

Abstract
Aims
Develop a population pharmacokinetics model of tacrolimus in organ transplant recipients receiving twice‐daily, immediate‐release (IR‐T; Prograf) and/or once‐daily, prolonged‐release (PR‐T; Advagraf or Astagraf XL) tacrolimus.
Methods
Tacrolimus concentration–time profiles were analysed from 8 Phase II studies in adult and paediatric liver, kidney and heart transplant patients receiving IR‐T and/or PR‐T. A tacrolimus population pharmacokinetic model, including identification of significant covariates, was developed using NONMEM.
Results
Overall, 23,176 tacrolimus concentration records were obtained from 408 patients. A 2‐compartment model with first‐order absorption and elimination described the concentration–time profiles. Tacrolimus absorption rate was 50% slower with PR‐T vs IR‐T. Tacrolimus apparent oral clearance was 44.3 L/h in Whites and 59% higher in Asians. Tacrolimus central volume of distribution was 108 L in males and 55% lower in females; trough concentrations were similar between formulations. Tacrolimus relative bioavailability was similar between formulations (geometric mean ratio PR‐T:IR‐T 95%, 90% confidence intervals: 89%, 101%). Asians had 83% and 51% higher relative bioavailability than Whites and Blacks, respectively, for IR‐T and PR‐T. Whites had 49% and 77% higher relative bioavailability than Blacks for PR‐T and IR‐T, respectively. Blacks had 52% lower relative bioavailability than Whites and Asians for IR‐T and PR‐T. Type of organ transplanted and patient population (adult/paediatric) did not have a significant effect on tacrolimus pharmacokinetics.
Conclusions
This population pharmacokinetic model described data from transplant recipients who received IR‐T and/or PR‐T. Tacrolimus trough concentrations and relative bioavailability were similar between formulations, supporting 1 mg:1 mg conversion from Prograf to Advagraf/Astagraf XL in clinical practice.
Keywords: immunosuppression, NONMEM, pharmacokinetics, transplantation
What is already known about this subject
Tacrolimus is a mainstay of immunosuppressive therapy after solid‐organ transplantation.
Tacrolimus is available as twice‐daily, immediate‐release (IR‐T) and once‐daily, prolonged‐release (PR‐T) formulations.
Tacrolimus has a narrow therapeutic range, with target whole blood trough concentrations of 5–20 ng/mL.
Tacrolimus trough concentrations are highly correlated with drug exposure and, consequently, with clinical transplant outcomes.
Few population pharmacokinetics studies have characterized the pharmacokinetics of both immediate‐ and prolonged‐release tacrolimus.
What this study adds
Although tacrolimus absorption rates differed between the immediate‐ and prolonged‐release formulations, interpatient variability in tacrolimus trough concentrations was similar, as was relative bioavailability.
Racial differences in relative bioavailability were noted (Asians>Whites>Blacks), independent of tacrolimus formulation.
The results support a 1 mg:1 mg conversion factor for switching patients from immediate‐release tacrolimus (Prograf) to prolonged‐release tacrolimus (Advagraf or Astagraf XL) in clinical practice.
1. INTRODUCTION
Preventing graft rejection is one of the most important challenges facing clinicians in organ transplantation, and patients are generally required to adhere to lifelong immunosuppression. Tacrolimus is the cornerstone of immunosuppressive therapy for the prophylaxis and treatment of allograft rejection in liver, kidney and heart transplantation. The prolonged‐release formulation of tacrolimus comprises a single, daily morning dose, providing a simpler treatment option than the twice‐daily, immediate‐release formulation.1, 2
Following oral administration, immediate‐release tacrolimus is rapidly absorbed compared with prolonged‐release tacrolimus (time to reach maximum plasma concentration of 2.9 vs 5.0 hours, following liver transplantation).3 Tacrolimus is extensively distributed in the body, as indicated by a large steady‐state volume of distribution, with a 20:1 distribution ratio of whole blood/plasma concentrations,1 and is highly bound to plasma proteins (approximately 99%), predominantly serum albumin (ALB).1, 2, 4 Metabolism of systemically available tacrolimus occurs in the liver, primarily by cytochrome P450 3A4 (CYP3A4) and CYP3A5, with predominantly faecal elimination.2, 5 However, there is also evidence of presystemic gastrointestinal metabolism in the intestinal wall by CYP3A4 and CYP3A5, which reduces the oral bioavailability of tacrolimus.2, 5 After conversion from immediate‐ to prolonged‐release tacrolimus, mean systemic exposure to tacrolimus (area under the concentration–time curve [AUC]) with the prolonged‐release formulation is approximately 10% lower than the immediate‐release formulation at equivalent doses.6, 7, 8 However, following dose adjustment, exposure is similar at steady state.9, 10, 11
Tacrolimus administration is complicated by a narrow therapeutic range, and inter‐ and intrapatient pharmacokinetic (PK) variability.12 Tacrolimus trough concentrations are monitored to guide dose adjustment, as trough concentration is highly correlated with tacrolimus AUC and subsequently clinical outcomes.13, 14 The relationship between tacrolimus trough concentrations and AUC is similar between prolonged‐ and immediate‐release tacrolimus in both liver and kidney transplant patients;15, 16 therefore the same therapeutic drug monitoring approach can be used with both formulations. Tacrolimus whole‐blood trough concentrations are generally maintained within the range of 5–20 ng/mL.17
Several studies have characterized the PK of immediate‐release tacrolimus;18, 19, 20, 21 however, there are few population PK studies characterizing the PK of both immediate‐ and prolonged‐release formulations.22 This modelling study was undertaken to characterize the population PK of immediate‐ and prolonged‐release tacrolimus in liver, kidney and heart transplant recipients, as well as identify the demographic and covariate factors that have a significant influence on tacrolimus PK.
2. METHODS
2.1. Patients and studies
Data were obtained for liver, kidney and heart transplant recipients from 8 Phase II studies with immediate‐release tacrolimus (Prograf, Astellas Pharma Ltd, Chertsey, UK) and prolonged‐release tacrolimus (Advagraf or Astagraf XL, Astellas Pharma Europe BV, The Netherlands)11, 15, 23, 24, 25, 26, 27, 28 (Table 1). The data presented here were derived from the internal databases of each Phase II study; 6 studies assessed tacrolimus PK in stable transplant recipients converted from immediate‐ to prolonged‐release tacrolimus.15, 23, 24, 25, 26, 27 Of these, 3 studies assessed adult kidney transplant recipients (02–0‐131, FG‐506E‐12‐02, and FJ‐506E‐KT01),15, 23, 24 1 assessed adult liver recipients (02–0‐152),25 1 assessed paediatric (mean age 9 years) liver recipients (03–0‐160)27 and 1 assessed adult heart recipients (FG‐506‐15‐02).26 The other 2 studies compared the PK profile of immediate‐ and prolonged‐release tacrolimus in de novo kidney or liver transplant recipients (FG‐506E‐12‐01 and FG‐506‐11‐01, respectively).11, 28 Full details of the methodology used in these studies have been reported previously.11, 15, 23, 24, 25, 26, 27, 28
Table 1.
Brief summary of tacrolimus phase II clinical studies
| Study protocol number | Population | Patients | Type of study | PKAS/FAS, n a |
|---|---|---|---|---|
| 02–0‐131 23 | Adult | Stable kidney transplant | Conversion from immediate‐ to prolonged‐release tacrolimus | 57 (PPS)/68 |
| FG‐506E‐12‐02 15 | Adult | Stable kidney transplant | Conversion between immediate‐ and prolonged‐release tacrolimus | 60/69 |
| FJ‐506E‐KT01 24 | Adult | Stable kidney transplant (Japanese) | Conversion from immediate‐ to prolonged‐release tacrolimus | 35/37 |
| 02–0‐152 25 | Adult | Stable liver transplant | Conversion between immediate‐ and prolonged‐release tacrolimus | 51 (PPS)/70 |
| FG‐506‐15‐02 26 | Adult | Stable heart transplant | Conversion from immediate‐ to prolonged‐release tacrolimus | 45/85 |
| 03–0‐160 27 | Paediatric | Stable paediatric liver transplant | Conversion from immediate‐ to prolonged‐release tacrolimus | 17/19 |
| FG‐506E‐12‐01 11 | Adult | Primary kidney transplant | Comparative study of prolonged‐ vs immediate‐release tacrolimus | 66/119 |
| FG‐506‐11‐01 28 | Adult | Primary liver transplant | Comparative study of prolonged‐ vs immediate‐release tacrolimus | 77/129 |
Contains previously unpublished study data. FAS, full‐analysis set; PKAS, pharmacokinetics‐analysis set; PPS, per‐protocol set.
The study protocols were reviewed and approved by the institutional review board or independent ethics committee of participating institutions, and the studies were conducted in accordance with Good Clinical Practice regulations and the Declaration of Helsinki. Written informed consent was obtained from each patient (or legal guardian) prior to enrolment.11, 15, 23, 24, 25, 26, 27, 28
2.2. Tacrolimus blood collection and assay in pharmacokinetic studies
Samples to assay tacrolimus whole‐blood trough concentrations were collected throughout the PK treatment period. On the days of PK assessment, blood samples were collected before oral administration of immediate‐ (first dose) or prolonged‐release tacrolimus, and 0.5, 1, 2, 3, 4, 6, 8, 12, 12.5, 13, 14, 15, 16, 18, 20 and 24 hours post dose. The sampling strategies used across the studies were similar, with an intraday precision of 2.4–7.9%, and an interday precision of 3.0–12.1%.
At each PK sampling time point, ≥1 mL of whole blood was collected, and samples frozen at −20°C until analysis. Concentrations of tacrolimus were determined in whole blood (ethylenediaminetetraacetic acid anticoagulant) using high‐performance liquid chromatography (LC) with tandem mass spectrometry (MS/MS), with the exception of study FJ‐506E‐KT01, which utilized an immunoassay. Briefly, internal standard (FR900520) and tacrolimus were extracted from whole blood using protein precipitation followed by solid phase extraction. Compounds of interest were eluted from the solid phase cartridge with methanol and then dried under a stream of nitrogen (40°C). The residue was reconstituted with 50:50 (v/v) acetonitrile:water and injected onto the LC/MS/MS system, where separation occurred on a reversed phase high‐performance LC column, and was detected with positive electrospray ionization MS/MS. This method was validated for tacrolimus determination in whole blood. Peak area ratios (compound/internal standard) were fitted to a weighted 1/concentration least squares linear regression analysis to calculate the line of best fit from the data. The equations of the calibration curves were then used to calculate the concentrations of tacrolimus in the whole blood samples from their measured peak area ratios. The lower limit of quantitation was 0.1 ng/mL.
2.3. Population pharmacokinetic analysis
2.3.1. Software
All models were developed in NONMEM29 version 7.3 within a Windows environment using gFortran Compiler (ICON Development Solutions, Ellicott City, MD, USA). SAS version 9.3 (SAS Institute Inc., Cary, NC, USA), R version 3.3.1 (The R Foundation), and S‐plus version 8.2 (TIBCO Corporation, Seattle, WA, USA) were used for modelling and simulation, data preparation, graphical analysis, model diagnostics, and statistical summaries. XPOSE4 and Perl‐speaks‐NONMEM30 (Department of Pharmaceutical Bioscience, Uppsala University, Sweden) was used for model diagnostics, model evaluation, and automated procedures (if needed). All available PK data (as previously described) from the 8 studies were included in the analysis. The first‐order conditional estimation with interaction between interpatient and residual random effects method in NONMEM was employed for all model runs. The NONMEM code for the final PK model is shown in Supporting information Appendix A.
2.3.2. Base model development
Initially, exploratory data analyses of individual whole‐blood tacrolimus concentrations were constructed by study and formulation to qualitatively explore the suitability of different base PK models. Zero‐order absorption and 1‐ and 2‐compartmental models with different absorption assumptions were tested. The interpatient variability and necessary interoccasion variability were modelled as a lognormal distribution. Residual variability was assessed for different assays, using residual error models, including additive, proportional, and combined (i.e. additive and proportional) models, and a residual additive error model with log‐transformed tacrolimus concentration data.
Model development and selection were driven by the data and were based on goodness‐of‐fit indicators.31 These included comparisons based on the minimum objective function value (OFV), successful minimization, completion of the covariance step (if possible), precision and plausibility of parameter estimates, and adequate goodness of fit based on visual inspection of diagnostic plots (e.g. observed vs predicted concentrations, conditional weighted residual vs predicted concentration or time, correlations of interpatient random effects). In addition, the condition number of the correlation matrix of the parameter estimates (i.e. the ratio of the largest to smallest eigenvalues) was assessed to ensure values <1000. Values >1000 could be indicative of an ill‐conditioned model. At the end stage of base model development, the stability of the base model was tested by varying the initial estimates of the parameters by 10–15%.
2.3.3. Covariate analysis
Covariate analysis was based on the previously developed parsimonious base model, and included covariates that affect population mean PK parameters, with hypothesis testing of statistical significance. PK parameter–covariate relationships were preceded by exploratory graphical analysis of the posterior Bayes estimates of random effects produced by the POSTHOC step of NONMEM vs all available covariates. Stepwise regression was used for the covariate analysis, with forward selection and backward elimination of covariates. First, the effect of each covariate was examined in a univariate manner by adding 1 covariate at a time to the base model. The covariate that resulted in the greatest statistically significant decrease in the value of the objective function was added to the base model, and the procedure was repeated stepwise until all significant covariates were included.
Once all covariate relationships for the PK parameters had been defined from the forward selection step, backward elimination of the covariates was performed one‐by‐one to determine if any covariates should be removed from the full model. The model for each relevant parameter–covariate relationship was prepared and tested using a stepwise covariate model approach implemented in Perl‐speaks‐NONMEM. Stepwise forward or backward comparisons, based on the likelihood ratio test and a prespecified level of significance, were made across nested multivariate models, each expressing different covariate–parameter combinations. According to the likelihood ratio test, the difference in −2 log likelihood from nested models was assumed to be asymptotically χ2 distributed with degrees of freedom (df) equal to the difference in the number of model parameters. Significance of covariate effect was determined at α = 0.001 (or 10.8 of change in NONMEM OFV with df = 1) at the forward selection and backward elimination steps.
The impact of baseline demographic characteristics, including weight, body mass index, lean body mass, age, race, sex, population (adult vs paediatric) and type of organ transplanted (kidney vs liver vs heart) were determined for tacrolimus absorption (absorption rate constant [Ka]; relative bioavailability [F1]), volume of distribution (central, Vc; peripheral, Vp), and elimination parameters (clearance [CL]). Race was self‐reported using standardized 7‐category classification (Asian, White, Black, American‐Indian or Alaskan native, or native Hawaiian or other Pacific islander). It should be noted that Asian patients in this analysis represent a heterogeneous population comprising Japanese, Chinese and other Far East groups. Given the involvement of the liver in tacrolimus metabolism, alanine aminotransferase (ALT) and aspartate aminotransferase (AST), ALB and total bilirubin were also evaluated as liver‐function‐related covariates for tacrolimus absorption, distribution, and elimination. Race was coded as a 3‐category covariate in the analysis: White, Black or Asian. Covariates related to patient renal function were not evaluated in the current analysis because urinary excretion accounts for <2% of the tacrolimus dose.2 The relationship between continuous covariates (COV) and the typical value of PK parameters (TVP) was primarily modelled using power models:
θTVP and θCOV are the fixed‐effect parameters and Typical_COV represents the approximate median of the general population. The relationship between categorical covariates (CAT) and the typical value of PK parameters was modelled as a linear proportional model:
θTVP and θCAT are fixed‐effect parameters and CAT represents the categorical covariates, which could be equal to 1 or 0, dependent on the category of the covariates. The lower‐bound value for θCAT was constrained to be >−1.
2.3.4. Model evaluation
The final full covariate model was evaluated using study‐stratified non‐parametric bootstrap and prediction‐corrected visual predictive checks (pcVPCs).32, 33 For the non‐parametric bootstrap procedure, 1000 replicate bootstrap data sets were obtained by random resampling using the patient as the sampling unit, with replacement from the original data set, and were fitted with the same model to obtain parameter estimates for each replicate. There were 491 runs with minimization terminated and 8 runs with estimates near a boundary, which were skipped. Empirical 95% confidence intervals (CIs) were constructed by obtaining the 2.5th and 97.5th percentiles of the resulting parameter distributions for those bootstrap runs with successful convergence. The final model parameter estimates were compared with the bootstrap median parameter estimates to evaluate the final model performance. The predictive performance of the final model was assessed with pcVPC. Simulation of 1000 new data sets was carried out using the final model with the estimated fixed‐ and random‐effects model parameters. As the tacrolimus dose was different in each patient, the pcVPC was based on dose‐normalized concentrations. The concentration–time profiles were plotted for the 50th percentile and the 5th and 95th percentiles (presenting the 90% prediction interval) of the simulated data and were overlaid with observed data.
2.3.5. Model simulation
The final model for tacrolimus with estimated fixed‐ and random‐effects parameters was applied to simulate tacrolimus trough concentrations under different covariate scenarios in order to determine whether any covariates retained in the final model had a significant impact on tacrolimus trough concentrations. The reference was the normal value of the covariate against which the covariate effect was assessed. Tacrolimus trough concentrations (12 and 24 hours post‐dose for immediate‐ and prolonged‐release tacrolimus, respectively) for 500 patients were simulated for immediate‐release (5 mg, twice daily) and prolonged‐release tacrolimus (10 mg, once daily) at steady state. Parameter estimates included only interpatient variability. Overall, 500 patients were re‐sampled from the observed data to provide a plausible combination of covariate values. The impact of each covariate was summarized using box plots. Liver function groups were categorized with AST as normal (25 IU/L), with mild elevation (100 IU/L), and with moderate elevation (400 IU/L), fixing ALB to a normal value of 39 g/L; or were categorized with ALB as normal (39 g/L) or hypoalbuminaemia (20 g/L), fixing AST to a normal value of 25 IU/L.
3. RESULTS
Overall, 23,176 tacrolimus concentration records were obtained from 408 patients. The baseline characteristics for patients in the final PK data set are summarized in Table 2. The study included 276 males and 132 females with a median (range) age of 48 years (5–71 years; 17 patients were paediatric) and body weight of 74 kg (18.5–148.5 kg). White (n = 340), Black (n = 24) or Asian (n = 44) were the only self‐reported races in the data set.
Table 2.
Summary of population demographics by studya
| Parameter | 02–0‐131 23 | FG‐506E‐12‐02 15 | FJ‐506E‐KT01 24 | 02–0‐152 25 | FG‐506‐15‐02 26 | 03–0‐160 27 | FG‐506E‐12‐01 11 | FG‐506‐11‐01 28 | All |
|---|---|---|---|---|---|---|---|---|---|
| Median age (range), yr | 47 (22–71) | 43.5 (20–64) | 56 (30–65) | 33 (20–55) | 52 (32–68) | 9 (5–13) | 45.5 (21–65) | 51 (26–65) | 48 (5–71) |
| Median weight (range), kg | 80.6 (45.4–148.5) | 74.5 (56–119) | 60 (42.2–77.1) | 85.5 (50.9–136.8) | 85.5 (62–137) | 31.8 (18.5–62.2) | 72 (42–115) | 79 (40–142) | 74 (18.5–148.5) |
| Sex, n | |||||||||
| Male | 38 | 42 | 21 | 29 | 39 | 5 | 44 | 58 | 276 |
| Female | 19 | 18 | 14 | 22 | 6 | 12 | 22 | 19 | 132 |
| Race, n | |||||||||
| White | 45 | 51 | 0 | 49 | 41 | 11 | 66 | 77 | 340 |
| Black | 11 | 1 | 0 | 2 | 4 | 6 | 0 | 0 | 24 |
| Asian | 1 | 8 | 35 | 0 | 0 | 0 | 0 | 0 | 44 |
Contains previously unpublished study data.
3.1. Base model
After testing the performance of different structural models, a 2‐compartment disposition model with first‐order elimination, first‐order absorption and an absorption lag time provided the best fit for the immediate‐ and prolonged‐release tacrolimus whole‐blood concentration–time profiles. The model was parameterized in terms of apparent oral clearance (CL/F), apparent intercompartmental clearance (Q/F), apparent central volume of distribution (Vc/F), apparent peripheral volume of distribution (Vp/F), Ka and absorption lag time (Lag). For immediate‐ and prolonged‐release tacrolimus formulations, the data supported different Ka but comparable interpatient variability for Ka. Interpatient variability was estimated for all structural PK model parameters except Lag, and the interoccasion variability was also estimated for relative bioavailability between immediate‐ and prolonged‐release tacrolimus. Different residual error models were tested, and the residual additive error model with the log‐transformed tacrolimus concentration data was found to best describe the data. Diagnostic plots for the base model showed adequate fit to the data, with no apparent trends of residuals over time or model predictions (data not shown).
3.2. Covariate model
Changes in OFV for key iterative models are shown in Supporting information 1. Results from the final model indicated that the absorption rate of prolonged‐release tacrolimus was 50% slower than the immediate‐release formulation. Tacrolimus CL/F was 44.3 L/h in Whites and 59% higher in Asians. Due to the lack of precision of the effect of Black race on tacrolimus CL/F, this parameter was not included in the final model. The effect of log‐transformed AST (LAST) on the clearance of tacrolimus was modelled as a power model normalized at 3.15 IU/L. The model predicted that if LAST increased by about 2.7‐fold, CL/F would decrease by about 30% (exp–0.318), Vc/F would increase by about 5.6‐fold (exp1.73), Vp/F would decrease by about 60% (exp–0.945), and F1 would increase by about 2.1‐fold (exp0.74). If ALB increased by about 2.7‐fold, both Vc/F (exp1.03) and F1 (exp1.04) would increase by about 2.8‐fold. Vc of tacrolimus in females was 55% less than in males.
The inclusion of covariate effects on tacrolimus PK all resulted in statistically significant decreases in OFV compared with the base model (p < 0.001). The type of organ transplanted (kidney vs liver vs heart) had no significant effect on principal PK parameters. As only 4.2% of the studied population were paediatric, there was insufficient power to evaluate the impact of population (adult vs paediatric) on tacrolimus PK. The final population PK model was described by the following equations:
Posthoc analysis of the empirical Bayes estimates for relative bioavailability showed limited clinical difference between formulations: the geometric mean ratio for prolonged‐release:immediate‐release tacrolimus was 95% [90% CI: 89%, 101%]). However, subgroup analysis revealed racial differences in relative bioavailability (Asians>Whites>Blacks). Asians had 83% (90% CI: 59%, 210%) and 51% (90% CI: 32%, 74%) higher relative bioavailability than Whites and Blacks, respectively, for both prolonged‐ and immediate‐release tacrolimus. Whites had 49% (90% CI: 128%, 175%) and 77% (90% CI: 51%, 208%) higher relative bioavailability for both prolonged‐ and immediate‐release tacrolimus, respectively, than Blacks. Blacks had 52% (90% CI: 43%, 59%) lower relative bioavailability than Whites and Asians for both prolonged‐ and immediate‐release tacrolimus.
All parameter estimates were identified with good precision, as standard errors of the parameter estimates were ≤50% of the estimated value. Goodness‐of‐fit plots (Figure 1) indicated a good fit of the model for most data where the observed tacrolimus concentrations satisfactorily matched the predicted population concentrations (PRED) or individual PRED. The distribution of the conditional weighted residuals was unbiased with respect to time or population predictions.
Figure 1.
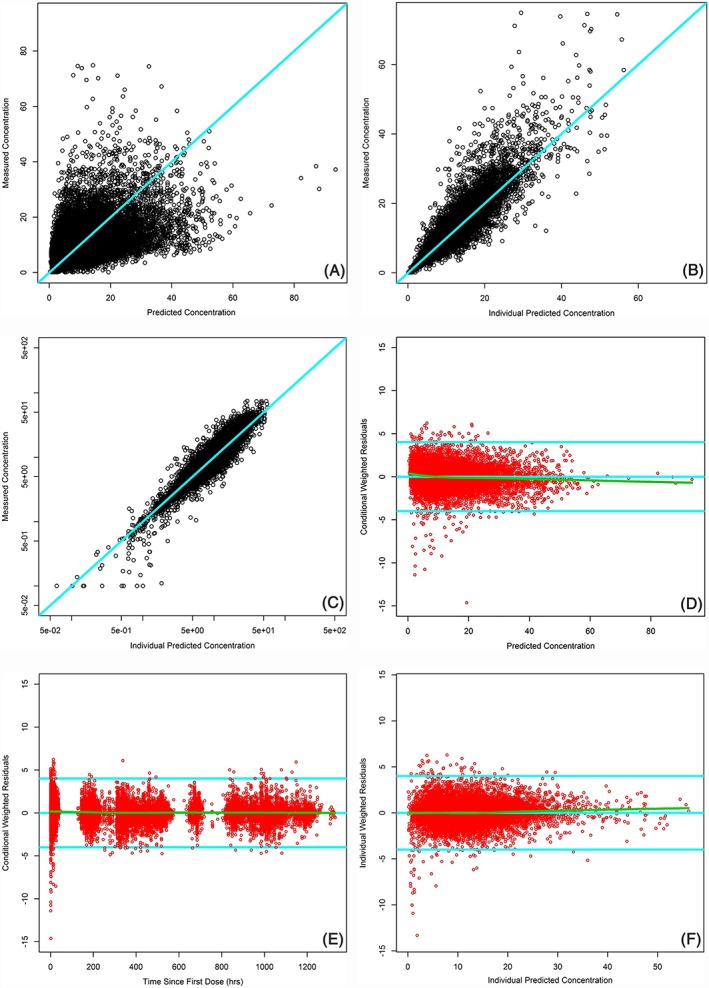
Basic goodness‐of‐fit graphs for the final tacrolimus population pharmacokinetics model. A, Observed vs population predicted tacrolimus concentrations. B, Observed vs individual predicted tacrolimus concentrations. C, Observed vs individual predicted tacrolimus concentrations in log scale. D, Conditional weighted residual error vs population predicted tacrolimus concentrations. E, Conditional weighted residual error vs time. F, Individual weighted residual error vs individual predicted tacrolimus concentrations. Note: the solid line in cyan is the line of identity or horizontal line, and the green line is the locally estimated scatterplot smoothing line
3.3. Model evaluation
The median values of parameters and 95% CIs obtained from the converged bootstrap runs for tacrolimus are presented in Table 3. The median values of parameters were in close agreement with the population estimates in the final models, suggesting that the NONMEM parameter estimates of the model were unbiased.
Table 3.
Parameter estimates of the final tacrolimus population pharmacokinetic covariate model
| Parameter | Value | Eta‐shrinkage, % b | %CV | Bootstrap median (n = 501) | Bootstrap 95% CI (n = 501) |
|---|---|---|---|---|---|
| CL/F, L/h | 44.3 | – | 3.43 | 44.154 | 41.47, 47.48 |
| Asian race on CL/F | 0.59 | – | 16.53 | 0.573 | 0.394, 0.791 |
| AST on CL/F | −0.318 | – | −44.97 | −0.329 | −0.6804, −0.0515 |
| V c /F, L | 110 | – | 10.55 | 109.958 | 87.78, 134.31 |
| Sex on V c /F | −0.446 | – | −15.63 | −0.444 | −0.56, −0.29 |
| AST on V c /F | 1.73 | – | 27.92 | 1.702 | 0.55, 2.54 |
| ALB on V c /F | 1.03 | – | 41.17 | 0.936 | 0.087, 1.782 |
| Q/F, L/h | 131 | – | 5.42 | 129.886 | 119.29, 143.85 |
| V p /F, L | 3180 | – | 7.39 | 3163.63 | 2756.26, 3709.92 |
| AST on V p /F | −0.945 | – | −16.19 | −0.937 | −1.275, −0.647 |
| K a , h −1 | 0.375 | – | 4.48 | 0.375 | 0.341, 0.404 |
| Prolonged‐release tacrolimus on K a | 0.499 | – | 3.61 | 0.498 | 0.465, 0.535 |
| F1 | 1.51 | – | 2.96 | 1.505 | 1.44, 1.58 |
| Asian race on F1 | 0.25 | – | 41.6 | 0.242 | 0.03, 0.43 |
| Black race on F1 | −0.433 | – | −10.83 | −0.431 | −0.52, −0.32 |
| AST on F1 | 0.74 | – | 26.89 | 0.726 | 0.228, 1.085 |
| ALB on F1 | 1.04 | – | 19.33 | 1.024 | 0.568, 1.439 |
| ALAG1 | 0.44 | – | 1.17 | 0.439 | 0.427, 0.452 |
| IPV‐CL, % | 30.9 | 29 | 7.32 | 30.98 | 26.08, 35.78 |
| IPV‐V c , % | 106 | 9.4 | 5.76 | 105 | 93.1, 117.8 |
| IPV‐Q, % | 39.3 | 29 | 12.07 | 38.47 | 28.55, 48.06 |
| IPV‐V p , % | 99 | 15.8 | 4.55 | 98.79 | 89.61, 109.5 |
| IPV‐K a , % | 35.5 | 31.7 | 6.63 | 34.93 | 29.83, 39.87 |
| IPV‐F1, % | 30.5 | 35 | 11.05 | 30.98 | 23.17, 38.9 |
| BOV‐F1, % | 59.9 | – | 3.84 | 59.75 | 55.59, 64.19 |
| RV1 a , % | 21.1 | – | 2.57 | 21.1 | 20.3, 22 |
| RV2 a , % | 15.8 | – | 5.51 | 15.8 | 14.2, 17.6 |
| Epsilon‐shrinkage, % b | 6.9 | – | – | – | – |
Residual variability was parameterized by the fixed‐effect parameter (θ) for different assays.
Eta‐ and epsilon‐shrinkages were estimated only for covariates included in the final model. Interpatient variability and residual variability were expressed as %CV; %CV was expressed as 100 × (standard error of the estimate/point estimate). Median and 95% CI were estimated from non‐parametric bootstrap estimates stratified by study.
ALAG1, absorption lag time; ALB, albumin; AST, aspartate aminotransferase; BOV, between‐occasion variability; CI, confidence interval; CL, clearance; CL/F, apparent oral clearance; CV, coefficient of variation; F1, relative bioavailability; IPV, interpatient variability; Ka, absorption rate; Q, intercompartmental clearance; Q/F, intercompartmental oral clearance; RV, residual variability; Vc, central volume of distribution; Vp, peripheral volume of distribution; Vc/F, apparent central volume of distribution; Vp/F, apparent peripheral volume of distribution.
Results from the dose‐normalized pcVPC analysis with the final parameter estimates in the tacrolimus PK model are shown in Figure 2. The pcVPC analysis suggests that the models can predict the distribution of observed tacrolimus concentrations for both immediate‐ and prolonged‐release tacrolimus. The calculated median (based on 1000 simulated data sets) represented the trend of the observed data. There were 295 (1.3%) and 522 (2.3%) data points below and above the prediction intervals, respectively. Most observed concentrations were within the 95% prediction interval, indicating that the predicted variability did not exceed the observed variability.
Figure 2.
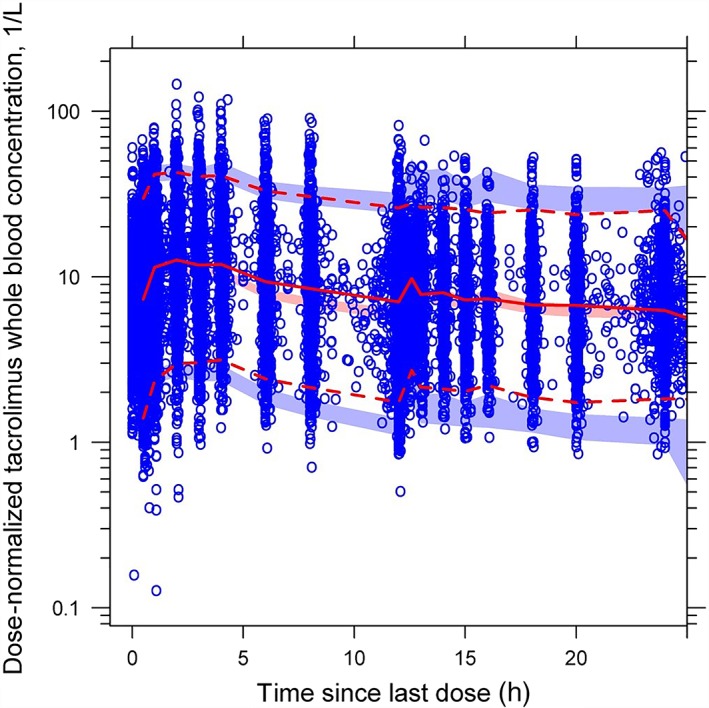
Prediction‐corrected visual predictive check graphs based on the final tacrolimus population pharmacokinetics model. The solid red line represents the median observed plasma tacrolimus concentration, and the semi‐transparent red field represents a simulation‐based 95% CI for the median. The observed 5th and 95th percentiles are presented with dashed red lines, and the 95% CIs for the corresponding model predicted percentiles are shown as semitransparent blue fields. The observed plasma concentrations are represented by blue circles. CI, confidence interval
3.4. Model simulation
Simulations were undertaken to compare trough concentrations for all identified covariates (Figure 3A–C). Tacrolimus trough concentration was lowest for Blacks and highest for Whites, and similar with prolonged‐ vs immediate‐release tacrolimus. There was no observed difference in trough concentration between males and females. Most tacrolimus trough concentrations from both formulations were within the clinical therapeutic window (5–20 ng/mL). Tacrolimus trough increased with greater AST activity and increasing ALB concentrations; however, trough concentrations of immediate‐ and prolonged‐release tacrolimus mostly fell below the therapeutic window when hypoalbuminaemia was present in Blacks, Whites or Asians (Figure 3C). Interpatient variability in tacrolimus trough concentrations was similar with the immediate‐ and prolonged‐release formulations throughout the simulations (Figure 3D).
Figure 3.
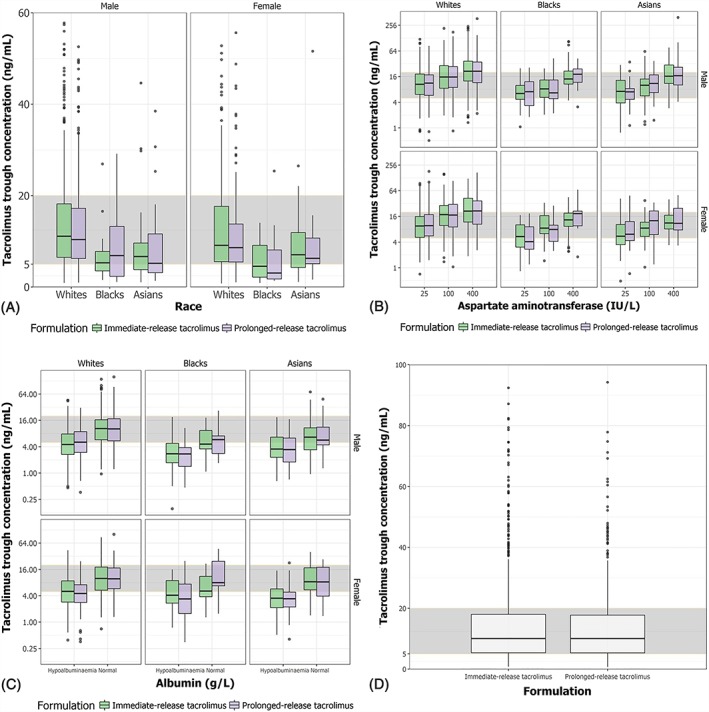
Simulated tacrolimus trough concentration for all covariates identified. A, Tacrolimus trough concentration stratified by race (White, Black, Asian), tacrolimus formulation (immediate‐release tacrolimus, prolonged‐release tacrolimus) and sex (male, female). B, Tacrolimus trough concentration stratified by aspartate aminotransferase (normal = 25 IU/L, mild elevation = 100 IU/L, moderate elevation = 400 IU/L), race, sex and tacrolimus formulation. C, Tacrolimus trough concentration stratified by albumin, race, sex and tacrolimus formulation. D, Interpatient variability in tacrolimus trough concentrations (concentration immediately prior to dosing across multiple doses) for immediate‐ and prolonged‐release tacrolimus. The shaded grey area represents the therapeutic window for tacrolimus trough concentrations (5–20 ng/mL). The box, solid line, and whiskers represent the interquartile range, median, and 5th/95th percentiles, respectively, of simulated tacrolimus trough concentrations
4. DISCUSSION
In this analysis, a 2‐compartmental model with first‐order elimination, first‐order absorption, and an absorption lag time adequately described the PK of immediate‐ and prolonged‐release tacrolimus in kidney, liver and heart transplant recipients. Prolonged‐release tacrolimus had slower absorption than immediate‐release tacrolimus, and clearance of tacrolimus was 59% higher in Asians than Whites. The PK of tacrolimus was similar between adult and paediatric populations in this study, and there was no effect of organ type; however, tacrolimus bioavailability differed between races. Interpatient variability in tacrolimus trough concentrations was similar for the immediate‐ and prolonged‐release formulations.
Typically, the estimated absorption rate of prolonged‐release tacrolimus was 50% slower compared with immediate‐release tacrolimus, due to the extended‐release properties of the prolonged‐release formulation. However, the absorption rate of prolonged‐release tacrolimus showed similar variability (35.5%) to immediate‐release tacrolimus. The absorption phase was similar across transplanted organ type, and between adult vs paediatric patients. During the first 6 weeks after transplantation, paediatric kidney transplant recipients require higher tacrolimus weight‐normalized starting doses than adults.34 Although 1 PK study of stable paediatric liver transplant recipients converted from immediate‐ to prolonged‐release tacrolimus was included in this analysis,27 there were only 17 paediatric patients in the study, compared with 391 adult patients. As such, there may be insufficient power to detect a significant age effect on principal PK parameters (CL, Ka, Vc, Vp and F1).
Following absorption, the median steady‐state volume of distribution was 3290 L for males and 3241 L for females, indicating that tacrolimus is extensively distributed in the body. Notably, while interpatient random variability and between‐occasions variability in the final model was moderate for most PK parameters, apparent volume of distribution exhibited large variability, estimated to be approximately 100%. The reason for this is unclear, but it may warrant further investigation.
Tacrolimus Vc was higher with increased vs lower concentrations of AST or ALB, which is consistent with tacrolimus usually being highly bound to plasma proteins, predominantly serum ALB.4 Other confounders that could affect the level of bound tacrolimus include the haematocrit value.35 The positive correlation between AST and Vc and the negative correlation between AST and Vp could be due to decreased hepatic clearance, indicated by elevated blood values of AST. As systemically available tacrolimus is cleared by hepatic metabolism, elevated AST could result in reduced tacrolimus clearance. Indeed, the covariate search identified this effect of AST on tacrolimus clearance.
A bioequivalence‐type comparison of the posthoc empirical Bayes estimates for relative bioavailability between immediate‐ and prolonged‐release tacrolimus revealed limited differences. This supports current clinical practice to convert from twice‐daily, immediate‐release tacrolimus (Prograf) to once‐daily, prolonged‐release tacrolimus (Advagraf or Astagraf XL) on a 1 mg:1 mg total‐daily‐dose basis, with continuous trough concentration monitoring to ensure adequate drug exposure. Notably, bioequivalence analyses revealed racial differences in the relative bioavailability of tacrolimus, with Asians having greater relative bioavailability than Whites, who had greater relative bioavailability than Blacks. This suggests, paradoxically, that some Asian patients may have lower tacrolimus dose requirements than Whites, while Blacks may need higher doses than Asians and Whites.
Whether Asians need lower doses of tacrolimus than Whites and Blacks in order to achieve therapeutic concentrations has not been explicitly examined in clinical studies. In this study, the tacrolimus clearance was higher in Asians than in Whites, which could result in lower tacrolimus exposure‐related trough concentrations in this population; however, it should be considered that the Asian population in this study was a heterogeneous group of Japanese, Chinese and Far East patients. Moreover, in a previous PK modelling study of tacrolimus in Asian liver transplant recipients, the estimated CL/F was 18.4 L/h,36 which is lower than the clearance observed for Whites in our study (44.3 L/h). This indicates the need for further studies to confirm the dose requirements for Asian compared with White patients.36
Lower relative bioavailability of tacrolimus in Blacks is consistent with earlier reports that African–Americans require higher doses of tacrolimus in order to achieve therapeutic drug concentrations.37, 38 Tacrolimus is extensively metabolized by CYP3A4 and CYP3A5, with CYP3A5 polymorphism expressed at a higher frequency in African–Americans,39 which could be responsible for the estimated racial effect. Presystemic metabolism by gastrointestinal CYP3A4 and P‐glycoprotein (P‐gp) has also been implicated in limiting oral bioavailability of tacrolimus. Conceivably, Blacks might express higher concentrations of P‐gp in the gut and intestine, thereby decreasing tacrolimus bioavailability;40 however, in the current study, CYP3A4 and P‐gp variants were not included as covariates. In a retrospective study that formed part of the immediate‐release tacrolimus development programme, simulated trough concentrations of tacrolimus were lower for Black vs White kidney transplant recipients, and Blacks required higher immediate‐release tacrolimus doses to attain similar trough tacrolimus concentrations to those in Whites.41 Similar findings were reported for a subset of heart transplant patients receiving immediate‐release tacrolimus‐based therapy in an observational parallel‐group study.40
In the current simulations, patients were converted from immediate‐release (Prograf) to prolonged‐release (Advagraf or Astagraf XL) tacrolimus on a 1 mg:1 mg total‐daily‐dose basis. The final model predicted that overall median tacrolimus trough concentrations were similar and within the therapeutic window with both formulations, supporting the 1 mg:1 mg conversion factor between immediate‐ and prolonged‐release tacrolimus recommended in clinical practice.2 There was no significant effect of sex on tacrolimus trough concentration; indeed a sex effect was only identified for Vc in this study. Tacrolimus trough concentrations increased with elevated concentrations of AST due to decreased clearance. However, in the presence of hypoalbuminaemia, trough concentrations were outside the therapeutic window with both formulations, irrespective of race. This suggests that dose adjustments may be required for patients with hypoalbuminaemia.
It is not possible to explain why sex had an effect on volume of distribution, whereas weight did not. However, haematocrit and time after transplant are considered important covariates affecting tacrolimus PK, and a limitation of the study was the lack of inclusion of these covariates in the model. A further limitation was the observed high variability of the data and large size of the sample across different studies, which reduced the ability of the VPC for the model to predict the 5th and 95th percentiles.
In conclusion, the tacrolimus population PK model adequately described the tacrolimus PK data observed in transplant recipients. Of the assessed covariates, the PK of immediate‐ and prolonged‐release tacrolimus was only affected by race, sex and liver function. The model confirmed that patients can be converted from immediate‐release (Prograf) to prolonged‐release (Advagraf or Astagraf XL) tacrolimus on a 1 mg:1 mg total‐daily‐dose basis, and showed that the population studied achieved clinical tacrolimus trough concentrations within the therapeutic range. However, for safety, it is important that following conversion from immediate‐ to prolonged‐release tacrolimus, trough concentrations should be monitored and dose adjustments made to maintain exposure on an individual patient basis.
COMPETING INTERESTS
Z.L. is employed by Astellas and has received a salary from Astellas; Peter Bonate is employed by Astellas and has received a salary from Astellas; James Keirns is a former employee of Astellas and has received a salary from Astellas.
CONTRIBUTORS
All authors made substantial contribution to design, acquisition, analysis and interpretation of data, and were involved in drafting and revising the manuscript.
Supporting information
Table S1 Key iterative models during the development of population PK covariate model.
Appendix S1 NONMEM code for the final PK model.
ACKNOWLEDGEMENTS
James Wallis, MRes, from Cello Health MedErgy provided medical writing and editorial support throughout the development of the manuscript under the direction of the authors. Medical writing and editorial support was funded by Astellas Pharma, Inc.
Lu Z, Bonate P, Keirns J. Population pharmacokinetics of immediate‐ and prolonged‐release tacrolimus formulations in liver, kidney and heart transplant recipients. Br J Clin Pharmacol. 2019;85:1692–1703. 10.1111/bcp.13952
REFERENCES
- 1. Astellas Pharma Europe Ltd . Advagraf 0.5mg, 1mg, 3mg and 5mg prolonged‐release hard capsules summary of product characteristics. 2015.
- 2. Astellas Pharma Europe Ltd . Prograf 0.5mg, 1mg, 5mg hard capsules summary of product characteristics. 2015.
- 3. Fischer L, Trunecka P, Gridelli B, Roy A, Vitale A, Valadivieso A. Pharmacokinetics for once‐daily versus twice‐daily tacrolimus formulations in de novo liver transplantation: a randomized, open‐label trial. Liver Transpl. 2011;17(2):167‐177. [DOI] [PubMed] [Google Scholar]
- 4. Staatz CE, Tett SE. Clinical pharmacokinetics and pharmacodynamics of tacrolimus in solid organ transplantation. Clin Pharmacokinet. 2004;43(10):623‐653. [DOI] [PubMed] [Google Scholar]
- 5. Barbarino JM, Staatz CE, Venkataramanan R, Klein TE, Altman RB. PharmGKB summary: cyclosporine and tacrolimus pathways. Pharmacogenet Genomics. 2013;23(10):563‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beckebaum S, Iacob S, Sweid D, et al. Efficacy, safety, and immunosuppressant adherence in stable liver transplant patients converted from a twice‐daily tacrolimus‐based regimen to once‐daily tacrolimus extended‐release formulation. Transpl Int. 2011;24(7):666‐675. [DOI] [PubMed] [Google Scholar]
- 7. Sabbatini M, Garofalo G, Borrelli S, et al. Efficacy of a reduced pill burden on therapeutic adherence to calcineurin inhibitors in renal transplant recipients: an observational study. Patient Prefer Adherence. 2014;8:73‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guirado L, Cantarell C, Franco A, et al. Efficacy and safety of conversion from twice‐daily to once‐daily tacrolimus in a large cohort of stable kidney transplant recipients. Am J Transplant. 2011;11(9):1965‐1971. [DOI] [PubMed] [Google Scholar]
- 9. Krämer BK, Charpentier B, Bäckman L, et al. Tacrolimus once daily (ADVAGRAF) versus twice daily (PROGRAF) in de novo renal transplantation: a randomized phase III study. Am J Transplant. 2010;10(12):2632‐2643. [DOI] [PubMed] [Google Scholar]
- 10. Albano L, Banas B, Klempnauer J, Glyda M, Viklicky O, Kamar N. OSAKA trial: a randomized, controlled trial comparing tacrolimus QD and BD in kidney transplantation. Transplantation. 2013;96(10):897‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wlodarczyk Z, Squifflet JP, Ostrowski M, et al. Pharmacokinetics for once‐ versus twice‐daily tacrolimus formulations in de novo kidney transplantation: a randomized, open‐label trial. Am J Transplant. 2009;9(11):2505‐2513. [DOI] [PubMed] [Google Scholar]
- 12. Musuamba FT, Mourad M, Haufroid V, Delattre IK, Verbeeck RK, Wallemacq P. Time of drug administration, CYP3A5 and ABCB1 genotypes, and analytical method influence tacrolimus pharmacokinetics: a population pharmacokinetic study. Ther Drug Monit. 2009;31(6):734‐742. [DOI] [PubMed] [Google Scholar]
- 13. Mathew BS, Fleming DH, Jeyaseelan V, et al. A limited sampling strategy for tacrolimus in renal transplant patients. Br J Clin Pharmacol. 2008;66(4):467‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miura M, Satoh S, Niioka T, et al. Early phase limited sampling strategy characterizing tacrolimus and mycophenolic acid pharmacokinetics adapted to the maintenance phase of renal transplant patients. Ther Drug Monit. 2009;31(4):467‐474. [DOI] [PubMed] [Google Scholar]
- 15. Van Hooff J, Van der I, Kallmeyer J, et al. Pharmacokinetics in stable kidney transplant recipients after conversion from twice‐daily to once‐daily tacrolimus formulations. Ther Drug Monit. 2012;34(1):46‐52. [DOI] [PubMed] [Google Scholar]
- 16. Florman S, Alloway R, Kalayoglu M, et al. Conversion of stable liver transplant recipients from a twice‐daily Prograf‐based regimen to a once‐daily modified release tacrolimus‐based regimen. Transplant Proc. 2005;37(2):1211‐1213. [DOI] [PubMed] [Google Scholar]
- 17. Wallemacq P, Armstrong VW, Brunet M, et al. Opportunities to optimize tacrolimus therapy in solid organ transplantation: report of the European consensus conference. Ther Drug Monit. 2009;31(2):139‐152. [DOI] [PubMed] [Google Scholar]
- 18. Antignac M, Hulot JS, Boleslawaki E, et al. Population pharmacokinetics of tacrolimus in full liver transplant patients: modelling of the post‐operative clearance. Eur J Clin Pharmacol. 2005;61(5‐6):409‐416. [DOI] [PubMed] [Google Scholar]
- 19. Fukatsu S, Yano I, Igarashi T, et al. Population pharmacokinetics of tacrolimus in adult recipients receiving living‐donor liver transplantation. Eur J Clin Pharmacol. 2001;57(6‐7):479‐484. [DOI] [PubMed] [Google Scholar]
- 20. Macchi‐Andanson M, Charpiat B, Jelliffe RW, Ducerf C, Fourcade N, Baulieux J. Failure of traditional trough levels to predict tacrolimus concentrations. Ther Drug Monit. 2001;23(2):129‐133. [DOI] [PubMed] [Google Scholar]
- 21. Sam WJ, Tham LS, Holmes MJ, et al. Population pharmacokinetics of tacrolimus in whole blood and plasma in Asian liver transplant patients. Clin Pharmacokinet. 2006;45(1):59‐75. [DOI] [PubMed] [Google Scholar]
- 22. Woillard J, de Winter BCM, Kamar N, Marquet P, Rostaing L, Rousseau A. Population pharmacokinetic model and Bayesian estimator for two tacrolimus formulations—twice daily Prograf® and once daily Advagraf®. Br J Clin Pharmacol. 2011;71(3):391‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alloway R, Steinberg S, Khalil K, et al. Two years postconversion from a Prograf‐based regimen to a once‐daily tacrolimus extended‐release formulation in stable kidney transplant recipients. Transplantation. 2007;83(12):1648‐1651. [DOI] [PubMed] [Google Scholar]
- 24. Oh CK, Huh KH, Lee JS, Cho HR, Kim YS. Safety and efficacy of conversion from twice‐daily tacrolimus to once‐daily tacrolimus one month after transplantation: randomized controlled trial in adult renal transplantation. Yonsei Med J. 2014;55(5):1341‐1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Florman S, Alloway R, Kalayoglu M, et al. Once‐daily tacrolimus extended release formulation: experience at 2 years postconversion from a Prograf‐based regimen in stable liver transplant recipients. Transplantation. 2007;83(12):1639‐1642. [DOI] [PubMed] [Google Scholar]
- 26. Alloway R, Vanhaecke J, Yonan N, et al. Pharmacokinetics in stable heart transplant recipients after conversion from twice‐daily to once‐daily tacrolimus formulations. J Heart Lung Transplant. 2011;30(9):1003‐1010. [DOI] [PubMed] [Google Scholar]
- 27. Heffron TG, Pescovitz MD, Florman S, et al. Once‐daily tacrolimus extended‐release formulation: 1‐year post‐conversion in stable pediatric liver transplant recipients. Am J Transplant. 2007;7(6):1609‐1615. [DOI] [PubMed] [Google Scholar]
- 28. Fischer L, Trunečka P, Gridelli B, et al. Pharmacokinetics for once‐daily versus twice‐daily tacrolimus formulations in de novo liver transplantation: a randomized, open‐label trial. Liver Transpl. 2011;17(2):167‐177. [DOI] [PubMed] [Google Scholar]
- 29. Bauer RJ. NONMEM Users Guide: Introduction to NONMEM 7.3.0. Gaithersburg; 2015.
- 30. Lindbom L, Ribbing J, Jonsson EN. Perl‐speaks‐NONMEM (PsN) – a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85‐94. [DOI] [PubMed] [Google Scholar]
- 31. Ette EI, Williams PJ, Kim YH, Lane JR, Liu M‐J, Capparelli EV. Model appropriateness and population pharmacokinetic modeling. J Clin Pharmacol. 2003;43(6):610‐623. [PubMed] [Google Scholar]
- 32. Ette EI. Stability and performance of a population pharmacokinetic model. J Clin Pharmacol. 1997;37(6):486‐495. [DOI] [PubMed] [Google Scholar]
- 33. Post TM, Freijer JI, Ploeger BA, Danhof M. Extensions to the visual predictive check to facilitate model performance evaluation. J Pharmacokinet Pharmacodyn. 2008;35(2):185‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andrews LM, Hesselink DA, van Gelder T, et al. A population pharmacokinetic model to predict the individual starting dose of tacrolimus following pediatric renal transplantation. Clin Pharmacokinet. 2018;57(4):475‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Størset E, Holford N, Midtvedt K, Bremer S, Bergan S, Åsberg A. Importance of hematocrit for a tacrolimus target concentration strategy. Eur J Clin Pharmacol. 2014;70(1):65‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu YX, Su QH, Wu KH, et al. A population pharmacokinetic study of tacrolimus in healthy Chinese volunteers and liver transplant patients. Acta Pharm Sin B. 2015;36(2):281‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mancinelli LM, Frassetto L, Floren LC, et al. The pharmacokinetics and metabolic dispostion of tacrolimus: a comparison across ethnic groups. Clin Pharmacol Ther. 2001;69(1):24‐31. [DOI] [PubMed] [Google Scholar]
- 38. Vadivel N, Garg A, Holt DW, Chang RW, MacPhee IA. Tacrolimus dose in black renal transplant recipients. Transplantation. 2007;83(7):997‐999. [DOI] [PubMed] [Google Scholar]
- 39. Kuehl P, Zhang J, Lin Y, et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001;27(4):383‐391. [DOI] [PubMed] [Google Scholar]
- 40. Mehra MR, Uber PA, Scott RL, Park MH. Ethnic disparity in clinical outcome after heart transplantation is abrogated using tacrolimus and mycophenolate mofetil‐based immunosuppression. Transplantation. 2002;74(11):1568‐1573. [DOI] [PubMed] [Google Scholar]
- 41. Fitzsimmons WE, Bekersky I, Dressler D, Raye K, Hodosh E, Mekki Q. Demographic considerations in tacrolimus pharmacokinetics. Transplant Proc. 1998;30(4):1359‐1364. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Key iterative models during the development of population PK covariate model.
Appendix S1 NONMEM code for the final PK model.