions, the percentage of genes with a proximal enhancer was lower than the average for total genes (28.7%) (Figure 3B, D, and F). Gene ontology (GO) analysis showed that genes in TE regions with proximal enhancers are mostly enriched in categories of DNA replication, integration, nucleotide binding, etc. (Figure S8).
STARR-seq enhancers overlap poorly with DHSs
DNase I hypersensitivity has been associated with DNA sequences with different types of activity, one of which is enhancing gene transcription [32]. To examine the endogenous accessibility of STARR-seq enhancers, we analyzed the DNase I hypersensitivity across the rice genome using previously published data [33]. Surprisingly, only 8.7% (839) of STARR-seq enhancers were found overlapping with DHSs (Figure 4A, Figure S9). Actually, the low overlap between DHSs and STARR-seq enhancers was also reported in humans that only about 13.6% of human STARR-seq enhancers co-localize with DHSs (Figure S9) [20]. Quite different in Drosophila, 48.5% of STARR-seq enhancers co-localize with DHSs [16] (Figure S9). These results together suggest that the hypersensitivity to DNase I digestion may not be an inseparable characteristic for functionally identified enhancers, which seems true at least in the rice and human genomes.
Figure 4.

Epigenetic marks associated with enhancers in rice genome
A. Numbers of identified enhancers that overlap with DHSs. B. Expression of genes in two biological replicates with proximal enhancers overlapping with DHS (open) or not (close) (****, P < 10−10, Wilcoxon rank-sum test). In this analysis, STARR-seq enhancers uniquely identified in each replicate were included. C. Epigenetic signals for enhancers overlapping with DHSs (red) or not (blue). D. Epigenetic signal for enhancers in non-TE (red) or TE (blue) regions, respectively. The horizontal gray dotted line shows the relative enrichment of the examined epigenetic signal of random genomic sites as a control. DHS, DNase I hypersensitive site.
Chromatin of high accessibility is frequently associated with active transcription [34]. For genes with proximal enhancers overlapping with DHSs (open chromatin), their expression levels are indeed generally higher than those of genes with non-DHS enhancers (close chromatin) in proximity (Figure 4B).
H3K4me1 is underrepresented at enhancers identified in the rice genome
H3K4me1 has been frequently used as a mark for enhancer prediction, which is enriched at enhancers identified by STARR-seq in both Drosophila and human genomes [16], [20]. However, in rice, H3K4me1 is obviously underrepresented at the endogenous sites of enhancers, independent of their overlapping state with DHS (Figure 4C) or location in TE or non-TE regions (Figure 4D). This surprising observation raises a question over the role of H3K4me1 as a reliable enhancer mark in the rice genome.
H3K27ac is enriched at rice STARR-seq enhancers
H3K27ac is another histone modification used for the prediction of active enhancers, which is also enriched at enhancers identified by STARR-seq in both Drosophila and human genomes [16], [20]. Similarly in rice, H3K27ac is also enriched although only at a selective portion of STARR-seq enhancers, specifically those associated with DHS (red curves, Figure 4C) or located in non-TE regions (red curves, Figure 4D). It is of note that even enhancers not associated with DHSs show slight enrichment of H3K27ac (blue curves, Figure 4C). Quite different from H3K4me1, H3K27ac seems to be a more conserved enhancer mark across plant and animal kingdoms.
Active transcription mark H3K4me3 is enriched at STARR-seq enhancers
H3K4me3 is generally associated with actively transcribed genes in animal models [35]. In the rice genome, H3K4me3 is enriched preferentially at STARR-seq enhancers that overlap with DHSs or are located in non-TE regions (Figure 4C and D). These observations could be explained by the fact that a significant portion of enhancers are located inside genes at the 5′UTR and in coding sequences (Figure 2D), where the H3K4me3 is detected if these genes are actively transcribed. However, these observations also suggest that enhancers can possibly be genes themselves in rice genome.
Repressive histone mark H3K27me3 colocalizes with STARR-seq enhancers
H3K27me3 is mostly associated with repressed chromatin state [35]. Surprisingly, we found that identified enhancers were enriched with H3K27me3 (Figure 4C and D; also see sC3, sC4, and sC7 in Figure 5A and B), which is not enriched for enhancers identified in either Drosophila or human genomes [16], [20]. Interestingly, many of these enhancers (sC3 and sC4) are also enriched with H3K4me3 and H3K27ac (Figure 5A and B). Whether these enhancers are poised or actively regulate gene expression is difficult to determine at this time. One possibility could be that the same genomic loci are modified differentially in different subpopulations of cells in culture. Different from H3K27me3, H3K9me3 is nearly completely absent from identified enhancers (Figure 5A).
Figure 5.

Epigenetic clustering reveals combinatorial association of active and repressive histone modifications with a subset of enhancers
A. STARR-seq enhancers are clustered over the signals of DHSs and five histone modifications as indicated at the top of the panel. The number and percentage of enhancers for each cluster are shown on the side. B. The level of enrichment of epigenetic marks for each cluster of enhancers is shown in different color. Signal density of clusters is ranked as absent (dark gray), low (light blue), medium (orange), and high (red). The signal density is determined by calculating the percentage of enhancers carrying an examined epigenetic mark over the total number of enhancers in a specific cluster (see Figure S10A for details). A cluster is designated to be absent of an epigenetic mark if ≤10% of the elements in that cluster carried the epigenetic mark examined, or designated as low, medium, or high if the percentage of elements in a specific cluster carrying an epigenetic mark examined was between 10%–30%, 30%–60%, or >60%. Number of enhancers in each cluster is shown at the bottom. C. Percentage of identified enhancers in each cluster with (red) or without (gray) a specific epigenetic mark. D. Percentage of enhancers in each cluster present in non-TE (red) or TE (gray) regions. E. Expression of genes with proximal enhancers of different cluster in TE (blue) or in non-TE (red) regions. Genes with enhancers in non-TE regions of nearly all clusters (except sC7) show significantly (*, P < 0.01, Wilcoxon rank-sum test) higher expression level than in those in TE regions.
Epigenetic clustering of identified enhancers
To find out if there is any unique combination of histone modifications, we clustered all identified enhancers into eight groups (sC1–8, s standing for STARR-seq enhancers) based on the signal strength of multiple epigenetic marks including DHS, H3K4me1, H3K4me3, H3K27ac, H3K27me3, and H3K9me3 (Figure 5A, Figure S10A). Clusters 1–7 (sC1–7) together contain only 37.9% (3650) of total enhancers (Figure 5A). And cluster sC8 alone contains 62.1% (5992) of the total enhancers, which carries negligible level of any analyzed epigenetic mark (Figure 5A and B).
In fact, only 334 sites (sC2) show strong signal for the presence of H3K4me1 (Figure 5A and B), even fewer than the enhancers overlapping with DHS (839) (Figure 4A). H3K4me3 is enriched for 4 clusters of enhancers (sC1 and sC3–5), for which different levels of H3K27ac are enriched as well (Figure 5A and B). Overall, H3K4me3 and H3K27ac are two mostly enriched histone marks, DHS and H3K27me3 are moderately enriched, whereas H3K4me1 and H3K9me3 are least enriched for enhancers in sC1–7 (Figure 5A–C).
STARR-seq enhancers in rice are slightly enriched in TE regions where gene activity is low. To reveal if enhancers of different clusters follow the same distribution pattern of total STARR-seq enhancers, we reexamined the distribution of each cluster of enhancers in the TE and non-TE regions. Quite different from total STARR-seq enhancers, averagely 83% of sC1–7 enhancers are mapped in non-TE regions (Figure 5D). Accordingly, the majority of sC8 enhancers (73%) were associated with TE regions (Figure 5D).
Enhancer activity of each cluster was also examined. Except sC7, genes associated with enhancers of all other clusters (sC1, sC2–6, and sC8) are expressed at significantly higher levels in non-TE regions than in TE regions (P < 0.01, Wilcoxon’s test; Figure 5E). Enhancers of sC7 are mostly enriched with repressive H3K27me3, different from other H3K27me3 enriched clusters (sC3 and sC4) that are also enriched with active H3K4me3 and H3K27ac (Figure 5A and B). Overall, the median expression levels of genes associated with proximal STARR-seq enhancers (sC1–2 and sC3–6) of active histone marks are significantly higher than the median level of total genes (Figure 5E). These results show that STARR-seq enhancer activity is well correlated with histone modifications and genomic sequence composition.
DHS-predicted enhancers differ from STARR-seq enhancers
Enhancers have been predicted based on chromatin accessibility in Arabidopsis [23]. To test if STARR-seq enhancers overlap with DHS-predicted enhancers, we followed the previous report [23] and defined a DHS site as enhancer if it is located >1.5 kb upstream of TSS but not in a gene body. According to this criterion, 13,770 out of total 37,168 DHSs were predicted to be enhancers (Figure 6A, Table S5). Only 20% of 13,770 sites are in TE regions, whereas the remaining 80% are located in non-TE regions (Figure 6A), indicating an under-representation and over-representation in TE and intergenic regions, respectively (Figure 6B). This distribution patterns is different from that of the STARR-seq enhancers (Figure 2C). In fact, DHS-predicted enhancers barely overlap with STARR-seq enhancers. There are only 220 overlapping sites, accounting for <2.3% and 1.6% of total STARR-seq and DHS-predicted enhancers, respectively (Figure 6C). These results show that DHS-predicted enhancers are a group of DNA elements different from STARR-seq enhancers.
Figure 6.

Predicted enhancers based on DHS location
A. Number of DHS-predicted enhancers (13,770) and non-enhancer DHSs (23,398) and percentage of these sites in different functional genomic regions are shown below. B. Relative enrichment/depletion of DHS-predicted enhancers and non-enhancer DHSs in different genomic regions. C. Overlap analysis among STARR-seq enhancers, DHS-predicted enhancers, and non-enhancer DHSs. D. DHS-predicted enhancers are clustered over the signal of five histone modifications. E. The level of enrichment of epigenetic mark for each cluster of enhancers is shown in different color (see Figure S10B for details). F. Expression of genes with proximal DHS-predicted enhancers in TE (blue) or non-TE (red) regions. G. Non-enhancer DHSs are clustered over the signal of five histone modifications as for STARR-seq enhancers. H. The level of enrichment of epigenetic mark for each cluster of enhancers is shown in different color (see Figure S10C for details). For panels E and H, signal density of clusters is ranked as absent (dark gray), low (light blue), medium (orange), and high (red). The signal density is determined by the percentage of enhancers in a specific cluster carrying the examined epigenetic mark. Number of enhancers in each cluster is shown at the bottom. A cluster is designated to be absent of an epigenetic mark if ≤10% of the elements in that cluster carried the epigenetic mark examined, or designated as low, medium, or high if the percentage of elements in a specific cluster carrying an epigenetic mark examined was between 10%–30%, 30%–60%, or >60%. I. Expression of genes with proximal non-enhancer DHSs in TE (blue) or non-TE (red) regions.
To reveal the epigenetic modification patterns, we carried out clustering analysis according to the histone modifications at DHS-predicted enhancers (Figure 6D, Figure S10B). Although DHSs indicate open chromatin possibly enriched with active histone marks, clustering analysis showed that only 16.9% of total DHS-predicted enhancers are enriched with any examined histone modifications including repressive H3K27me3 (Figure 6D and E). Interestingly, H3K4me1 is also absent from nearly all DHS-predicted enhancers (Figure 6D and E). Similar to STARR-seq enhancers, H3K4me3 is highly enriched for most clusters (dC1–5, d standing for DHS-predicted enhancers) (Figure 6D and E). Interestingly, H3K27me3 and H3K27ac are also enriched for two (dC3 and dC7) and three (dC1, dC4, and dC6) different clusters, respectively (Figure 6D and E), but different from STARR-seq enhancer clusters (sC3 and sC4), in which these two modifications are enriched for about 20% sites (data not shown).
To test if DHS-predicted enhancers increase proximal gene expression, similar analysis was conducted as for STARR-seq enhancers. As many as four clusters (dC1 and dC3–dC5) in TE or non-TE regions differ little in their effect on the expression level of proximal genes (P > 0.05, Wilcoxon test; Figure 6F). Moreover, 5 out of 8 clusters of enhancers (dC1, dC3–5, and dC7) show no positive effect on proximal gene expression compared to the median expression level of all genes (Figure 6F). These results together suggest that enhancer prediction based on the location of DHSa is less successful in screening out regulatory elements that are positively correlated with higher gene expression or positive histone marks at endogenous loci.
DHSs not predicted as enhancers may function as promoters
Since DHS-predicted enhancers are not well correlated with histone modification and proximal gene activity, we wonder if the remaining DHSs not predicted as enhancers (non-enhancer DHSs, 23,398 sites) (Figure 6A, Table S6) behave similarly. First, we examined the distribution of non-enhancer DHSs, which was found to differ sharply from that of the DHS-predicted enhancers. Non-enhancer DHSs are obviously enriched in the 5′ upstream regions of genes, TSS flanking regions, and 5′UTR. They are also slightly overrepresented in first introns and 3′UTRs (Figure 6B). We further clustered non-enhancer DHSs as for DHS-predicted enhancers. A little more than half of non-enhancer DHSs (50.5%) are grouped into several clusters, carrying different types of histone modifications (nC1–7, n standing for non-enhancer DHSs) (Figure 6G and H, Figure S10C). For nC1–7, the histone modification patterns are similar to those of dC1–7, except that there are many more enhancers found in nC1–7 than in dC1–7 (Figure 6E and H). The expression levels of genes with non-enhancer DHSs in proximity were also examined. Interestingly, expression levels of genes with most clusters of non-enhancer DHSs are actually higher than the median expression levels of all genes (Figure 6I). Considering the locations of non-enhancer DHSs, it is reasonable to expect that genes with these sites carrying active histone modifications are active and expressed at a decent level. The fact that most of 9140 non-enhancer DHSs (nC1–5) are enriched with H3K4me3, a mark of active transcription, suggests that these sites may be promoters rather than enhancers.
We further compared the distribution of non-enhancer DHSs, STARR-seq enhancers, and DHS-predicted enhancers. These three groups of elements show strikingly different distribution patterns relative to the TSS (Figure 7A). DHSs are mostly positioned close to TSSs, with 91% and 81% of all DHSs mapped within the regions ranging from 5 kb upstream of TSS to 5 kb downstream of TSS in the rice and Drosophila genomes, respectively (Figure S11). DHS-predicted intergenic enhancers (13,770, 37% out of total 37,168 DHSs genome-wide) are located at least 1.5 kb upstream of the TSS and outside of a gene body (Figure 7A, middle) as defined. Accordingly, non-enhancer DHSs (23,398, 63% out of total 37,168 DHSs genome-wide) (Figure 7A, right) are located within gene body and the regions 1.5 kb 5′upstream of TSSs. Specifically, these DHS sites are overrepresented in sequences <200 bp upstream of TSS, TSS ±50 bp, and the 5′UTR regions (Figure 6B), dramatically different from the distributions of STARR-seq enhancers, which are mostly enriched within the gene body favoring the 5′ end (Figure 7A, left). Only 619 out of 23,395 of non-enhancer DHSs overlap with STARR-seq enhancers (Figure 6C). Our analysis shows that at least a portion of the non-enhancer DHSs are functional enhancers, suggesting that enhancer prediction arbitrarily based on DHS location may also lead to loss of real functional elements.
Figure 7.

Distribution of three types of DNA elements and proposed enhancer function model in rice genome
A. Relative enrichment of STARR-seq enhancers, DHS-predicted enhancers, and non-enhancer DHS sites. Red and blue lines show sites in non-TE and TE regions, respectively; the dashed line indicates the location of TSS. B. The proposed model for enhancer distribution and functional mechanisms in rice genome. Gene body is shown as light blue rectangle box. Red boxes indicate STARR-seq enhancers. DHS-predicted enhancers and non-enhancer DHSs are shown as arrows in dark red and dark blue, respectively. Density of STARR-seq enhancers gradually decreases within the gene body from the 5′TSS to the 3′TTS. Genes can be self-regulated by enhancers located within themselves. DHS-predicted enhancers are mostly located not far away from gene bodies. Non-enhancer DHSs are mostly enriched at promoter regions. Enhancers in one gene may activate other genes separated far away linearly but close in three-dimensional space. The start and orientation of transcription are indicated using an orange arrow.
Discussion
Histone modifications have been used for enhancer identification [6]. H3K4me1 is the primary modification used to predict if a genomic locus is a potential enhancer at all. Other histone modifications have also been employed to identify enhancers of different states. For example, H3K27ac is associated with active and super enhancers, and the coexistence of H3K27me3 and H3K4me1 is believed to be indicative of poised enhancers [12], [11]. However, such methods may fail to predict enhancers in genomic loci devoid of histone modifications. Moreover, enhancers predicted based on epigenetic modifications still need to be validated using genetic methods which is time- and labor-consuming, if large number of predicted sites need to be tested [36].
STARR-seq measures enhancer activity of candidate sequences independent of their endogenous chromatin context and epigenetic state, which has been successfully applied to enhancer analysis in both Drosophila and human genomes [16], [20]. To improve our understanding of the nature and functional mechanisms of plant enhancers, we employ STARR-seq and report here the first functional enhancer mapping genome-wide in the important model plant rice.
Previous work predicting enhancers by chromatin sensitivity to DNase I digestion arbitrarily exclude DHSs located <1.5 kb upstream of the TSS and within a gene body [23]. In Drosophila, the majority of STARR-seq enhancers are actually located within a gene body or in proximity to genes [16]. Similarly, our STARR-seq analysis of the rice genome also shows that the majority of enhancers are localized within the gene body. The consistent observation of enhancer enrichment in the gene body in two evolutionarily far-separated genomes may suggest that most genes could be regulated by DNA elements built into their sequences (Figure 7B). Furthermore, it will be interesting to see whether enhancers in one gene can also activate other genes separated by long distances linearly. Capture Hi-C in the plant genome, using identified enhancers as anchors, may be used to reveal whether genes separated far away can be co-regulated by enhancers located within gene bodies.
Our analysis also reveals several unexpected features of enhancers in the rice genome. Firstly, the majority of STARR-seq enhancers do not overlap with DHSs. DNase I hypersensitivity can be associated with any open and relaxed chromatin region, including insulators and other protein binding sites [32]. Predicting enhancers relying solely on the location of DHSs relative to genes may fail to rule out the possibilities that many DHSs predicted as enhancers may actually be of other non-enhancer functions, and many DHSs not predicted as enhancers may in fact function as real enhancers.
Secondly, although H3K4me1 has been used to predict enhancers in mammals [37], we find that H3K4me1 is absent from most STARR-seq enhancers and DHSs independent of their locations in the rice genome. These observations suggest that H3K4me1 may not necessarily be a conservative enhancer mark in the rice genome. However, at the same time, H3K4me3 is found enriched at many identified enhancers. Whether H3K4me3 is a real unique enhancer mark in plant requires more experimental evidence.
Third, many STARR-seq enhancers are enriched with repressive H3K27me3, majority of which are co-enriched with active chromatin marks of H3K4me3. Enhancers with both repressive H3K27me3 and H3K4me3 could be bivalent elements [38]. Surprisingly, H3K27ac and H3K27me3 co-exist at about 20% of STARR-seq enhancers in clusters sC3–5 (Figure 5A and B). These two modifications are mutually exclusive at the same location on histone H3 tail [39]. Currently, we cannot rule out the possibilities that histones at these enhancers may be modified differentially in different subgroups of cells, or even differentially on different allele in the same cell. In either case, further careful analysis is warranted to reveal the underlying causes of this intriguing observation.
In summary, we present a comprehensive enhancer activity map generated by quantitative measurement using STARR-seq for an important model plant. Successful characterization of enhancers in different cell types will help to improve our understanding of the tissue-specific selection of enhancers during development and shed new lights on the elusive functional mechanisms of enhancers at large.
Materials and methods
STARR-seq reporter plasmid construction and library preparation
To perform STARR-seq in rice cells, we constructed a screening vector (Figure S12) using the backbone of plasmid pBI221 by introducing a CMV 35S mini promoter, an intron and a GFP sequence, which are arranged sequentially and their sequences are shown in Table S7. Linear vector plasmid pBI221 was obtained by PCR amplification.
We constructed reporter library as previously described [16] with some modifications. Briefly, we extracted genomic DNA from the 2-week-old rice seedlings. DNA was fragmented by sonication (30% power, 5 s on, 5 s off, repeat 30 times in a volume of 600 µl) (Scientz II-D, Ningbo Scientz Biotechnology, Ningbo, China). DNA fragments (500–800 bp in length) were repaired and ligated to VAHTS Adapters for Illumina with VAHTS Mate Pair Library Prep Kit for Illumina® (Catalog No. ND104, Vazyme Biotech, Nanjing, China) following the manufacturer’s protocol. We then cloned the adaptor-ligated genomic DNA into linearized vector using the ClonExprress II One Step Cloning Kit (Catalog No. C112, Vazyme Biotech). Ligated product was used to transform Trans1-T1 Phage Resistant Chemically Competent Cell (CD501, TransGen Biotech, Beijing, China) according to the manufacturer’s protocol. Transformed bacterial cells were cultured and reporter plasmids were purified using E.Z.N.A.® Endo-Free Plasmid Maxi Kit (Catalog No. D6926, Omega Bio-tek, Norcross, GA) and quantified on NanoDrop ONE (Thermo Fisher Scientific, Waltham, MA).
Protoplast transfection
Protoplasts from rice stem were isolated and transfected as described [40] with minimal modification. For transfection, 30–40 μg of reporter plasmid DNA was mixed with 100 μl of protoplasts (∼1 × 106 cells) in a tube containing 110 μl of freshly prepared solution of polyethylene glycol (40%, W/V) (Catalog No. 807490, Sigma–Aldrich Biotech, St. Louis, MO). The protoplasts transfected were cultured at 28 °C in the dark.
Construction of reporter cDNA and input plasmid libraries for Illumina sequencing
mRNA and plasmid DNA in transfected protoplasts were recovered after 16 h of transfection using TransZol Up Plus RNA Kit (Catalog No. ER501, TransGen Biotech, Beijing, China) and poly(A)+ RNA fraction was isolated using VAHTS mRNA Capture Beads (Catalog No. N401, Vazyme Biotech). 5 U of DNase I (Catalog No. M0303S, New England Biolabs, Ipswich, MA) was used to digest DNA at 37 °C for 20 min. Synthesis of first strand cDNA was carried out using TransScript One-Step gDNA Removal and cDNA synthesis SuperMix (Catalog No. AU311, TransGen Biotech). The total reporter cDNA was amplified for Illumina sequencing with a 2-step nested PCR strategy using the TransStart® FastPfu Fly DNA Polymerase (Catalog No. AP231, TransGen Biotech). First-round PCR products were purified using GeneJET PCR Purification Kit (Catalog No. K0702, Thermo Fisher Scientific) and was used as template for the second-round PCR amplification with VAHTS™ DNA Adapters for Illumina® (Catalog No. N302, Vazyme Biotech). Second-round PCR products were purified using GeneJET PCR Purification Kit and eluted in 20–30 μl of the elution buffer (EB).
After the capture of poly(A)+ RNA, the left aqueous solution was treated with 10 μl of RNase A (Catalog No. GE 101, TransGen® Biotech) to remove any RNA in solution before plasmid DNA was purified using GeneJET PCR Purification Kit and eluted in 50 μl of EB. Purified plasmid DNA was amplified with the TransStart® FastPfu Fly DNA Polymerase and VAHTSTM DNA Adapters for Illumina® to enrich the inserted sequences cloned in reporter plasmids. DNA of reporter inserts was purified using GeneJET PCR Purification Kit and eluted in 20–30 μl of EB.
Both cDNA and reporter inserts DNA libraries were sequenced on Illumina HiSeq X Ten platform.
Mapping and STARR-seq enhancer identification
We used Bowtie2 [41] to map the sequencing data to the Nipponbare reference genome (IRGSP1.0). Mapped reads were filtered with SAMtools [42] and only uniquely mapped reads were kept. The reproducibility of two independent experiments was evaluated and the Pearson correlation coefficient was calculated by multiBamSummary and plotCorrelation in deepTools [43]. Genome coverage of reporter insert DNA was calculated by BEDTools [44].
STARR-seq enhancer identification was carried out as described [16] using R package BasicSTARRseq and Bonferroni correction was performed to adjust P values. Genomic region was identified as enhancer if the enrichment of cDNA over input plasmid insert is ≥1.3 fold and the adjusted P value is <0.001. Only enhancers found in both replicates were kept for further analysis. Overlapping enhancers from two replicates were merged using BEDTools merge [44]. The distance between enhancer and the proximal TSS was computed using BEDTools closest command.
Comparison between STARR-seq enhancers and DHS-predicted enhancers
DHS data previously generated [33] were used to predict enhancers according to their location relative to genes in the rice genome following the definition previously described for Arabidopsis [23]. Genomic regions sensitive to DNase I digestion were identified as DHSs using MACS1.4 [45]. BEDTool intersect was used to filter the DHSs in intergenic regions. If the middle point of a STARR-seq enhancer fell within the sequence of a DHS-predicted enhancer, then these two elements are considered overlapping.
AgriGO V2.0 [46] was used for GO analysis for enhancer proximal genes.
Enhancer cluster analysis on epigenetic modification
We downloaded dataset of H3K4me3 and H3K27me3 (accession No. GSE19602) [47], H3K27ac and H3K9me3 (accession No. GSE79033 [48], as well as DHSs (accession No. GSE26734) [35] from the Gene Expression Omnibus (GEO). Data of H3K4me1 (accession No. PRJCA000387) were downloaded from GSA [49].
Genomic regions with enriched histone modification were called using MACS1.4 [45]. We used 10,000 randomly selected regions of 700 bp in length as control, and repeated for at least 10 times to calculate the mean value of analyzed features as explained in Figure legend. R package EnrichedHeatmap [50] was used to plot the enrichment of histone modifications (H3K4me1, H3K4me3, H3K27ac, H3K27me3, and H3K9me3) and DHSs with the center of analyzed elements positioned at middle point and extended upstream and downstream up to 5 kb. K-means in Cluster3.0 [51] was used to cluster STARR-seq enhancers, DHS-predicted enhancers, and non-enhancer DHSs. We submitted the sequences of STARR-seq enhancers, DHS-predicted enhancers, and non-enhancer DHSs to the MEME-ChIP web server19 for de novo motif finding in the JASPAR CORE (2018) 20 plant database (http://jaspar.genereg.net/collection/core/). The motifs identified are enclosed in Table S8.
We used R for all statistical analysis.
Data availability
The sequencing datasets in this study can be accessed at the Gene Expression Omnibus (GEO) as GEO: GSE121231.
Authors’ contributions
CH and LN designed and supervised the experiments. JS constructed the reporter library and validated the identified sites. YZ participated in cell preparation, transfection, and sequencing library preparation. NH carried out bioinformatics analysis. WS processed the raw data and participated in bioinformatics analysis. YZ helped with reporter library construction. LL advised on data analysis. CH wrote the manuscript with input from all authors. All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (Grant Nos. 31571347 to CH and 31771430 to LL), Guangdong Science and Technology Department, China (Grant No. 2016A030313642 to CH), Shenzhen Science and Technology Innovation Commission, China (Grant No. JCYJ20150529152146478 to CH), Huazhong Agricultural University Scientific & Technological Self-innovation Foundation, China to LL and the Thousand Youth Talents Plan of China to CH. We thank Drs. Shengtao Hou and Andrew P. Hutchins for manuscript editing.
Handled by Lei Li
Footnotes
Peer review under responsibility of Beijing Institute of Genomics, Chinese Academy of Sciences and Genetics Society of China.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2018.11.003.
Contributor Information
Longjian Niu, Email: niulj@mail.sustc.edu.cn.
Chunhui Hou, Email: houch@sustc.edu.cn.
Supplementary material
The following are the Supplementary data to this article:
Supplementary Figure S1.
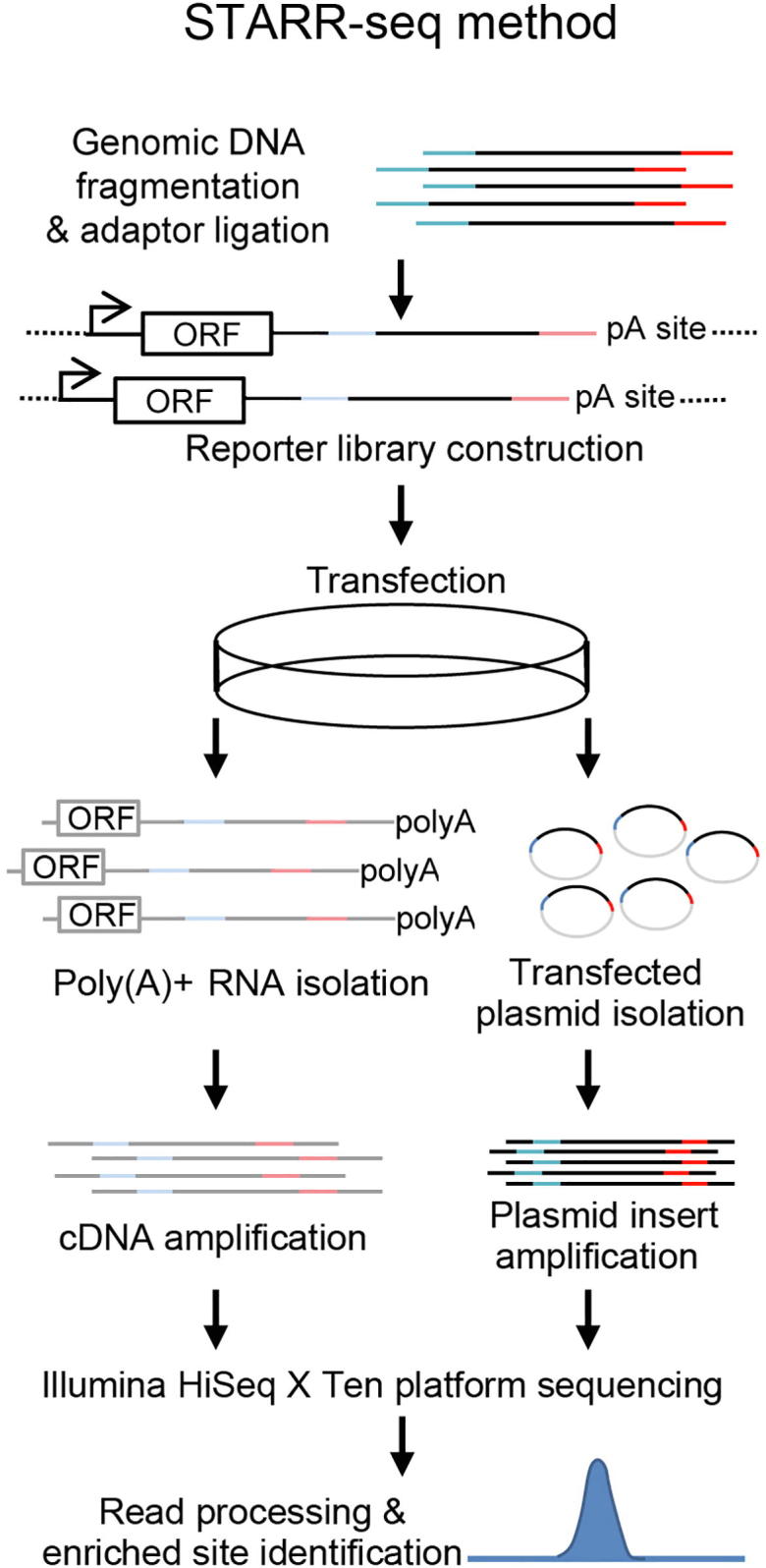
STARR-seq work flow Briefly, genomic DNA was fragmented, repaired, and ligated with proper adaptors and then cloned into reporter vector plasmid. Then the DNA of reporter plasmids was transfected into cultured cells. RNA and plasmid DNA were isolated after 16 h and converted into libraries for sequencing on Illumina HiSeq X Ten platform. A CMV 35S mini promoter was used to drive basal expression of a reporter gene. ORF, open-reading frame; pA site, polyadenylation site.
Supplementary Figure S2.

Input plasmid and cDNA libraries Fragment sizes of STARR-seq input plasmid library (A) and cDNA library (B), with the median fragment sizes indicated. Coverage of the rice non-TE regions by independent fragments (cumulative) for input plasmid library (C) and cDNA library (E). Coverage of the rice TE regions by independent fragments (cumulative) for input plasmid library (D) and cDNA library (F).
Supplementary Figure S3.

GC content analysis for the input plasmid and cDNA libraries The genome sequence was arbitrarily divided into 100 bp windows and binned together according to their GC content. Each boxplot shows the read depth of all positions within the respective windows for STARR-seq input plasmid library (A) and cDNA library (B). For each GC content bin, if a genomic window was covered by more fragments than 50% of genomic windows of the same GC content, then the number of fragments covering this genomic window was set as the median value.
Supplementary Figure S4.

Correlation analysis of all sequenced libraries Correlations of reads from all sequenced libraries were calculated. Number of reads was counted for 500 bp bins for each library. If the read numbers from two analyzed libraries for a same bin are both zero, this bin is then excluded from correlation analysis.
Supplementary Figure S5.

Correlation analysis of enhancers from two replicates Correlation of the strength of all enhancers identified by two independent biological replicates was determined using Pearson’s correlation coefficient analysis (r = 0.604). Enhancer activity was calculated based on fold change of cDNA read number over input plasmid read number for 600 bp bins across the whole genome. If the fold change is above 1.3, or at least 30% increase in cDNA than plasmid inserts, then that region is considered as an enhancer.
Supplementary Figure S6.
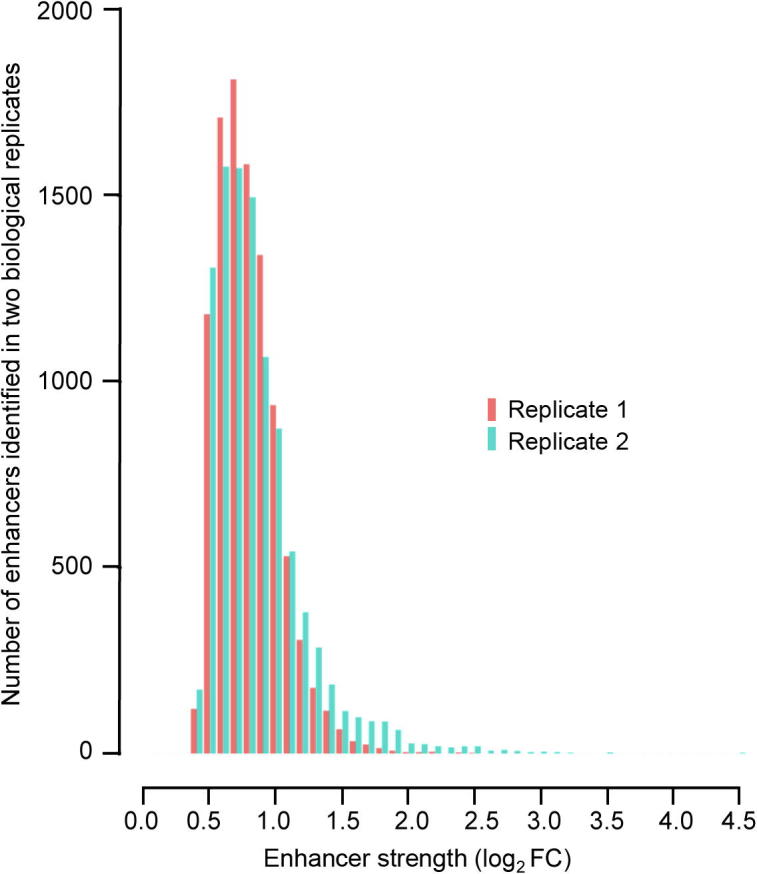
STARR-seq enhancers show a wide range of strengths Distribution of STARR-seq strength values for identified enhancers in two biological replicates.
Supplementary Figure S7.
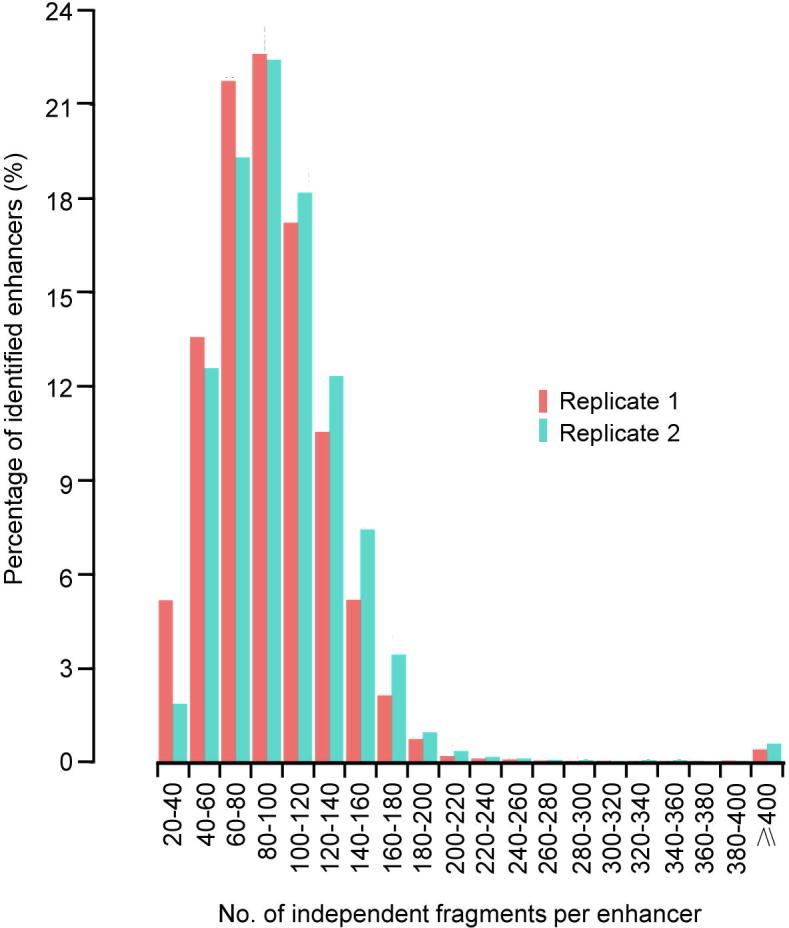
Number of unique fragments supporting the enhancer identification The percentage of enhancers versus the number of independent genomic fragments is shown. The X axis shows the number of unique fragments in the region corresponding to an identified STARR-seq enhancer. The Y axis shows the percentage of enhancers supported by indicated numbers of unique fragments.
Supplementary Figure S8.

GO analysis of genes with proximal promoters in TE regions Gene Ontology analysis for genes with proximal enhancer in TE regions was performed. The top ten categories of enriched genes are shown.
Supplementary Figure S9.
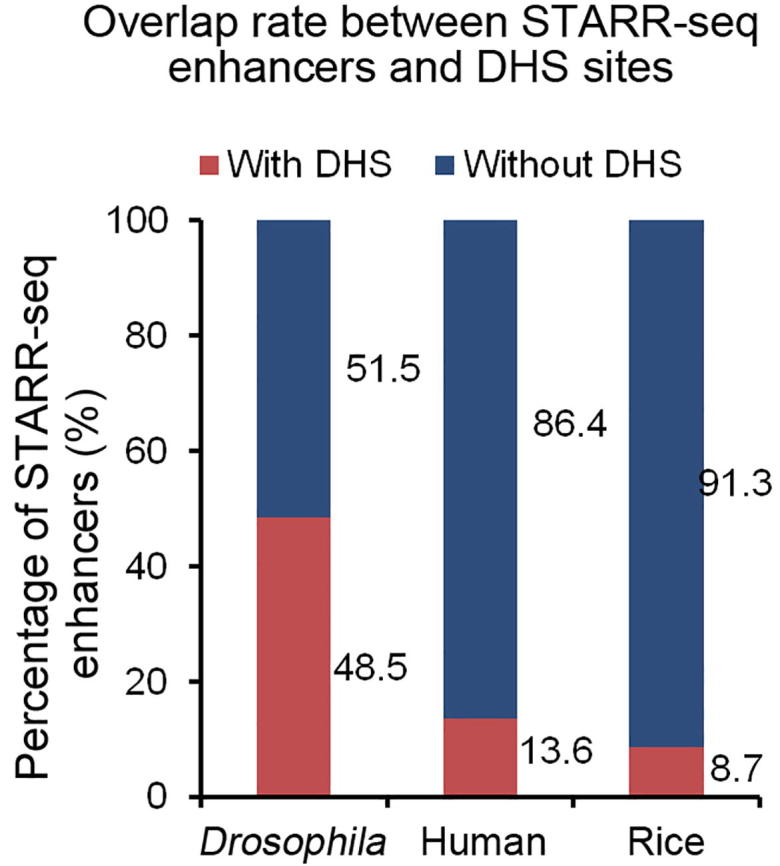
Overlap rate between STARR-seq enhancers and DHS sites The Y axis shows the percentage of STARR-seq enhancers overlapping (red) or not overlapping (blue) with DHSs in three different organisms.
Supplementary Figure S10.

Percentage of elements in clusters carrying specific epigenetic mark STARR-seq enhancers (A), DHS-predicted enhancers (B), and non-enhancer DHSs (C) were clustered into eight groups. The percentage of elements carrying the corresponding mark was calculated and shown as red bar within each cluster. A cluster is designated to be absent of an epigenetic mark if ≤10% of the elements in that cluster carrying the epigenetic mark examined, or designated as low, medium, or high if the percentage of elements in a specific cluster carrying an epigenetic mark examined between 10%–30%, 30%–60%, and >60%.
Supplementary Figure S11.

Distribution of DHSs relative to TSSs The relative DHS enrichment in Drosophila and rice genomes were calculated and normalized against the total number of bins at each specific position. The moving bin size is 500 bp. B. The percentage of DHSs in TSS flanking regions in rice and Drosophila genomes.
Supplementary Figure S12.
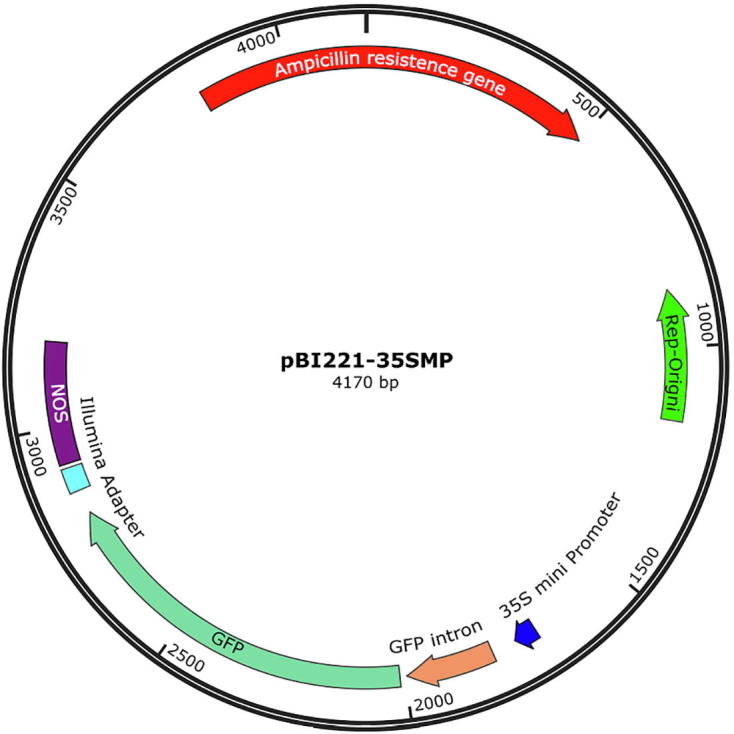
Diagram of reporter vector Plasmid pBI221 was used as reporter vector. A 35S mini promoter was used as basic driver of reporter gene expression. Locations of NOS (plant polyadenylation site), GFP, intron, replication origin, and antibiotic resistant genes are shown in the diagram.
References
- 1.Marand A.P., Zhang T., Zhu B., Jiang J. Towards genome-wide prediction and characterization of enhancers in plants. Biochim Biophys Acta Gene Regul Mech. 2017;1860:131–139. doi: 10.1016/j.bbagrm.2016.06.006. [DOI] [PubMed] [Google Scholar]; Marand AP, Zhang T, Zhu B, Jiang J. Towards genome-wide prediction and characterization of enhancers in plants. Biochim Biophys Acta Gene Regul Mech 2017;1860:131–9. [DOI] [PubMed]
- 2.Bulger M., Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011;144:327–339. doi: 10.1016/j.cell.2011.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell 2011;144:327–39. [DOI] [PMC free article] [PubMed]
- 3.Li G., Ruan X., Auerbach R.K., Sandhu K.S., Zheng M., Wang P. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012;148:84–98. doi: 10.1016/j.cell.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, et al. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 2012;148:84–98. [DOI] [PMC free article] [PubMed]
- 4.Kieffer-Kwon K.R., Tang Z., Mathe E., Qian J., Sung M.H., Li G. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell. 2013;155:1507–1520. doi: 10.1016/j.cell.2013.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kieffer-Kwon KR, Tang Z, Mathe E, Qian J, Sung MH, Li G, et al. Interactome maps of mouse gene regulatory domains reveal basic principles of transcriptional regulation. Cell 2013;155:1507–20. [DOI] [PMC free article] [PubMed]
- 5.Tang Z., Luo O.J., Li X., Zheng M., Zhu J.J., Szalaj P. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell. 2015;163:1611–1627. doi: 10.1016/j.cell.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, et al. CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 2015;163:1611–27. [DOI] [PMC free article] [PubMed]
- 6.Heintzman N.D., Stuart R.K., Hon G., Fu Y., Ching C.W., Hawkins R.D. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]; Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 2007;39:311–8. [DOI] [PubMed]
- 7.Visel A., Blow M.J., Li Z., Zhang T., Akiyama J.A., Holt A. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]; Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 2009;457:854–8. [DOI] [PMC free article] [PubMed]
- 8.Heintzman N.D., Hon G.C., Hawkins R.D., Kheradpour P., Stark A., Harp L.F. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–112. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]; Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009;459:108–12. [DOI] [PMC free article] [PubMed]
- 9.Visel A., Rubin E.M., Pennacchio L.A. Genomic views of distant-acting enhancers. Nature. 2009;461:199–205. doi: 10.1038/nature08451. [DOI] [PMC free article] [PubMed] [Google Scholar]; Visel A, Rubin EM, Pennacchio LA. Genomic views of distant-acting enhancers. Nature 2009;461:199–205. [DOI] [PMC free article] [PubMed]
- 10.Kim T.K., Hemberg M., Gray J.M., Costa A.M., Bear D.M., Wu J. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 2010;465:182–7. [DOI] [PMC free article] [PubMed]
- 11.Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]; Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 2010;107:21931–6. [DOI] [PMC free article] [PubMed]
- 12.Zentner G.E., Tesar P.J., Scacheri P.C. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011;21:1273–1283. doi: 10.1101/gr.122382.111. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 2011;21:1273–83. [DOI] [PMC free article] [PubMed]
- 13.Visel A., Taher L., Girgis H., May D., Golonzhka O., Hoch R.V. A high-resolution enhancer atlas of the developing telencephalon. Cell. 2013;152:895–908. doi: 10.1016/j.cell.2012.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; Visel A, Taher L, Girgis H, May D, Golonzhka O, Hoch RV, et al. A high-resolution enhancer atlas of the developing telencephalon. Cell 2013;152:895–908. [DOI] [PMC free article] [PubMed]
- 14.Whyte W.A., Orlando D.A., Hnisz D., Abraham B.J., Lin C.Y., Kagey M.H. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]; Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013;153:307–19. [DOI] [PMC free article] [PubMed]
- 15.Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-Andre V., Sigova A.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell 2013;155:934–47. [DOI] [PMC free article] [PubMed]
- 16.Arnold C.D., Gerlach D., Stelzer C., Boryn L.M., Rath M., Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013;339:1074–1077. doi: 10.1126/science.1232542. [DOI] [PubMed] [Google Scholar]; Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 2013;339:1074–7. [DOI] [PubMed]
- 17.Inoue F., Ahituv N. Decoding enhancers using massively parallel reporter assays. Genomics. 2015;106:159–164. doi: 10.1016/j.ygeno.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]; Inoue F, Ahituv N. Decoding enhancers using massively parallel reporter assays. Genomics 2015;106:159–64. [DOI] [PMC free article] [PubMed]
- 18.Vanhille L., Griffon A., Maqbool M.A., Zacarias-Cabeza J., Dao L.T., Fernandez N. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat Commun. 2015;6:6905. doi: 10.1038/ncomms7905. [DOI] [PubMed] [Google Scholar]; Vanhille L, Griffon A, Maqbool MA, Zacarias-Cabeza J, Dao LT, Fernandez N, et al. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nat Commun 2015;6:6905. [DOI] [PubMed]
- 19.Dao L.T.M., Galindo-Albarran A.O., Castro-Mondragon J.A., Andrieu-Soler C., Medina-Rivera A., Souaid C. Genome-wide characterization of mammalian promoters with distal enhancer functions. Nat Genet. 2017;49:1073–1081. doi: 10.1038/ng.3884. [DOI] [PubMed] [Google Scholar]; Dao LTM, Galindo-Albarran AO, Castro-Mondragon JA, Andrieu-Soler C, Medina-Rivera A, Souaid C, et al. Genome-wide characterization of mammalian promoters with distal enhancer functions. Nat Genet 2017;49:1073–81. [DOI] [PubMed]
- 20.Liu Y., Yu S., Dhiman V.K., Brunetti T., Eckart H., White K.P. Functional assessment of human enhancer activities using whole-genome STARR-sequencing. Genome Biol. 2017;18:219. doi: 10.1186/s13059-017-1345-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu Y, Yu S, Dhiman VK, Brunetti T, Eckart H, White KP. Functional assessment of human enhancer activities using whole-genome STARR-sequencing. Genome Biol 2017;18:219. [DOI] [PMC free article] [PubMed]
- 21.Liu S., Liu Y., Zhang Q., Wu J., Liang J., Yu S. Systematic identification of regulatory variants associated with cancer risk. Genome Biol. 2017;18:194. doi: 10.1186/s13059-017-1322-z. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu S, Liu Y, Zhang Q, Wu J, Liang J, Yu S, et al. Systematic identification of regulatory variants associated with cancer risk. Genome Biol 2017;18:194. [DOI] [PMC free article] [PubMed]
- 22.Muerdter F., Boryn L.M., Woodfin A.R., Neumayr C., Rath M., Zabidi M.A. Resolving systematic errors in widely used enhancer activity assays in human cells. Nat Methods. 2018;15:141–149. doi: 10.1038/nmeth.4534. [DOI] [PMC free article] [PubMed] [Google Scholar]; Muerdter F, Boryn LM, Woodfin AR, Neumayr C, Rath M, Zabidi MA, et al. Resolving systematic errors in widely used enhancer activity assays in human cells. Nat Methods 2018;15:141–9. [DOI] [PMC free article] [PubMed]
- 23.Zhu B., Zhang W., Zhang T., Liu B., Jiang J. Genome-wide prediction and validation of intergenic enhancers in Arabidopsis using open chromatin signatures. Plant Cell. 2015;27:2415–2426. doi: 10.1105/tpc.15.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhu B, Zhang W, Zhang T, Liu B, Jiang J. Genome-wide prediction and validation of intergenic enhancers in Arabidopsis using open chromatin signatures. Plant Cell 2015;27:2415–26. [DOI] [PMC free article] [PubMed]
- 24.Yang W., Jefferson R.A., Huttner E., Moore J.M., Gagliano W.B., Grossniklaus U. An egg apparatus-specific enhancer of Arabidopsis, identified by enhancer detection. Plant Physiol. 2005;139:1421–1432. doi: 10.1104/pp.105.068262. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yang W, Jefferson RA, Huttner E, Moore JM, Gagliano WB, Grossniklaus U. An egg apparatus-specific enhancer of Arabidopsis, identified by enhancer detection. Plant Physiol 2005;139:1421–32. [DOI] [PMC free article] [PubMed]
- 25.Clark R.M., Wagler T.N., Quijada P., Doebley J. A distant upstream enhancer at the maize domestication gene tb1 has pleiotropic effects on plant and inflorescent architecture. Nat Genet. 2006;38:594–597. doi: 10.1038/ng1784. [DOI] [PubMed] [Google Scholar]; Clark RM, Wagler TN, Quijada P, Doebley J. A distant upstream enhancer at the maize domestication gene tb1 has pleiotropic effects on plant and inflorescent architecture. Nat Genet 2006;38:594–7. [DOI] [PubMed]
- 26.McGarry R.C., Ayre B.G. A DNA element between At4g28630 and At4g28640 confers companion-cell specific expression following the sink-to-source transition in mature minor vein phloem. Planta. 2008;228:839–849. doi: 10.1007/s00425-008-0786-1. [DOI] [PubMed] [Google Scholar]; McGarry RC, Ayre BG. A DNA element between At4g28630 and At4g28640 confers companion-cell specific expression following the sink-to-source transition in mature minor vein phloem. Planta 2008;228:839–49. [DOI] [PubMed]
- 27.Schauer S.E., Schluter P.M., Baskar R., Gheyselinck J., Bolanos A., Curtis M.D. Intronic regulatory elements determine the divergent expression patterns of AGAMOUS-LIKE6 subfamily members in Arabidopsis. Plant J. 2009;59:987–1000. doi: 10.1111/j.1365-313X.2009.03928.x. [DOI] [PubMed] [Google Scholar]; Schauer SE, Schluter PM, Baskar R, Gheyselinck J, Bolanos A, Curtis MD, et al. Intronic regulatory elements determine the divergent expression patterns of AGAMOUS-LIKE6 subfamily members in Arabidopsis. Plant J 2009;59:987–1000. [DOI] [PubMed]
- 28.Raatz B., Eicker A., Schmitz G., Fuss E., Muller D., Rossmann S. Specific expression of LATERAL SUPPRESSOR is controlled by an evolutionarily conserved 3' enhancer. Plant J. 2011;68:400–412. doi: 10.1111/j.1365-313X.2011.04694.x. [DOI] [PubMed] [Google Scholar]; Raatz B, Eicker A, Schmitz G, Fuss E, Muller D, Rossmann S, et al. Specific expression of LATERAL SUPPRESSOR is controlled by an evolutionarily conserved 3' enhancer. Plant J 2011;68:400–12. [DOI] [PubMed]
- 29.Zhao D., Ferguson A.A., Jiang N. What makes up plant genomes: the vanishing line between transposable elements and genes. Biochim Biophys Acta. 2016;1859:366–380. doi: 10.1016/j.bbagrm.2015.12.005. [DOI] [PubMed] [Google Scholar]; Zhao D, Ferguson AA, Jiang N. What makes up plant genomes: the vanishing line between transposable elements and genes. Biochim Biophys Acta 2016;1859:366–80. [DOI] [PubMed]
- 30.Hirsch C.D., Springer N.M. Transposable element influences on gene expression in plants. Biochim Biophys Acta Gene Regul Mech. 2017;1860:157–165. doi: 10.1016/j.bbagrm.2016.05.010. [DOI] [PubMed] [Google Scholar]; Hirsch CD, Springer NM. Transposable element influences on gene expression in plants. Biochim Biophys Acta Gene Regul Mech 2017;1860:157–65. [DOI] [PubMed]
- 31.Zhang J., Luo W., Zhao Y., Xu Y., Song S., Chong K. Comparative metabolomic analysis reveals a reactive oxygen species-dominated dynamic model underlying chilling environment adaptation and tolerance in rice. New Phytol. 2016;211:1295–1310. doi: 10.1111/nph.14011. [DOI] [PubMed] [Google Scholar]; Zhang J, Luo W, Zhao Y, Xu Y, Song S, Chong K. Comparative metabolomic analysis reveals a reactive oxygen species-dominated dynamic model underlying chilling environment adaptation and tolerance in rice. New Phytol 2016;211:1295–310. [DOI] [PubMed]
- 32.Gross D.S., Garrard W.T. Nuclease hypersentitive sites in chromatin. Ann Rev Biochem. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]; Gross DS, Garrard WT. Nuclease hypersentitive sites in chromatin. Ann Rev Biochem 1988;57:159–97. [DOI] [PubMed]
- 33.Zhang W., Wu Y., Schnable J.C., Zeng Z., Freeling M., Crawford G.E. High-resolution mapping of open chromatin in the rice genome. Genome Res. 2012;22:151–162. doi: 10.1101/gr.131342.111. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhang W, Wu Y, Schnable JC, Zeng Z, Freeling M, Crawford GE, et al. High-resolution mapping of open chromatin in the rice genome. Genome Res 2012;22:151–62. [DOI] [PMC free article] [PubMed]
- 34.Klemm S.L., Shipony Z., Greenleaf W.J. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet. 2019;20:207–220. doi: 10.1038/s41576-018-0089-8. [DOI] [PubMed] [Google Scholar]; Klemm SL, Shipony Z, Greenleaf WJ. Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 2019;20:207–20. [DOI] [PubMed]
- 35.Greer E.L., Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]; Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet 2012;13:343–57. [DOI] [PMC free article] [PubMed]
- 36.Kellis M., Wold B., Snyder M.P., Bernstein B.E., Kundaje A., Marinov G.K. Defining functional DNA elements in the human genome. Proc Natl Acad Sci U S A. 2014;111:6131–6138. doi: 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, et al. Defining functional DNA elements in the human genome. Proc Natl Acad Sci U S A 2014;111:6131–8. [DOI] [PMC free article] [PubMed]
- 37.Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]; Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell 2007;129:823–37. [DOI] [PubMed]
- 38.Vastenhouw N., Schier A.F. Bivalent histone modifications in early embryogenesis. Curr Opin Cell Biol. 2012;24:374–386. doi: 10.1016/j.ceb.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vastenhouw N, Schier AF. Bivalent histone modifications in early embryogenesis. Curr Opin Cell Biol 2012;24:374–86. [DOI] [PMC free article] [PubMed]
- 39.Tie F., Banerjee R., Stratton C.A., Prasad-Sinha J., Stepanik V., Zlobin A. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development. 2009;136:3131–3141. doi: 10.1242/dev.037127. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tie F, Banerjee R, Stratton CA, Prasad-Sinha J, Stepanik V, Zlobin A, et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 2009;136:3131–41. [DOI] [PMC free article] [PubMed]
- 40.Zhang Y., Su J., Duan S., Ao Y., Dai J., Liu J. A highly efficient rice green tissue protoplast system for transient gene expression and studying light/chloroplast-related processes. Plant Methods. 2011;7:30. doi: 10.1186/1746-4811-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhang Y, Su J, Duan S, Ao Y, Dai J, Liu J, et al. A highly efficient rice green tissue protoplast system for transient gene expression and studying light/chloroplast-related processes. Plant Methods 2011;7:30 [DOI] [PMC free article] [PubMed]
- 41.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]; Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed]
- 42.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N. The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]; Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence Alignment/Map format and SAMtools. Bioinformatics 2009;25:2078–9. [DOI] [PMC free article] [PubMed]
- 43.Ramirez F., Ryan D.P., Gruning B., Bhardwaj V., Kilpert F., Richter A.S. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–W165. doi: 10.1093/nar/gkw257. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 2016;44:W160–5. [DOI] [PMC free article] [PubMed]
- 44.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]; Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841–2. [DOI] [PMC free article] [PubMed]
- 45.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9:R137. [DOI] [PMC free article] [PubMed]
- 46.Tian T., Liu Y., Yan H., You Q., Yi X., Du Z. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017;45:W122–W129. doi: 10.1093/nar/gkx382. [DOI] [PMC free article] [PubMed] [Google Scholar]; Tian T, Liu Y, Yan H, You Q, Yi X, Du Z, et al. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res 2017;45:W122–9. [DOI] [PMC free article] [PubMed]
- 47.He G., Zhu X., Elling A.A., Chen L., Wang X., Guo L. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell. 2010;22:17–33. doi: 10.1105/tpc.109.072041. [DOI] [PMC free article] [PubMed] [Google Scholar]; He G, Zhu X, Elling AA, Chen L, Wang X, Guo L, et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 2010;22:17–33. [DOI] [PMC free article] [PubMed]
- 48.Fang Y., Wang X., Wang L., Pan X., Xiao J., Wang X.E. Functional characterization of open chromatin in bidirectional promoters of rice. Sci Rep. 2016;6:32088. doi: 10.1038/srep32088. [DOI] [PMC free article] [PubMed] [Google Scholar]; Fang Y, Wang X, Wang L, Pan X, Xiao J, Wang XE, et al. Functional characterization of open chromatin in bidirectional promoters of rice. Sci Rep 2016;6:32088. [DOI] [PMC free article] [PubMed]
- 49.Pan X., Fang Y., Yang X., Zheng D., Chen L., Wang L. Chromatin states responsible for the regulation of differentially expressed genes under 60Co∼γ ray radiation in rice. BMC Genomics. 2017;18:778. doi: 10.1186/s12864-017-4172-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; Pan X, Fang Y, Yang X, Zheng D, Chen L, Wang L, et al. Chromatin states responsible for the regulation of differentially expressed genes under 60Co∼γ ray radiation in rice. BMC Genomics 2017;18:778. [DOI] [PMC free article] [PubMed]
- 50.Gu Z., Eils R., Schlesner M., Ishaque N. EnrichedHeatmap: an R/Bioconductor package for comprehensive visualization of genomic signal associations. BMC Genomics. 2018;19:234. doi: 10.1186/s12864-018-4625-x. [DOI] [PMC free article] [PubMed] [Google Scholar]; Gu Z, Eils R, Schlesner M, Ishaque N. EnrichedHeatmap: an R/Bioconductor package for comprehensive visualization of genomic signal associations. BMC Genomics 2018;19:234. [DOI] [PMC free article] [PubMed]
- 51.de Hoon M.J., Imoto S., Nolan J., Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]; de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics 2004;20:1453–4. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing datasets in this study can be accessed at the Gene Expression Omnibus (GEO) as GEO: GSE121231.