

Abstract
Aims
Navoximod (GDC‐0919, NLG‐919) is a small molecule inhibitor of indoleamine‐2,3‐dioxygenase 1 (IDO1), developed to treat the acquired immune tolerance associated with cancer. The primary objectives of this study were to assess navoximod's absolute bioavailability (aBA), determine the mass balance and routes of elimination of [14C]‐navoximod, and characterize navoximod's metabolite profile.
Methods
A phase 1, open‐label, two‐part study was conducted in healthy volunteers. In Part 1 (aBA), subjects (n = 16) were randomized to receive oral (200 mg tablet) or intravenous (5 mg solution) navoximod in a crossover design with a 5‐day washout. In Part 2 (mass balance), subjects (n = 8) were administered [14C]‐navoximod (200 mg/600 μCi) as an oral solution.
Results
The aBA of navoximod was estimated to be 55.5%, with a geometric mean (%CV) plasma clearance and volume of distribution of 62.0 L/h (21.0%) and 1120 L (28.4%), respectively. Mean recovery of total radioactivity was 87.8%, with 80.4% detected in urine and the remainder (7.4%) in faeces. Navoximod was extensively metabolized, with unchanged navoximod representing 5.45% of the dose recovered in the urine and faeces. Glucuronidation was identified as the primary route of metabolism, with the major glucuronide metabolite, M28, accounting for 57.5% of the total drug‐derived exposure and 59.7% of the administered dose recovered in urine.
Conclusions
Navoximod was well tolerated, quickly absorbed and showed moderate bioavailability, with minimal recovery of the dose as unchanged parent in the urine and faeces. Metabolism was identified as the primary route of clearance and navoximod glucuronide (M28) was the most abundant metabolite in circulation with all other metabolites accounting for <10% of drug‐related exposure.
Keywords: Bioavailability, Drug metabolism, Clinical pharmacology, Oncology, Pharmacokinetics
What is already known about this subject
Navoximod (also known as GDC‐0919 and NLG‐919) is a potent and selective small‐molecule inhibitor of IDO1 for treatment of local and peripheral immune tolerance that might arise in association with cancer.
What this study adds
The study provided definitive information regarding the absolute bioavailability and the metabolism, elimination and clearance pathways of navoximod.
Identification of glucuronidation as the major metabolic pathway of navoximod and M28 as the major circulating metabolite.
1. INTRODUCTION
Indoleamine‐2,3‐dioxygenase 1 (IDO1, EC 1.13.11.52) is a cytosolic enzyme that catalyses the oxidation of the essential amino acid, L‐tryptophan, into kynurenine.1 The IDO1 pathway plays an important role in the local control of inflammation and acquired immune peripheral tolerance.1, 2, 3 IDO1 is expressed in tumour and host immune cells in patients with various tumour types and its expression level is associated with significantly worse clinical outcomes.4, 5, 6, 7, 8 Given the important role of IDO1 in immune tolerance and overexpression in many human cancers, it is a prime target for cancer immunotherapy.
Navoximod (also known as GDC‐0919 and NLG‐919) is a potent and selective small‐molecule inhibitor of IDO1 and an investigational drug candidate for the treatment of local and peripheral immune tolerance that might arise in association with cancer. Nonclinical pharmacology studies indicated that navoximod inhibits IDO1 with a potency of 75–90 nM in cell‐based assays.9 In preclinical models, navoximod in combination with anti‐PD‐L1 (programmed death‐ligand 1), enhances the activity of intratumoral CD8+ T cells and inhibits tumour growth compared to either treatment alone.10
Clinically, navoximod has favourable safety, tolerability and pharmacokinetic profiles when given as a monotherapy or in combination with atezolizumab, a PD‐L1 inhibitor.11, 12 Navoximod is rapidly absorbed and demonstrates linear pharmacokinetics with dose‐proportional increases in exposure over the dose range of 50–1000 mg. Navoximod has a half‐life of approximately 12 hours, supporting a twice daily (BID) dosing regimen. The mean accumulation index of navoximod is approximately 1.7, which is consistent with the half‐life and BID dosing regimen.
The objectives of the current study were to determine the absolute bioavailability, characterize the pharmacokinetics of navoximod following oral and IV administration, determine the mass balance and routes of excretion of navoximod, and characterize the metabolites of navoximod in plasma, urine and faeces. Overall, the data from this study provided a comprehensive understanding of the disposition of navoximod in humans, which was applied to interpret its oral bioavailability and total metabolism.
2. METHODS
2.1. Study design
This was a Phase 1, single‐centre, open‐label, two‐part absolute bioavailability (aBA) and absorption, metabolism and excretion (AME) study, conducted at the Covance Clinical Research Unit (CRU) in Madison, Wisconsin. Part 1 was a randomized, two treatment cross‐over study to determine the aBA of navoximod following separate oral administration of navoximod (one 200 mg tablet) and a single intravenous (IV) dose of navoximod (5 mg solution, pushed over 3–5 minutes) under fasted conditions, to 16 healthy volunteers. Each study participant received both treatments separated by 5 days and the order of treatment was determined by randomization. Subjects participating in Part 1 were confined continuously at the CRU from time of check‐in (Day −1) until clinic discharge (Day 11).
Part 2 was a single‐arm AME study to investigate the mass balance of [14C]‐navoximod administered as a single 200 mg/600 μCi oral dose. Subjects (n = 8) participating in Part 2 were confined continuously at the CRU from the time of check‐in (Day −1) until clinic discharge, for a minimum stay of 5 days post‐dose (Day 6) and a maximum confinement of 14 days post‐dose (Day 15). Clinic discharge was based on subjects meeting the discharge criteria that blood and plasma radioactivity levels were below the limit of quantitation for two consecutive samples and ≥90% of the dose had been recovered; or total radioactivity in excreta reached ≤1% of the administered dose for two consecutive collections.
2.2. Subjects
Healthy volunteers that were eligible for inclusion in this study were males (Parts 1 and 2) or females (Part 1 only) between 18 and 55 years of age, with a body mass index of 18.5–32 kg/m2. Subjects were in good health, as determined from a medical history, vital signs, 12‐lead electrocardiogram and clinical laboratory evaluations. Female subjects of child‐bearing potential were not pregnant or lactating and were willing to use non‐hormonal contraceptive methods from the time of signing the Informed Consent Form (ICF) or 10 days prior to check‐in (Day −1) until at least 28 days after the last study drug administration.
Participants were excluded if any of the following criteria were applicable: significant history or clinical manifestation of any metabolic, allergic, dermatological, hepatic, renal, haematological, pulmonary, cardiovascular, gastrointestinal and/or neurological/psychiatric disorder; history of autoimmune disease, idiopathic pulmonary fibrosis, pneumonitis, food/drug hypersensitivity, stomach/intestinal surgery that would alter drug absorption, alcoholism/drug addiction and/or tuberculosis; prior allogeneic bone marrow or solid organ transplantation; major surgical procedure within 28 days; severe infection within 4 weeks; prior exposure to immune‐modulating oncology therapeutics; use of prescription medications/products within 14 days; use of non‐prescription medications/products within 7 days; participation in a previous radiolabel study within 12 months (Part 2 only); exposure to significant radiation within 12 months (Part 2 only).
The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. All participants provided written informed consent prior to any study‐related procedures. The clinical study protocol, informed consent forms, and any information given to the subjects and relevant supporting information were submitted for review and approval by the institutional review board (IRB) of Covance prior to study screening.
2.3. Dose rationale
Single and twice daily oral doses (up to the maximum assessed dose of 1000 mg) of navoximod monotherapy are well tolerated in clinical studies in cancer patients;11, 12 therefore, a single 200 mg oral dose was considered suitable for this study. A 5 mg intravenous (IV) dose was selected based on pharmacokinetic simulations, with a predicted C max below the median C max of a 200 mg oral dose (assuming a conservative absolute bioavailability of 0.5).
Given the short half‐life of navoximod (~12 hours), a radioactivity dose of 600 μCi for the AME portion of this study was selected to ensure there was sufficient radioactivity in circulation for metabolite profiling using conventional radio‐detection methods. The radiation burden of a single 600 μCi oral dose of 14C‐navoximod was estimated to be 0.56% of the 3000–5000 mrem limits specified by the United States Code of Federal Regulations (CFR) based on dosimetry calculations from tissue distribution data obtained in rats.
2.4. Safety assessments
In Parts 1 and 2, safety assessments (physical examinations, 12‐lead electrocardiograms [ECGs], vital signs, How Do You Feel? [HDYF?] inquiries, and clinical laboratory evaluations) were performed at screening, during the study and/or at study completion/clinic discharge/early termination (ET). All adverse events (AEs), whether volunteered, elicited or noted on physical examination, were recorded throughout the study.
2.5. Materials
Navoximod was prepared by Wuxi AppTec Co., Ltd (Shanghai, China) and the film‐coated 200 mg navoximod tablet was prepared by Hoffman‐LaRoche (Basel, Switzerland). [14C]‐navoximod (specific activity 10.6 mCi/mmol) was synthesized by Selcia (Essex, UK).
2.6. Radiolabelled and IV dose preparation
The IV drug product was an aqueous solution containing 5 mg of navoximod in 5 mL of normal saline (0.9% sodium chloride in water for injection). The oral aqueous solution contained 200 mg of [14C]‐navoximod (22.2 MBq; 600 μCi) mixed in 20 mL water. The solubility of navoximod in water and other physiological relevant media is 200 mg/mL. Prior formulation studies determined that the IV and oral solution formulations were appropriate to obtain the target drug concentrations using high‐performance liquid chromatography (HPLC) analysis. In addition, the IV and oral solution appearances were clear and colourless prior to dosing.
2.7. Sample collection
In Part 1 of the study, blood samples for plasma concentrations of navoximod were collected from subjects at pre‐dose, end of IV push (IV dose only), 0.083 (IV dose only), 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 18, 24, 36, 48 and 72 h post‐dose. In Part 2 of the study, blood samples for plasma concentrations of navoximod and total radioactivity in the plasma and blood were collected at pre‐dose and 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 18, 24, 36, 48, 72 and 96 h post‐dose, and continued until the radioactivity counts were below the detection limit or a maximum confinement of 15 days was reached. Urine was collected pre‐dose and at 0–4 h, 4–8 h, 8–12 h and 12 to 24 h collection intervals; and then at 24 h intervals until the subject was discharged from the clinical site. Faecal samples were collected pre‐dose and at 24 h intervals until the subject was discharged from the clinical site.
2.8. Analytical methods
2.8.1. Bioanalysis of unlabelled navoximod in plasma
Plasma concentrations of navoximod were determined using one of two validated liquid chromatographic‐tandem mass spectrometry (LC–MS/MS) methods with a detection range of 0.05–50 ng/mL or 1–1000 ng/mL for low and high concentration samples, respectively. In both assays, samples were processed using protein precipitation extraction procedures and [13C, 15N2]‐navoximod was used as the internal standard. Chromatography of navoximod was achieved with an Atlantis T3 (5 μm, 50 × 4.6 mm, Waters Corporation, Milford, MA) column. The mobile phase A was 1 mM ammonium acetate in acetonitrile:water:formic acid (5:95:0.025) and the mobile phase B was 1 mM ammonium acetate in acetonitrile:water:formic acid (95:4.975:0.025). The gradient was 10% B for the first 0.4 minutes, linearly increased to 25% B between 0.4 and 2.0 minutes, further increased to 90% B between 2.0 and 2.3 minutes, maintained at 90% B between 2.3 to 3.2 minutes, decreased to 10% B between 3.2 and 3.4 minutes, and maintained at 10% B between 3.4 and 4.0 minutes. The flow rate was 1.8 mL/min and the cycle time was approximately 4.7 minutes. A Sciex API 4000 (detection range: 1–1000 ng/mL) or Sciex 5500 (detection range: 0.05–50 ng/mL) with an electrospray source in the positive ion multiple‐reaction monitoring (MRM) mode was used for detection of navoximod in plasma samples. The MRM transition for navoximod and the internal standard, [13C, 15N2]‐navoximod, was m/z 317.3 → 189.2 and m/z 320.2 → 192.1, respectively. The intra‐run and inter‐run precision of both assays was within 7.2% and accuracy was within ±12.2% of the nominal concentration values. Navoximod was found to be stable in human plasma over five freeze–thaw cycles, at least for 110 days at −10°C to −30°C and −60°C to −80°C, and for 24 h at room temperature.
2.8.2. Determination of radioactivity
Blood and faecal homogenates were combusted using a Model 307 Sample Oxidizer (Packard Instrument Company, Meriden, CT) and the resulting 14CO2 was trapped in Carbo‐Sorb and mixed with Perma Fluor. Oxidation efficiency was evaluated on each day of sample combustion by analysing a commercial radiolabelled standard both directly in scintillation cocktail and by oxidation. Acceptance criteria were combustion recoveries of 95 to 105%. Ultima Gold XR scintillation cocktail was used for samples analysed directly. All samples were analysed for radioactivity in Model 2900TR or 2910TR liquid scintillation counters (Packard Instrument Company) for at least 5 minutes or 100 000 counts. The LLOQ for radioactivity analysis by liquid scintillation counting is defined as three‐fold above blank matrix counts. Scintillation counting data (dpm) were automatically corrected for background counts. Each sample was homogenized or mixed before radioanalysis (unless the entire sample was used for analysis) and analysed in duplicate.
2.9. Metabolite profiling and identification
2.9.1. Sample preparation and chromatographic separations
Plasma radioprofiles were collected at 0.5, 1, 2, 4, 6, 8, 12, 24, 48, 72, 96, 120, 144 and 168–336 h post‐dose as pooled samples across the eight subjects to obtain the pharmacokinetics of the abundant metabolites. Plasma samples were also pooled across 0–48 h based on AUC for individual subjects to assess inter‐subject variability. Pooled plasma samples were extracted twice with three volumes of acetonitrile (ACN). The extracts from each step were combined and concentrated under vacuum, then re‐dissolved with water: ACN (2:1, v:v) for radiometric analysis. The pooled urine (0–48 h) represented >90% of the total radioactivity excreted in urine and was directly injected for LC–MS radioprofile analysis following centrifugation. The pooled faecal homogenates (0–96 or 0–120 h) also represented >90% of the total radioactivity excreted in the faeces. Faecal homogenates were extracted twice with three volumes of ACN. The supernatants were combined and evaporated to dryness under vacuum and the residues were reconstituted with water: ACN (2:1, v:v) for LC–MS radioprofiling.
Chromatographic separations were performed using an UltiMate 3000 UPLC system (Thermo Scientific, San Jose, CA) using a Kinetex EVO C18 column (5 μm, 4.6 × 50 mm, Phenomenex, Torrance, CA) and mobile phases A (10 mM ammonium acetate in H2O, adjust to pH 5) and B (10 mM ammonium acetate in MeOH/H2O [v/v, 90/10]). Gradient was 5% B for 2 minutes, increased to 10% B over 2 minutes, increased to 30% over 29 minutes, increased to 45% B over 13 minutes, increased to 65% over 3 minutes, rapidly to 95% in 0.5 minutes. The column was flushed at 95% B for 5 minutes, then returned to 5% B over 0.5 minutes and re‐equilibrated to these conditions for 5 minutes. The total run time was 60 minutes. For radioprofiling, the column flow was split 3:1 with the minor component directed to the mass spectrometer and the major component directed to a fraction collector for collection to DeepWell LumaPlate 96 well plates (Perkin Elmer, Waltham, MA) based on time (0.22 min/fraction). Following fraction collection, the plates were dried under vacuum. The radioactivity in each fraction was measured using a MicroBeta Scintillation and Luminescence Counter (Perkin Elmer, Shelton, CT) for 5 minutes.
Radiochromatograms were reconstructed using Laura Evaluation software (LabLogic, Brandon, FL) and the radiopeaks were integrated to determine the percent distribution of radioactivity in each sample.
2.9.2. Metabolite structure identification
Mass spectra were obtained with an LTQ Orbitrap Fusion Lumos high‐resolution mass spectrometer equipped with an electrospray ionization source from Thermo Scientific (San Jose, CA). The electrospray ion source voltage was 3.5 kV. The heated capillary temperature was 300°C and vaporizer temperature was 250°C. The scan‐event cycle consisted of a full‐scan mass spectrum at a mass resolving power of 120 000 (at m/z 400) and the corresponding data‐dependent MS/MS and MS3 CID and HCD were acquired at a resolving power of 15 000. Accurate mass measurements were performed using internal calibration using fluoranthene (M+ = 202.0777).
2.10. Pharmacokinetic analyses
Mean plasma concentration–time profiles and total radioactivity for plasma, whole blood, urine and faeces data were plotted using GraphPad Prism Version 7.04. PK parameters for unlabelled navoximod in plasma, and total radioactivity in plasma and whole blood were determined by standard non‐compartmental methods, using Phoenix WinNonlin v. 6.4 (Pharsight Corporation, Mountain View, CA). Samples below the LLOQ were set to zero for all analyses unless they occurred between two quantifiable concentrations in a profile, in which case they were set to missing. The following PK parameters were calculated for navoximod and total radioactivity in plasma and whole blood: maximum observed concentration (C max), area under the concentration–time curve extrapolated to infinity (AUC0‐∞), area under the concentration–time curve from time 0 to the last measurable concentration (AUC0–last), time to maximum observed concentration (t max), apparent elimination half‐life (t 1/2) calculated as 0.693/λz, absolute bioavailability (F) of navoximod using the ratio of dose‐normalized oral AUC0‐∞ to dose normalized IV AUC0‐∞, systemic clearance of navoximod following IV administration (CL), and apparent oral clearance of navoximod (CL/F), volume of distribution of navoximod following IV administration (Vz) and apparent volume of distribution of navoximod (V/F).
2.11. Statistics
The sample size of Part 1 (aBA) was selected to minimize exposure of navoximod to healthy volunteers, but was adequate to characterize the safety, tolerability and pharmacokinetics of navoximod (not based on formal sample size calculations). For Part 2 (hAME), the sample size was based on practical considerations needed to meet the objectives of an ADME study,13 and was not powered on the basis of statistical hypothesis testing.
No formal statistical analyses were performed in this study. Plasma, blood, urine and faecal concentration, and radioactivity–time curves are presented as mean and standard deviation. PK parameters are summarized by descriptive statistics. Dose‐normalized geometric mean ratios and 90% confidence intervals (CIs) were computed for PK parameters for oral versus IV dosing to estimate absolute bioavailability.
2.12. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,14 and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18.15
3. RESULTS
3.1. Subjects
Sixteen healthy volunteers were enrolled in Part 1 of the study; 13 (81.3%) were male and three (18.8%) were female with an average (range) age of 36 (19–51) years, weight of 81.5 (59.4–96) kg, and BMI of 26.2 (21.7–31.2). Of the subjects included, 10 (62.5%) self‐identified as white, five (31.3%) as black or African American, and one (6.3%) as Asian. All subjects in Part 1 completed the study and were included in all analyses.
Eight healthy male volunteers, independent from Part 1 of the study, were enrolled in Part 2 of the study. Subjects had an average (range) age of 36 (23–46) years, weight of 81.7 (60.5–102) kg, and BMI of 25.6 (19.6–30.5). Of the subjects included, six (75%) identified as white, and two (25%) as black or African American. Seven subjects completed Part 2 of the study; one discontinued on Day 13 (withdrawal by subject).
3.2. Pharmacokinetics
3.2.1. Part 1: Absolute bioavailability
Plasma pharmacokinetic parameters for navoximod following IV (5 mg, push over 3–5 min) and oral (200 mg tablet) administration are presented in Table 1 and mean concentration–time profiles are presented in Figure 1. The geometric mean exposure ratio (oral/IV) for dose normalized AUC0‐∞, as the measure of absolute bioavailability, was 0.555 (geometric % coefficient of variation [%CV]: 22.3%). The geometric mean (range) terminal elimination half‐life was consistent for oral (11.0 [6.96–14.3] hours) and IV (12.6 [9.22–20.4] hours) dosing. The geometric mean (%CV) navoximod plasma clearance and volume of distribution following IV infusion were 62.0 L/h (21.0%) and 1120 L (28.4%), respectively. The geometric mean (%CV) apparent clearance (CL/F) and apparent volume of distribution (V/F) for orally administered navoximod were 112 L/h (31.4%) and 1780 L (31.9%).
Table 1.
Summary of pharmacokinetic parameters of navoximod following oral and IV administration (part 1)
| Parameter | Oral: 200 mg tablet (n = 16) | IV: 5 mg solution (n = 16) |
|---|---|---|
| AUC(0–∞) (h.ng/mL) | 1790 (31.4) | 80.6 (21.0) |
| AUC(0–72) (h.ng/mL) | 1770 (31.3) | 79.1 (21.3) |
| C max (ng/mL) | 683 (42.2) | 114 (70.7) |
| t max (h)a | 0.5 (0.25–1.02) | 0.0833 (0.0833–0.583) |
| t 1/2 (h)b | 11.0 (6.96–14.3) | 12.6 (9.22–20.4) |
| CL/F or CL (L/h) | 112 (31.4) | 62.0 (21.0) |
| Vz/F or Vz(L) | 1780 (31.9) | 1120 (28.4) |
| F | 0.555 (22.3) | ‐‐ |
Geometric mean (geometric CV%) results are presented unless otherwise indicated.
Median (minimum, maximum) results are presented for tmax.
Geometric mean (range) results are presented for t1/2.
Figure 1.
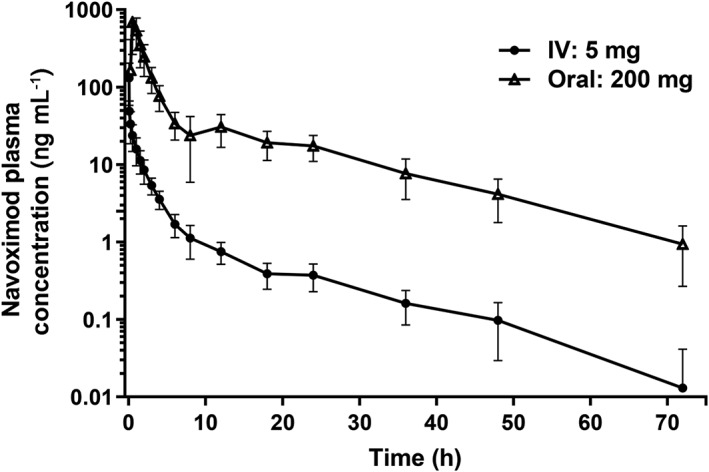
Log‐linear plot of mean (± SD) plasma navoximod concentration following administration of a 200 mg oral tablet (open triangles) or a 5 mg IV solution (closed circles)
3.2.2. Part 2: Mass balance
The single mass dose of [14C]‐navoximod ranged from 191 to 192 mg and the radioactive dose of [14C]‐navoximod ranged from 600 to 604 μCi.
Pharmacokinetic parameters for unlabelled navoximod and total radioactivity in plasma and whole blood are summarized in Table 2; mean concentration–time profiles are presented in Figure 2. Following oral administration of a 200 mg/600 μCi solution of [14C]‐navoximod, median t max for navoximod in the plasma and total radioactivity in the blood was 0.5 hours, while t max for total radioactivity in the plasma was 0.75 h. Thereafter, plasma concentrations of navoximod declined with a geometric mean (range) half‐life of 9.41 (7.54–12.8) hours, while total radioactivity in the plasma and whole blood had substantially longer half‐lives of 143 (79.6–190) and 206 (64.3–336) hours, respectively. Total plasma radioactivity concentrations were significantly higher than navoximod plasma concentrations. Geometric mean C max and AUC0‐∞ ratios indicated that navoximod represented 10.6% and 4.21%, respectively, of plasma total radioactivity, suggesting the presence of circulating metabolites.
Table 2.
Mean pharmacokinetic parameters for radioactivity in plasma and whole blood following administration of 200 mg/600 μCi [14C]‐navoximod
| Parameter | Plasma navoximod (n = 8) | Plasma total radioactivity (n = 8) | Whole blood total radioactivity (n = 8) |
|---|---|---|---|
| AUC(0‐∞) (h.ng/mL)a | 1540 (31.3) | 36,700 (32.0) | 38,900 (55.4) |
| AUC(0‐last) (h.ng/mL)b | 1510 (31.6) | 31,000 (27.2) | 22,500 (39.9) |
| C max (ng/mL)a | 464 (33.1) | 4400 (30.8) | 2340 (28.7) |
| t max (h)c | 0.5 (0.25–1.5) | 0.75 (0.5–1.5) | 0.5 (0.5–1.5) |
| t 1/2 (h)d | 9.41 (7.54–12.8) | 143 (79.6–190) | 206 (64.3–336) |
| CL/F (L/h) | 124 (31.4) | 5.22 (32.0) | 4.92 (55.4) |
| Vz/F (L) | 1680 (29.7) | 1080 (19.7) | 1460 (27.5) |
Geometric mean (geometric CV%) results are presented unless otherwise indicated.
Units for total radioactivity are h.ng equivalents/g for AUC(0‐last) and AUC(0‐∞), and ng equivalents/g for C max.
AUC(0‐last): Last measurable concentration is 336 h unless the subject discontinued early or fulfilled discharge criteria prior to 336 h.
Median (minimum, maximum) results are presented for t max.
Geometric mean (range) results are presented for t 1/2.
Figure 2.
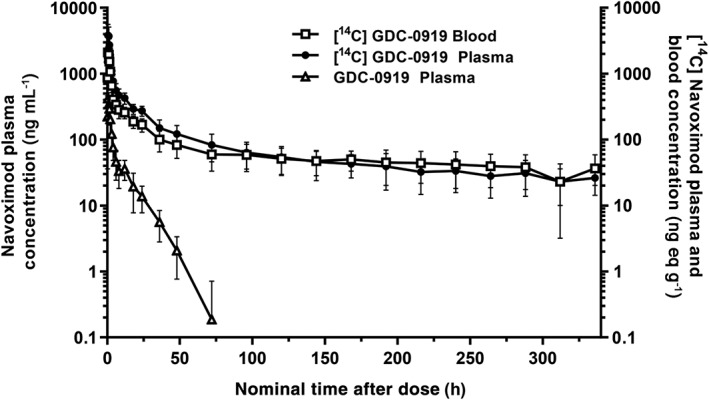
Log‐linear mean (± SD) plasma concentration time profile of navoximod (open triangles), and mean (± SD) plasma (closed circle) and whole blood (open square) concentration profiles of [14C]‐navoximod following oral administration of 200 mg/600 μCi [14C]‐navoximod
The blood‐to‐plasma ratio was determined from the total radioactivity, which included both parent and metabolites. The geometric mean whole blood to plasma AUC0‐∞ ratio for total radioactivity was 0.965. Blood‐to‐plasma ratios increased from 0.5 to 1.6 over the duration of the study, indicating slightly higher association of [14C]‐radioactivity towards blood cells with time. Since the half‐life of total radioactivity is more than 10 times longer than the half‐life of navoximod, the changes of the blood‐to‐plasma ratio over time is likely related to metabolites rather than navoximod.
Cumulative recovery of total radioactivity in urine and faeces following administration of [14C]‐navoximod is presented in Figure 3. The mean total recovery of the radioactive dose in excreta up to 336 h post dose was 87.8%, with 85.0% of the total radioactivity recovered within the first 72 h. The majority of the total radioactivity was eliminated in the urine, with mean ± SD recoveries of 80.4% ± 5.53% and 7.44% ± 3.82% in the urine and faeces, respectively.
Figure 3.
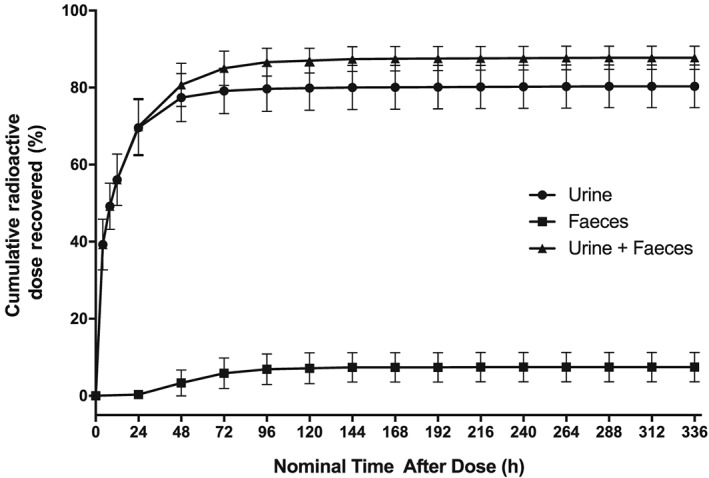
Mean (± SD) cumulative recovery of total [14C] radioactivity following oral administration of 200 mg/600 μCi [14C]‐navoximod expressed as a percentage of the administered [14C] dose in excreta, urine + faeces (triangles), urine (circles), and faeces (squares)
3.3. Extraction recovery of metabolite profiling samples
The extraction recoveries of the radioactivity from plasma samples decreased over time, with 95.0% of the radioactivity extracted from the 0.5‐hour post‐dose plasma sample and 52.5% of the radioactivity extracted from the 48‐hour post‐dose plasma sample. The majority of the circulating radioactivity at ≥72 hours post‐dose was not extractable (extraction recoveries were 27–38%). The extraction recoveries of the AUC0–48 hour pooled plasma samples among the eight subjects were 76.1–86.5%. The plasma samples were extracted multiple times until the radioactivity in the supernatant was below the detection limit of the liquid scintillation counter. Therefore, the unextracted radioactivity was presumed to be tightly associated with plasma proteins. The extraction recoveries of faecal homogenate were 83.3–109% (n = 8).
3.4. Navoximod metabolite profiling and identification
Representative HPLC radiochromatograms of pooled 0–48 h plasma, plasma at 72 h and 120 h, pooled urine and pooled faeces are presented in Figure S1. The protonated molecular ion [M + H]+ and major product ions observed by high resolution full scan and MS2 experiments for navoximod and its metabolites are listed in Table S1. The discussion of MS/MS fragment interpretation of navoximod and its metabolites is also provided in the supplemental information.
The relative abundances and exposures of navoximod and its metabolites in plasma, and their percentage of dose detected in urine and faeces are provided in Table 3. The proposed biotransformation pathways are presented in Figure 4. In the pooled 0–48 h plasma, unchanged parent, navoximod, was a minor component in circulation, with an AUC0–48 of 4.40 μM.h (7.00% of the total exposure at 0–48 h). M28 (glucuronide conjugate of navoximod on the hydroxyethyl moiety) was the most prominent metabolite up to 48 h post‐dose, with an AUC0–48 of 36.4 μM.h (57.5% of the drug‐derived exposure). M19 (glucuronide of navoximod on hydroxyl of cyclohexane) was detected in circulation with an AUC0–48 2.36 μM.h (3.14% of the total 0–48 h exposure). M1 was identified as thiocyanate, with an AUC0–48 of 3.12 μM.h (5.54% of the exposure). Thiocyanate was likely formed through oxidative ring opening of imidazole followed by release of cyanide, which was converted to thiocynate in vivo. As thiocyanate is an endogenous material in humans, the levels of thiocyanate reported here were based on radioactivity and only accounted for the portion from navoximod.
Table 3.
Navoximod and its abundant metabolites in human plasma, urine, and feces after a single oral administration of 200 mg/600 μCi [14C]‐navoximod to healthy subjects
| Analyte | Biotransformation | Plasma | Urine | Feces | Total | |
|---|---|---|---|---|---|---|
| % of TRA | AUC0–48‐h (μM/h)a | % of doseb | % of doseb | % of doseb | ||
| Navoximod | Unchanged parent | 7.00 | 4.40 | 1.66 | 3.79 | 5.45 |
| M1 (thiocyanate) | Ring cleavage | 5.54 | 3.54 | 0.614 | 0.138 | 0.752 |
| M17 | Oxidation (+ 34 Da) | D | D | 1.78 | 1.54 | 3.32 |
| M19 | Glucuronidation | 3.14 | 2.36 | 8.74 | ND | 8.74 |
| M28 | Glucuronidation | 57.5 | 36.4 | 59.7 | ND | 59.7 |
| M31 | Oxidation (+16 Da) | D | D | 1.71 | D | 1.71 |
| M38 | Glucuronidation | D | D | 1.76 | ND | 1.76 |
D, only detected by mass spectrometry; ND, not detected; TRA, total radioactivity.
Mean values from AUC 0–48 hour of pooled plasma (n = 8) adjusted with extraction recovery. The unextracted radioactivity accounted for 20.4% of the total radioactivity in the plasma sample.
Mean values from eight subjects.
Figure 4.
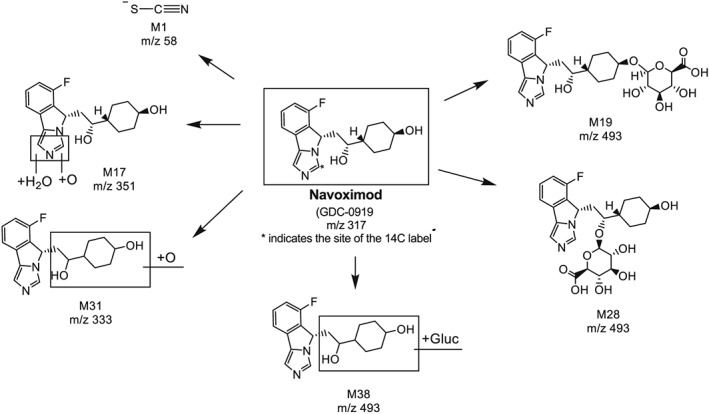
Proposed biotransformation pathways of navoximod
As total radioactivity had a relatively long half‐life, plasma samples at 72, 96, 120, 144 and 168–336 hours post‐dose as pooled samples across the eight subjects were analysed to investigate the nature of the long‐lasting radioactivity in circulation. M1 accounted for most of the extractable radioactivity at 72 and 96 hours post‐dose, and was the only detectable peak in the radioprofiles ≥120 hour post‐dose. M1 reached a maximal plasma concentration of 0.264 μM at 1 h followed by a slow decline that contributed to the apparent long‐lived circulating radioactivity.
Urine was the major excretion route of navoximod‐related radioactivity and accounted for 80.4% of the administered dose. The metabolite profiles of pooled urine were very similar among the eight subjects. Unchanged navoximod was a minor component in urine and represented only 1.66% of the dose. M28 was the most prominent urinary metabolite, accounting for 59.7% of the dose. M19 was also an abundant metabolite, representing 8.74% of the dose. All other urinary metabolites were minor, each representing less than 2% of the administered dose.
Only 7.44% of the administered radioactivity was recovered in faeces, indicating that faecal excretion was a minor route of elimination for navoximod‐related radioactivity. The metabolite profiles from the pooled faeces were similar among the eight subjects. Unchanged navoximod represented 3.79% of the dose. M17 (oxidation, +34 Da) accounted for 1.54% of the dose. All other metabolites in faeces represented less than 1% of the administered dose.
3.5. Safety and tolerability
All IV and oral doses in both parts of the study were generally well tolerated and all adverse events were mild in severity. There were no clinically significant changes in laboratory test parameters or in blood pressure, pulse rate, electrocardiographic or physical examination findings.
4. DISCUSSION
This study investigated the absolute bioavailability of navoximod, determined the mass balance and routes of elimination of [14C]‐navoximod, and characterized the metabolite profile. Results from this study demonstrate that navoximod has a fast, systemic clearance, relative to hepatic blood flow, and high volume of distribution, with an absolute bioavailability following a single oral dose of 200 mg navoximod of 55.5%. Approximately 87.8% of the radioactive dose was recovered in excreta up to 336 h post dose, most of which (85.0%) was recovered within the first 72 h. Most of the dose (80.4%) was eliminated in the urine while only 7.44% of the dose was excreted through faeces, indicating that the oral dose had been well absorbed. Full metabolite profiling of plasma, urine and faecal samples showed that navoximod is extensively metabolized. The major metabolic pathway of navoximod in humans was glucuronidation, with oxidation as a minor metabolic pathway.
M28 (navoximod glucuronide) was the most abundant metabolite in circulation, accounting for most of the drug‐derived exposure. All the other circulating metabolites represented less than 10% of the total drug‐related exposure for all subjects. One metabolite with long half‐life was M1 (thiocyanate), reaching a maximal plasma concentration of 0.264 μM at 1 h, followed by a slow decline, contributing to the apparent long‐lived circulating radioactivity but accounting for <0.1% of the administered dose and was eliminated in urine and faeces as a minor metabolite (<1% of the dose). The level of 14C‐thiocyanate (M1) in circulation from subjects dosed with 200 mg of navoximod is negligible compared to the endogenous thiocyanate levels in humans, which exist at significantly higher concentrations in the range of 50–150 μM.16, 17, 18 The extractable component of the sample decreased at later time points as the total circulating radioactivity decreased such that after 72 h post‐dose most of the radioactivity was not extracted. This unextracted radioactivity at late time points was relatively low (e.g., 83.1 ng • equivalents/mL at 72 h) and represented <0.1% of the administered dose.
The disposition of navoximod and its metabolites following a single oral dose of 200 mg navoximod to healthy volunteers is presented in Figure 5. The fraction of the dose absorbed (Fa) was estimated to be 0.841 using the sum of radioactivity recovered in urine (80.4%) and radioactivity in faeces that was characterized as metabolites (3.65%). Fa could be higher than 0.841 because part of the unchanged navoximod recovered in faeces could come from glucuronide metabolites (M19, M28, and M38) that were reverted to parent by glucosidase in the gastrointestinal tract or from the absorbed parent drug through biliary excretion. The hepatic first pass extraction was determined to be ~34%. With 55.5% of the oral dose entering systematic circulation (corresponding to F), only 1.66% was recovered as the unchanged navoximod in urine. Approximately 82.4% of the dose was recovered as metabolites with the majority in urine (78.7%). The fraction of metabolism (fm) was estimated to be 0.98.
Figure 5.
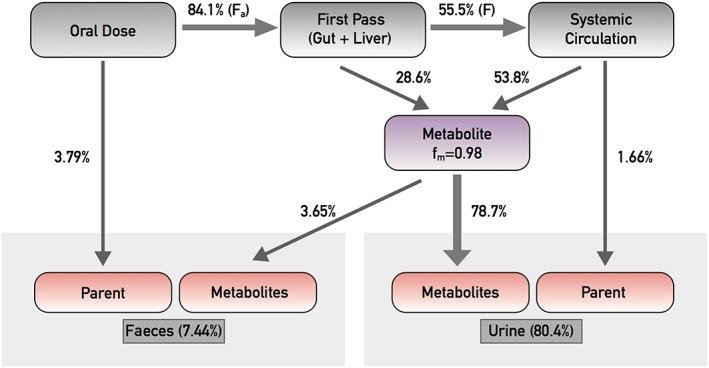
Model of mass balance, excretion and fraction of metabolism (fm) for an oral dose of navoximod
It is interesting to note that total IV clearance was high (62 L/hour, ~71% of blood flow), but bioavailability was moderate (0.555). The observed bioavailability was higher than anticipated if all clearance mechanisms were attributed to hepatic clearance, suggesting significant extra‐hepatic clearance pathway involved in the elimination of navoximod in humans. Bioavailability is the product of Fa, Fg and Fh; therefore, Fh * Fg is 0.66. UGT reaction phenotyping experiments identified UGT1A9 and 2B7 as the two major UGTs responsible for navoximod metabolism (unpublished results). UGT1A9 is highly expressed in kidney relative to the liver and UGT2B7 is predominately expressed in the kidney and liver with trace amounts in the gastrointestinal tract. Therefore, it is reasonable to assume first‐pass gut metabolism is negligible (Fg = 1), and Fh is approximately 0.66. The hepatic clearance is then 29.6 L/hour (CLhep = [1 − Fh] * Qh). As the renal clearance is low (CLr = [cumulative amount of unchanged navoximod excreted in urine over the sampling interval]/AUC0‐∞ = 1.22 L/hour), the extrahepatic clearance is estimated to be 31.2 L/h. Since glucuronide metabolites detected in urine accounted for 70.2% of dose and UGT1A9 and 2B7 are mainly expressed in kidney, it is possible that metabolism through glucuronidation in the kidney could play a significant role in the clearance of navoximod.
In conclusion, this study determined the absolute bioavailability, mass balance, elimination routes, and metabolite profiling and characterization of navoximod in healthy subjects. Navoximod was well tolerated and showed moderate oral bioavailability. Navoximod was quickly absorbed and extensively metabolized. The major metabolic pathway of navoximod was glucuronidation.
COMPETING INTERESTS
S.M., J.S., E.Y., E.Yost, X.L., R.Z., R.M., S.R.‐J., L.S. and K.M. are employees of Genentech, Inc. and shareholders of F. Hoffmann La Roche, Ltd. At the time the research was conducted, H.L., A.P. and J.A.W. were employees of Genentech, Inc. and shareholders of F. Hoffmann La Roche, Ltd. N.S. and L.J. are employees of Covance, Inc., which has received funds from Genentech in connection with this study. This work was supported by Genentech, Inc.
CONTRIBUTORS
All authors contributed to study design, data analysis and/or interpretation. All authors critically reviewed the manuscript and contributed to content development.
Supporting information
Table S1 Molecular ion and major product ions observed for navoximod and its metabolites in humans
Figure S1 Representative HPLC radiochromatograms of (a) 0–48‐h pooled plasma, (b) plasma at 72 h, (c) plasma at 120 h, (d) 0–48‐h pooled urine, and (e) 0–96‐h pooled feces
ACKNOWLEDGEMENTS
The authors would like to thank the healthy volunteers for their contribution to this clinical study and the research staff at the Covance Clinical Research Unit (Madison, WI) for conducting the clinical study. Additionally, the authors thank Drs. Cyrus Khojasteh and Cornelis Hop for review and valuable inputs into the study design and analyses. This work was presented in part at the 2018 American Society of Clinical Pharmacology and Therapeutics annual meeting (Abstract PII‐125).
Ma S, Suchomel J, Yanez E, et al. Investigation of the absolute bioavailability and human mass balance of navoximod, a novel IDO1 inhibitor. Br J Clin Pharmacol. 2019;85:1751–1760. 10.1111/bcp.13961
PI Statement: The authors confirm that the Principal Investigator for this paper is Nicholas Siebers and that he had direct clinical responsibility for patients.
REFERENCES
- 1. Munn DH, Mellor AL. IDO in the tumor microenvironment: Inflammation, counter‐regulation, and tolerance. Trends Immunol. 2016;37(3):193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Muller AJ, DuHadaway JB, Donover PS, Sutanto‐Ward E, Prendergast GC. Inhibition of indoleamine 2,3‐dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat Med. 2005;11(3):312–319. [DOI] [PubMed] [Google Scholar]
- 3. Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34(3):137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Godin‐Ethier J, Hanafi LA, Piccirillo CA, Lapointe R. Indoleamine 2,3‐dioxygenase expression in human cancers: Clinical and immunologic perspectives. Clin Cancer Res. 2011;17(22):6985–6991. [DOI] [PubMed] [Google Scholar]
- 5. Brody JR, Costantino CL, Berger AC, et al. Expression of indoleamine 2,3‐dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle. 2009;8(12):1930–1934. [DOI] [PubMed] [Google Scholar]
- 6. Curti A, Pandolfi S, Valzasina B, et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25‐ into CD25+ T regulatory cells. Blood. 2007;109:2871–2877. [DOI] [PubMed] [Google Scholar]
- 7. Witkiewicz A, Williams TK, Cozzitorto J, et al. Expression of indoleamine 2,3‐dioxygenase in metastatic pancreatic ductal adenocarcinoma recruits regulatory T cells to avoid immune detection. J Am Coll Surg. 2008;206:849–854. discussion 54‐6 [DOI] [PubMed] [Google Scholar]
- 8. Brochez L, Chevolet I, Kruse V. The rationale of indoleamine 2,3‐dioxygenase inhibition for cancer therapy. Eur J Cancer. 2017;76:167–182. [DOI] [PubMed] [Google Scholar]
- 9. Mautino M LC, Vahanian N, Adams J, Van Allen C, Sharma MD, Johnson TS, Munn DH. Synergistic antitumor effects of combinatorial immune checkpoint inhibition with anti‐PD‐1/PD‐L1 antibodies and the IDO pathway inhibitors NLG919 and indoximod in the context of active immunotherapy. AACR Annual Meeting, San Diego, CA, 2014: Abstract 5023.
- 10. Spahn JPJ, Lorenzana E, Kan D, et al. Improved anti‐tumor immunity and efficacy upon combination of the IDO1 inhibitor GDC‐0919 with anti‐PD‐L1 blockade versus anti‐PD‐L1 alone in preclinical tumor models. J Immuno Therapy Cancer. 2015;3(Suppl 2):P303. [Google Scholar]
- 11. Jung KH, LoRusso PM, Burris HA, et al. Phase I study of the indoleamine 2,3‐dioxygenase 1 (IDO1) inhibitor navoximod (GDC‐0919) administered with PD‐L1 inhibitor (atezolizumab) in advanced solid tumors. Clin Cancer Res. 2019. 10.1158/1078-0432.CCR-18-2740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nayak‐Kapoor A, Hao Z, Sadek R, et al. Phase Ia study of the indoleamine 2,3‐dioxygenase 1 (IDO1) inhibitor navoximod (GDC‐0919) in patients with recurrent advanced solid tumors. J Immunother Cancer. 2018;6(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Penner N, Klunk LJ, Prakash C. Human radiolabeled mass balance studies: Objectives, utilities and limitations. Biopharm Drug Dispos. 2009;30(4):185–203. [DOI] [PubMed] [Google Scholar]
- 14. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, Gray AJG, Bruce L, Alexander SPH, Anderton S, Bryant C, Davenport AP, Doerig C, Fabbro D, Levi‐Schaffer F, Spedding M, Davies JA. NC‐IUPHAR. The IUPHAR/ Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018;46:D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Faccenda E, et al. CGTP Collaborators. The Concise Guide to PHARMACOLOGY 2017/: Enzymes. Br J Pharmacol 2017;174:S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Foss OP, Lund‐Larsen PG. Serum thiocyanate and smoking: Interpretation of serum thiocyanate levels observed in a large health study. Scand J Clin Lab Invest. 1986;46(3):245–251. [DOI] [PubMed] [Google Scholar]
- 17. Shin YGMH, van Heuveln FH, Wieling J, Halladay J, Sahasranaman S, Hop CECA. Validation of a method for the determination of thiocyanate in human plasma by UV/VIS spectrophotometry and application to a phase I clinical trial of GDC‐0425. Transl Clin Pharmacol. 2015;23(2):59–65. [Google Scholar]
- 18. Takahashi RH, Halladay JS, Siu M, et al. Novel mechanism of decyanation of GDC‐0425 by cytochrome P450. Drug Metab Dispos. 2017;45(5):430–440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Molecular ion and major product ions observed for navoximod and its metabolites in humans
Figure S1 Representative HPLC radiochromatograms of (a) 0–48‐h pooled plasma, (b) plasma at 72 h, (c) plasma at 120 h, (d) 0–48‐h pooled urine, and (e) 0–96‐h pooled feces