

Abstract
Protozoans of the genus Cryptosporidium are the causative agent of the gastrointestinal disease, cryptosporidiosis, which can be fatal in immunocompromised individuals. Cryptosporidium hominis (C. hominis) bifunctional thymidylate synthase-dihydrofolate reductase (TS-DHFR) is an essential enzyme in the folate biosynthesis pathway and a molecular target for inhibitor design. Previous studies have demonstrated the importance of the ChTS-DHFR linker region “crossover helix” to the enzymatic activity and stability of the ChDHFR domain. We conducted a virtual screen of a novel non-active site pocket located at the interface of the ChDHFR domain and crossover helix. From this screen we have identified and characterized a noncompetitive inhibitor, compound 15, a substituted diphenyl thiourea. Through subsequent structure activity relationship studies, we have identified a time-dependent inhibitor lead, compound 15D17, a thiol-substituted 2-hydroxy-N-phenylbenzamide, which is selective for ChTS-DHFR, and whose effects appear to be mediated by covalent bond formation with a non-catalytic cysteine residue adjacent to the non-active site pocket.
Keywords: Cryptosporidium hominis, DHFR, Non-active site, Allosteric, Virtual screen, Glide
Graphical Abstract
Cryptosporidiosis is a gastrointestinal illness, which in humans is primarily caused by two Cryptosporidium parasites, Cryptosporidium hominis (C. hominis) and Cryptosporidium parvum (C. parvum).1 Cryptosporidium is capable of surviving without a host for many months and at a wide range of temperatures, and are well known to infect large groups of people when food or water supplies become contaminated.2, 3 Healthy individuals experience gastrointestinal distress that can last for two or more weeks, while infection rates are higher and more severe in children, the elderly, and especially in immunocompromised individuals.4, 5 For example, people with HIV/AIDS are more likely to develop severe symptoms, including persistent, debilitating and possibly fatal diarrhea and wasting.6 In addition, a major impact of cryptosporidiosis is also felt in developing countries, where it is increasingly recognized as a contributor to the morbidity and mortality of children.2 Currently, nitazoxanide is the only FDA-approved drug for the treatment of cryptosporidiosis.5 However, the efficacy of nitazoxanide is variable in immunocompetent patients, limited in children, and nonexistent in immunocompromised patients, indicating a pressing need for improved therapies.4, 5
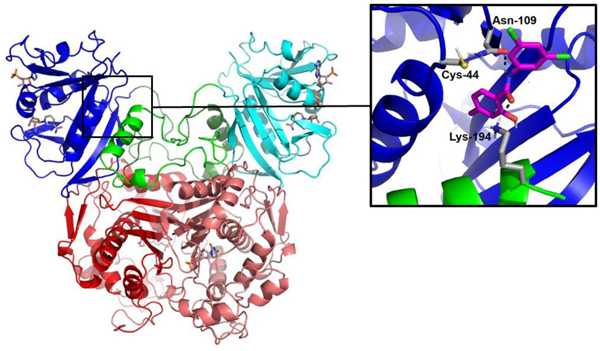
Our current understanding of nucleotide metabolism in Cryptosporidium has made it possible to identify numerous proteins which serve essential roles in the life cycle of the parasite.8 One such protein is thymidylate synthase-dihydrofolate reductase (TS-DHFR), a bifunctional enzyme which is necessary for the production of thymidine monophosphate and folate in Cryptosporidium. TS-DHFR has been extensively characterized and shown to be a promising target for the development of inhibitors.9–12 Whereas TS and DHFR exist as monofunctional enzymes in humans and in bacteria, in Cryptosporidium the two enzymes are linked together on the same polypeptide chain and form a dimer via a TS-TS interface.12–14 Interestingly, the DHFR domains of TS-DHFR also form dimer interactions in a manner which is unique to this class of enzymes. As revealed by the crystal structure of C. hominis TS-DHFR (ChTS-DHFR), the linker region extends from each DHFR domain of the dimer, forms mostly hydrophobic interactions by way of the “crossover helix” motif with the opposite DHFR domain, and crosses back to the original monomer to complete the DHFR and TS domains (Figure 1A).14 Recognizing that these interactions are unique to ChTS-DHFR led us to investigate whether such features could be explored to develop parasite-specific inhibitors.
Figure 1.

Structure of TS-DHFR from Cryptosporidium hominis. (A) The dimeric ChTS-DHFR protein: DHFR (blue/teal) and TS (red/pink) are shown with active site ligands colored in gray. The magnified insets show a surface representation of a non-active site pocket formed between active site Helix B (blue) and the Crossover helix (green). (B) A magnified view of the pocket without any rotation relative to the diagram in 1A. (C) The same pocket, this time rotated along the vertical axis by 90° to reveal the opening of the pocket. (PDB ID: 1QZF)7
Contacts formed between the crossover helix and the DHFR domain have been shown by mutational analyses to be necessary for full catalysis and domain stability of ChDHFR.14, 15 Earlier studies showed a decrease in catalytic activity when mutating phenylalanine 207 at the base of the crossover helix to alanine.14 The resulting crystal structure of the ChTS-DHFR mutant revealed a small but significant shift in the position of the crossover helix which appears to weaken its interactions with the Helix B of the DHFR catalytic domain, explaining the observed decrease in catalytic activity for this mutant.14 With this information in mind, it is conceivable that disrupting crossover helix interactions in ChTS-DHFR with small molecules may lead to allosteric inhibition of the enzyme.
In all available crystal structures of ChTS-DHFR, a non-active site pocket which may accommodate small molecules is formed just above the crossover helix and adjacent to the DHFR domain of the bifunctional protein (Figure 1B). Residues from both the crossover helix and Helix B of the DHFR catalytic domain form a significant part of the pocket (Figure 1C). The pocket was originally identified using the web-based application Q-SiteFinder 16 and confirmed as a potential small molecule binding pocket using the SiteMap17 tool in Schrödinger (SiteScore ≥ 0.800). Visual inspection of the pocket reveals a concave surface of about 59 Å2 with multiple amino acid residues in position to form hydrogen-bonding interactions. The pocket also includes a phenylalanine at position 111 which is inaccessible to solvent, but capable of forming pi-stacking interactions with an aromatic group from an incoming ligand. We turned to an in silico approach to search for compounds which can potentially maximize interactions within the proposed pocket.
Computational virtual screening of large chemical libraries provides an inexpensive way to search for new leads targeting novel binding pockets. To aid in our efforts, the program Glide from the 2014–2 release of the Schrödinger Suite was accessed through the Structural Biology Grid and used to perform virtual screening of 14,400 commercially-available, drug-like small molecules from the Maybridge Hitfinder library. Input protein and ligand structures used for docking were prepared using Schrödinger’s Protein Preparation Wizard18 and LigPrep19 tools, respectively. Ligands were allowed to sample different binding orientations while the proposed binding pocket in ChTS-DHFR (PDB ID: 1QZF, chain B from the A/B complex) was kept rigid. Docking was performed in successive standard precision (SP) and extra precision (XP) modes. Structures from the SP screen were ranked according to their Glide Score, and the top 15% (~3000 structures) were subject to a second virtual screen using the XP mode in Glide. The results were ranked according to their Glide XP Scores, and the top 100 poses were inspected. Filtering was done manually to remove poses which did not contain at least two hydrogen bonds between the docked ligand and the crossover and/or catalytic Helix B of the DHFR domain. From 44 matching compounds, 15 were purchased for testing.
The compounds were ranked based on the Glide XP Score, as shown in Supplementary Table 1. All 15 compounds were subject to a radiometric assay previously used in our lab to evaluate their ability to inhibit DHFR steady-state activity in recombinant ChTS-DHFR (Figure 2A).14 Compound 15 demonstrated greater than 50 % inhibition of DHFR at 500 μM (Figure 2, Supplementary Table 1). In a separate experiment we evaluated the inhibition of compound 15 against TS activity and observed little inhibition of the TS reaction (data not shown), which is not surprising given the large distance separating the proposed non-active site pocket from the TS domain. Although poor solubility at concentrations beyond 500 μM precluded determination of an IC50 value, it is clear from the radiometric experiments that compound 15 exhibits dose-dependent inhibition of ChDHFR (Figure S1). Two additional compounds, 11 and 14, also inhibited ChDHFR at 500 μM by 32.6 % and 21 % respectively, while the remaining compounds were not effective inhibitors (Figure 2A, Supplementary Table 1).
Figure 2.

Glide screen compound ChDHFR inhibition screen. (A) ChDHFR percent activity in the presence of 500 µM Glide screen compounds. (B) Chemical structure of compound 15, which inhibited ChDHFR by 52.2 %.
In order to determine the mode of inhibition for compound 15, we conducted a steady-state rate profile of ChDHFR with its substrate, dihydrofolate, at varying concentrations of compound 15 using the radiometric assay. In theory, noncompetitive inhibition would suggest the binding of compound 15 to an allosteric binding site unique from the catalytic binding site of dihydrofolate. Indeed, compound 15 displayed noncompetitive (mixed) inhibition for dihydrofolate at 0, 100, and 250 μM of the compound, as observed by a concentration-dependent decrease in kcat, 2.17 ± 0.04 s−1, 1.90 ± 0.05 s−1, and 1.20 ± 0.04 s−1, respectively, while the change in Km was insignificant at 1.2 ± 0.2 μM, 1.3 ± 0.3 μM and 1.4 ± 0.4 μM, respectively (Figure 3A and 3B). One possible explanation for the noncompetitive mechanism observed is that compound 15 is displacing NADPH, the cofactor in the ChDHFR-catalyzed reduction of dihydrofolate to tetrahydrofolate. If this is the case, then the activity of compound 15 is mediated by binding to the ChDHFR active site and not the proposed binding pocket in Figure 1C. In order to rule out binding to the active site, we repeated the steady-state rate profile of ChDHFR at varying concentrations of compound 15 and NADPH, this time keeping the dihydrofolate concentration constant. Due to the limitations of our assay, we were only able to determine the observed rate with respect to 100 µM NADPH. Compound 15 appears to display noncompetitive inhibition with respect to NADPH, which is evident by a decrease in kobs, 1.98 ± 0.09 s−1, 1.60 ± 0.06 s−1, and 1.33 ± 0.08 s−1 at 0, 100, and 250 μM of compound 15, respectively.
Figure 3.

Kinetic analysis of compound 15. (A) Steady-state competition assay with varying concentrations of dihydrofolate: 5 μM, 10 μM, 25 μM, 50 μM, and 100 μM. Data fit to a hyperbolic equation. (B) Lineweaver-Burk plot for data in (A). For all graphs, data points with circle (⬤) denote the reaction with no inhibitor, data points with square (⬛) denote the reaction with 100 μM compound 15, and data points with diamond (◆) denote the reaction with 250 μM compound 15. Steady-state competition assay with respect to 100 µM NADPH at concentrations of 0 µM, 100 µM and 250 µM of compound 15 indicated noncompetitive inhibition (data not shown).
Upon closer inspection of subsequent docking models of compound 15 with ChTS-DHFR, we observed two types of predicted binding configurations between the compound and the non-active site pocket. In the first configuration, the 1-(4-bromo-2-methylphenyl)-3-phenylthiourea moiety of compound 15 is directed into the pocket (Figure 4A), while the 2-hydroxy-N-phenylbenzamide moiety is embedded in the pocket in the second configuration (Figure 4B). Furthermore, each configuration forms unique contacts with residues in the non-active site pocket. We evaluated these observations by conducting a structure activity relationship (SAR) study utilizing commercially available compounds derived from the structures of the two different embedded moieties of compound 15.
Figure 4.

Two docked poses for compound 15 (magenta) with the non-active site pocket. The residues that make putative interactions with compound 15 are shown in gray. (A) The 1-(4-bromo-2-methylphenyl)-3-phenylthiourea moiety, highlighted in green in the 2D structure above, is embedded into the pocket. (B) The opposite 2-hydroxy-N-phenylbenzamide moiety, highlighted in blue above, is embedded into the pocket. Hydrogen bonds (black) are shown. (PDB ID: 1QZF)7
At 500 µM of compound, derivatives of the 1-(4-bromo-2-methylphenyl)-3-phenylthiourea moiety produced no greater than 30 % inhibition of ChDHFR (data not shown). On the other hand, several derivatives of the 2-hydroxy-N-phenylbenzamide moiety demonstrated significant inhibitory activity (Supplementary Table 2). In particular, we observed that compounds containing halogen substituents para to the hydroxyl moiety of the phenolic ring (Figure 5A, red circle) demonstrated greater inhibitory activity against ChDHFR. Moreover, compounds containing substituents larger than a methyl group meta to the hydroxyl moiety of the phenolic ring (Figure 5A, blue circle) displayed little ChDHFR inhibition. Finally, we observed that compounds containing chlorine substituents meta to the amide linker of the second aromatic ring (Figure 5A, green circles) displayed greater levels of inhibition against ChDHFR, while most other substituents to this ring resulted in compounds with little or no inhibitory activity (Figure 5A, Supplementary Table 2). These observations are consistent with the structure of compounds 15D8, 15D10, 15D12, 15D13, and 15D15 (Figure 5B), which, at 500 μM, inhibited ChDHFR by 49 %, 61 %, 61 %, 54 %, and 47 %, respectively. Due to poor solubility at concentrations greater than 500 µM, we were not able to determine IC50 values for compounds 15D8, 15D10, 15D12, 15D13, and 15D15, however these compounds do exhibit similar dose-dependent inhibition of ChDHFR as observed for the parent compound 15 (Figure S2).
Figure 5.

Structure activity relationship (SAR) analysis of 2-hydroxy-N-phenylbenzamide moiety derivatives. (A) Derivative compounds containing a methyl substituent at position indicated by the blue circle or a halogen substituent at the position indicated by the red circle displayed inhibitory activity when combined with chlorine substituents at the positions indicated by the green circles. (B) Chemical structure of compound 15D8, 15D10, 15D12, 15D13, and 15D15. (C) Docked pose for derivative 15D10 (magenta), a representative model for the binding of the SAR compounds with the non-active site pocket of ChTS-DHFR. The residues that make putative interactions with 15D10 are shown in gray. Hydrogen bonds (black) are shown. In most models, the phenolic moiety of 15D10 is positioned within the non-active site pocket, while exposing a chlorine substituent to solvent. In some models of 15D10 and other derivatives, a chlorine substituent is positioned near residue Cys44, located at the C-terminal end of the DHFR B helix. In this model, there is a 5.1 Å distance between Cys44 and the nearest chlorine substituent of 15D10. (PDB ID: 1QZF)7
In order to derive a better understanding of the results from our SAR study, we utilized Glide to model the binding of the SAR compounds with the non-active site pocket of ChTS-DHFR shown in Figure 1C. The majority of models position the phenolic moiety of these compounds within the non-active site pocket, while exposing a chlorine substituent to solvent. In some models, the chlorine substituent is positioned near residue Cys44, located at the C-terminal end of the DHFR B helix. A representative model for the binding of the SAR compounds with the non-active site pocket of ChTS-DHFR is shown in Figure 5C. Because of the close proximity of this cysteine residue to the non-active site pocket, compounds 15D17 and 15D18 were designed to covalently target Cys44 with a sulfhydryl group (Figure 5B and 5C, Supplementary Table 2). Initially, 500 µM of compound 15D17 and 15D18 demonstrated modest inhibitory activity, about 14 % and 21 %, respectively, using the original assay conditions in which each compound was incubated for 10 min with ChDHFR. Because the formation of a disulfide bond between the covalent compounds and Cys44 may take longer than 10 min to occur, we incubated the potential covalent inhibitors with ChTS-DHFR for 1 h, 16 h, 48 h and 72 h. We observed that both covalent inhibitors demonstrated time-dependent inhibition, with almost complete inhibition at 72 hours for 15D17 and 48 hours for 15D18 (Figure 6). DMSO controls incubated simultaneously with compound-containing samples did not suffer a loss of enzymatic activity, indicating that compound 15D17 and 15D18 are responsible for the observed inhibition.
Figure 6.
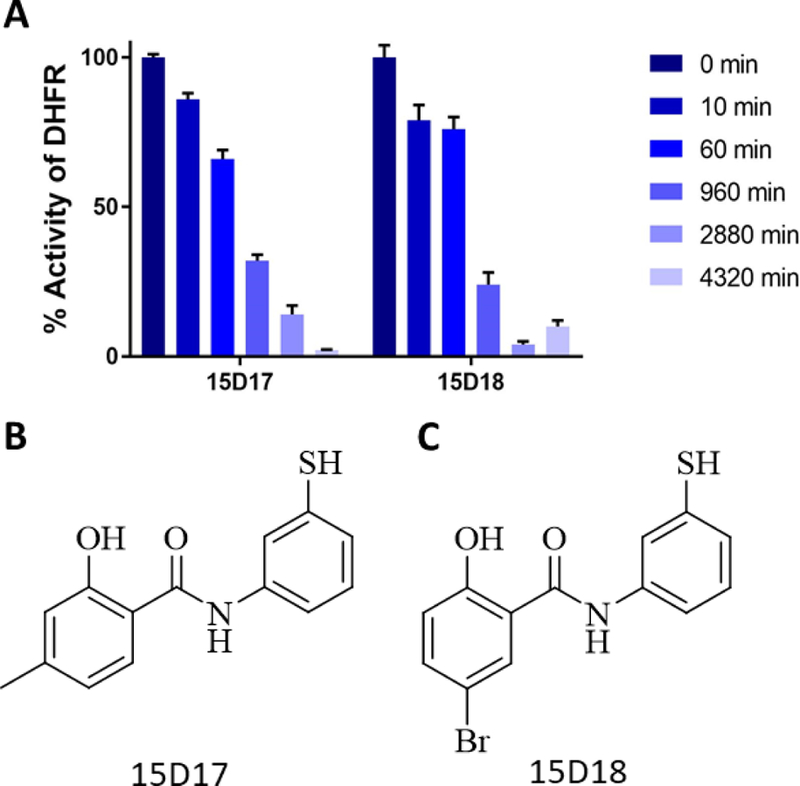
Time-dependent inhibition of ChDHFR (A) Time-dependent inhibition of ChDHFR by covalent inhibitors 15D17 and 15D18. There was no significant change in activity of ChDHFR for DMSO controls incubated for same time periods. (Data not shown) (B) Chemical structure of compound 15D17. (C) Chemical structure of compound 15D18.
We prepared a Cys44Ser mutant ChTS-DHFR to further evaluate whether compound 15D17 and 15D18 bind within the non-active site pocket and form a covalent interaction with residue Cys44 of the protein. Both compounds were pre-incubated with Cys44Ser mutant ChTS-DHFR for 16 h prior to initiation of the reaction. Compound 15D17 displayed a 2-fold decrease in inhibition between wild-type and Cys44Ser ChDHFR, about 68 % and 38 %, respectively (Figure 7). This significant decrease in inhibition would suggest that 15D17 does indeed bind within our non-active site pocket, forming a disulfide bond with residue Cys44. We did not, however, observe a discernable change in ChDHFR inhibition for compound 15D18 (Figure 7). As a negative control, we also incubated compound 15D10, the parent compound of 15D17 which does not contain a sulfhydryl group, with both wild-type and Cys44Ser mutant ChTS-DHFR. We observed no significant change in inhibition in the control experiment, indicating that, unlike 15D10, the activity of 15D17 is mediated by disulfide bond formation.
Figure 7.
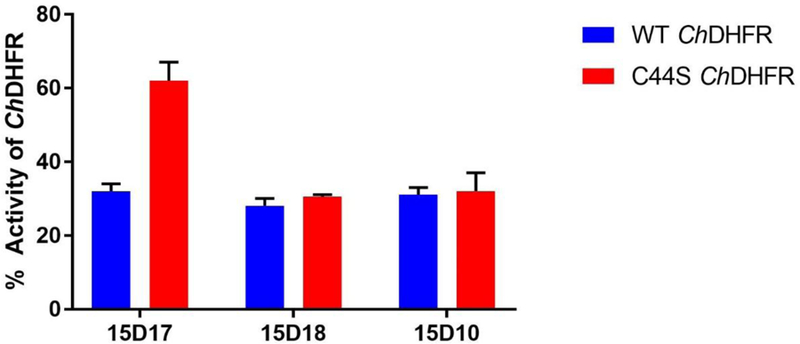
Inhibition of ChDHFR and Cys44Ser ChDHFR by covalent inhibitors 15D17 and 15D18. Covalent compounds and enzyme were incubated for 16 h before the reaction was initiated. 15D10, the parent compound of 15D17, was used as a negative control.
When 10 mM DTT was added to the reaction mixture before incubation of enzyme and covalent compounds, 15D17 inhibition was abolished, while 15D18 inhibited ChDHFR by ~31 % with no change in inhibition for 15D10 (Figure 8). This suggest that DTT in the preincubation solution sequesters compound 15D17 and prevents it from interacting with the enzyme. Additionally, this data suggests that 15D17 does not inhibit ChDHFR by interacting with the active site or an alternative cysteine residue in the ChDHFR domain, or through aggregation. While DTT does negate some of 15D18 inhibition of ChDHFR, this data would suggest that 15D18 may be binding in more than one location. There is no such effect observed for the control compound 15D10, as expected given the lack of a sulfhydryl group.
Figure 8.
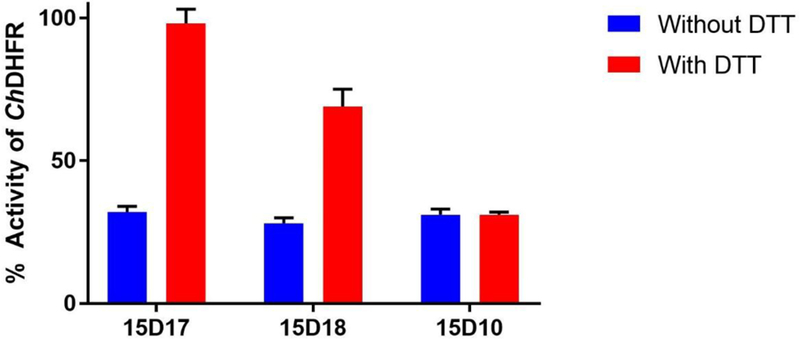
ChDHFR inhibition in the presence of 10 mM DTT. Covalent compounds 15D17 and 15D18 and enzyme were incubated for 16 h with 10 mM DTT before the reaction was initiated. 15D10, the parent compound of 15D17, was used as a negative control.
We next investigated the specificity of compounds 15D17 and 15D18 for ChTS-DHFR over human DHFR, which does not have a cysteine residue corresponding to Cys44 of ChTS-DHFR (Figure S3). Both compounds were incubated with human DHFR for 16 hours to evaluate any inhibitory effects. Compound 15D17, was found to be highly selective for ChDHFR, having negligible effect on human DHFR. This is in dramatic contrast to 15D18, which inhibited human DHFR by about 95 % (Figure 9).
Figure 9.

Inhibition of ChDHFR and human DHFR by covalent inhibitors 15D17 and 15D18. Covalent compounds and enzyme were incubated for 16 h before the reaction was initiated.
The results for compound 15D17 and 15D18 suggest that while both compounds demonstrate time-dependent inhibition of ChDHFR, 15D17 appears to bind the non-active site pocket, and demonstrates high specificity for ChDHFR. The decrease in inhibition observed for 15D17 between wild-type and Cys44S ChTS-DHFR, suggests that the formation of a disulfide bond is required for 15D17 to bind with the non-active site pocket. Furthermore, our preincubation of 15D17 with enzyme and 10 mM DTT data suggests that, 15D17 does not indiscriminately inhibit ChDHFR by binding to the active site or randomly interacting with the enzyme. On the other hand, the time-dependent inhibition demonstrated by 15D18 suggests that this compound may be targeting a different cysteine residue. ChDHFR contains two additional cysteine residues in addition to Cys44, Cys113 and Cys164 (Figure S4) Cys113 resides in the active site pocket, while Cys164 is located near the interface between the TS and DHFR domains. Perhaps the time-dependent inhibition is due to 15D18 targeting one of these residues. Since this inhibition is only reversed by 31 % for ChDHFR when 15D18 was preincubated with enzyme and 10 mM DTT, this suggests an alternate inhibition pathway may also be operative. The observed time dependence inhibition of 15D18 may occur through another mechanism, possibly due to the para-bromo phenol ring of the compound being unstable and susceptible towards oxidation. This could in turn produce a quinonoid type electrophilic species. Similar time dependent inhibition results are observed for the human DHFR and 15D18.
Previous studies from our lab looked at the effect of interfering with crossover helix interactions in ChTS-DHFR. One study utilized peptide mimetics of the crossover helix to inhibit wild-type ChTS-DHFR noncompetitively, and with an IC50 of 230 μM.13 A second study used a virtual screening approach to identify small molecules capable of binding in a non-active site pocket below the crossover helix, adjacent to the TS domain.12 In the latter example, the compound Flavin Mononucleotide (FMN) was also found to inhibit ChDHFR activity noncompetitively, though with an improved IC50 value of 55 μM.12 Here too, we demonstrate that ChDHFR activity can be inhibited by disrupting interactions between the crossover helix and ChDHFR with small molecules aimed at a novel non-active site pocket. Collectively, these efforts help underscore the importance of crossover helix interactions for ChDHFR catalysis. Additionally, our findings lend support to the use of virtual screening and structure-guided modeling approaches in the discovery of compounds targeting novel binding pockets. Computational modeling proved especially helpful in instructing the design of, compound 15D17, which is the first example of a covalent inhibitor designed to target a non-catalytic pocket in ChTS-DHFR. Targeting non-catalytic cysteine residues is a promising strategy for lead generation and optimization, and is yet to be fully explored in drug discovery efforts aimed at developing Cryptosporidium-specific inhibitors.20
In conclusion, we have conducted a structure-based virtual screen of a novel non-active site pocket of ChTS-DHFR. Our screen identified compound 15, which inhibits ChDHFR by 52 %, displays mixed noncompetitive inhibition with respect to dihydrofolate, and is not competitive with respect to NADPH. Furthermore, we conducted an SAR study utilizing derivatives of compound 15 which led to the development of covalent compounds designed to target Cys44. Compound 15D17 demonstrated time-dependent inhibition through covalent interaction via a disulfide bond and selectively inhibits ChDHFR, while mutation of Cys44 interferes with binding of 15D17 with the non-active site pocket. Work is currently underway to obtain co-crystal structures in order to validate our modeling and obtain a better understanding of the interactions between compound 15D17 and the proposed ChTS-DHFR non-active site pocket. The discovery of 15D17 offers an excellent starting point for further lead optimization of covalent inhibitors to the DHFR allosteric site.
Supplementary Material
Acknowledgments
This work is supported by NIH Grant (AI083146) to K.S.A., Training Grant (5T32AI007404–23) to D.J.C.
References
- 1.Mercado R, Buck GA, Manque PA, Ozaki LS, Cryptosporidium hominis infection of the human respiratory tract, Emerg Infect Dis, 13 (2007) 462–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laurent F, Lacroix-Lamande S, Innate immune responses play a key role in controlling infection of the intestinal epithelium by Cryptosporidium, Int J Parasitol, 47 (2017) 711–721. [DOI] [PubMed] [Google Scholar]
- 3.Ryan U, Fayer R, Xiao L, Cryptosporidium species in humans and animals: current understanding and research needs, Parasitology, 141 (2014) 1667–1685. [DOI] [PubMed] [Google Scholar]
- 4.Checkley W, White AC Jr., Jaganath D, Arrowood MJ, Chalmers RM, Chen XM, Fayer R, Griffiths JK, Guerrant RL, Hedstrom L, Huston CD, Kotloff KL, Kang G, Mead JR, Miller M, Petri WA Jr., Priest JW, Roos DS, Striepen B, Thompson RC, Ward HD, Van Voorhis WA, Xiao L, Zhu G, Houpt ER, A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium, Lancet Infect Dis, 15 (2015) 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borad A, Ward H, Human immune responses in cryptosporidiosis, Future Microbiol, 5 (2010) 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ivanetich KM, Santi DV, Bifunctional thymidylate synthase-dihydrofolate reductase in protozoa, FASEB J, 4 (1990) 1591–1597. [DOI] [PubMed] [Google Scholar]
- 7.O’Neil RH, Lilien RH, Donald BR, Stroud RM, Anderson AC, Phylogenetic classification of protozoa based on the structure of the linker domain in the bifunctional enzyme, dihydrofolate reductase-thymidylate synthase, J Biol Chem, 278 (2003) 52980–52987. [DOI] [PubMed] [Google Scholar]
- 8.Zhu G, Guo F, Cryptosporidium Metabolism, in: Cacciò SM, Widmer G (Eds.) Cryptosporidium: parasite and disease, Springer Vienna, Vienna, 2014, pp. 361–379. [Google Scholar]
- 9.Greenwood BM, Bojang K, Whitty CJ, Targett GA, Malaria, Lancet, 365 (2005) 1487–1498. [DOI] [PubMed] [Google Scholar]
- 10.Yuthavong Y, Kamchonwongpaisan S, Leartsakulpanich U, Chitnumsub P, Folate metabolism as a source of molecular targets for antimalarials, Future Microbiology, 1 (2006) 113–125. [DOI] [PubMed] [Google Scholar]
- 11.Kumar VP, Cisneros JA, Frey KM, Castellanos-Gonzalez A, Wang Y, Gangjee A, White AC Jr., Jorgensen WL, Anderson KS, Structural studies provide clues for analog design of specific inhibitors of Cryptosporidium hominis thymidylate synthase-dihydrofolate reductase, Bioorg Med Chem Lett, 24 (2014) 4158–4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martucci WE, Udier-Blagovic M, Atreya C, Babatunde O, Vargo MA, Jorgensen WL, Anderson KS, Novel non-active site inhibitor of Cryptosporidium hominis TS-DHFR identified by a virtual screen, Bioorg Med Chem Lett, 19 (2009) 418–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martucci WE, Rodriguez JM, Vargo MA, Marr M, Hamilton AD, Anderson KS, Exploring novel strategies for AIDS protozoal pathogens: alpha-helix mimetics targeting a key allosteric protein-protein interaction in C. hominis TS-DHFR, Medchemcomm, 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Atreya CE, Anderson KS, Kinetic characterization of bifunctional thymidylate synthase-dihydrofolate reductase (TS-DHFR) from Cryptosporidium hominis: a paradigm shift for ts activity and channeling behavior, J Biol Chem, 279 (2004) 18314–18322. [DOI] [PubMed] [Google Scholar]
- 15.Vargo MA, Martucci WE, Anderson KS, Disruption of the crossover helix impairs dihydrofolate reductase activity in the bifunctional enzyme TS-DHFR from Cryptosporidium hominis, Biochem J, 417 (2009) 757–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laurie AT, Jackson RM, Q-SiteFinder: an energy-based method for the prediction of protein-ligand binding sites, Bioinformatics, 21 (2005) 1908–1916. [DOI] [PubMed] [Google Scholar]
- 17.SiteMap, version 3.1, Schrodinger, LLC, New York, NY, 2014. [Google Scholar]
- 18.Schrodinger Suite 2014–2 Protein Preparation Wizard; Epik version 2.8, Schrodinger, LLC, New York, NY, 2014. [Google Scholar]
- 19.LigPrep, version 3.0, Schrodinger, LLC, New York, NY, 2014. [Google Scholar]
- 20.Hallenbeck KK, Turner DM, Renslo AR, Arkin MR, Targeting Non-Catalytic Cysteine Residues Through Structure-Guided Drug Discovery, Curr Top Med Chem, 17 (2017) 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.