

SUMMARY
Hox genes pattern the anterior-posterior axis of animals, and are posited to drive animal body plan evolution, yet their precise role in evolution has been difficult to determine. Here, we identified evolutionary modifications in the Hox gene Abd-B that dramatically altered its expression along the body plan of Drosophila santomea. Abd-B is required for pigmentation in Drosophila yakuba, the sister species of D. santomea, and changes to Abd-B expression would be predicted to make large contributions to the loss of body pigmentation in D. santomea. However, manipulating Abd-B expression in current-day D. santomea does not affect pigmentation. We attribute this epistatic interaction to four other genes within the D. santomea pigmentation network, three of which have evolved expression patterns that do not respond to Abd-B. Our results demonstrate how body plans may evolve through small evolutionary steps distributed throughout Hox regulated networks. Polygenicity and epistasis may hinder efforts to identify genes and mechanisms underlying macroevolutionary traits.
Graphical Abstract
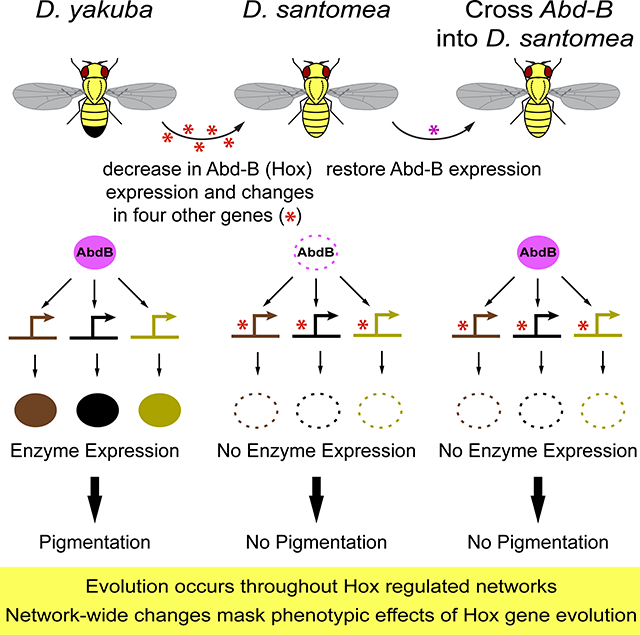
Liu et al. identify five genes that have evolved regulatory mutations during the loss of pigmentation in Drosophila santomea. While changes they find in the Hox gene Abd-B would be predicted to have substantial effects on phenotype, they show that modifications throughout its downstream network epistatically mask the effects of Hox gene evolution.
INTRODUCTION
A major challenge for evolutionary biology is to understand how phenotypic evolution results from genomic divergence. Hox genes, a family of Homeobox-containing transcription factors that regulate differential development along the anterior-posterior animal axis, have been singled out as particularly important for body plan evolution [1, 3]. Across a wide variety of taxa, correlations exist between Hox gene expression patterns and the specific morphology of serially homologous organs, such as vertebrae, limbs, or body segments [5–9]. These studies imply that changes in Hox expression patterns have driven body plan evolution, and some experimental evidence supports this conclusion [10–12]. However, this model likely overemphasizes the importance of Hox genes relative to other loci during the evolution of body plans. Unbiased approaches, such as genetic linkage studies often find that morphological traits are polygenic [13], and these studies have rarely implicated Hox genes as likely contributors to morphological variation. It is thus unclear precisely how Hox gene evolution contributes to differences in animal body plans.
We have explored this problem by studying the evolution of a Hox-regulated polygenic trait in two species that display a dramatic phenotypic difference in cuticular pigmentation: D. yakuba males have darkly pigmented posterior abdominal segments (Figure 1B), which reflects the ancestral state for this clade [14], whereas D. santomea evolved loss of most abdominal pigmentation (Figure 1C). In insects, the Hox gene Abdominal-B (Abd-B) specifies identity of the posterior abdominal segments, and is required for male-specific posterior body segment pigmentation in the clade containing Drosophila melanogaster and D. yakuba [14, 15]. Previous quantitative trait locus (QTL) mapping identified four genomic regions contributing to the abdominal pigmentation difference between D. yakuba and D. santomea [4] (Figure 1A). Further studies identified mutations in a cis-regulatory element of tan as the likely cause of one of these QTLs [16, 17], however the genes underlying other QTLs remained uncharacterized.
Figure 1. The Hox gene Abd-B is positioned within one of the abdominal pigmentation QTLs of D. santomea and is differentially expressed.

(A) Summary of QTLs [4], candidate genes, and introgression lines studied here and previously [17]. (B-C) The adult abdomen of D. yakuba (B) and D. santomea (C) males. (D-E) Top panels: Immunostaining of Abd-B in pupal abdomens of D. yakuba (D) and D. santomea (E) males at mid-late pupal stages (63–68 hAPF). Bottom panels show magnified images of the A5 regions outlined (red). See also Figure S1.
We identified a total of five genes contributing to the D. santomea phenotype, including changes that alter the regulatory region and expression of Abd-B. We elucidate how evolving interactions among these genes epistatically mask the phenotypic effects of Hox gene evolution in modern-day D. santomea. The polygenicity and epistasis uncovered in this evolving Hox-regulated network highlights important challenges in resolving the molecular and genetic causes of macroevolution.
RESULTS
Evolution of the D. santomea Abd-B locus
Because Abd-B falls within a published pigmentation QTL [4] (Figure 1A) and regulates the pigmentation gene network of D. melanogaster, we assessed its potential role in D. santomea. We found no differences in the predicted protein sequences encoded by Abd-B of D. yakuba and D. santomea (Table S1), but observed dramatic differences in its expression between these species. While Abd-B was expressed in the tergites of abdominal segments 5 and 6 (A5 and A6) of D. yakuba (Figure 1D), it was present only in the A6 tergite of D. santomea pupae (Figure 1E). We did not detect any differences in Abd-B expression between D. yakuba and D. santomea at other stages of development, nor of the Hox proteins encoded by Ubx and abd-A, which are adjacent to Abd-B in the genome (Figure S1).
We next employed a reciprocal hemizygosity test [18] to determine whether evolution of Abd-B contributed to the loss of abdominal pigmentation in D. santomea. In this test, two sets of hybrid progeny are generated in which one or the other parental allele is mutated, while its homolog is left intact (Figure 2B). The two resulting hemizygotes are therefore genetically equivalent, with the sole exception of the single mutated locus. Thus, any phenotypic differences between reciprocal hemizygotes can be attributed to an evolutionary difference between the parental wild-type alleles. To this end, we introduced mutations into a ~1 kb initiator element named IAB5, which drives Abd-B expression in the A5 segment [19]. We replaced the IAB5 initiator element in D. yakuba and D. santomea with an eye-directed red fluorescent protein (RFP) marker cassette [20] using CRISPR/Cas9 assisted homology directed repair [21] (Figure 2A, Figure S2A). D. yakuba individuals heterozygous for the IAB5Δ deletion exhibited reduced pigmentation in the A5 body segment (Figure S2B–C). Between reciprocal hemizygotes, we observed that male hybrids with a functional D. yakuba IAB5 allele showed significantly darker A5 pigment intensity in a larger area than male hybrids with a functional D. santomea IAB5 allele (Figure 2C, Figure S2E, F and L). Furthermore, we confirmed that Abd-B expression differs among the two classes of reciprocal hemizygote progeny (Figure S2J–K). These results indicate that evolution of Abd-B expression was mediated by changes in its cis-regulatory sequences, and that changes in Abd-B have phenotypic effects in the hybrid background.
Figure 2. Evolution of a cis regulatory element of Abd-B contributes to the D. santomea phenotype.

(A) Map of the genomic region between abd-A and Abd-B, with the iab-5 region highlighted in gray. The 1 kb IAB5 initiator element (red) was replaced by an eye-directed RFP cassette (orange triangle). (B) Outline of the reciprocal hemizygosity test of IAB5, showing phenotypes of representative hemizygotes. (C) Quantification of A5 segment pigmented area in male progeny of the crosses shown in (B) (Student’s t-test, ****: p<0.0001). (D, E) in situ hybridization to detect GFP expression of D. yakuba and D. santomea iab-5 GFP reporter transgenic flies at late pupal stages (93–94 hAPF). See also Figure S2.
To localize function-altering changes in Abd-B, we tested the ~15 kb iab-5 region, which contains the IAB5 initiator element, and which has been implicated in the maintenance of Abd-B activation in the A5 and more posterior segments [22]. We generated GFP reporter constructs containing the iab-5 region from either D. yakuba or D. santomea (Figure 2A), which were inserted into a common landing site in the D. melanogaster genome. The D. yakuba reporter construct drove GFP mRNA in A5 and A6 segments of late stage pupae (Figure 2D), but the D. santomea reporter construct did not (Figure 2E). Both constructs drove apparently identical expression patterns in embryonic stages (Figure S2R–T). To test whether the critical evolved differences in iab-5 function resided within or outside of the IAB5 initiator element, we reciprocally transplanted the 1kb IAB5 initiator elements between D. yakuba and D. santomea constructs. In each case, expression resembled that of the background into which the 1kb IAB5 element was placed (Figure S2R–X). Thus, mutations altering the function of iab-5 reside outside of its initiator element, in a presumptive maintenance element.
Epistasis masks Abd-B alleles in D. santomea
To examine the contribution of Abd-B to the D. santomea phenotype on its own, we introduced the D. yakuba Abd-B allele into a mostly D. santomea genetic background by introgression. We backcrossed offspring from a D. yakuba/D. santomea hybrid cross for seven generations to D. santomea and mapped introgressed DNA breakpoints [2]. We identified one line that contained the D. yakuba Abd-B gene (Figure 1A, Figure S1K), and consistent with our reporter gene results, Abd-B was expressed in a D. yakuba-like pattern within this line (Figure 3A). Despite the dramatic increase in A5 Abd-B expression in these animals (c.f. Figure 3A and Figure 1D, E), they did not show increased A5 pigmentation (Figure 3D–F). The contrast between the absence of phenotypic effects of the D. yakuba Abd-B locus in a D. santomea background, as in this introgression line, compared with its effect together with other D. yakuba alleles, as in the reciprocal hemizygosity test, indicates that epistatic interactions mask the phenotypic effects of the D. yakuba Abd-B allele in a D. santomea background.
Figure 3. Epistatic interactions mask the phenotypic effects of a D. yakuba Abd-B allele in D. santomea.

(A-C) Expression patterns of Abd-B in introgression lines containing the D. yakuba Abd-B gene (A), pdm3 gene (B), or ebony gene (C) at mid-late pupal stages (~68 hAPF). Bottom panels show magnified images of the A5 regions outlined by red dashed lines. (D, E) Comparison of the Abd-B introgression phenotype (E) to wildtype D. santomea (D). (F) Quantification of A5 segment intensity of Abd-B introgression individuals compared to D. santomea (ns = not significant, Student’s t-test). See also Figure S3.
To identify genes that epistatically mask the effect of Abd-B, we examined the expression of additional genes in the pigmentation gene network—yellow, tan, and ebony—whose genomic locations are consistent with previously-identified QTLs (Figure 1A). yellow and tan promote dark melanic pigmentation, whereas ebony promotes yellow-colored cuticle, suppressing dark pigment formation [23, 24]. In D. yakuba, yellow and tan are expressed in posterior abdominal segments [16], while ebony transcripts accumulate only in anterior abdominal segments (Figure 4B–D). In D. yakuba IAB5Δ mutants, which display reduced A5 pigmentation (Figure 4G), yellow and tan expression were strongly reduced in A5 (Figure 4H, I), and ebony expression expanded into the A5 segment (Figure 4J). These changes in expression caused by the Abd-B allele mirror the differences we observe for these genes in D. santomea (Figure 4M–P). Thus, in D. yakuba, Abd-B directly or indirectly upregulates yellow and tan and downregulates ebony. In contrast, in the Abd-B introgression line, the expression of all three genes show a D. santomea pattern (Figure S3H, M and R), suggesting that these genes have become insensitive to AbdB. Below, we examine how changes throughout the pigmentation network of D. santomea caused genes in this network to function largely independently of Abd-B regulation.
Figure 4. An Abd-B-regulated network is differentially expressed in D. santomea.

(A-F) Expression patterns of yellow (B), tan (C), ebony (D), and Pdm3 (E) in D. yakuba (A), and summarized in (F). (G-L) Phenotype of an IAB5Δ homozygote (G), and corresponding expression of yellow (H), tan (I), ebony (J), and Pdm3 (K) are summarized in (L). Animal in panel (H) is heterozygous for IAB5Δ, and (I-K) are homozygotes. (M-R) Expression of yellow (N), tan (O), ebony (P), and Pdm3 (Q) in D. santomea are summarized in (R). See also Figure S4.
Enhanced suppression of pigmentation by Pdm3
The D. santomea abdomen shows strong reductions in yellow expression (Figure 4N) that can be attributed to changes in upstream factors [16]. However, the cis-regulatory changes we observed at Abd-B cannot fully explain the reduction in yellow expression, because the D. santomea A6 body segment expresses high levels of Abd-B protein (Figure 1E). Thus, we assessed yellow expression in our other introgression lines (Figure S3). We identified one introgression line that substantially increased abdominal pigmentation and yellow expression in A6 (Figure S3D, I) and included D. yakuba DNA from a region within the chromosome II QTL (Figure 1A). This introgression includes the POU domain transcription factor pdm3 (Figure S1N), which suppresses pigmentation in D. melanogaster [25] and is associated with natural variation in abdominal pigmentation in species of the Drosophila montium clade [26]. We could not identify other candidate genes within this introgression and therefore tested whether evolved changes at pdm3 contribute to the pigmentation difference between D. yakuba and D. santomea. We generated null alleles of pdm3 in D. yakuba and D. santomea by CRISPR/Cas9 genome editing to perform a reciprocal hemizygosity test (Figure S4A). Compared to the D. yakuba allele of pdm3, the D. santomea allele conferred reduced pigmentation in males mothered by D. santomea females (Figure 5A, Figure S4A–E) and in female reciprocal hemizygotes (Figure S4I–K). However, an effect was not observed in male progeny from crosses mothered by D. yakuba (Figure S4F–H). Because males are hemizygous for the X chromosome, the two classes of male progeny differ by which maternal X chromosome is present. We estimate that when the D. santomea pdm3 allele and the D. santomea X chromosome are present together, the A6 segment of males is almost two-fold lighter than expected under additivity (4.1 units, P <1e-4 by ANOVA).
Figure 5. Changes throughout the Abd-B regulated network contributed to the D. santomea phenotype.

(A-D) Reciprocal hemizygosity tests showing the intensity of the A6 segment in hemizygous progeny. (A) Test for pdm3 was carried out in males mothered by D. santomea. Tests for yellow (B) and tan (C) were performed using female progeny. (D) Test for ebony measured males of a background heterozygous for a fragment of the D. yakuba genome which contains ebony (“e intr”) in an otherwise D. santomea background. (*: p<0.05; ****: p<0.0001, Student’s t-test). See also Figure S5 and S6.
To localize the evolutionary changes at pdm3 that explain its phenotypic effects, we first examined the predicted protein sequence encoded by pdm3 in multiple strains of each species and found no amino-acid differences that were fixed (Table S1). We next compared the distribution of Pdm3 protein expression (Figure 4E, Q). In both species, Pdm3 is broadly expressed during early stages of pupal development (Figure S4N, O), but declines during later stages in D. yakuba (Figure S4Q, S and U). In D. santomea, Pdm3 expression in both A5 and A6 persists into late pupal development (Figure 4Q, Figure S4R, T and V). Although Pdm3 is highly expressed in segments that express Abd-B, its expression is unchanged in the Abd-B introgression line (Figure S4P). Thus, Pdm3 acts as a suppressor of pigmentation genes, including yellow, and has evolved increased A5 and A6 expression levels in D. santomea, contributing to its light-colored phenotype.
Reduced sensitivity of yellow and tan to Abd-B
Although changes at Abd-B and pdm3, when combined, should greatly reduce yellow expression, a QTL near the telomere of the X chromosome includes yellow (Figure 1A)[4]. We therefore investigated whether evolution of yellow also contributes to loss of pigmentation in D. santomea. We generated null alleles of yellow in both species for reciprocal hemizygosity testing (Figure S5A, C and E). Since yellow is located on the X chromosome, we could examine only reciprocal hemizygote females, which display weaker pigmentation than males. Nonetheless, these individuals showed a quantitative difference in pigmentation (Figure 5B, Figure S5F–G), revealing that evolution of yellow contributed to the loss of pigmentation in D. santomea. We found no fixed differences that would alter the amino acid sequence of yellow between the two species (Table S1). Furthermore, expression of yellow mRNA in reciprocal hemizygotes was correlated with the difference in pigmentation (Figure S5H, I), indicating that yellow function has evolved through cis-regulatory mutations. To localize function-altering cis-regulatory changes at yellow, we generated reporter constructs of its upstream regulatory region [27] from each species and compared their activities (Figure S6). In both males and females, the D. santomea reporters were expressed at much lower levels (Figure S6B–I), and the effect was most pronounced in the male A5 segment, where expression was reduced to background levels (Figure S6I). Similar results were obtained for a smaller construct containing the known abdominal enhancer of yellow (Figure S6A, S6J–O). These results indicate that the D. santomea abdominal enhancer of yellow has reduced activity and is apparently less responsive to positive regulation from Abd-B.
Previous studies strongly suggested that tan is the causative gene underlying a QTL of large effect on the X chromosome [16, 17] (Figure 1A). We confirmed the phenotypic contribution of tan by reciprocal hemizygosity testing (Figure 5C, S5J-P). The strength of pigmentation difference in the hemizygotes is similar to the effect of a small introgression line encompassing tan (Figure 1A and [17]). Our previous studies of tan also revealed that the D. santomea abdominal enhancer was inactivated [16, 17], leading to the loss of abdominal expression (Figure 4O). As this enhancer is downstream of Abd-B [28], changes at tan also likely contributed to epistatic interactions observed with Abd-B. Indeed, tan expression is not noticeably altered in introgression backgrounds containing D. yakuba Abd-B or pdm3 (Figure S3R, S).
A transposon insertion expands ebony expression
The gene ebony is located within the chromosome III QTL interval (Figure 1A), and its expression has expanded into the A5 and A6 tergites of D. santomea (Figure 4P). To determine whether evolution of ebony has contributed to the pigmentation difference, we identified a third chromosome introgression that includes ebony, but excludes Abd-B (Figure S1O). To test whether the phenotype of this introgression is caused by ebony itself, we performed reciprocal hemizygosity tests with null alleles (Figure S7A–C) generated by CRISPR/Cas9 targeted mutagenesis. Similar to our results with pdm3, we found epistatic interactions between ebony and the X chromosome (Figure S7), consistent with the fact that Ebony acts in the same biochemical pathway as Yellow and Tan, which are encoded by genes that reside on the X chromosome. To measure the phenotypic contribution of ebony to pigmentation in isolation, we generated an ebony null allele in the introgression line carrying the D. yakuba ebony allele. This new mutation allowed us to perform a reciprocal hemizygosity test in a background in which all other characterized loci affecting abdominal pigmentation carry D. santomea alleles. The reciprocal hemizygotes with a functional D. yakuba ebony allele produced darker A5 and A6 pigmentation than flies with the functional D. santomea ebony allele (Figure 5D and Figure S7D–F), confirming that evolution at ebony contributes to the evolved pigmentation patterns.
We next sought the molecular cause of the evolved change in ebony function. Comparisons of multiple strains from each species revealed three fixed substitutions that alter its predicted amino acid sequence (Data S1); only one change (A50S) is derived in D. santomea and it occurs within its nonribosomal peptide synthetase domain [29]. This amino acid change may alter Ebony function, but we have not yet investigated it further. We also found that the ebony introgression line showed reduced A6 expression of ebony (Figure S3O) mirroring the phenotype of this line, and recapitulating the expression pattern of the D. yakuba ebony gene contained within its introgressed segment (Figure 4D). We therefore tested whether changes in the cis-regulatory region of ebony can explain its altered expression pattern.
We assayed the genomic region that includes all known ebony regulatory elements [30] from both D. yakuba and D. santomea in transgenic reporter assays in D. melanogaster (Figure 6A). The D. yakuba and D. santomea constructs drove patterns of reporter protein expression that resembled the normal patterns of abdominal ebony expression in D. yakuba and D. santomea, respectively (Figure 6C, D). Chimeric reporter constructs localized the functional changes underlying this difference to the ebony upstream region (Figure S7N–Q).
Figure 6. A transposon insertion at ebony confers increased expression in the D. santomea abdomen.

(A) Map of the ebony locus indicating the position of a transcription-activating abdominal enhancer (“act”) and two repressive elements, “male rep” and the intronic “stripe rep” element. Schematic of ebony full regulatory GFP reporter construct (eFR-GFP), which contains the entire upstream and intronic regions of ebony. Red triangle indicates the position of a ~500 bp helitron transposon present in D. santomea. (B) Phylogeny showing a single origin of the D. santomea ebony helitron. Genomic helitrons represent top matches in the D. yakuba, D. santomea, and D. teissieri genomes. (C-E) GFP expression patterns of transgenic reporters in the posterior body segments A5 and A6 measured 6 hours post-eclosion. (C) D. yakuba eFR-GFP. (D) D. santomea eFR-GFP. (E) D. yakuba eFR+TE-GFP construct, in which the helitron element from D. santomea was precisely cloned into the D. yakuba 5’ regulatory region. Middle and bottom images show magnified regions outlined in the top images by red and blue dashed lines. See also Figure S7 and Data S2.
To identify candidate molecular changes, we sequenced the ebony 5’ regulatory region in 128 strains of D. santomea and 103 strains of D. yakuba (Data S2) and identified a 481 bp partial transposable element of the helitron class [31] in all strains of D. santomea (Figure 6A, B). By comparison, only two D. yakuba strains carried an insertion related to the D. santomea helitron element in this genomic position (Figure 6B, Data S2), and the phenotypic effects of this insertion may be masked in these two strains due to interactions with other pigmentation loci. This insertion is located in a known repressive regulatory element required to prevent ebony expression in the posterior segments of males (Figure 6A), suggesting that the helitron insertion may have disrupted the repressive regulatory element. To test this, we generated a reporter construct containing the D. yakuba ebony full regulatory region with the D. santomea helitron insertion. This construct drove strong expression in A5 and A6 (Figure 6E, Figure S7M), which confirms that this insertion contributed to the evolved increase in expression of the D. santomea ebony allele, rendering ebony independent from Abd-B regulation.
DISCUSSION
Results from evolutionary developmental biology have suggested that Hox genes drive the evolution of animal body plans [5–9, 32]. We have identified evolutionary modifications in one such Hox gene, Abd-B, that modified its expression pattern along the anterior-posterior axis in D. santomea. However, our results suggest that previous candidate gene studies have inflated the perceived contribution of Hox genes to body plan evolution. We found that evolutionary changes at Abd-B comprise only one of at least five genes that have evolved incrementally to modify the output of an entire regulatory network (Figure 7). Changes throughout this network underlie epistatic interactions with Abd-B. Our findings add to a growing list of examples that have implicated Hox genes as contributing to polygenic traits [10, 33, 34]. The polygenic architecture and epistatic interactions underlying this evolved phenotypic difference may have implications for other studies of phenotypic evolution, including studies at the macroevolutionary level.
Figure 7. Genes throughout a Hox-regulated network contribute to a major shift in phenotype.

The pigmentation network of D. yakuba is compared to D. santomea. Stars represent cis-regulatory changes that altered network topology. Long arrows or bars between genes show activating or repressing interactions, respectively. A solid line indicates a direct interaction. Dashed lines represent indirect or undetermined connections. Arrows after gene names indicate up or down regulation in D. santomea.
We cannot currently distinguish whether the changes at Abd-B were phenotypically visible when they initially appeared. If these mutations arose early during the evolution of the D. santomea phenotype, we expect that they would have caused dramatic phenotypic effects when they arose, as in our D. yakuba Abd-B mutants (Figure 4G, Figure S2B–D). Alternatively, the changes at Abd-B may have arisen later in this process, perhaps to make this phenotypic change more genetically robust [35]. In either case, the effects of these changes are now phenotypically silent in modern-day D. santomea. All of the genes we characterized are members of a single interconnected network; yellow, tan, and ebony are downstream of Abd-B in the ancestral network in D. yakuba (Figure 4H–J). The inactivation of Abd-B-responsive cisregulatory elements in all three genes masks the effects of Abd-B evolution in D. santomea. These observations suggest that the temporal order of fixation of alleles at different loci within this regulatory network would have altered the phenotypic visibility of these alleles as the lightly-colored abdomen of D. santomea evolved. Similar patterns of serial epistasis may mask the contributions that many genes make to the ultimate differences between species [36], leading scientists to overlook their historical role in generating phenotypes. Uncovering the topology of ancestral and derived networks can thus help predict such epistatic interactions [37].
Our observations motivate a revised microevolutionary model of body plan evolution, building upon Akam’s prescient hypothesis [38] of Hox-gene evolution, in which small changes in individual Hox genes accumulate to generate large differences in body plans. We envision that macroevolution of body plans results from changes in Hox gene expression patterns, together with changes throughout Hox-regulated networks. Over long periods of time, the combined effects of changes in multiple nodes of Hox regulated networks may generate the dramatic differences in body plans that we now observe across animals. Changes to the structure of these networks will govern epistatic interactions, and our resulting ability to detect the phenotypic consequences of individual genes. Given sufficiently long expanses of time, we anticipate that most genes within a single regulatory network may evolve to contribute to differences in network function. Hence, we should seek the causes of body plan evolution not just in Hox genes themselves, but in changes throughout Hox-regulated developmental networks.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mark Rebeiz (rebeiz@pitt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila strains and culture
Stocks were maintained at room temperature on standard cornmeal agar media. D. yakuba Ivory Coast (14021–0261.00) and D. santomea STO.4 (14021–0271.00) were obtained from Drosophila Species Stock Center (http://blogs.cornell.edu/drosophila/). D. santomea STO CAGO1482 was obtained from the Andolfatto lab. A D. melanogaster yellow white (yw) strain that was isogenized for eight generations was used for crosses to normalize the backgrounds of GFP reporter transgenes. A total of 103 D. yakuba and 128 D. santomea iso-female lines were used to test for the presence of the ~500 bp transposable element insertion in the D. santomea ebony 5’ region by PCR screening (Data S2).
METHOD DETAILS
Generation and mapping of introgression lines
D. yakuba strain Ivory Coast and D. santomea strain STO.4 were crossed and fertile hybrid females were backcrossed to males of D. santomea for 7 generations. During the process of backcrossing, we identified individual males with distinctive abdominal pigmentation patterns and crossed these to D. santomea females. Offspring of these crosses were inbred in an attempt to generate recombinant inbred strains. Once these strains produced similar phenotypes in all male progeny, DNA was prepared from individual males and genome-wide ancestry estimates were generated using multiplexed shotgun genotyping [2]. The pdm3 introgression was generated by crossing the YFP-marked D. yakuba strain #1694 [18] to STO.4 for several generations and selecting animals with the YFP marker. The D. yakuba genomic fragment introgressed in this line is marked by a YFP marker [39] that is inserted into a large intron of the pdm3 gene itself. Because this may affect pdm3 function, we restricted the analyses of pdm3 to pure D. yakuba and D. santomea specimens.
in situ hybridization
in situ hybridization was performed as described [16] with small modifications. In brief, following puparium formation, samples were aged to differing extents for each probe, dissected in cold PBS, and fixed in PBS containing 4% paraformaldehyde (E.M.S. Scientific) and 0.1% Triton X-100. PCR was performed to generate RNA probe templates that had a T7 promoter appended through primer design. See Table S2 for the list of probe primers. Digoxigenin-labeled probes were generated using a 10X Dig labeling mix (Roche Diagnostics) and T7 RNA polymerase (Promega). Dissected samples were probed using an in situ hybridization robot (Intavis). Because D. yakuba and D. santomea are very closely related, spatial differences in pigmentation gene expression could reliably be detected with the same probe made from one species. To ensure the maximal ability to detect expression, we used probes from the species predicted to have the lowest expression level. Therefore, the yellow probe was made from D. santomea, while the ebony probe was generated from D. yakuba. To compare the expression patterns and levels among D. yakuba, D. santomea and the introgression lines, samples were treated with the same concentration of the probe under identical experimental procedures. For GFP in situ hybridization, the probe template was amplified from our transgenic S3aG vector. Samples were timed at different stages for different genes. For D. yakuba, D. santomea, introgression lines, D. yakuba IAB5Δ CRISPR and reciprocal hemizygotes, yellow in situ abdominal samples were dissected at mid-late pupal stages, which is 72–78 hours after pupal formation (hAPF). tan in situ samples were prepared at late pupal stages (90–94 hAPF). For ebony, samples were collected within half an hour after eclosion. For GFP in situ experiments, samples were from late pupal stages (93–95 hAPF).
GFP transgenic reporter assays
D. yakuba and D. santomea ebony and the 1 kb initiator element IAB5 reporter constructs were PCR amplified and cloned by enzyme digestion into the S3AG vector [40], which contains a basal promoter driving GFP, flanked by SF1 and gypsy insulators. The full iab-5 reporter constructs were generated by the assembly of multiple fragments using the Infusion cloning system (Clontech), inserting between Asc I and Sbf I sites upstream of GFP. See Tables S3 and S4 for primers used to clone regulatory constructs. Primers were designed using the GenePalette Software tool [41, 42]. Restriction sites (Asc I and Sbf I) were appended to primers (Integrated DNA Technologies) for insertion into the S3AG multicloning site. D. melanogaster transformant lines were generated by phiC31 mediated site specific recombination into the 51D insertion site on the second chromosome [43].
Of note, in the case of yellow reporter constructs, previous studies using transgenic reporter constructs found no evidence that the yellow abdominal cis-regulatory element had diverged between these species [14]. However, those experiments employed randomly inserted P-element transgenes, which have limited power to detect small differences in activity. We therefore cloned both the body element and wing+body elements of D. yakuba and D. santomea yellow into GFP reporter constructs (Figure S6A), and inserted these into a common landing site 51D on the second chromosome. The larger w+b element reporter contains regulatory sequences necessary for activity in both the abdomen, and wing [27], and generally drives a much closer recapitulation of abdominal expression than the body element alone.
As for ebony reporter constructs, the ebony regulatory region contains one activating abdominal enhancer and two repressive silencer elements [30]. The abdominal enhancer (Figure 6A, “act”) drives expression throughout the abdomen, even in locations that normally lack ebony expression. A promoter-proximal element (Figure 6A, “male rep”) is required to prevent ebony expression in the posterior segments of males. A silencer located within the large first intron of ebony (Figure 6A, “stripe rep”) is required to prevent expression from the posterior edges of tergites. Therefore, tests of ebony regulatory sequence function were carried out using constructs that place its 5’ regulatory region upstream of GFP and its large intron downstream of GFP (Figure 6A). Insertion of the ebony intron downstream of GFP was performed using the Infusion cloning kit (Clontech), placing intron sequences into a Spe I site downstream of GFP.
In the case of iab-5 reporter constructs, transgenes were also inserted into the attP2 site on the third chromosome [44].
Transgenic animals were mounted on slides in halocarbon oil and imaged on an Olympus Fluoview 1000 confocal microscope. For IAB5 and iab-5 reporter constructs, flies were mounted at late pupal stages (93–95 hAPF) in halocarbon oil 27 and imaged, or dissected for in situ hybridization to detect GFP mRNA. For yellow reporter constructs, flies were mounted at 91 hAPF in halocarbon oil 27 and imaged. For ebony reporters, samples were aged for 6 hours after eclosion before mounting in halocarbon oil 700 and imaging. Samples were imaged with standard settings in which the brightest samples were not saturated.
Imaging of GFP reporters in embryos
D. yakuba and D. santomea IAB5 and iab-5 transgenic GFP reporter embryos were collected at stage 11 from grape agar plates, treated with fresh 50% bleach for 3 minutes, followed by rinsing with deionized H2O containing 0.1% Triton. Embryos were then transferred into a mixture of 3mL heptane and 3mL PBS fix (PBS fix: PBS containing 4% paraformaldehyde (E.M.S. Scientific)) and were rotated on an orbital shaker set to 200–250 rpm at room temperature for 20 minutes. To remove the vitelline membranes, the bottom layer (PBS fix) was removed and 3mL of methanol was added, followed by vigorous shaking by hand for 1 minute. The devitellinized embryos were transferred into a 1.5 mL Eppendorf tube and the supernatant was removed and replaced with PBT (PBS containing 0.1% Triton). Methanol exposure was minimized during the process to reduce its negative effects on GFP. The embryos were suspended in glycerol mounting solution (80% glycerol, 0.1M Tris, pH 8.0) and mounted on slides. Imaging was performed with an Olympus Fluoview 1000 confocal microscope.
Survey of amino acid changing differences in D. yakuba and D. santomea populations
The D. yakuba strains used for the amino acid polymorphism and divergence analysis in candidate genes include twenty partially inbred strains from Rogers et al. [45], and ten synthetic diploid individuals derived from pairwise crosses of twenty isofemale strains (unpublished), and the D. santomea strains include sixteen synthetic diploid individuals derived from pairwise crosses of thirty-six isofemale strains (unpublished). All synthetic diploid individuals were pair-end 150nt sequenced on an Illumina HiSeq to >30X depth coverage. All sequences were mapped to their respective reference genomes with BWA-MEM[46]: D. santomea STOCAGO 1482 and D. yakuba NY73PB (unpublished). SNPs were called and filtered using a custom pipeline (https://github.com/YourePrettyGood/PseudoreferencePipeline) using SAMtools [47] for variant calling, producing an updated and masked FASTA sequence for each individual. Genotype calls for the partially inbred D. yakuba strains were collapsed into a single haplotype for all inbred regions. For each gene of interest, the longest protein isoform that encompasses all the exons of that gene was identified if possible, and the corresponding coding sequences were extracted. In the case of Abd-B, no single isoform encompasses all the exons, so two isoforms that in unison include all the exons were analyzed. Coding sequences extracted from D. teissieri GT53w reference genome (unpublished) were used to identify the ancestral state at divergent sites (Data S1, Table S1). The extracted coding sequences were translated into protein sequences using EMBOSS transeq [48], and if necessary, the protein sequences of the three species were aligned to each other using a combination of PRANK [49] and manual alignment. Polymorphic and divergent sites between D. santomea and D. yakuba were identified and polarized with the D. teissieri state using a custom Python script.
Generation of CRISPR/Cas9 mutants
The null mutations of ebony and yellow were generated by CRISPR/Cas9-mediated nonhomologous end joining using gRNA targets indicated in the Table S5. Embryos of D. yakuba and D. santomea were injected by Rainbow Transgenics with a mixture of 100 ng/μL in vitro transcribed Cas9 mRNA and 200 ng/μL in vitro transcribed gRNA. In vitro transcribed Cas9 mRNA was generated using a plasmid containing a T7 RNA polymerase promoter driving a Drosophila codon-optimized Cas9 (T7::Cas9). The plasmid was linearized 3’ of the Cas9 gene and RNA was generated using the mMESSAGE mMACHINE T7 Transcription Kit (Invitrogen). In vitro transcribed gRNAs were generated for all experiments by first performing PCR to generate a dsDNA template including a T7 promoter and gRNAs were then generated by performing in vitro transcription using the MEGAscript T7 Transcription kit (Invitrogen). Mutated alleles of ebony and yellow were identified by screening offspring of injected animals for mutant phenotypes. Mutations were confirmed by PCR and sequencing of targeted regions.
The IAB5Δ, pdm3, and tan mutations were induced by CRISPR/Cas9 mediated homologous recombination [50]. Targeting plasmids for each locus, constructed using Gibson assembly [51], contained approximately 1kb of genomic DNA from the targeted locus (homology arms) flanking a 3XP3::DsRed cassette. Alleles containing successful homologous recombination events were identified by the expression of DsRed in the eyes of the progeny of injected animals. In the case of pdm3, expression was not detected in the eyes, but instead was detected in neurons in the neck. It is possible that regulatory enhancers in pdm3 suppress the ability of the 3XP3 promoter to drive strong eye expression, but instead drive expression in a limited number of neurons that are visible in the neck. Integration into the correct genomic location for each locus was confirmed by PCR and sequencing. The guide RNA sequences and genomic targets for homology directed repair construct are listed in Tables S5 and S6, respectively.
Reciprocal hemizygosity tests
Following the scheme that is illustrated in Figure 2B, the reciprocal hemizygosity test compares the progeny of two separate crosses. The first group includes offspring from D. yakuba wildtype crossed to D. santomea CRISPR mutants (Figure 2B, left panel). The second group includes offspring from D. santomea wildtype crossed to D. yakuba CRISPR mutants (Figure 2B, right panel). For tan, IAB5 and yellow, we performed D. yakuba-mothered crosses, which readily generate offspring. For pdm3 and ebony, to decrease the strong influence of the D. yakuba X chromosome (containing yellow and tan), we performed D. santomea-mothered crosses. For ebony, we also performed a test using mutant introgression lines, which provides a more sensitive background to detect phenotypic differences among progeny. Instead of using D. yakuba as a parent, we used the ebony introgression line that contains a D. yakuba copy of the ebony gene.
Imaging of adult Drosophila abdomens
The eclosed progeny from the reciprocal hemizygosity test (RHT) crosses were directly transferred into new vials roughly every seven days. After culturing at room temperature for specific time periods, flies were selected on a CO2 pad and mounted for imaging. For IAB5 RHTs, samples were images after incubation for 7–11 days. pdm3 D. santomea maternal RHT samples were imaged at 7–8 days. pdm3 D. yakuba maternal RHT flies were imaged at 6–16 days. ebony introgression RHT samples were imaged at 7–9 days. ebony RHT samples were aged 6–16 days. For tan and yellow RHTs, samples were imaged at 7–8 days. The parental stocks carrying mutations in ebony, yellow and tan are homozygous viable, thus all progeny from reciprocal crosses could be phenotyped. The IAB5 and pdm3 mutant alleles were marked by 3xP3::DsRed (RFP) and these stocks were maintained in the heterozygous state. For reciprocal hemizygosity test crosses using these lines, only offspring expressing DsRed were imaged. Abdomens were mounted on slides covered with double-sided sticky tape and imaged using a Leica M205C Stereo Microscope with a DFC425C camera. The Leica Montage package was used to generate an extended focus brightfield image.
Immunohistochemistry
Immunohistochemistry of the pupal abdominal epidermis was performed as previously described [52]. For Abd-B, flies were dissected in cold PBS and fixed in PBS containing 4% paraformaldehyde (E.M.S. Scientific) and 0.1% Triton-X-100 (PBT-fix) at mid-late pupal stages (which were 63–66 hAPF for D. yakuba, D. yakuba IAB5 CRISPR lines and IAB5 reciprocal hemizygotes and 68–71 hAPF for D. santomea and introgression lines respectively) since D. yakuba and D. santomea develop at different rates. Abdominal, larval or embryonic samples were incubated at 4°C in 1:100 diluted primary antibody (Developmental Studies Hybridoma Bank #1A2E9) in PBS containing 0.1% Triton-X-100 (PBT) overnight, followed by three 15-minute PBT washes at room temperature. The samples then were transferred into 1:500 diluted secondary antibody (Alexa Fluor 488 donkey anti-mouse IgG, Invitrogen Thermo Fisher Scientific Life Technologies, stock concentration 2mg/mL) in PBT and incubated in room temperature for 2.5h, followed by three washes in PBT, each for 15 minutes. Finally, the samples were mounted in glycerol mounting solution (80% glycerol, 0.1M Tris, pH 8.0) and imaged.
For the Abd-B immunostainings of the IAB5 reciprocal hemizygosity (RH) cross offspring, the parental homozygous viable CRISPR lines are difficult to obtain and the fluorescence during pupal stages is difficult to assess. We therefore screened for animals carrying the IAB5 deletion based on the fluorescence of the 3xP3::DsRed cassette in the larval nervous system. Fluorescent larvae were allowed to grow on grape agar plates and were aged to mid-late pupal stages (65 hAPF) upon the formation of white pre-pupa to ensure the proper staging of samples.
For stainings involving Pdm3, we dissected samples at several different mid-late (65–66 hAPF in Figure 4E, K, Q and Figure S4N–P, 69–72 hAPF in Figure S4Q, R) and late pupal stages (72–75 hAPF in Figure S4S,T and 84–90 hAPF in Figure S4U–V) and incubated the samples in the primary Guinea pig anti-Pdm3 antibody [53] at a 1:100 dilution at 4°C for 36h (instead of overnight). The secondary antibody (Alexa Fluor 594 goat anti-guinea pig IgG antibody from Invitrogen Thermo Fisher Scientific Life Technologies, stock concentration 2mg/mL) was used at 1:500 dilution and incubated for 6h before washing.
For abd-A, samples were prepared at mid-late pupal stages (~68 hAPF). We used a Guinea Pig anti-abd-A antibody[54] at a 1:500 dilution, detected by Alexa Fluor 647 anti-guinea pig IgG (H+L) antibody from Life Technologies (#A-21450) at 1:500 dilution.
For Ubx, samples were collected at mid-late pupal stages (~68 hAPF). We used a mouse antiUbx 5’ exon antibody (DSHB # FP3.38) at a 1:100 dilution at 4°C overnight, followed by 1:500 diluted Alexa Fluor 488 donkey anti-mouse IgG (Invitrogen Thermo Fisher Scientific Life Technologies, stock concentration 2mg/mL).
Analysis of helitron sequences at ebony
Thirty-one D. santomea F1 synthetic diploid flies were generated by crossing two unrelated isofemale lines. DNA was extracted from single female flies and the ebony helitron region was isolated through PCR, barcoded, and sequenced using PacBio. Consensus haplotypes were generated using SMRT Analysis protocol RS_Long_Amplicon_Analysis.1, and the top two haplotypes in terms of haplotype length and depth of coverage were kept for each diploid individual. After filtering for full-length consensus haplotypes with high depth of coverage, fifty-eight sequences were included in the analysis as the D. santomea ebony helitron sequences. The D. yakuba ebony helitron sequences for cy17C and cy04B were generated by sequencing a PCR product that covers the ebony helitron region. Genomic DNA was extracted from single male flies for cy17C and cy04B. Forward (GAGAACATTGTTGCCGACAAGC) and reverse (TGCCAGCCGTCATGTTGTGCTTC) primers were used for amplifying the PCR product. The sequencing primer is CTCAATGTGGTCCCATTTGCATTCG. To identify the genomic helitrons in D. santomea, D. yakuba, and D. teissieri, the repeat name, class, and subclass of the ebony helitron (DNAREP1_DYak, RC, and Helitron respectively) were identified from a RepeatMasker [55] run on the D. santomea reference genome STOCAGO 1482 (unpublished), and the consensus sequence for this helitron was extracted from Repbase [56]. The Repbase consensus sequence was then queried in the D. santomea reference, D. yakuba reference genome NY73PB (unpublished), and D. teissieri reference genome GT53w (unpublished) using BLAST 2.4.0 [57]. For D. santomea and D. yakuba, the top three hits from each of the five major chromosome arms were included as genomic helitrons, and for D. teissieri the top hit from each of the top fifteen scaffolds were included. All sequences were then aligned using MUSCLE 3.8.31 [58], and the phylogenetic tree was generated using the neighbor joining algorithm excluding columns from the alignment with any missing data using the R package APE [59].
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of relative fluorescent intensity
Relative fluorescent intensity was measured using a method previously described [16, 30] with minor modifications.
For ebony reporters, the light intensity (L) of the A5 or A6 segment was measured as the mean value of hemisegments outlined by the free hand selection tool in Adobe Photoshop. The background intensity (B) of A5 or A6 was measured as the average value of the intensities from three different square regions from the midline of the corresponding body segment that lacked epithelial cell or bristle fluorescence. The absolute light intensity (AL) of each segment was obtained from L − B. The relative percentage of fluorescent intensity of each segment was calculated as:
For quantifying the fluorescent intensity of the yellow reporter in fly abdomens, the absolute light intensity (AL) was calculated by measuring the light intensity (L) of A5 or A6 segments and subtracting the background intensity (B) from A4 from each sample. The relative percentage of fluorescent intensity of each segment was measured as:
Quantifications of Drosophila abdominal pigmentation
Average intensity of segment pigmentation average intensity
Images were converted into grayscale in Adobe Photoshop. The light value (L) was recorded as the mean value on a 0–255 scale, using the free hand selection of the segment of interest. The segment intensity was calculated as follows:
Quantification of segment pigmentation area percentage
The pigmentation area value (P) and the whole segment area value (W) were recorded in Image J by outlining the pigmentation area and the whole segment area of A5 segment, using the free hand selection tool. The segment pigmentation area percentage was calculated as follows:
A two-tailed Student’s t-test was used when comparing two sets of quantitative data. The box plots in the figures were drawn using the boxplot server: http://shiny.chemgrid.org/boxplotr/. Data points were plotted as jittered plots. Center lines represent the medians. Box borders show the 25th and 75th percentiles (Q1 and Q3) as determined in R. Whiskers extend 1.5 times the interquartile range = Q3-Q1. Data points are shown as open circles.
Epistasis was quantified using a standard ANOVA model implemented in R that includes an interaction term between allelic states at pdm3 or ebony and the parent of origin of the X chromosome.
DATA AND SOFTWARE AVAILABILITY
Gene diagrams and primer design
GenePalette software [41, 42] was used for primer design, and visualization and manipulation of genomic sequences is available at http://www.genepalette.org/
Supplementary Material
Data S1. Ebony protein variants among D. yakuba and D. santomea lines. Related to STAR Methods. Red color indicates D. yakuba lines. Black color shows D. santomea lines. The sequence of D. teissieri (D. tei) is used as the inferred ancestral state.
Data S2. D. yakuba and D. santomea lines used for ebony TE insertion screening. Related to Figure 6.
* indicates the presence of a transposable element insertion related to the helitron element fixed in D. santomea.
Expression changes in a Hox gene correlate with loss of pigmentation in D. santomea
Restoring Hox expression to D. santomea does not restore pigmentation phenotype
Changes throughout a downstream Hox-regulated network mask effects of Hox evolution
Hox-regulated traits evolve by small steps distributed throughout entire networks
Acknowledgments
We would like to recognize the kindness and generosity of Sean B. Carroll in whose lab part of this work was started. We thank Cheng-Ting Chien, Brian Gebelein, and the DSHB for the generous provision of antibodies used in this work, Daniel Matute for providing D. yakuba, D. santomea, and D. teissieri lines, and Jessica Cande for sharing an unpublished introgression line. This research was supported by funding from the National Institutes of Health (GM114093 to MR and PA), the National Science Foundation (IOS-1555906 to TMW and MR).
Footnotes
Declaration of Interests: Authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carroll SB (1995). Homeotic genes and the evolution of arthropods and chordates. Nature 376, 479–485. [DOI] [PubMed] [Google Scholar]
- 2.Andolfatto P, Davison D, Erezyilmaz D, Hu TT, Mast J, Sunayama-Morita T, and Stern DL (2011). Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res. 21, 610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gellon G, and McGinnis W (1998). Shaping animal body plans in development and evolution by modulation of Hox expression patterns. BioEssays 20, 116–125. [DOI] [PubMed] [Google Scholar]
- 4.Carbone MA, Llopart A, DeAngelis M, Coyne JA, and Mackay TFC (2005). Quantitative trait loci affecting the difference in pigmentation between Drosophila yakuba and D. santomea. Genetics 171, 211–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Averof M, and Patel NH (1997). Crustacean appendage evolution associated with changes in Hox gene expression. Nature 388, 682–686. [DOI] [PubMed] [Google Scholar]
- 6.Khila A, Abouheif E, and Rowe L (2009). Evolution of a novel appendage ground plan in water striders is driven by changes in the Hox gene Ultrabithorax. PLoS Genet. 5, e1000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahfooz NS, Li H, and Popadic A (2004). Differential expression patterns of the hox gene are associated with differential growth of insect hind legs. Proc. Natl. Acad. Sci 101, 4877–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogers BT, Peterson MD, and Kaufman TC (1997). Evolution of the insect body plan as revealed by the Sex combs reduced expression pattern. Development 124, 149–157. [DOI] [PubMed] [Google Scholar]
- 9.Burke AC, Nelson CE, Morgan BA, and Tabin C (1995). Hox genes and the evolution of vertebrate axial morphology. Development 121, 333–346. [DOI] [PubMed] [Google Scholar]
- 10.Stern DL (1998). A role of Ultrabithorax in morphological differences between Drosophila species. Nature 396, 463–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavlopoulos A, Kontarakis Z, Liubicich DM, Serano JM, Akam M, Patel NH, and Averof M (2009). Probing the evolution of appendage specialization by Hox gene misexpression in an emerging model crustacean. Proc. Natl. Acad. Sci. U. S. A 106, 13897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibson G, and Hogness DS (1996). Effect of polymorphism in the Drosophila regulatory gene Ultrabithorax on homeotic stability. Science 271, 200–3. [DOI] [PubMed] [Google Scholar]
- 13.Mackay TFC, Stone EA, and Ayroles JF (2009). The genetics of quantitative traits: challenges and prospects. Nat. Rev. Genet 10, 565–577. [DOI] [PubMed] [Google Scholar]
- 14.Jeong S, Rokas A, and Carroll SB (2006). Regulation of body pigmentation by the Abdominal-B Hox protein and its gain and loss in Drosophila evolution. Cell 125, 1387–1399. [DOI] [PubMed] [Google Scholar]
- 15.Sánchez-Herrero E, Vernós I, Marco R, and Morata G (1985). Genetic organization of Drosophila bithorax complex. Nature 313, 108–13. [DOI] [PubMed] [Google Scholar]
- 16.Jeong S, Rebeiz M, Andolfatto P, Werner T, True J, and Carroll SBB (2008). The evolution of gene regulation underlies a morphological difference between two Drosophila sister species. Cell 132, 783–793. [DOI] [PubMed] [Google Scholar]
- 17.Rebeiz M, Ramos-Womack M, Jeong S, Andolfatto P, Werner T, True J, Stern DLL, and Carroll SBB (2009). Evolution of the tan locus contributed to pigment loss in Drosophila santomea: a response to Matute et al et al. Cell 139, 1189–1196. [DOI] [PubMed] [Google Scholar]
- 18.Stern DL (2014). Identification of loci that cause phenotypic variation in diverse species with the reciprocal hemizygosity test. Trends Genet. 30, 547–54. [DOI] [PubMed] [Google Scholar]
- 19.Busturia A, and Bienz M (1993). Silencers in abdominal-B, a homeotic Drosophila gene. EMBO J. 12, 1415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horn C, Jaunich B, and Wimmer EA (2000). Highly sensitive, fluorescent transformation marker for Drosophila transgenesis. Dev. Genes Evol 210, 623–9. [DOI] [PubMed] [Google Scholar]
- 21.Gratz SJ, Ukken FP, Rubinstein CD, Thiede G, Donohue LK, Cummings AM, and O’Connor-Giles KM (2014). Highly Specific and Efficient CRISPR/Cas9-Catalyzed Homology-Directed Repair in Drosophila. Genetics 196, 961–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mihaly J, Barges S, Sipos L, Maeda R, Cléard F, Hogga I, Bender W, Gyurkovics H, and Karch F (2006). Dissecting the regulatory landscape of the Abd-B gene of the bithorax complex. Development 133, 2983–2993. [DOI] [PubMed] [Google Scholar]
- 23.Wittkopp PJ, True JR, and Carroll SB (2002). Reciprocal functions of the Drosophila yellow and ebony proteins in the development and evolution of pigment patterns. Development 129, 1849–1858. [DOI] [PubMed] [Google Scholar]
- 24.True JR, Yeh S-D, Hovemann BT, Kemme T, Meinertzhagen IA, Edwards TN, Liou S-R, Han Q, and Li J (2005). Drosophila tan Encodes a Novel Hydrolase Required in Pigmentation and Vision. PLoS Genet. 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogers WA, Grover S, Stringer SJSJ, Parks J, Rebeiz M, and Williams TMTM (2014). A survey of the trans-regulatory landscape for Drosophila melanogaster abdominal pigmentation. Dev. Biol 385, 417–32. [DOI] [PubMed] [Google Scholar]
- 26.Yassin A, Delaney EK, Reddiex AJ, Seher TD, Bastide H, Appleton NC, Lack JB, David JR, Chenoweth SF, Pool JE, et al. (2016). The pdm3 Locus Is a Hotspot for Recurrent Evolution of Female-Limited Color Dimorphism in Drosophila. Curr. Biol 26, 2412–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wittkopp PJ, Vaccaro K, and Carroll SB (2002). Evolution of yellow gene regulation and pigmentation in Drosophila. Curr Biol 12, 1547–1556. [DOI] [PubMed] [Google Scholar]
- 28.Camino EMEM, Butts JCJC, Ordway A, Vellky JEJE, Rebeiz M, and Williams TMTM (2015). The Evolutionary Origination and Diversification of a Dimorphic Gene Regulatory Network through Parallel Innovations in cis and trans. PLoS Genet. 11, e1005136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richardt A, Kemme T, Wagner S, Schwarzer D, Marahiel MA, and Hovemann BT (2003). Ebony, a novel nonribosomal peptide synthetase for beta-alanine conjugation with biogenic amines in Drosophila. J Biol Chem 278, 41160–41166. [DOI] [PubMed] [Google Scholar]
- 30.Rebeiz M, Pool JE, Kassner VA, Aquadro CF, and Carroll SB (2009). Stepwise modification of a modular enhancer underlies adaptation in a Drosophila population. Science (80-.). 326, 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kapitonov VV, and Jurka J (2007). Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet. 23, 521–529. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka K, Barmina O, Sanders LE, Arbeitman MN, and Kopp A (2011). Evolution of Sex-Specific Traits through Changes in HOX-Dependent doublesex Expression. PLoS Biol. 9, e1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Signor SA, Liu Y, Rebeiz M, and Kopp A (2016). Genetic Convergence in the Evolution of Male-Specific Color Patterns in Drosophila. Curr. Biol 26. [DOI] [PubMed] [Google Scholar]
- 34.Tian L, Rahman SR, Ezray BD, Franzini L, Strange JP, Lhomme P, and Hines HM (2019). A homeotic shift late in development drives mimetic color variation in a bumble bee. Proc. Natl. Acad. Sci. U. S. A, 201900365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Budd GE (1999). Does evolution in body patterning genes drive morphological change-or vice versa? BioEssays 21, 326–332. 6%3A%3AAID-BIES9%3E3.0.CO%3B2–0 [Accessed April 8, 2018]. [Google Scholar]
- 36.Stern DL (2010). Evolution, Development, and the Predictable Genome 1st ed. (W. H. Freeman; ). [Google Scholar]
- 37.Phillips PC (2008). Epistasis--the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet 9, 855–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akam M (1998). Hox genes, homeosis and the evolution of segment identity: no need for hopeless monsters. Int. J. Dev. Biol 42, 445–51. [PubMed] [Google Scholar]
- 39.Stern DL, Crocker J, Ding Y, Frankel N, Kappes G, Kim E, Kuzmickas R, Lemire A, Mast JD, and Picard S (2017). Genetic and Transgenic Reagents for Drosophila simulans, D. mauritiana, D. yakuba, D. santomea, and D. virilis. G3:Genes|Genomes|Genetics 7, 1339–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams TM, Selegue JE, Werner T, Gompel N, Kopp A, and Carroll SB (2008). The regulation and evolution of a genetic switch controlling sexually dimorphic traits in Drosophila. Cell 134, 610–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rebeiz M, and Posakony JW (2004). GenePalette: a universal software tool for genome sequence visualization and analysis. Dev Biol 271, 431–438. [DOI] [PubMed] [Google Scholar]
- 42.Smith AF, Posakony JW, and Rebeiz M (2017). Automated tools for comparative sequence analysis of genic regions using the GenePalette application. Dev. Biol 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bischof J, Maeda RK, Hediger M, Karch F, and Basler K (2007). An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A 104, 3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Groth AC, Fish M, Nusse R, and Calos MP (2004). Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics 166, 1775–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rogers RL, Cridland JM, Shao L, Hu TT, Andolfatto P, and Thornton KR (2014). Landscape of standing variation for tandem duplications in Drosophila yakuba and Drosophila simulans. Mol. Biol. Evol 31, 1750–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv, 1303.3997. [Google Scholar]
- 47.Li H (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rice P, Longden I, and Bleasby A (2000). EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–7. [DOI] [PubMed] [Google Scholar]
- 49.Löytynoja A (2014). Phylogeny-aware alignment with PRANK. In Methods in molecular biology (Clifton, N.J.), pp. 155–170. Available at: http://www.ncbi.nlm.nih.gov/pubmed/24170401 [DOI] [PubMed]
- 50.Yu Z, Ren M, Wang Z, Zhang B, Rong YS, Jiao R, and Gao G (2013). Highly Efficient Genome Modifications Mediated by CRISPR/Cas9 in Drosophila. Genetics 195, 289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–5. [DOI] [PubMed] [Google Scholar]
- 52.Gompel N, and Carroll SB (2003). Genetic mechanisms and constraints governing the evolution of correlated traits in drosophilid flies. Nature 424, 931–935. [DOI] [PubMed] [Google Scholar]
- 53.Chen C-K, Chen W-Y, and Chien C-T (2012). The POU-domain protein Pdm3 regulates axonal targeting of R neurons in the Drosophila ellipsoid body. Dev. Neurobiol 72, 1422–32. [DOI] [PubMed] [Google Scholar]
- 54.Li-Kroeger D, Witt LM, Grimes HL, Cook TA, and Gebelein B (2008). Hox and Senseless Antagonism Functions as a Molecular Switch to Regulate EGF Secretion in the Drosophila PNS. Dev. Cell 15, 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smit A, Hubley R, and Green P (2017). RepeatMasker Open-4.0.6 2013–2015. http://www.repeatmasker.org.
- 56.Jurka J (1998). Repeats in genomic DNA: mining and meaning. Curr. Opin. Struct. Biol 8, 333–7. [DOI] [PubMed] [Google Scholar]
- 57.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, and Lipman DJ (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paradis E, Claude J, and Strimmer K (2004). APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20, 289–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Ebony protein variants among D. yakuba and D. santomea lines. Related to STAR Methods. Red color indicates D. yakuba lines. Black color shows D. santomea lines. The sequence of D. teissieri (D. tei) is used as the inferred ancestral state.
Data S2. D. yakuba and D. santomea lines used for ebony TE insertion screening. Related to Figure 6.
* indicates the presence of a transposable element insertion related to the helitron element fixed in D. santomea.
Data Availability Statement
Gene diagrams and primer design
GenePalette software [41, 42] was used for primer design, and visualization and manipulation of genomic sequences is available at http://www.genepalette.org/