

Abstract
The tumor suppressor protein p53 is a powerful regulator of the embryo’s susceptibility to diverse teratogenic stimuli, functioning both as a teratogenesis inducer and suppressor. Targets which p53 engages to fulfill its functions remains largely undefined, though. We asked whether the miR-34 family, identified as one of the main targets of p53, mediates its function as a teratogenesis inducer. For this, pregnant ICR, p53 and miR-34a heterozygous mice and Spargue-Dowley rats were exposed to 5-aza-2’-deoxycytidin (5-aza), a teratogen inducing limb reduction anomalies (LRA) of the hindlimbs in mice and LRA of either the hindlimbs or forelimbs in rats. Using Hind- and forelimb buds of 5-aza-exposed embryos, we found that the miR-34 family members are the most upregulated miRNAs in mouse and rat limb buds, with their expression level being significantly higher in limb buds destined to exhibit LRA. It was also observed that Met and Gas1 are targeted by miR-34a in embryonic limb buds and that p53 mediates the 5-aza-induced transcriptional miR-34 family activation and miR-34a targets suppression. Finally, we showed that p53 regulates the teratogenic response to 5-aza acting as a teratogenesis inducer and that miR-34a gene deletion do not affect the susceptibility of mice to 5-aza. Overall, this study supports earlier observations demonstrating miR-34 family as a regulatory target of p53 but suggests for the first time that miR-34 family play a redundant function in the pathway(s) engaged by p53 acting as a teratogenesis inducer.
Keywords: miRNA, microRNA, miR-34, limbs, development, p53, teratogens
Introduction
A variety of roles have been shown for the p53 protein to date [1,2]. Among these is its role as a powerful regulator of the embryo’s susceptibility to harmful maternal stimuli and environmental embryopathic stresses (teratogens) [3, 4]. Yet, relatively little is known about the molecular mechanisms through which p53 regulates the response to teratogenic stimuli and additional in vivo studies in embryos are needed to reveal these mechanisms. Indeed, it has been well demonstrated that the set of genes engaged by p53 depends on the cell type and the nature and intensity (dose) of the stress [1,2]. This observation itself strongly suggests that a p53 signaling pathway formed in embryos exposed to stress may differ significantly from that formed in adults. Furthermore, evidence that p53 is capable of functioning both as a teratogenesis suppressor and inducer [5] indicates considerable diversity in the pathways through which p53 regulates the teratogenic response. In these conditions, it is worth noticing studies suggesting microRNAs (miRNAs) as important components of diverse signaling pathways formed by p53 to fulfill its functions [6–11].
miRNAs are a group of small, about 22 nt long non-coding RNAs derived from longer RNA precursors (pre-miRNA) of ~70 to 100 nt, which are processed from primary transcripts (pri-miRNA). miRNAs guide post-transcriptional repression of protein-coding genes by base-pairing with the 3’ untranslated region (3’ UTR) of target mRNAs [12]. One possible mechanism is that miRNAs regulate their gene targets in a fine-tuning manner, while controlling the expression of a large proportion of mammalian genes [13–15]. During recent years, compelling evidence have been collected suggesting that miR-34 family members (miR-34a, b and c) are among p53 downstream targets which p53 engages in order to regulate apoptosis [16–18]. Meanwhile, apoptosis in embryos is tightly regulated and the failure of this regulation inevitably entails maldevelopment [19]. Together, the above data motivated us to ask whether the miR-34 family is involved in p53-mediated response to a teratogenic insult which activates apoptosis.
Pregnant mice and rats exposed to such a teratogen as 5-aza-2’-deoxycytidin (5-aza) were used as models in this work. The choice of the models was based on in vivo studies demonstrating that in mice 5-aza induces limb reduction anomalies (LRA) of the hindlimbs whereas in rats it is able to induce LRA of either the hindlimbs or forelimbs, depending on the development stage at the time of exposure [20–23]. Thus, by using 5-aza we could monitor the expression of miRNAs and their target genes in embryonic structures destined to be malformed simultaneously with those that resist teratogen effect and develop normally within the same embryo. Besides, the above studies have also provided evidence suggesting that 5-aza-induced excessive apoptosis and suppression of cell proliferation are key intermediate steps in the pathogenesis of 5-aza-induced limb anomalies. In parallel, the possibility that p53 may be involved in mediating those cell responses has been suggested by experiments in different cell cultures demonstrating that 5-aza treatment activates p53 and induces p53-dependent apoptosis as well as inhibits cell proliferation via p53-dependent activation of p21 (Waf1/Cip1) [24, 25].
In this study, four basic experimental models were used (see Figure 1): (i) mice treated on day 10 of gestation (GD10, Carnegie stages ~11-12) exhibiting hindlimb LRA and intact forelimbs (Figure 1A); (ii) rats treated on GD 12 (Carnegie stage ~12) also exhibiting hindlimb LRA and intact forelimbs (Figure 1B); (iii) mice treated on GD9 (Carnegie stages ~9-10) exhibiting intact limbs (Figure 1C); and, (iv) rats treated on GD 11 (Carnegie stage ~10) exhibiting forelimb LRA and intact hindlimbs (Figure 1C).
Figure 1: 5-aza-induced LRA models.
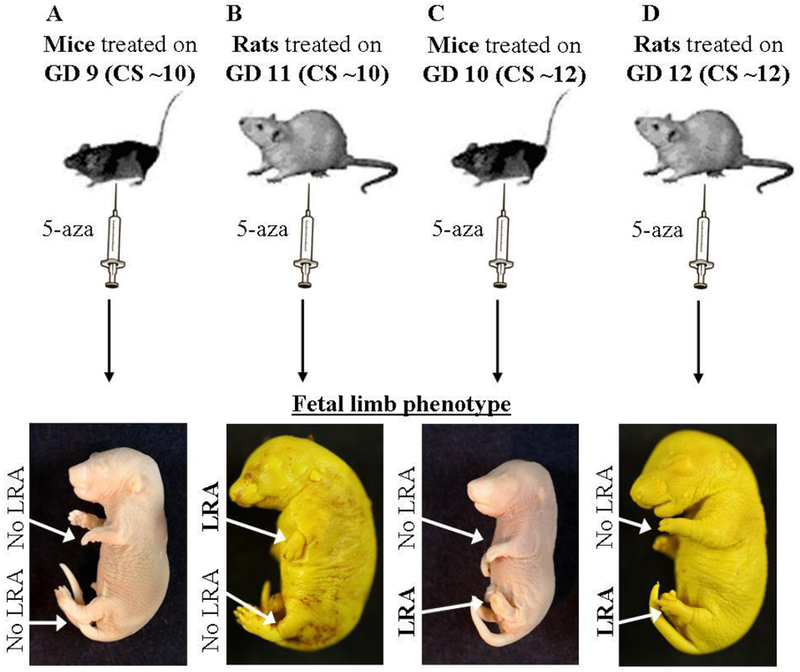
(A-D) Illustration (top) and images (bottom) of the mice and rats limb development models following 5-aza administration at the indicated developmental time points. Images were taken on GD 18. GD = Gestation Day; CS = Carnegie Stage.
Using these models, as well as p53 and miR-34a knockout mice, we asked: (i) how does exposure to 5-aza affect the global expression pattern of miRNAs in embryonic limb buds; after narrowing down to a specific miRNA family, we moved on to ask: (ii) are 5-aza-induced alterations in miR-34 family expression organ- and species-specific; (iii) which mechanisms does 5-aza engage to alter miR-34 family expression and are these p53-dependent; (iv) which are the miR-34 family targets in limb buds of 5-aza exposed embryos; and, (v) how does the limb phenotype correlate with the expression of p53, miR-34 famly and its targets.
Materials and Methods
Animals
Six-to-eight-week-old ICR mice and Spargue-Dowley rats were obtained from the Tel Aviv university animal facility. Breeding pairs of p53 knockout mice [26] and miR-34a knockout mice [27] were received from Prof. Moshe Oren (Weizmann Institute of Science, Israel) and Dr. Lin He (University of California, Berkeley), respectively. A colony of p53 and miR-34a heterozygous mice is being maintained in Tel Aviv University Animal Facility.
The animals were maintained on a 14-h light/10-h dark cycle with food and tap water ad libitum. To obtain pregnancy, females were caged with males for 3h, from 7 to 10 a.m. (darkness), and the presence of a vaginal plug (for mice) or spermatozoids in a vaginal smear (for rats) at 11 a.m. was designated as day 0 of pregnancy (GD 0).
Genotyping of the p53 knockout mice was performed as described elsewhere [28]. Briefly, DNA was extracted from tails and PCR was performed using 3 primers (Sigma): 5′-ACAGCGTGGTGGTACCTTAT-3′, 5′-TATACTCAGAGCCGGCCT-3′, and 5′-CTATCAGGACATAGCGTTGG-3’ under the following conditions: 94°C for 3 min, 40 cycles of (94°C for 30 sec, 55°C for 30 sec, 72°C for 1 min), 72°C for 5 min. Genotyping of the miR-34a knockout mice was performed as for p53 knockout mice, with these exceptions: The presence of a miR-34a allele was identified using the primers (Sigma): Forward: 5’-CCAGCTGTGAGTAATTCTTTG-3′, Reverse: 5 ‘-ACAATGTGCAGCACTTCTAG-3‘ while a 102 bp band could be visualized on an Agarose gel. The presence of miR-34a knockout allele was identified by the presence of a lacZ sequence using the primers: Forward: 5‘-CGTCACACTACTTCTGAACGTCG-3‘, Reverse: 5’-CAGACGATTCATTGGCACCATGC-3’.
5-Aza-2′-deoxycytidin (5-aza) treatment and testing of limb phenotype
The treatment schedules were chosen based on results of our and other studies addressing 5-aza teratogenesis [22, 23, 29]. These studies revealed that in mice and rats, 5-Aza induces limb reduction anomalies (LRA) of hindlimbs if it is injected on GD 10 and GD 12, at approximately Carnegie stage 12, respectively. Yet, in rats 5-Aza induces also LRA of the forelimbs being injected on GD 11, at approximately Carnegie stage 10, whereas no LRA was recorded in mice exposed to 5-AZA on GD9 paralleling the same Carnegie stage in rats. These results were reproduced in our preliminary experiments with mice and rats maintained in our animal facility (Figure 1). Correspondingly, in this study ICR mice were intraperitoneally injected with 5-Aza-2’-deoxycytidin (Sigma) (5-aza) at a dose of 0.5 mg/kg (a teratogenic dose) or 0.15 (a sub-threshold teratogenic dose) (in 0.2 ml saline/20 g body weight) on GD9 or GD10. Rats were injected with 5-aza at a dose of 1.0 mg/kg (in 1.0 ml saline/100g body weight) on GD 11 or GD 12. The p53 and miR-34a heterozygous females were injected with 5-aza on GD 10 at doses 0.5 or 1 mg/kg (in 0.2 ml saline/20g body weight). Pregnant females injected with saline were used as a control throughout the study. To compare the susceptibility of the different p53 or miR-34a genotypes’ embryos to 5-aza-induced teratogenic insult, female mice were sacrificed by cervical dislocation on GD 18, the uteri were removed and implantation sites, resorptions and live fetuses were recorded. Then, fetuses were stained using Alizarin red and Alcian blue protocol [30] and long bones of the hindlimbs (femur, tibia and fibula) were examined for absence, misshaping, incomplete or non-ossification [31]. Animal experiments were approved by the Ethics Committee for Animal Use of Tel Aviv University.
RNA extraction
Hind and forelimbs were dissected at the indicated times following 5-aza treatment on GD 11,12 for rats and GD 9,10 for mice. Only forelimbs were dissected for mice treated on GD 9 since the hindlimb buds are hardly visible at this stage and thus cannot be separated from the torso.
Total RNA was purified using RNeasy kit (Qiagen) for dissected limbs or Trizol (Invitrogen) for C3H10T1/2 cells. RNA quality was assessed using NanoDrop ND-1000 spectrophotometer (Thermo Scientific).
MiRNA profiling and screening
First-strand cDNA was synthesized from total RNA using Megaplex reverse transcriptase reaction with the High Capacity cDNA kit (Applied Biosystems). cDNA and TaqMan Universal PCR Master Mix (No AmpErase UNG; Applied Biosystems) was then transferred into a loading port on Rodent TLDA card A according to the manufacturer’s instructions. PCR amplification was carried using ABI Prism 7900HT Sequence Detection System under the following conditions: 2 min at 50°C, 10 min at 95°C, 40 cycles of (30 sec at 95°C and 1 min at 60°C). miRNA relative levels were calculated based on the comparative threshold cycle (Ct) method. The RQs were calculated using the equation: RQ = 2−ΔΔCt.
Gene expression microarray analysis
Gene expression analysis was performed using Affymetrix GeneChip Mouse Gene 1.0 ST arrays according to the instruction manual, as described in the Affymetrix website (28,853 genes across 770,317 distinct probes; http://www.affymetrix.com). A total of eight arrays were carried out in biological duplicates. Gene profiling array was carried out on CEL files using Partek Genomics Suite TM, version 6.5 (http://www.partek.com). Data were normalized and summarized with the robust multi-average method [32], followed by analysis of variance (ANOVA).
MiR-34 target screening
Putative miR-34 family targets were retrieved by screening for those found in the overlap of the following gene lists: (i) bioinformatics target prediction: putative targets of the mouse miR-34 family according to TargetScan [33]; (ii) relation of the gene function to one of the gene ontology terms enriched in gene expression assays in mice hindlimbs that are destined to exhibit LRA (24 hours following 5-aza treatment): skeletal system development, regulation of developmental process, cell adhesion and regulation of cell differentiation (according to ‘DAVID Bioinformatics Resources’ [34, 35] gene-annotation enrichment analysis; data not shown); (iii) miRNA-target anti-correlative expression. Genes which are downregulated in the hindlimbs by more then 1.3 fold.
MiRNA expression analysis by Real-Time Polymerase Chain Reaction (PCR)
First-strand cDNA was synthesized from total RNA using a MultiScribe reverse transcriptase reaction with the High Capacity cDNA kit (Applied Biosystems) and TaqMan MicroRNA Assay RT primer (Applied Biosystems) for specific miRNA or U6-snRNA. Mixtures containing cDNA, TaqMan Universal PCR Master Mix (No AmpErase UNG; Applied Biosystems) and TaqMan MicroRNA Assay Real Time probe (Applied Biosystems) for each miRNA, were loaded on 96 well plates while PCR amplification and results analysis were done as described under ‘MiRNA profiling and screening’ in Materials and Methods (Thermal cycler conditions: 2 min at 50°C, 10 min at 95°C, 40 cycles of (15 sec at 95°C and 1 min at 60°C).
mRNA and pri-miRNA expression analysis by Real-Time PCR
cDNA synthesis of RNA isolated from limb buds or cells was performed using a MultiScribe reverse transcriptase reaction with the High Capacity cDNA kit (Applied Biosystems). Mixtures containing cDNA, specific primers (Sigma) (see below) and Power SYBR green PCR master mix (Applied Biosystems) were loaded on 96 well plates and PCR amplification was performed as described under ‘miRNA profiling and screening’ in Materials and Methods (Thermal cycler conditions: 2 min at 50°C, 10 min at 95°C, 40 cycles of (15 sec at 95°C and 1 min at 60°C) and dissociation curve cycle of 15 sec at 95°C, 15 sec at 60°C and 15 sec at 95°C). Result analysis was performed as described under ‘miRNA profiling and screening’, using Gapdh as endogenous control.
The primers used were: Gapdh(mouse & rat)-Fwd: 5′ AAA GTG GAT GTC GTC GCC ATC AAT GAT 3′, Gapdh-Rev: 5′ CTG GAA GAT GGT GAT GGG ATT TCC ATT 3′; Met(mouse)-Fwd: 5′ CCA AGC AGT TCA GCA CGT AG 3’, Met-Rev: 5’ ATG ATA GAC ACA GCC AAA ATG C 3’; Met(rat)-Fwd: 5’ TCT GAA GCT TTG TTG TGT ACG G 3′, Met-Rev: 5′ TTG AAG AGA CTG CTT GCT TCC 3′; Gas1(mouse & rat)-Fwd: 5′ GCG AAT CGG TCA AAG AGA AC 3′, Gas1-Rev: 5′ CTTC GTC GTA GTA GTC GTC CAG 3′; Papss2-(mouse & rat)-Fwd: 5’ GAG GTG GCC AGG CTC TTT 3′, Papss2-Rev: 5′ GGG CAT TCT CAC GAT CCT T 3′; Pdgfra(mouse)-Fwd: 5’ TCT GGT CCT CAG CTG TCT CC 3′, Pdgfr-Rev: 5′ TCA TTC TCG TTT GGG AGG AT 3′; Pdgfra(rat)-Fwd: 5′ TCG AAG GCA GGC ACA TTT AT 3′, Pdgfra-Rev: 5′ AAT GAC TAA AGA ATC CGT GC 3′; pri-miR-34 primers from [6]: mmu-pri-mir-34a-Fwd: 5’ CTG TGC CCT CTT GCA AAA GG 3’, mmu-pri-mir-34a-Rev: 5’ GGA CAT TCA GGT GAG GGT CTT G 3’; mmu-pri-mir-34b c-Fwd: 5’ GGC AGG AAG GCT CCA GAT G 3’, mmu-pri-mir-34b/c-Rev: CCT CAC TGT TCA TAT GCC CAT TC 3’.
Bisulfite sequencing analysis
Bisulfite sequencing analysis was performed as described previously [36]. Genomic DNA was extracted from hindlimb buds dissected from GD 10 mice and 2 ug of it was treated with Bisulphite using EpiTect Bisulphite Kit (QIAGEN). This kit converts unmethylated cytosine to uracil, whereas methylated cytosine are unaffected. Enzymatically methylated control DNA (CpGenome Universal Methylated DNA, Chemicon) was used as positive control. For mmu-miR-34a promoter analysis, Bisulphite-treated genomic DNA was used as a template to amplify fragments of 539 bp covering the 469 bp long CpG island (see Supp. Figure 1) using the primers (cytosine converted to uracil in italics): Fwd: TTG TTT GTT ATA GGT TTA TTT AGG TTT, Rev: ACA AAC CCAAAC ACA ACC CCT AC. For mmu-miR-34b/c promoter, amplification was performed for two fragments of 337 bp and 395 bp, together covering the 648 bp long CpG island. For all reactions FastStart Taq DNA Polymerase (Roche) was used as follows: 95°C for 5min, 30 cycles of (95°C for 30sec, 55°C for 40sec and 64°C for 1min) and 64°C for 7min. Gel-purified PCR products were sub-cloned into a TOPO-TA vector (Invitrogen). For each PCR fragment, 10-12 individual clones were sequenced on both strands and methylation status of each CpG pair was examined.
5-aza treatment and miRNA transfection of C3H10T1/2
For 5-aza treatment, murine mesenchymal cell line C3H10T1/2 [37] was seeded in 12-well plates (0.7*10Λ5 cells) in DMEM supplemented with 10% FBS and 1% Pen/Strep (penicillin/streptomycin). 5-Aza-2′-deoxycytidin (100 uM; Sigma) was added at 90-100% confluence. For miR-34a transfection, cells were seeded in 12-well plates (0.7-0.9*10Λ5 cells) in DMEM supplemented with 10% FBS. Transfection of pre-miR-34a (Ambion) was performed 24 hours later using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer’s instructions, with 20 pmol (~28 nM; low transfection) / 80 pmol (~115 nM; High transfection) or a scrambled RNA. In all C3H10T1/2 experiments, cells were harvested for RNA purification 24 hours later.
Gas1 3’ UTR plasmid
Fragments of ~400 bp of Gas1 3’ UTR spanning miR-34 binding site was cloned into the XhoI-NotI restriction site downstream of the Renilla Luciferase Reporter gene of the psiCHECK-2 plasmid (Promega) that also contains a Firefly Luciferase Reporter (used as control) under a different promoter. For this purpose, the 3’ UTR fragments were PCR-amplified from mice genomic DNA and XhoI-NotI restriction sites were added (italics), using the primers: Fwd: 5’ AC A CTC GAG TCC ATC GGT AAT GCT CAG TG 3’, Rev: 5’ AAG GTCAAG CGG CCG CTA CAA AGT AC A GCA ACT GGT A 3’. The miRNAs binding sites was site-directed mutated (4 bases in the seed region) by PCR reaction of the plasmid using the enzyme PfuUltra II Fusion HS DNA Polymerase (Genex), and the PCR reaction: 1)95C for 2min, 2)(95°C for 20sec, 58°C for 20sec, 72°C for 2min)X16, 3)72°C for 3min. The primers used for mutagenesis were the one indicated below (target nucleotides in italics) and a complementary Reverse primer: 5’ CCT AAA GCT CGG TAC CAA TAT CTA GGA AAA CCT C 3’. Products were then incubated with DPNI (New England BioLabs) to digest the methylated source plasmid and the mutated plasmid was sequenced to confirm mutagenesis prior to use.
Dual Luciferase assay
HEK293T cells were seeded in 24-well plates in DMEM supplemented with 10% FBS and 1% Pen/Strep (penicillin/streptomycin). 24 hours later cells were transfected with pre-miR-34a or scrambled RNA (Ambion) using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer’s instructions and 24 hours later with the plasmid psiCHECK-2 containing the desired 3’ UTR with or without site-directed mutations using the TransIT-LT1 Transfection Reagent (Mirus), according to the manufacturer’s instructions. 24 hours later firefly and renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System kit (Promega) and a Veritas microplate luminometer, according to Promega’s instructions.
Western blotting
Mice limbs were collected and lysed in IGEPAL lysis buffer (Sigma Aldrich), quantified using Bio-Rad protein assay (Bio-Rad Laboratories). Sixty μg protein samples were then resolved on 10% SDS PAGE gels, transferred to nitrocellulose membrane (Whatman), blocked with 5% milk in TBS-tween or 1% BSA- and 10% Sodium Azide in TBS-tween, and incubated in primary antibody diluted in blocking solution at 4°C overnight. The antibodies that used were: Actin (Clone C4 (MAB1501, Millipore) and phospho-p53 (Ser15) (9284, Cell Signaling Technology), with the corresponding HRP-conjugated secondary antibodies. Detection was performed using the ECL method (Thermo scientific) and developed using X-ray film (Kodak). Band intensities were quantified using Image-J while relative expression levels were normalized to the expression of Actin.
Statistical analysis
The expression of tested miRNAs/mRNAs was evaluated in three to four samples of fore- and hindlimbs prepared from embryos taken from different litters. The GT2-method for multiple comparisons [38] was used to statistically analyze the data obtained. For the statistical analysis of data characterizing 5-aza induced hindlimb anomalies in p53+/− and miR-34a+/− mice, an embryo was used as an independent variable and the chi-square test was run to look for an association across treatment groups and embryonic genotypes as described [39]. The two-tailed level of significance of differences was equal to 0.05 for all tested parameters.
Results
5-aza modifies miRNA expression in the fore-and hindlimb buds
The expression profile of a comprehensive set of miRNAs was first tested in the hindlimb and forelimb buds of mouse and rat embryos. RNA was collected 24 hours following 5-aza treatment at teratogenic doses 0.5 and 1.0 mg/kg on GD 10 and GD 12, respectively (Supp. Table 1). This treatment regime induces LRA of the hindlimbs but not of the forelimbs [22, 23]. We observed that out of hundreds of miRNAs, miR-34a and miR-34c stood out as the two most upregulated miRNAs in hindlimb buds of both mice and rats (Figure 2A), i.e., in the limbs destined to be malformed. At the same time, in the teratologically resistant forelimb buds, upregulation of miR-34a was far less prominent than that in the hindlimb buds and no upregulation of miR-34c was detected (Figure 2A). When exposing mice to a sub-threshold dose of 5-aza, 0.15 mg/kg, we observed that the extent to which 5-aza affects the expression of miR-34a and c is dose-dependent (Figure 2B).
Figure 2: miRNA profilings indicate miR-34 family elevation in limbs destined to be malformed.
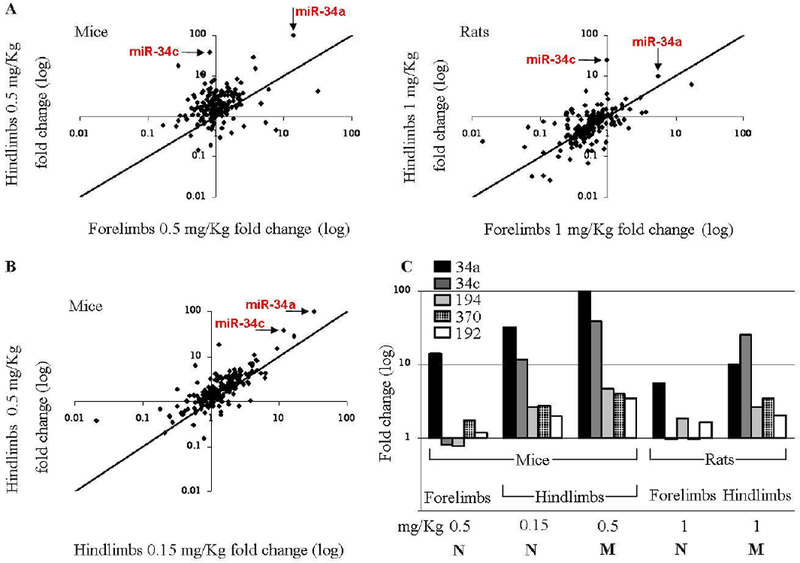
miRNA dysregulation is mostly identified in limb buds destined to be malformed compared to resistant limb buds: (A) 24 hours following injection with 5-aza on GD 10/12 (mice [left] and rats [right], respectively); (B) 24 hours following GD 10 injection with a teratogenic dose (0.5 mg/Kg) versus a sub-threshold dose (0.15 mg/Kg). (C) Expression of miRNAs that showed a dose-dependant upregulation in mice and rats hindlimbs, with either limited or no upregulation in the forelimbs. ‘M’/’N’ indicates Malformed/Normal limb development.
In the above experiments, along with miR-34a and c, three additional miRNAs (miR-194, miR-370 and miR-192 ) showed a dose-dependant upregulation in mice and rats hindlimbs, (Figure 2C). Yet, 5-aza-induced alterations in the expression level of these miRNAs were far less prominent than those registered for miR-34a and c. Additionally, unlike miR-34a and c, the expression level of these miRNAs did not differ significantly in the hind- and forelimb buds although seemingly tended to be higher in the former.
We thus further focused on the miR-34 family members miR-34a and c as well as on miR-34b. We note that miR-34b was absent from the miRNA profiling platform we used, yet given its potential functional role, we tested it using other methods in subsequent experiments.
The expression of miR-34 family is higher in an embryonic structure destined to be malformed
Whereas in mice 5-aza induces only LRA of the hindlimbs, in rats it can also induce LRA of the forelimbs. These observations allowed us to examine whether the extent of 5-aza-induced miR-34 family activation in limb buds is associated with the occurrence of LRA or is it organ- or species-type dependent. We have thus examined the expression of miR-34 family in the two additional models: rats exposed to 5-aza on GD 11 which exhibit LRA of the forelimbs but not of the hindlimbs and mice exposed to 5-aza on approximately the same developmental stage (on GD 9) exhibiting no LRA. As expected, the level of miR-34a expression was higher in limb buds destined for LRA than in limb buds destined to develop normally, in both mice and rats (Figure 3A). Remarkably however, the extent of miR-34a elevation was comparable between all models, for the limb buds destined to be malformed as well as for those that develop normally.
Figure 3: miR-34 dose elevation is correlated to the limb developmental fate.
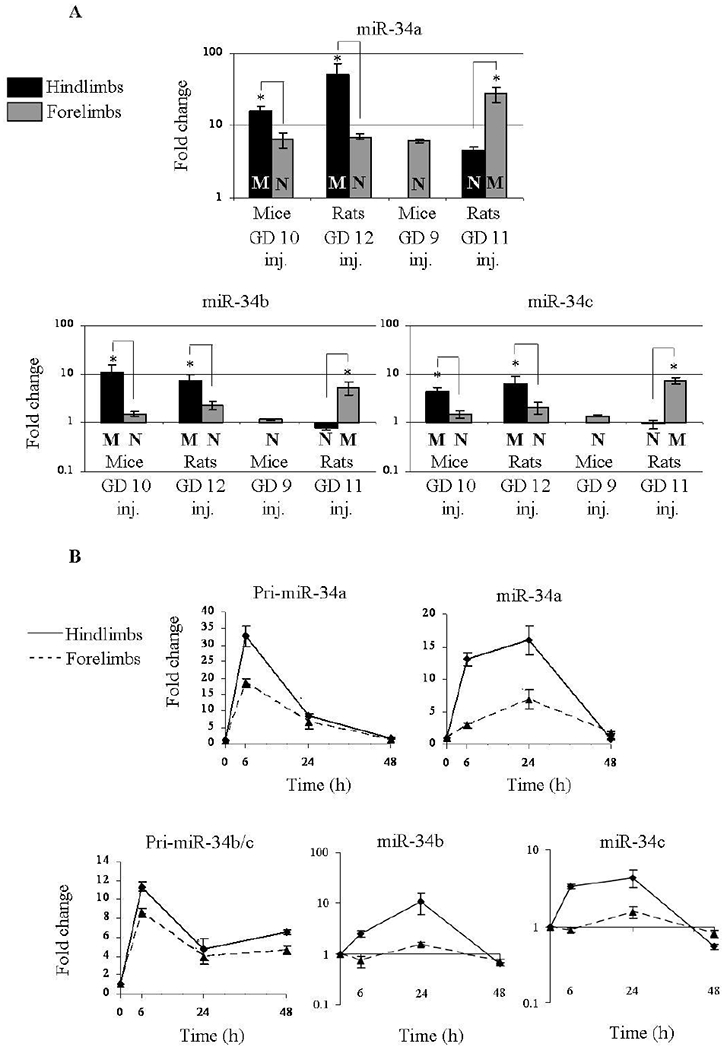
(A) Distinction between miR-34a, b, and c expression in the four models of limb development toxicity. Expression was determined 24 hours following 5-aza treatment. ‘M’/’N’ indicates Malformed/Normal limb development. (B) Expression kinetics of miR-34a, b and c and their pri-miRNA forms in mice hindlimb buds destined to be malformed and resistant forelimb buds in mice exposed to 5-aza on GD 10. (A+B) Fold change values were calculated relative to control animals and U6-snRNA as endogenous control. Results were analyzed statistically using the GT2 test for multiple comparisons and presented as 95% comparison intervals for the means. Means with intervals that do not overlap are significantly different (indicated by *). Means with intervals, which do not reach 1 (the level of expression in controls), differ significantly from controls.
The expression level of miR-34b and c (Figure 3B) was found to be far lower than that of miR-34a. Yet, the expression pattern of these miRNAs in fore-and hindlimb buds of rats and mice did not generally differ from that demonstrated by miR-34a.
Thus, the above results demonstrate that miR-34a is the member of the miR-34 family mostly affected by exposure to 5-aza and will thus be mainly addressed in further experiment. These results also suggest that the extent to which 5-aza activates miR-34 family in the embryo is dependent more on the susceptibility of the tested organ to 5-aza-induced teratogenic stimuli at the moment of exposure than on organ or species specificity.
miR-34 genes are transcriptionaly induced by 5-aza
Given that each miRNA is a derivative of its transcribed primary transcript (pri-miRNA), we measured the level of the RNAs processed to yield the miR-34 family mature miRNAs following exposure to 5-aza. We observed that the expression of both the pri-miR-34a transcript and the transcript common for miR-34b and miR-34c was elevated in the mice limb buds following 5-aza treatment on GD 10. This elevation was of higher magnitude in the hindlimb buds destined to be malformed. (Figure 3B). Plausibly, the expression of these pri-miRNA forms preceded that of the mature forms of miR-34 family in the hindlimb buds as it picked at 6 hours following treatment and then decreased towards 24 and 48 hours whereas the elevation of miR-34 family mature forms in the hindlimb buds was observed as early as 6 hours following treatment but picked at 24 hours, and was abolished at 48 hours (Figure 3B).
Thus, this data suggests that miR-34 genes are transcriptionaly induced by 5-aza and are then processed to yield their mature active forms.
Met and Gas1 are targeted by miR-34a in embryonic limb buds
Each miRNA can potentially bind a large set of target genes. In order to deepen our understanding of the potential role of the miR-34 family in distorted or normal limb development, we explored its putative gene targets. We retrieved these genes by screening for those found in the overlap of the following gene lists: (i) miR-34 family bioinformatic putative prediction of target genes; (ii) genes that function in one of the relevant pathways associated with 5-aza-induced limb maldevelopment; and (iii) genes that showed a down-regulation at the same time when the miRNA levels were elevated. The last two gene lists were based on our broad gene expression profiling in the mice hindlimbs buds that are destined to be malformed, and in the forelimbs that develop normally, 24 hours following 5-aza treatment on GD 10 (data not shown). The overlap of the above datasets retrieved 51 genes (Figure 4A), of those, 25 possess a conserved miR-34 binding site on their 3’-UTRs. We subsequently used all four models described above to screen for the genes that showed a dysregulation in a limb phenotype-dependent manner. We observed that Gas1, Pdgfra, Met and Papss2 were downregulated in limb buds destined to be malformed. Furthermore, these genes were only modified to a lower or null extent in limb buds that develop normally (Figure 4B).
Figure 4: miR-34 family putative targets show an anti-correlative expression.
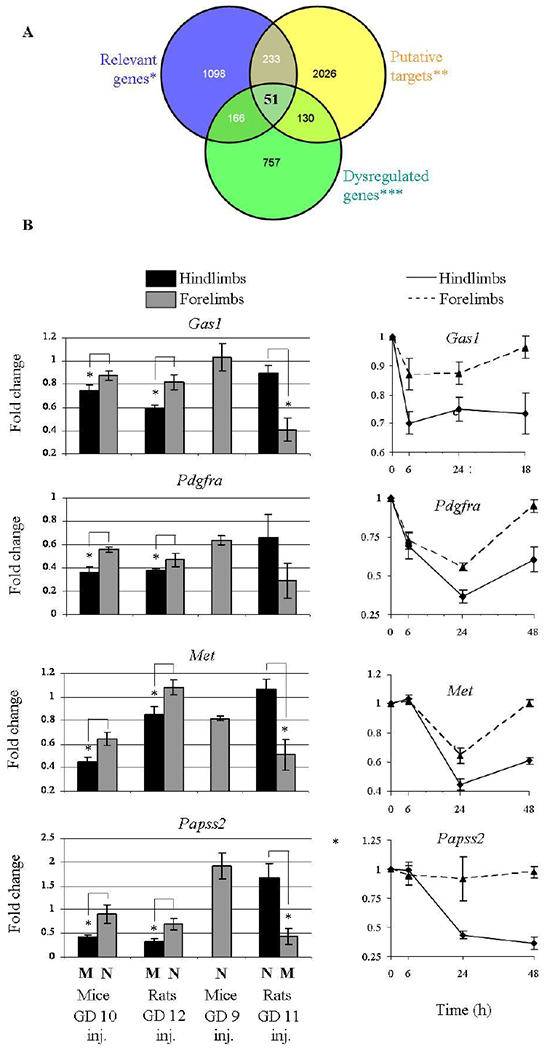
(A) A Venn diagram assisted target search by crossing data from ‘Relevant genes′ (genes with a relevant role for limb development), ′Putative targets′ for mice miR-34 according to TargetScan, and ′Dysregulated genes′ which are downregulated in mice hindlimbs destined to be malformed or upregulated in mice resistant forelimb buds (see Materials and Methods). (B) The expression of selected genes 24 hours following 5-aza treatment in the four models (left). Expression kinetics in mice hindlimb buds destined to be malformed together with resistant forelimb buds in mice exposed to 5-aza on GD 10. ‘M’/’N’ indicates Malformed/Normal limb development. The statistics methods were as described in Figure 3.
While only Met and Pdgfra were previously shown to be targeted by miR-34a [40, 41], we tested which of these genes are genuine targets of miR-34a in the limb development context. Using the murine C3H10T1/2 cell line that is used as a model of limb buds at this developmental stage [37], we observed that treatment of these cells with 5-aza results in miR-34a induction 24 hours later (data not shown). We have shown above that the extent of miR-34a elevation, induced by 5-aza, is associated with the organ susceptibility to 5-aza-induced teratogenic stimuli. Thus, we wanted to mimic this effect in the cell line and test the anti-correlation between the miRNA levels and its target genes. For this purpose we transfected miR-34a into C3H10T1/2 cells in two doses that resulted in an average over-expression of ~5 and ~20 fold (Figure 5 A). Out of the putative target genes from our screen (Gas1, Pdgfra, Met and Papss2), two, Met and Gas1, demonstrated a miR-34a dose-dependant down-regulation in our cell system (Figure 5A).
Figure 5: miR-34a regulates the expression of Met and Gas1 in a dose-dependant manner.
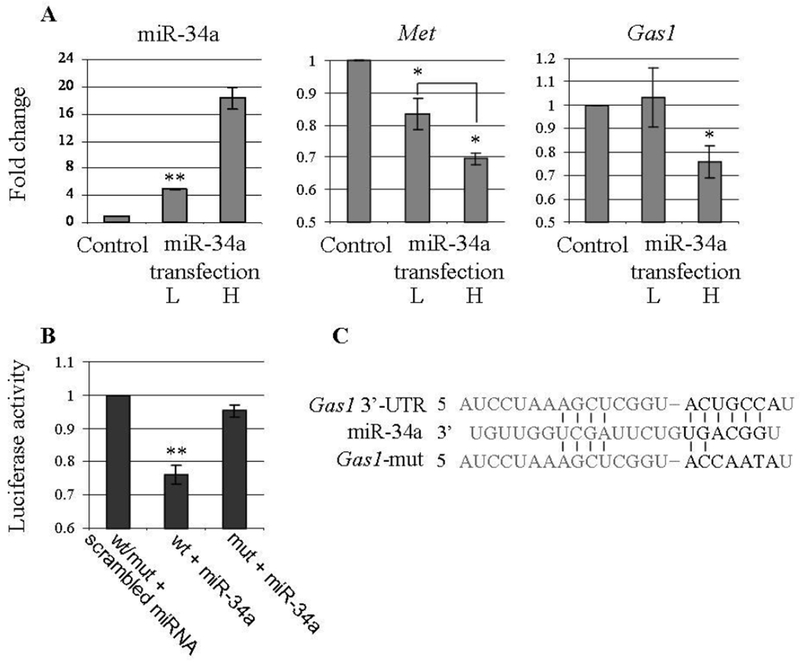
(A) miR-34a, Met and Gasl expression in C3H10T1/2 cells 24 hours following miR-34a low (L; 4.9 fold) and high (H; 18.3 fold) transfections. Fold change was calculated relative to a scrambled RNA transfection and U6-snRNA or Gapdh as endogenous controls. Values are presented as mean ± SEM (N>3). (B) A region from the mouse Gas1 3’-UTRs spanning the miR-34 binding site was cloned into the psiCHECK-2 plasmid downstream of a Renilla luciferase reporter gene. This plasmid or its mutated version was transfected 24 hours after miR-34a transfection into HEK293T cells. Renilla luciferase activity was measured 24 hours later and normalized to firefly luciferase activity. Similarly, transfections with a scrambled miRNA and a mutated version (mut) of miR-34a binding site we used as controls. Values are presented as mean ± SEM (N>3). (C) An illustration of miR-34a and its binding site on the mouse Gas1 3’-UTR (or its mutated version). P-value < 0.05, 0.002 is indicated by * / **, respectively (two-sided t-test).
While Met is an established target of miR-34a [40], we asked whether miR-34a regulation of Gas1 is also based on direct interaction of the miRNA and the gene transcript. For this purpose we employed the Luciferase reporter assay. A region of mouse Gas1 3’-UTR spanning miR-34 family binding site was cloned downstream to a Renilla Luciferase reporter gene. This reporter plasmid was transfected following miR-34a transfection into HEK293T cells. Relative expression of the Renilla Luciferase reporter was then measured compared to that of a Firefly Luciferase reporter, a control transfection and the 3’-UTRs mutated in the miRNA binding site. We observed that miR-34a reduces the Renilla Luciferase-Gas1 activity to 0.76 fold (Figure 5B and C; P-value < 0.002), indicating a direct regulation of miR-34a upon Gas1 expression.
p53 acts as an inducer of 5-aza teratogenesis
Earlier in vitro studies implied that p53 may be involved in the mechanisms underlying 5-aza teratogenesis [24, 25]. In our current work we used p53 heterozygous mice exposed to 5-aza on GD 10 (a model of 5-aza-induced LRA of the hindlimbs; reproductive performance in Supp. Table 2) to test this assumption. First, by using Western blot analysis we showed that the exposure to 5-aza increases the level of p53 protein in fore- and hindlimb buds of p53 +/+ embryos and that this increase is more prominent in the hindlimb buds (Figure 6A). Then, we evaluated the teratogenic response of p53+/− mice to 5-aza by examining the long bones of the hindlimbs (the femur, tibia and fibula) of GD 18 embryos and recorded abnormalities such as incomplete ossification, non-ossified bone, misshaped bone and absent of bone [31] (which indicate increasing levels of limb abnormalities). We observed that in females injected with a high dose of 5-aza (1 mg/kg), the level of postimplantation death of embryos exceeded 60%, being significantly higher compared with that registered in controls (7.9%) and females exposed to a low dose (0.5mg/kg) of 5-aza (10.5%) (Supp. Table 2). Yet, a departure from the Mendelian 25%:50%:25% genotype ratio was not observed in surviving fetuses. The pattern of 5-aza-induced anomalies and their incidence were found to be practically identical in p53 +/+ and p53 +/− fetuses (Figure 6B). Yet, the response of p53 −/− fetuses to 5-aza-induced teratogenic stimuli differed dramatically from that of their p53 positive counterparts. Indeed, when females were treated with 1.0 mg/kg 5-aza, more than 40% of p53 positive fetuses exhibited such anomalies as a non-ossified bone, a misshaped bone and an absent of bone. At the same time, all p53 knockout fetuses had a less severe anomaly such as incomplete ossification but other anomalies were absent. Furthermore, a lower dose of 5-aza, 0.5 mg/kg, turned out to be a threshold teratogenic dose for p53 −/− embryos, inducing incomplete ossification in single fetuses only. Yet, the proportion of their p53 positive counterparts exhibiting non-ossified or/and misshaped bone remained high enough (Figure 6B). These results strongly suggest that p53 acts as an inducer of 5-aza teratogenesis.
Figure 6: p53-null embryos are less sensitive to 5-aza-induced LRA.
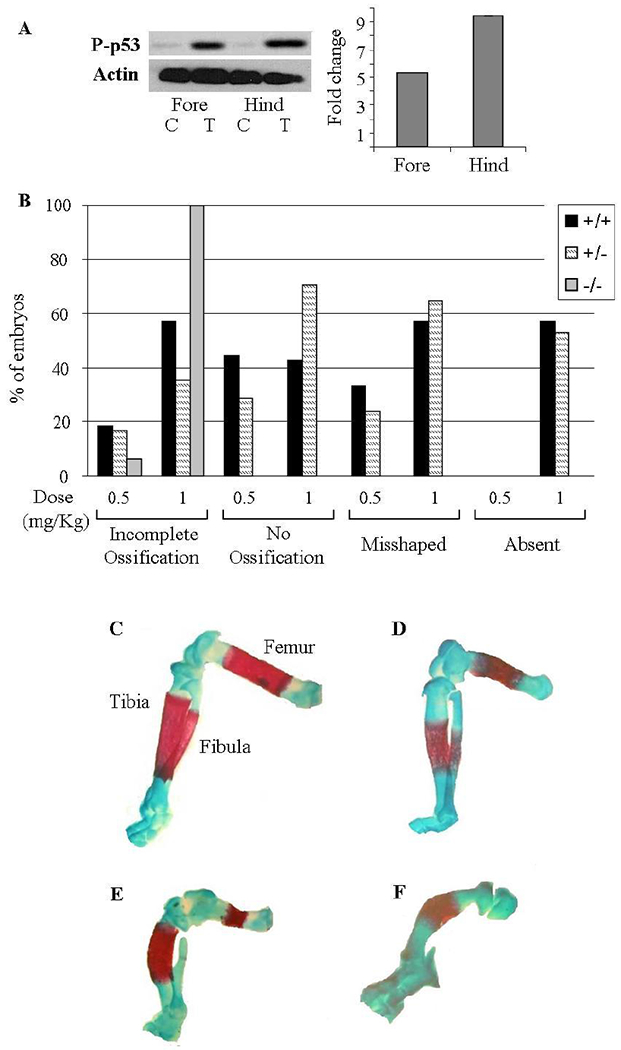
(A) Representative Western blot of phosphorylated p53 in 5-aza treated (‘T’) mice hind limbs destined to be malformed and resistant forelimbs and their controls (‘C’). Quantification was performed in the hind and forelimbs relative to their controls and the Actin levels. (B) Percentage of the different p53 genotypes’ fetuses suffering from the indicated anomalies following 5-aza treatment on GD 10. (B-E) Alizarin red and Alcian Blue staining of cartilage and bone. Limbs were examined on GD 18. (C) Normal long bones: Femur, Tibia and Fibula. (D-F) Representative long bones anomalies: (D) Incomplete ossification of all long bones; (E) All long bones are misshaped, incomplete ossification of Femur and no ossification of the Fibula; (F) Incomplete ossification of the Tibia while the Femur and Fibula are absent.
p53 mediates the 5-aza-induced miR-34 family activation and miR-34a targets suppression
We tested the expression of all members of the miR-34 family in limb buds of p53 positive and negative embryos, 24 hours following exposure to 5-aza. We observed that the level of miR-34 family expression in limb buds of p53 +/+ and p53 +/− embryos was practically identical, being higher, as expected, in the hindlimb than in forelimb buds (Figure 7A). The level of miR-34 family expression in limb buds of p53 −/− embryos was significantly lower than that recorded in their p53 +/+ and +/− counterparts. Remarkably however, it was equal in the forelimb and hindlimb buds and it was higher than that in controls. This data suggest that a p53-dependent mechanism is the sole mechanism underlying the different levels of the miR-34 family activation in limb buds of different fates. Nevertheless, a p53-independent mechanism through which 5-aza activates the miR-34 family in embryonic limb buds also exits. We noticed that the promoters of the mouse and rat miR-34 genes, in addition to p53 binding sites, contain CpG islands, similarly to the previously addressed human promoters [42–46] (Supp. Figure 1). 5-aza is a demethylating agent and has been shown to directly demethylate these CpG islands in humans, implying its potential to use this mechanism to activate miR-34 family in mouse limb buds. Yet, our experiments have demonstrated that the methylation of the miR-34 family promoters in mouse limb buds is very poor (Supp. Figure 1) seemingly eliminating the possibility of promoter methylation as the mode of p53-independent regulation of miR-34 (a,b and c).
Figure 7: miR-34 family elevation is p53-dependant while miR-34a targets suppression is p53 and miR-34a-dependant.
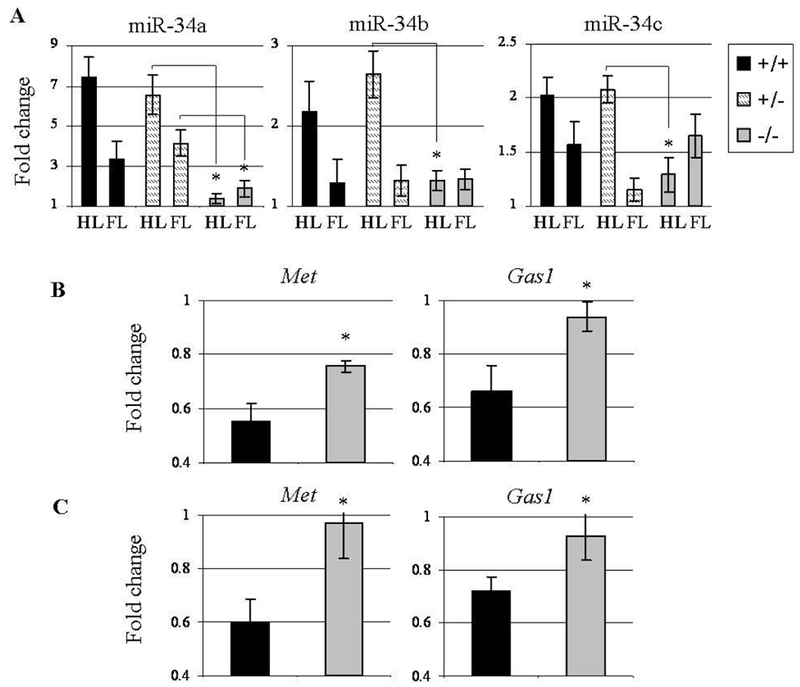
Expression was tested 24 hours following 5-aza treatment on GD 10. (A) miR-34a,b,c expression in hindlimbs (HL) and forelimbs (FL) of different p53 genotypes’ mice embryos. (B) Met and Gas1 expression in the hindlimbs of different p53 genotypes’ mice embryos. (C) Met and Gas1 expression in the hindlimbs of different miR-34a genotypes’ mice embryos. The statistics methods were as described in Figure 3.
Then, we tested the expression of miR-34a established targets, Met and Gas1 in the hindlimbs of p53 positive and negative embryos. We observed that the 5-aza-induced inhibition of these genes was apparent in p53 +/+ embryos but was partly (for Met) and almost completely (for Gas1) attenuated in p53 −/− embryos (Figure 7B).
miR-34a regulates Met and Gas1 expression but not 5-aza teratogenesis
The above data suggested that p53 mediates the 5-aza-induced suppression of Met and Gas1 in limb buds destined to be malformed via miR-34 family activation. In order to test this suggestion and whether miR-34 activation is a pathogenic event for 5-aza teratogenesis, we utilized miR-34a heterozygous mice [27] exposed to 5-aza on GD 10 (a model of 5-aza-induced LRA of the hindlimbs; Reproductive performance in Supp. Table 3). No external anomalies as well as anomalies of the long bones of the hindlimbs were observed in GD 18 miR-34a −/− fetuses of control mice. When mice were exposed to 0.5 mg/kg 5-aza, downregulation of miR-34a established targets, Met and Gas1, was detected in the hindlimb buds of miR-34a +/+ but not in −/− fetuses (Figure 7C), suggesting that miR-34a is a powerful regulator of these genes in the hindlimb buds of in vivo developing embryos. At the same time, we observed that the pattern and intensity of the long bone anomalies were practically identical in miR-34a +/+ and miR-34a −/− fetuses (Supp. Table 4), not differing from those registered in p53 +/+ fetuses of 5-aza treated p53 heterozygous females. This result suggests that miR-34a plays a redundant function in the pathway(s) which p53 engages to function as an inducer of 5-aza terateratogenesis.
Discussion
5-aza is a well-known teratogen inducing limb reduction anomalies (LRA) such as phocomelia and meromelia in mice and rats in an organ- and species-specific fashion. Our study showed that p53 regulates the teratogenic response to 5-aza acting as a teratogenesis inducer. While addressing how 5-aza affects the expression profile of miRNAs in the limb buds we observed that miR-34 family members are the most upregulated miRNAs in mouse and rat limb buds. Further experiments revealed that 5-aza activates miR-34 family by affecting their primary transcripts with a higher magnitude in the limb buds destined to be malformed. It has also been observed that 5-aza activates these miRNAs in both p53-dependent and p53-independent manner and, importantly, the activation was found to be much more prominent when it is mediated by p53.
Altogether, the above data support earlier observations demonstrating miR-34 family as a regulatory target of p53 [16–18]. Yet, in experiments in miR-34a heterozygous mice we observed that the teratogenic response of miR-34a knockout embryos did not differ from that of their miR-34a positive counterparts. This result indicates that miR-34a is dispensable for the function of p53 as a teratogenesis inducer.
The function of p53 as a teratogenesis inducer is firstly associated with its ability to activate the pro-apoptotic signal transduction pathway in response to teratogen-induced apoptotic stimuli [5]. The miR-34 family members are suggested to be key components of p53 networks controlling apoptosis [47]. Do the miR-34b and miR-34c genes, which are coactivated by 5-aza in p53-dependent manner, compensate for miR-34a loss or do they act as independent apoptosis inducers?
Obviously, our study does not allow excluding such scenarios. It is worth noticing, though, that as the positive feedback loops between p53 and miR-34a but not between p53 and miR-34b/c exist (miR-34a activates p53 via suppression of SIRT1 or via Myc-mediated pathway) [47], it is conceivable that miR-34a is a far more powerful apoptosis inducer than miR-34b/c. Additionally, the level of expression was suggested to be a factor influential enough in determining the ability of miR-34 to induce apoptosis, as shown for miR-34a [48]. Meanwhile, in our study, the expression level of miR-34b/c in the limb buds sensitive to 5-aza-induced teratogenic stimuli was significantly lower than that of miR-34a. In light of this evidence, it seems unlikely that miR-34b/c may fully compensate for mir-34a loss in embryos responding to 5-aza.
Thus, our results arguing against an essential role for miR-34 family in the function of p53 as an inducer of 5-aza teratogenesis seemingly conflicts with studies indicating the miR-34 family members as important components of the p53-controled pro-apoptotic network [47]. This discrepancy is eliminated, though, if we take in mind that these studies were performed mainly in in vitro cell-based models which are hardly suitable for studies addressing developmental toxicity due to their failure to adequately represent the full scope of developmental complexity. Interestingly, however, our results seem to be indirectly supported by a study performed recently in mice carrying targeted deletion of all three members of the miR-34 family. Indeed, ionizing radiation in high doses is a teratogen activating the p53-mediated pro-apoptotic signaling [5]. The study cited above has shown that miR-34 family function is not required for p53-mediated apoptosis induced by ionizing radiation in thymocytes of the miR-34 (a, b and c) knockout mice.
Furthermore, the set of genes targeted by any miRNA depends on the cellular context [47]. Therefore, it is hardly surprising that, whereas there is a plethora of genes regulating apoptosis detected as miR-34a targets in various cultured cells, in the limb buds of embryos exposed to 5-aza only two genes, Met and Gas1, were shown to be genuine miR-34a targets. Can these genes be involved in determining the response of embryos to 5-aza-induced apoptotic stimuli?
Such a possibility is suggested by studies in in vitro models demonstrating that both Met and Gas1 are able to counteract and promote apoptosis in a cell-context depended manner. At the same time, 5-aza-induced teratogenic insult causes not only suppression of Met and Gas1: we observed that it also activates many pro-apoptotic genes, including Myc, Bak1, Bax, phlda3 and Itgb3bp (data not presented) as well as caspase3 [29]. And noteworthy, both heterozygous Gas1 and Met mice demonstrate normal phenotype of the long bones Together, the above data suggest that miR-34a-mediated suppression of Met and Gas1 may not significantly affect the intensity of excessive apoptosis initiated by 5-aza in the hind limb buds.
In conclusion, although our study suggests no a functional role of miR-34a in teratogen-initiated p53-mediated apoptosis, the possibility that this miR-34 family may regulate the susceptibility of embryos to teratogens remains realistic enough. First, as mentioned above, p53 is able to act not only as a teratogenesis inducer but also as a protector and its protective role may firstly be associated with such processes as antioxidant defense and DNA repair [5]. It is conceivable that these p53 activities may be regulated by miR-34a via miR-34a-p53 feedback loops. Second, our current study as well our previous work with cyclophosphamide (CP) show that these teratogens are able to activate miR-34 family acting at a sub-threshold teratogenic dose, i.e. a dose inducing no structural anomalies. At the same time, it has been shown that distorted expression of miR-34 family in embryos results in the appearance of offspring having no external anomalies at birth but exhibiting trabecular bone architecture indicative of bone loss in adulthood. And our recent study has shown that a single exposure of pregnant mice to a sub-threshold dose of 5-aza also results in bone loss in adult offspring [29]. It is undoubtedly important to reveal whether other teratogens can affect offspring in this way and to what extent miR-34 family are involved in mediating this phenomenon. Third we observed that 5-aza and CP are able to activate miR-34 family in p53-independent fashion. This observation implies a scenario in which miR-34 family plays a regulatory role in the response to embryophatic stresses not activating the p53 pathway while the reality of this scenario deserves to be explored. It is obvious that in vivo models should be the first choice for such studies.
Supplementary Material
References
- 1.Lane D and Levine A, p53 Research: the past thirty years and the next thirty years. Cold Spring Harb Perspect Biol, 2010. 2(12): p. a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vousden KH and Prives C, Blinded by the Light: The Growing Complexity of p53. Cell, 2009. 137(3): p. 413–31. [DOI] [PubMed] [Google Scholar]
- 3.Torchinsky A, Fein A, and Toder V, Teratogen-induced apoptotic cell death: does the apoptotic machinery act as a protector of embryos exposed to teratogens? Birth Defects Res C Embryo Today, 2005. 75(4): p. 353–61. [DOI] [PubMed] [Google Scholar]
- 4.Mirkes PE, Cell death in normal and abnormal development. Congenit Anom (Kyoto), 2008. 48(1): p. 7–17. [DOI] [PubMed] [Google Scholar]
- 5.Torchinsky A and Toder V, Mechanisms of the embryo’s response to embryopathic stressors: a focus on p53. J Reprod Immunol, 2010. 85(1): p. 76–80. [DOI] [PubMed] [Google Scholar]
- 6.He L, et al. , A microRNA component of the p53 tumour suppressor network. Nature, 2007. 447(7148): p. 1130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raver-Shapira N, et al. , Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell, 2007. 26(5): p. 731–43. [DOI] [PubMed] [Google Scholar]
- 8.Chang TC, et al. , Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell, 2007. 26(5): p. 745–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tarasov V, et al. , Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle, 2007. 6(13): p. 1586–93. [DOI] [PubMed] [Google Scholar]
- 10.Bommer GT, et al. , p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol, 2007. 17(15): p. 1298–307. [DOI] [PubMed] [Google Scholar]
- 11.Corney DC, et al. , MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate In control of cell proliferation and adhesion-independent growth. Cancer Res, 2007. 67(18): p. 8433–8. [DOI] [PubMed] [Google Scholar]
- 12.Bartel DP, MicroRNAs: target recognition and regulatory functions. Cell, 2009. 136(2): p. 215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friedman RC, et al. , Most mammalian mRNAs are conserved targets of microRNAs. Genome Res, 2009. 19(1): p. 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hornstein E and Shomron N, Canalization of development by microRNAs. Nat Genet, 2006. 38 Suppl: p. S20–4. [DOI] [PubMed] [Google Scholar]
- 15.Krol J, Loedige I, and Filipowicz W, The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet, 2010. 11(9): p. 597–610. [DOI] [PubMed] [Google Scholar]
- 16.Raver-Shapira N and Oren M, Tiny actors, great roles: microRNAs in p53’s service. Cell Cycle, 2007. 6(21): p. 2656–61. [DOI] [PubMed] [Google Scholar]
- 17.Feng Z, et al. , Tumor suppressor p53 meets microRNAs. J Mol Cell Biol, 2011. 3(1): p. 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hermeking H, The miR-34 family in cancer and apoptosis. Cell Death Differ, 2010. 17(2): p. 193–9. [DOI] [PubMed] [Google Scholar]
- 19.Toder V, et al. , The role of pro- and anti-apoptotic molecular interactions in embryonic maldevelopment. Am J Reprod Immunol, 2002. 48(4): p. 235–44. [DOI] [PubMed] [Google Scholar]
- 20.Rogers JM, et al. , Cell death and cell cycle perturbation in the developmental toxicity of the demethylating agent, 5-aza-2’-deoxycytidine. Teratology, 1994. 50(5): p. 332–9. [DOI] [PubMed] [Google Scholar]
- 21.Branch S, et al. , Teratogenic effects of the demethylating agent 5-aza-2’-deoxycytidine in the Swiss Webster mouse. Toxicology, 1996. 112(1): p. 37–43. [DOI] [PubMed] [Google Scholar]
- 22.Branch S, et al. , 5-AZA-2’-deoxycytidine-induced dysmorphogenesis in the rat. Teratog Carcinog Mutagen, 1999. 19(5): p. 329–38. [PubMed] [Google Scholar]
- 23.Rosen MB and Chernoff N, 5-Aza-2’-deoxycytidine-induced cytotoxicity and limb reduction defects in the mouse. Teratology, 2002. 65(4): p. 180–90. [DOI] [PubMed] [Google Scholar]
- 24.Karpf AR, et al. , Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-aza-2’-deoxycytidine. Mol Pharmacol, 2001. 59(4): p. 751–7. [PubMed] [Google Scholar]
- 25.Zhu WG, et al. , 5-aza-2’-deoxycytidine activates thep53/p21Waf1/Cip1 pathway to inhibit cell proliferation. J Biol Chem, 2004. 279(15): p. 15161–6. [DOI] [PubMed] [Google Scholar]
- 26.Jacks T, et al. , Tumor spectrum analysis in p53-mutant mice. Curr Biol, 1994. 4(1): p. 1–7. [DOI] [PubMed] [Google Scholar]
- 27.Choi YJ, et al. , miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol, 2011. 13(11): p. 1353–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pekar O, et al. , p53 regulates cyclophosphamide teratogenesis by controlling caspases 3, 8, 9 activation and NF-kappaB DNA binding. Reproduction, 2007. 134(2): p. 379–88. [DOI] [PubMed] [Google Scholar]
- 29.Torchinsky A, et al. , Bone loss in adult offspring induced by low-dose exposure to teratogens. J Bone Miner Metab, 2012. 30(3): p. 270–80. [DOI] [PubMed] [Google Scholar]
- 30.Inouye M, Differential Staining of cartilage and Bone in Fetal Mouse Skeleton by Alcian Blue and Alizarin Red S. Congenital Anomolies, 1976. 16: p. 171–173. [Google Scholar]
- 31.Wise LD, et al. , Terminology of developmental abnormalities in common laboratory mammals (version 1). Teratology, 1997. 55(4): p. 249–92. [DOI] [PubMed] [Google Scholar]
- 32.Irizarry RA, et al. , Use of mixture models in a microarray-based screening procedure for detecting differentially represented yeast mutants. Stat Appl Genet Mol Biol, 2003. 2: p. Article1. [DOI] [PubMed] [Google Scholar]
- 33.Lim LP, et al. , Vertebrate microRNA genes. Science, 2003. 299(5612): p. 1540. [DOI] [PubMed] [Google Scholar]
- 34.Huang da W, Sherman BT, and Lempicki RA, Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res, 2009. 37(1): p. 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang da W, Sherman BT, and Lempicki RA, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc, 2009. 4(1): p. 44–57. [DOI] [PubMed] [Google Scholar]
- 36.Lodygin D, et al. , Inactivation of miR-34a by aberrant CpG methylation in multiple types of cancer. Cell Cycle, 2008. 7(16): p. 2591–600. [DOI] [PubMed] [Google Scholar]
- 37.Shea CM, et al. , BMP treatment of C3H10T1/2 mesenchymal stem cells induces both chondrogenesis and osteogenesis. J Cell Biochem, 2003. 90(6): p. 1112–27. [DOI] [PubMed] [Google Scholar]
- 38.Sokal RR and Rohlf FJ, Biometry: the principles and practice of statistics in biological research. 1995: New York (NY): Freeman, 3 1995. [Google Scholar]
- 39.Fleiss JL, Statistical methods for rates and proportions. 1981: New York: Wiley [Google Scholar]
- 40.Li N, et al. , miR-34a inhibits migration and invasion by down-regulation of c-Met expression in human hepatocellular carcinoma cells. Cancer Lett, 2009. 275(1): p. 44–53. [DOI] [PubMed] [Google Scholar]
- 41.Silber J, et al. , miR-34a repression in proneural malignant gliomas upregulates expression of its target PDGFRA and promotes tumorigenesis. PLoS One, 2012. 7(3): p. e33844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiroki E, et al. , MicroRNA-34b functions as a potential tumor suppressor in endometrial serous adenocarcinoma. Int J Cancer, 2012. 131(4): p. E395–404. [DOI] [PubMed] [Google Scholar]
- 43.Mazar J, et al. , Epigenetic regulation of microRNA genes and the role of miR-34b in cell invasion and motility in human melanoma. PLoS One, 2011. 6(9): p. e24922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kalimutho M, et al. , Epigenetically silenced miR-34b/c as a novel faecal-based screening marker for colorectal cancer. Br J Cancer, 2011. 104(11): p. 1770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nalls D, et al. , Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS One, 2011. 6(8): p. e24099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, et al. , CpG island methylation status of miRNAs in esophageal squamous cell carcinoma. Int J Cancer, 2012. 130(7): p. 1607–13. [DOI] [PubMed] [Google Scholar]
- 47.Okada N, et al. , A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014. March 1; 28(5):438–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamakuchi M, et al. , miR-34a repression of SIRTl regulates apoptosis. Proc Natl Acad Sci USA. 2008. September 9; 105(36):13421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.