

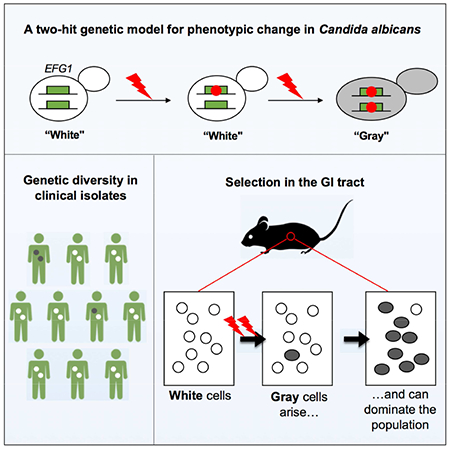
Summary
Candida albicans is a commensal fungus of human gastrointestinal and reproductive tracts, but also causes life-threatening systemic infections. The balance between colonization and pathogenesis is associated with phenotypic plasticity, with alternative cell states producing different outcomes in a mammalian host. Here, we reveal that gene dosage of a master transcription factor regulates cell differentiation in diploid C. albicans cells, as EFG1 hemizygous cells undergo a phenotypic transition inaccessible to ‘wildtype’ cells with two functional EFG1 alleles. Notably, clinical isolates are often EFG1 hemizygous and thus licensed to undergo this transition. Phenotypic change corresponds to high-frequency loss of the functional EFG1 allele via de novo mutation or gene conversion events. This phenomenon also occurs during passaging in the gastrointestinal tract with the resulting cell type being hypercompetitive for commensal and systemic infections. A ‘two-hit’ genetic model therefore underlies a key phenotypic transition in C. albicans that enables adaptation to host niches.
Keywords: Phenotypic switch, Candida albicans, phase variation, pathogenesis, gastrointestinal tract, transcription factor, population genetics
Graphical Abstract
eTOC Blurb
Microorganisms use diverse strategies to adapt to their environment. Liang et al. reveal a two-hit genetic mechanism underlies a high-frequency phenotypic transition in the diploid fungus Candida albicans. Clinical isolates heterozygous for a key transcription factor readily lose gene function triggering a transition that impacts both commensalism and pathogenicity.
Introduction
Microbial species utilize a wide range of mechanisms to adapt to their environment. These include ‘bet hedging’, in which pre-existing population heterogeneity promotes survival under fluctuating conditions (Ackermann, 2015; Martins and Locke, 2015). Diversity is also important in microbial communities as it can allow for a division of labor between specialized cell types (Norman et al., 2015). The mechanisms that generate phenotypic heterogeneity are of particular interest in pathogens as they often dictate host interactions. Processes such as phase variation, antigenic switching and phenotypic switching are observed across bacterial, fungal, and protozoan pathogens and involve a variety of genetic and epigenetic mechanisms (Deitsch et al., 2009; Noble et al., 2017; van der Woude and Baumler, 2004).
Phenotypic plasticity is particularly relevant to the lifestyle of the opportunistic fungal pathogen Candida albicans. This diploid species lives as a commensal within the gastrointestinal and reproductive tracts of humans (Noble et al., 2017), yet is also a leading cause of bloodstream infections worldwide (Brown et al., 2012; Pfaller and Diekema, 2007; Wisplinghoff et al., 2004). This fungus can grow as single-celled yeast or as filamentous cells (Sudbery, 2011) and can also adopt alternative yeast-like cell types; these are defined by distinct cell and colony morphologies and show striking differences in colonization and pathogenesis (Noble et al., 2017).
Phenotypic flexibility in C. albicans is exemplified by the white-opaque switch, in which heritable and reversible transitions occur between ‘white’ and ‘opaque’ states (Slutsky et al., 1987). This epigenetic switch is regulated by a transcription factor (TF) circuit that includes Efg1, which promotes white cell formation, and Wor1, which promotes opaque cell formation (Hernday et al., 2013; Huang et al., 2006; Sonneborn et al., 1999; Srikantha et al., 2006; Zordan et al., 2006; Zordan et al., 2007). Efg1 is a pleiotropic factor that regulates filamentation, metabolism, biofilm formation, and virulence, in addition to white-opaque switching (Doedt et al., 2004; Lo et al., 1997; Ramage et al., 2002; Stoldt et al., 1997). Two more recently characterized cell types are the ‘gray’ state and the ‘GUT’ state, which share overlapping transcriptional control with the white-opaque switch (Pande et al., 2013; Tao et al., 2014). Switching between multiple yeast-like cell types therefore contributes to phenotypic heterogeneity in C. albicans populations (Noble et al., 2017).
In this work, we reveal an unexpected genetic mechanism underlies switch-like behavior in C. albicans. We first show that diploid strains engineered to be EFG1 hemizygous undergo switching to a cell state that is inaccessible to ‘wildtype’ cells with two functional alleles. This phenomenon includes clinical isolates that are naturally EFG1 hemizygous and thus primed to undergo this transition. Surprisingly, high-frequency switching involves loss of the functional EFG1 allele thereby generating an efg1 null mutant. We also show that the gray state, previously thought to represent an epigenetic state, similarly arises via mutational loss of EFG1 in isolates that are naturally EFG1 heterozygous. Finally, we reveal that EFG1 heterozygotes passaged in the gastrointestinal tract lose EFG1 function and that the resulting cells exhibit increased fitness in both gastrointestinal and systemic infections. These experiments establish that EFG1 directs both genetic and epigenetic variation in C. albicans, and that stochastic loss of this gene frequently occurs with consequences for infection outcomes.
Results
EFG1 Gene Dosage Determines C. albicans Cell Fate Decisions
In C. albicans, white-opaque switching is regulated by TFs including Efg1 and Wor1, and by the mating-type like (MTL) locus as only MTL homozygous cells readily switch to the opaque state (Figure 1A)(Miller and Johnson, 2002). As EFG1 haploinsufficiency can impact cell properties (Glazier et al., 2017; Zavrel et al., 2012), we constructed heterozygous and homozygous EFG1 deletions in MTLa/a and MTLa/α backgrounds to examine the role of this TF in defining cell identity.
Figure 1. EFG1 hemizygosity enables C. albicans cells to form an alternative cell state.
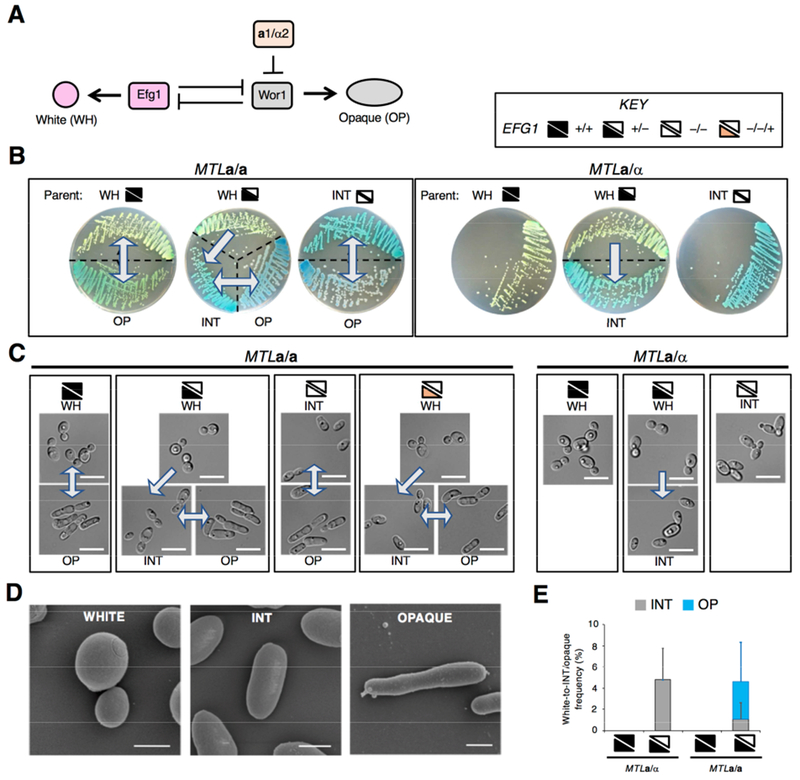
(A) White-opaque switching is regulated by Efg1 which promotes white cell formation and Wor1 which promotes opaque cell formation. MTLa1/α2 represses WOR1 and thus limits opaque formation by MTLa/α cells.
(B) SC5314-derived strains grown on CHROMagar (for 3 days at 22°C) exhibit three cell states, white, intermediate (INT) and opaque. Arrows indicate unidirectional or bidirectional switching. Key indicates wildtype (+/+), heterozygous (+/−), and homozygous (−/−) EFG1 genotypes, as well as an efg1 null into which EFG1 was restored (−/−/+).
(C) Microscopic images of cells from CHROMagar (3 days at 22°C). Scale bar, 10 μm.
(D) Scanning electron micrographs of SC5314 white, INT and opaque cells. Scale bars, 2 μm.
(E) Frequency of phenotypic switching on CHROMagar after 7 days at 22°C. Three independent experiments performed.
Strains are SC5314 derivatives: MTLa/a EFG1+/+; RBY717, MTLa/a EFG1+/−; CAY6970. MTLa/a efg1−/−; CAY6977, MTLa/α EFG1+/+; SN87, MTLa/α EFG1+/−; CAY7064, MTLa/α efg1−/−; TF156, MTLa/a efg1−/−::EFG1; CAY6911.
See also Figure S1.
As expected, C. albicans SC5314 a/a cells exhibited white-opaque switching whereas a/α cells were locked in the white state (Figure 1B and 1C). Surprisingly, however, deletion of one EFG1 allele resulted in EFG1+/− cells that underwent switching to a phenotypic state distinct from white or opaque (Figure 1B-1D). This ‘intermediate’ (INT) cell state arose regardless of which EFG1 allele was deleted (Figure S1A) and was observed in both a/a and a/α strains. Thus, EFG1 hemizygous a/α strains adopted two cell states, white and INT, whereas the corresponding a/a strains formed three cell states: white, INT, and opaque (Figure 1B-1D).
Multiple cell states were easily distinguished by growth on CHROMagar Candida medium (Odds and Bernaerts, 1994). Here, colonies formed by white, INT, and opaque cells were light green, turquoise, and blue/purple, respectively (Figure 1B). Microscopic analysis showed that INT cells were ovoid and distinct from that of rounder white cells or more elongated opaque cells (Figure 1C and 1D). The three cell states were also apparent on other media, with INT and opaque cells undergoing filamentation under different conditions than those supporting filamentation of white cells (Figure S1B and S1C).
A quantitative analysis of switching on CHROMagar established that ~5% of white EFG1+/− colonies showed sectoring to INT in a/α cells, whereas a/a colonies showed frequent switching to both INT and opaque states (Figure 1E). The frequency of white-to-INT switching was similar to that for white-to-opaque switching under standard culture conditions (Slutsky et al., 1987; Zordan et al., 2007). Loss of both EFG1 alleles in a/α cells locked cells in the INT state, whereas a/a efg1 null cells still showed reversible INT-opaque switching (Figure 1B and 1C). WOR1 was essential for formation of the opaque state, and deletion of both EFG1 and WOR1 in an a/a strain therefore locked cells in the INT state.
These experiments reveal that loss of one EFG1 allele enables white cells to transition to the INT state, whereas this state is inaccessible to wildtype cells. Formation of the INT state is dependent on EFG1 dosage yet independent of MTL or WOR1 control. Furthermore, efg1 null cells undergo reversible switching between INT and opaque states if MTL homozygous.
A Subset of Clinical Isolates Exhibit INT State Formation
We next examined the phenotypes and genotypes of sequenced C. albicans isolates. These included 20 isolates from commensal and pathogenic sources (Hirakawa et al., 2015) and 43 serial isolates from 11 oropharyngeal candidiasis (OPC) patients (Ford et al., 2015). Strikingly, 7 of the 63 isolates showed formation of the INT state; 5 presented in the white state but showed switching (2-10% colony sectoring) to the INT state (P37037, P75063, 1619, 2823 and 4018; Figure 2A and 2B), while two isolates presented in the INT state. The latter were isolate 945 which was MTLa/α and locked in the INT state, whereas P94015 was MTLa/a and underwent INT-opaque switching (Figure 2A and 2B).
Figure 2. Clinical isolates containing EFG1 polymorphisms form the ‘intermediate’ cell state.
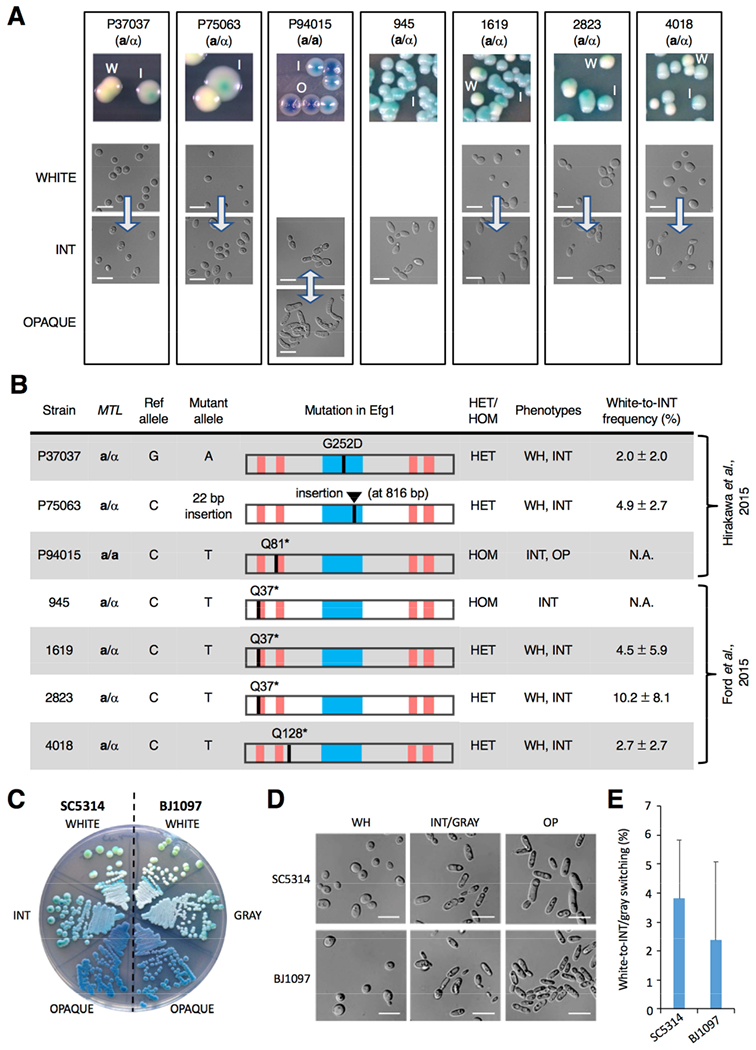
(A) Images of colony and cell phenotypes on CHROMagar (W, white; I, intermediate (INT); O, opaque) at 22°C. Scale bar, 10 μm. Arrows indicate transitions between states.
(B) Analysis of 63 clinical isolates revealed that 7 contained disruptive polymorphisms in EFG1. The APSES DNA-binding domain (blue box) and polyglutamine repeats (red bars) are shown. EFG1 mutations were heterozygous (HET) or homozygous (HOM), and cell states observed were white (WH), intermediate (INT) and opaque (OP). White-to-INT transition frequency was determined on YPD medium (22°C for 7 days) with three independent assays.
(C) White/INT/opaque phenotypes in SC5314 (EFG1+/− parental strain) v. white/gray/opaque phenotypes in BJ1097. Cells were grown on CHROMagar at 22°C for 3 days.
(D) White/INT/opaque phenotypes in SC5314 (EFG1+/− parental strain) v. white/gray/opaque phenotypes in BJ1097. Cells grown on YPD at 22°C for 3 days.
(E) Comparison of white-to-INT and white-to-gray transition frequencies in SC5314 and BJ1097, respectively, on YPD after 7 days at 22°C. Error bars, ± SD. Three independent assays performed.
See also Figure S2.
Analysis revealed that the 7 strains competent for INT state formation were disrupted for one or both EFG1 alleles (Figure 2B), whereas the other 56 isolates carried two intact EFG1 alleles. Isolates 945 and P94015 that presented in the INT state were homozygous efg1 null, whereas the 5 isolates that exhibited white-to-INT switching were EFG1 heterozygous. Disruptive EFG1 mutations included 3 nonsense mutations (Q37*, Q81* and Q128*), a frameshifting indel, and a missense mutation (G252D) within the DNA binding domain (Figure 2B, Figure S2A and S2B). Overall, 6 out of 31 individuals (19%) carried EFG1 defective isolates, with both a homozygous mutant (945) and a heterozygous mutant (1619) isolated from one individual.
All 63 isolates were also examined for ORF-disrupting mutations in the other 7 TFs that form the core white-opaque network. However, only 1 of these isolates contained a disruptive mutation in any of the 7 genes, with isolate P78048 carrying a heterozygous Q216* mutation in WOR3. Thus, loss-of-function mutations in EFG1 are more frequent than mutations in other TFs of the core white-opaque circuit.
To establish the impact of EFG1 gene dosage in different strains, we generated EFG1+/− derivatives of EFG1+/+ isolates and found that hemizygous strains formed white colonies that sectored to INT and, if MTL homozygous, also to opaque (Figure S2C). Conversely, an intact EFG1 allele was integrated into P75063, an MTLa/α strain with a single functional EFG1 allele. The native P75063 isolate formed both white and INT states, whereas P75063(+EFG1) exclusively formed the white state (Figure S2C). Disruptive EFG1 mutations therefore exist in a number of natural C. albicans isolates, and EFG1+/− hemizygous cells are competent for switching to INT independent of strain background.
C. albicans INT and Gray States Represent Equivalent Cell States
A subset of clinical C. albicans isolates were previously shown to undergo tristable white-gray-opaque switching in which gray cells represent a distinct epigenetic state (Tao et al., 2014). Given potential parallels with white-INT-opaque switching, we compared SC5314 EFG1+/− cells with BJ1097 cells in which white-gray-opaque switching was originally observed. Strikingly, the phenotypes of INT and gray states were similar at both the cellular and colony level, and frequencies of white-to-INT and white-to-gray transitions were also comparable (Figure 2C-2E). Moreover, analysis of BJ1097 white cells revealed that these cells were heterozygous for a nonsense mutation (Q61*) in EFG1. These results show that INT and gray states are phenotypically similar and that both arise in strains that are functionally EFG1 heterozygous. Given these parallels (and those described below) we refer to the INT state as the gray state from here on.
Genetic Disruption of EFG1 Underlies the White-to-Gray Transition
We initially presumed, as did others (Tao et al., 2014), that the white-to-INT/gray switch was epigenetic given its high frequency. However, we did not observe reverse gray-to-white switching and the EFG1 gene product was not detected in gray cells (Figure S3), potentially indicating a genetic mechanism. To examine EFG1 alleles, Sanger sequencing was performed following 30 independent white-to-gray switching events in SC5314 EFG1+/− cells. Strikingly, EFG1 could not be PCR amplified from half of the gray colonies. Furthermore, in gray cells still containing EFG1, we detected a frameshifting indel upstream of the DNA binding domain in each case (Figure 3A). Indels included 7 deletions and 3 insertions that occurred almost exclusively at trinucleotide (CAA/CAG)n repeats encoding for polyglutamine (polyQ) tracts (Figure 3A and Figure S4A).
Figure 3. Genetic loss of EFG1 underlies the white-to-gray transition in vitro.
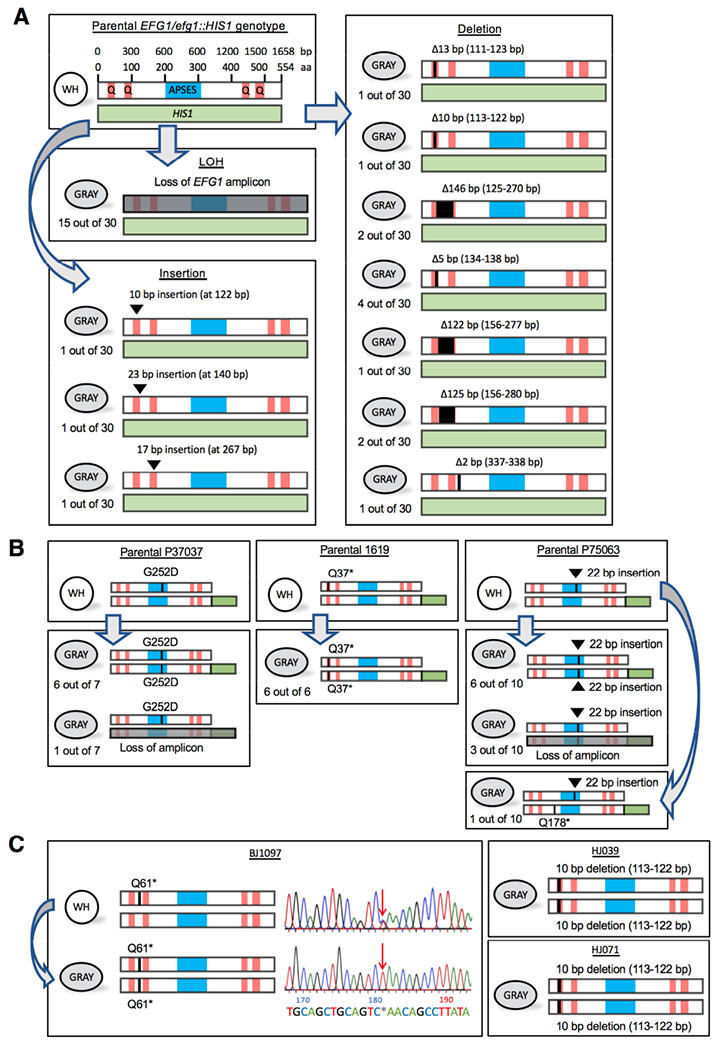
(A) Schematics show EFG1 mutations from white-to-gray switching. APSES, DNA binding domain. Q, polyglutamine tracts. The parental SC5314 strain is heterozygous for EFG1 (one allele replaced with HIS1) and mutational patterns are shown for 30 independent white-to-gray transitions.
(B) Schematics depicting EFG1 mutations in three clinical strains (P37037, 1619, and P75063) upon switching to the gray state. The intact EFG1 allele was tagged with GFP (green box) in the parental strain. Independent switching events are shown.
(C) Schematic of EFG1 mutations present in three clinical strains (BJ1097, HJ039, and HJ071) in which the gray state was first identified. For BJ1097, the chromatogram shows Sanger sequencing of EFG1 in white and gray cells. Arrow indicates a mutation (C181T) causing a premature stop codon (Q61*) that is heterozygous in white cells but homozygous in gray cells.
See also Figure S4 and S5.
To define potential gene conversion tracts, we examined 6 single nucleotide polymorphism (SNP) positions on Chr R using restriction fragment length polymorphism (SNP-RFLP) analysis (Forche et al., 2004). The 6 SNPs were heterozygous in parental white cells and had retained their heterozygosity in 3 gray lineages that had indels within the EFG1 ORF (Figure S4B). In contrast, analysis of 4 gray lineages that had lost the EFG1 ORF revealed that 3 had undergone loss of heterozygosity (LOH) for a single SNP positioned 1906 bp downstream to the ORF, whereas 1 had retained all 6 heterozygous positions (Figure S4B). These results indicate that localized gene conversion events were responsible for gray state formation in these 4 lineages.
For comparison, we examined the frequency with which ADE2 function was lost in an SC5314 ADE2+/− hemizygous strain. Single colonies were analyzed for the emergence of red (ade2 null) sectors, yet no sectoring was detected in >2,500 colonies examined (<0.04%). This is consistent with reports that mutation rates vary across the C. albicans genome (Ene et al., 2018; Forche et al., 2011) and suggests that EFG1 may represent a hotspot for gene conversion and/or mutational events.
Gray State Formation in Clinical Isolates Involves Loss of EFG1 Function
EFG1 was analyzed in three clinical isolates, 1619, P37037 and P75063, that are EFG1+/− and undergo the white-to-gray transition. Here, the functional EFG1 allele was fused to GFP to enable expression monitoring and genotyping. In each case, formation of the gray state resulted in loss of GFP expression (Figure S5A) due to mutational events. In 22 out of 23 independent switching events gene conversion generated a homozygous efg1 null, with 4 of these events also involving loss of the GFP-tagged ORF (Figure 3B). In the one other switching event, gray cells contained a new EFG1-disrupting mutation (Q178* in P75063; Figure 3B). The distribution of LOH tracts was evaluated by SNP-RFLP analysis. As in SC5314, most LOH events impacted only the EFG1 ORF together with the immediate flanking sequence (10 out of 12 LOH events were <10 kb in P37037 and P75063) although long-tract events were occasionally detected (2 LOH events in P75063 were >1 Mb) (Figure S4C).
We also evaluated EFG1 sequences in isolate BJ1097 (Tao et al., 2014). White cells contained a heterozygous Q61* mutation in EFG1, yet derived gray cells had undergone gene conversion creating a homozygous efg1 null (Figure 3C). EFG1 alleles from gray cells formed by HJ039 and HJ071 (Tao et al., 2014) were also disrupted due to a homozygous 10-bp deletion at the 5’ end of the gene (Figure 3C).
Together, these results establish that clinical isolates often contain polymorphisms that disrupt EFG1 function, and that the white-to-gray transition involves high-frequency loss of the functional allele in clinical EFG1+/− strains via either gene conversion or de novo mutation.
Gene Expression Profiling Reveals Cell State-Specific Transcriptomes
RNA sequencing was performed and revealed that white, gray and opaque cells exhibit cell state-specific transcriptomes (Figure S6A-C and Table S1). Gene Ontology (GO) analysis indicated that metabolic-associated processes were differentially regulated among the three states. Thus, white and gray cells showed expression differences in fatty acid/carboxylic acid catabolism, as well as in redox and cellular transport processes, whereas gray and opaque cells showed expression differences in carboxylic acid and lipid catabolism (Table S1). This is consistent with EFG1 regulating metabolic gene expression (Doedt et al., 2004; Lan et al., 2002; Pierce et al., 2013).
We also noted differences between EFG1+/+ and efg1−/− opaque cells as 826 genes were differentially expressed between these two cell types (Figure S6C and Table S1), and these cells generated distinct cell and colony phenotypes (Figure S7A and S7B). Furthermore, EFG1+/+ opaque cells switched to the white state en masse at 37°C, whereas efg1 null opaque cells partially retained this state at this temperature (Figure S7C). These results highlight how both genetic and epigenetic factors impact the C. albicans transcriptome and that efg1 null ‘opaque-like’ cells show distinct properties from conventional EFG1-expressing opaque cells.
Stochastic White-to-Gray Switching Enhances Fitness in the GI Tract
Different C. albicans cell states exhibit differential interactions with the mammalian host. White cells colonize the gastrointestinal tract (GI) and are virulent in disseminated infections (Noble et al., 2010; Pande et al., 2013), whereas gray cells are avirulent in the systemic model yet can colonize the mouse tongue ex vivo (Tao et al., 2014). Gray cells have not been directly evaluated in the GI tract, although efg1 null mutants were previously shown to be hypercompetitive in this niche (Pande et al., 2013; Pierce et al., 2013; Pierce and Kumamoto, 2012).
Here, we examined both the fitness and heritability of different cell types during GI colonization, initially using SC5314 a/α cells that cannot access the opaque state. First, a 50:50 mix of EFG1+/− white and efg1−/− gray cells was introduced into the GI and cell types evaluated for the presence of a genetic marker (‘genotype’) and for colony appearance (‘phenotype’) upon recovery from fecal pellets. We found that gray cells rapidly outcompeted white cells by 2 days post-infection (dpi) (Figure 4A), consistent with the heightened fitness of efg1 null mutants in this niche. We also performed an analogous experiment in which 99% white (EFG1+/+) cells were co-inoculated with 1% gray (efg1 null) cells. Despite being in the minority, gray cells again took over the population so that >90% of recovered cells were in the gray state by 12 dpi (Figure 4B).
Figure 4. MTLa/α gray cells exhibit a fitness advantage over white cells in a commensal GI model.
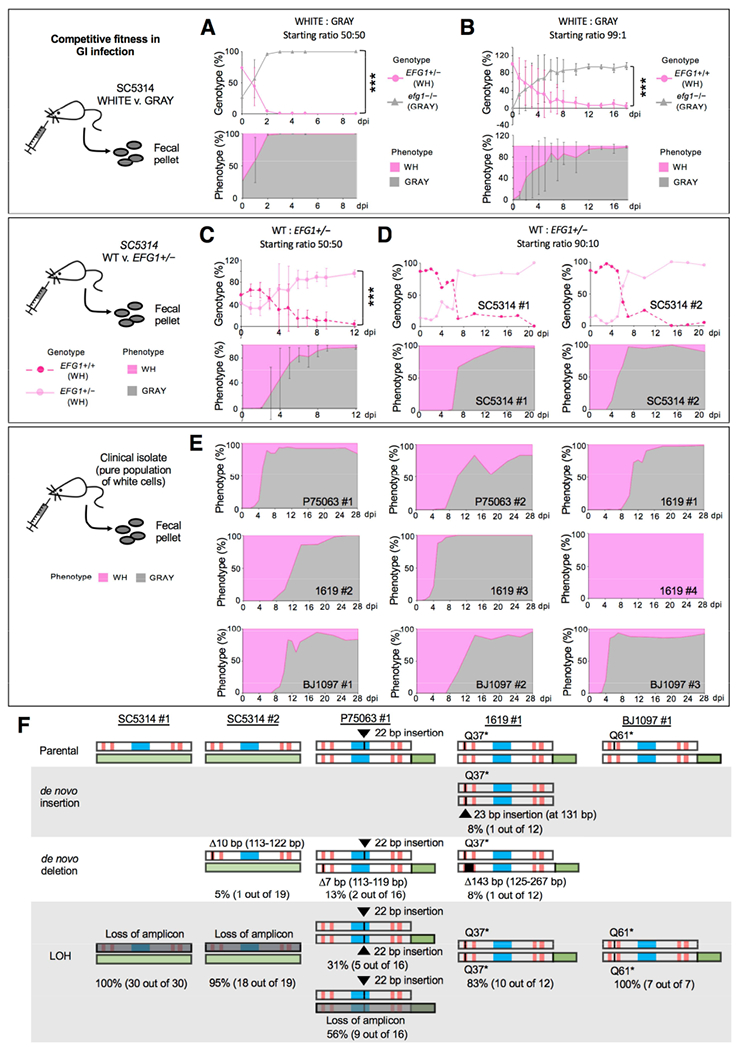
(A) Competition between SC5314 white (EFG1+/−) and gray (efg1−/−) states in a GI model. Cell types were co-inoculated 50:50 and analyzed upon recovery from fecal pellets. NATR v. NATS analysis was used to define strain type (genotype) and colony color used to define the phenotype. Strains are CAY7768/7065. n=3 cohoused mice. Mean values with SD. ***, P<0.001 by t test.
(B) Competition between SC5314 white (EFG1+/+) and gray (efg1−/−) cells inoculated in a 99:1 ratio (CAY8282 and 7769). n=3 mice housed separately. Mean values plotted with SD. ***, P<0.001 by t test.
(C) A 50:50 competition between SC5314 EFG1+/+ and EFG1+/− cells (both in the white state) in the GI model (Strains CAY7770 and 7064). n=3 cohoused mice. Mean values with SD. ***, P<0.001 by t test.
(D) A 90:10 competition between EFG1+/+ and EFG1+/− cells (strains CAY8282 and 7768). n=2 mice housed separately.
(E) Strains P75073, 1619, and BJ1097 (naturally EFG1+/−) were evaluated for GI colonization. Cells were introduced in the white state and recovered from fecal pellets. Strains with GFP-tagged EFG1 (CAY9159/9165/9195) or wildtype isolates were used. n=9 independent experiments with mice housed separately.
(F) Summary of EFG1 mutations arising during white-to-gray switching in the GI model. Genotypes examined by PCR and Sanger sequencing to determine mutations in EFG1.
See also Figure S5.
Next, a direct competition between white EFG1+/+ and EFG1+/− cells was performed. The two genotypes showed similar fitness levels for the first 48 h after which a subset of EFG1+/− cells switched to the gray state and largely swept the population (Figure 4C). Similar competitions were performed using EFG1+/+ and EFG1+/− cells mixed in a 90:10 ratio. White EFG1+/− cells again switched to the gray state in 2 out of 4 experiments and gray cells then took over the population (Figure 4D). In one experiment the EFG1+/− strain was lost (SC5314#3), while in another, both strains were retained out to 30 dpi and yet the EFG1+/− strain did not change its phenotype (SC5314#4) indicating the stochasticity of white-to-gray transitions in vivo (Figure S5C).
We also analyzed 3 clinical EFG1+/− strains for their behavior in the GI model. Strains 1619, BJ1097, and P75063 (all MTLa/α) were inoculated in the white state yet gray cells appeared (3 to 8 dpi) and subsequently took over the population in 8 out of 9 independent experiments (Figure 4E). In the majority of these cases white cells were still present at low frequencies even at 28 dpi. Together, these experiments establish that EFG1 hemizygosity provides an adaptive advantage to C. albicans as these cells stochastically convert to the gray state, and that this cell type exhibits increased fitness relative to white cells in the GI model.
Formation of the Gray State In Vivo Involves Mutational Loss of EFG1 Function
We examined gray cells arising during GI passage and found that EFG1-GFP expression was lost upon white-to-gray switching in multiple independent events (Figure S5B). Both de novo mutations and gene conversion events caused inactivation of EFG1. For example, gray cells arising in one experiment with SC5314 EFG1+/− cells had all undergone LOH at EFG1, whereas 95% of those in a second experiment had experienced LOH with the remaining 5% acquiring a de novo 10 bp deletion within EFG1 (Figure 4F). The three clinical isolates passaged in the GI also lost EFG1 expression upon forming the gray state (Figure S5B). This was often due to gene conversion at EFG1 (31/35 events), with frameshifting indels also observed (4/35 events, Figure 4F). In some cases, multiple events occurred within a single infecting population as seen with strain 1619 (expt. #1) where LOH (83%), an insertion (8%) and a deletion (8%) were all detected within EFG1 (Figure 4F). Indels again mapped to CAA/CAG repeats in EFG1 similar to those detected in vitro. These experiments establish that both engineered and natural EFG1 hemizygous strains undergo the white-to-gray transition in vivo due to gene conversion or de novo mutations that abolish EFG1 function, and that the resulting cells exhibit a significant fitness advantage in GI colonization over white cells.
Impact of Cell State Transitions on Fitness and Virulence in a Systemic Model
The relative fitness of white and gray states was also compared during disseminated infection. White EFG1+/− cells and gray efg1 null cells were co-inoculated (50:50) into the bloodstream and cells recovered from internal organs 7 dpi. We found that >90% of recovered cells in the kidney, spleen and liver were gray cells, whereas white and gray cells were present at equivalent numbers in the brain (Figure 5A). We also monitored host survival and found that white cells were more virulent than gray cells (Figure 5B) consistent with EFG1 representing a key virulence factor (Lo et al., 1997) and with an analysis of BJ1097 gray cells (Tao et al., 2014). These results suggest that the white-to-gray transition allows for a partial uncoupling of the ability of C. albicans cells to colonize most internal organs from the ability to cause morbidity.
Figure 5. Competitive fitness and virulence of MTLa/α white and gray cell states in disseminated infection.
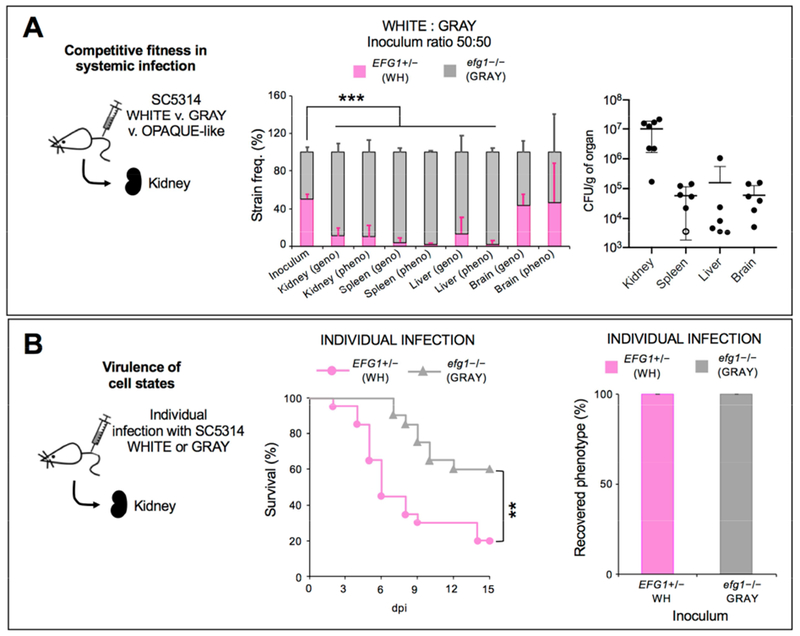
(A) Competitive fitness of C. albicans cells in a disseminated infection model. A 50:50 mix of white EFG1+/− and gray efg1−/− cells were co-inoculated into the murine tail vein and recovered 7 dpi from the kidney, spleen, liver and brain. Two independent experiments were performed using EFG1+/− NATR versus efg1−/− NATS cells (CAY7768 versus 7065, n = 3 mice) and efg1−/− NATR versus EFG1+/− NATS cells (CAY7769 versus 7064, n = 4 mice). ‘Geno’ indicates colonies were identified using the SAT1 marker and ‘pheno’ indicates the colony phenotype. Open circle indicates sample was excluded from genotype/phenotype analysis due to low CFUs (<10 colonies recovered). ***, P<0.001 by t test. Error bars, SD.
(B) Virulence and phenotypes of C. albicans in the disseminated infection model. Kaplan-Meier survival curves comparing the virulence of white and gray cells (CAY7064/7065). **, P <0.005 by log-rank test. n=20 mice each. Phenotypes of cells recovered from the kidney at 15 dpi (or when moribund). Error bars, SD.
Genetic and Epigenetic Mechanisms Act in Tandem to Control C. albicans Behavior In Vivo
MTLa/α strains are restricted from forming the opaque state and we therefore also assayed MTLa/a cells that can readily adopt this state. Opaque cells formed by efg1−/− cells will be referred to as opaque-like cells given their distinct properties (Figure S7A-C). Conventional EFG1+/+ opaque cells have been shown to exhibit reduced fitness in the GI, as well as instability and reduced virulence in the systemic model (Kvaal et al., 1997; Pande et al., 2013).
The fitness of MTLa/a white, gray, and opaque-like cells was compared by pairwise competitions in the GI tract. These experiments revealed that MTLa/a gray cells outcompeted MTLa/a white cells (Figure 6A) similar to the analogous MTLa/α cell types (Figure 4A). We also found that opaque-like cells converted to the gray state within 1-2 dpi and that these cells then rapidly outcompeted white cells (Figure 6B) or were equally competitive against cells inoculated in the gray state (Figure 6C).
Figure 6. Comparison of different MTLa/a cell states in infection.
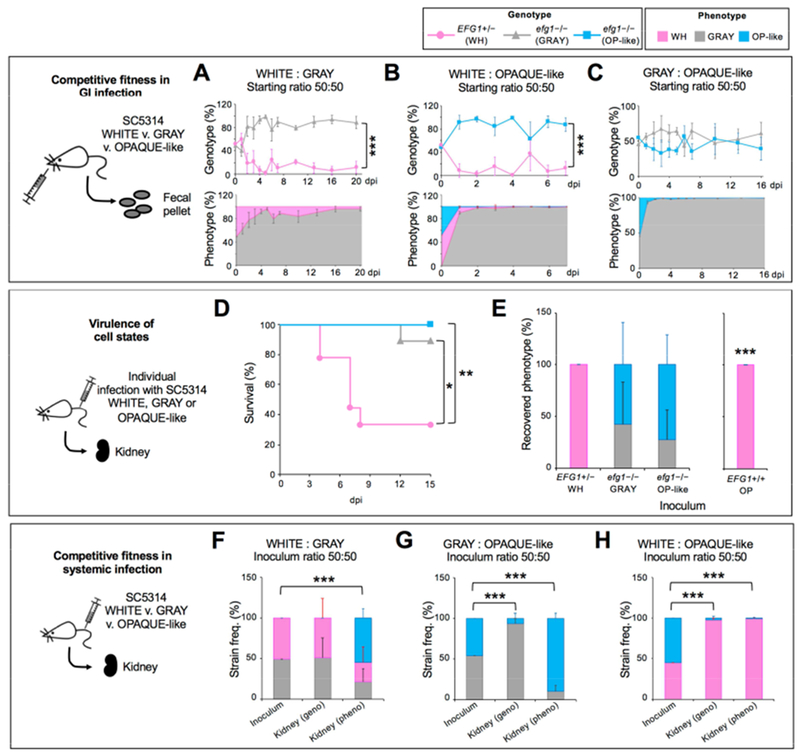
(A) Competitive fitness of C. albicans MTLa/a cells in a disseminated infection model. A 50:50 mix of white EFG1+/− and gray efg1−/− cells were co-inoculated in the GI (CAY6970/7869). Cells analyzed from fecal pellets. n=3 mice. ***, P<0.001 by t test.
(B) A 50:50 mix of white EFG1+/− and opaque-like efg1−/− cells were co-inoculated in the GI (CAY6970/7868) and cells analyzed from fecal pellets. n=4 mice. ***, P<0.001 by t test.
(C) A 50:50 mix of gray efg1−/− and opaque-like efg1−/− cells were co-inoculated in the GI (CAY7022/7868) and cells analyzed from fecal pellets. n=3 mice. ***, P<0.001 by t test.
(D) Kaplan-Meier survival curves comparing white, gray and opaque-like cells (CAY6970/7022/7023). *, P <0.05 and **, P <0.005 by log-rank test. n=9 mice each.
(E) Phenotypes of cells recovered from the kidney at 15 dpi (or when moribund) for EFG1+/− and efg1−/− strains and 3 dpi for EFG1+/+ isolate. n=7 mice for EFG1+/− and efg1−/− strains and n=4 mice for the EFG1+/+ isolate. Error bars, SD. ***, P<0.001 by t test compared to the inoculum.
(F) Competitive fitness of C. albicans cells in a disseminated model. A 50:50 mix of white EFG1+/− and gray efg1−/− cells (CAY6970/7869) were co-inoculated in the tail vein and recovered 7 dpi from the kidney. n=4 mice. ***, P<0.001 by t test.
(G) A 50:50 mix of gray efg1−/− and opaque-like efg1−/− cells (CAY7022/7868) were co-inoculated in the tail vein and recovered 7 dpi from the kidney. n=3 mice. ***, P<0.001 by t test.
(H) A 50:50 mix of white EFG1+/− and opaque-like efg1−/− cells (CAY6970/7868) were co-inoculated into the tail vein and recovered 7 dpi from the kidney. n=5 mice. ***, P<0.001 by t test.
In the tail-vein model, MTLa/a white cells were again the most virulent cell type, with little or no killing observed with gray or opaque-like cells (Figure 6D). White cells stably maintained this state, whereas gray and opaque-like cells underwent interconversion with one another (20-40% switching; Figure 6E). Pairwise competition assays revealed that white and gray cells were equally competitive with one another and both outcompeted opaque-like cells (see ‘genotypes’, Figure 6F-6H). Interestingly, however, cells inoculated in the gray state were mostly recovered in the opaque-like state, despite the low competitive fitness of opaque-like cells (Figure 6G). This could reflect high rates of gray-to-opaque-like switching in the kidney or that gray cells are favored early in infection and that opaque-like cells are favored later during infection.
Given the frequent recovery of opaque-like cells from the kidney, we also re-evaluated conventional (EFG1+/+) SC5314 opaque cells in the tail-vein model. In contrast to opaque-like cells, conventional opaque cells were highly unstable and completely reverted to the white state by 3 dpi (Figure 6E). These experiments establish that both genetic background (EFG1+/+ v. efg1−/−) and epigenetic cell state (white v. opaque or gray v. opaque-like) define C. albicans behavior in the host.
Discussion
In microbial species, the formation of different cellular subtypes can enable bet hedging, regulate virulence factor expression, promote immune evasion, and allow for a division of labor. Phenotypic heterogeneity is achieved via both genetic and epigenetic mechanisms, and the latter is exemplified by the C. albicans white-opaque switch in which transitions occur between two heritable cell types. However, C. albicans is also able to adopt a number of other phenotypic states for which there has been little mechanistic understanding. We reveal that formation of the ‘gray’ state is due to mutational events in isolates hemizygous for a key TF, establishing that genetic events also modulate switch-like behavior in this species. Furthermore, hemizygous strains undergo frequent transitions to the gray state in vivo, thereby impacting the commensal and pathogenic properties of C. albicans cells in the host.
Diversity in C. albicans Populations Underlies a Switch-Like Transition
We initially observed the white-to-gray transition in strains engineered to be EFG1 hemizygous and subsequently identified clinical isolates that are EFG1+/− and similarly undergo this switch. Multiple ORF-disrupting EFG1 mutations were identified in natural isolates, yet disruptive mutations in other white-opaque TFs were rare. Analysis of two collections of sequenced isolates revealed that 19% of infected individuals carried strains with disrupted EFG1 alleles, establishing that loss-of-function EFG1 mutations are frequent in natural populations. Recently, the genome sequences of 182 isolates were reported (Ropars et al., 2018) and a subset of these isolates were also found to carry disrupted EFG1 alleles, albeit at a lower frequency (4/182; see STAR Methods). Our results therefore establish that EFG1 is mutated in diverse collections of C. albicans isolates from around the world including those from commensal, OPC, and disseminated infections.
A Mutational Mechanism Underlies the White-to-Gray ‘Phenotypic Switch’
We initially presumed that the white-to-gray transition was epigenetic given its high frequency. Surprisingly, however, gray cells were found to arise via mutational loss of the functional EFG1 allele in strains hemizygous for this TF. This often involved de novo mutations within trinucleotide polyQ tracts, suggesting that DNA replication slippage or recombination contributes to this mutational pattern. Trinucleotide repeat instability is also associated with human disorders such as Huntington’s, where expansions in HTT gene repeats lead to neurological disease (Lopez Castel et al., 2010). The mutational pathways impacting gray state formation in C. albicans may therefore parallel those influencing the stability of polyQ tracts in higher eukaryotes.
While de novo mutations were responsible for a subset of white-to-gray transitions, the majority of events involved gene conversion. This is consistent with gene conversion being more frequent than de novo mutation in C. albicans (Forche et al., 2011) and that LOH can result in increased drug resistance in this species (Bennett et al., 2014; Coste et al., 2006; Ford et al., 2015; White, 1997). Mapping of LOH tracts often revealed localized gene conversion events at the EFG1 ORF. It is therefore possible that EFG1 represents a ‘fragile site’ that promotes DNA recombination, although fragile sites are typically located away from the centromere (Fungtammasan et al., 2012) whereas EFG1 is adjacent to the centromere on Chr R.
Parallel Mechanisms Generate Heterogeneity in Pathogens from Different Kingdoms
The mechanisms that promote adaptation in fungal pathogens include epigenetic switching, aneuploid formation, mutator states, and sexual/parasexual reproduction (Bennett et al., 2014; Calo et al., 2013; Heitman et al., 2014; Kwon-Chung and Chang, 2012; Noble et al., 2017). The current study establishes that C. albicans can also undergo high-frequency phenotypic change due to stochastic genetic events. We note that mutational mechanisms similarly promote variation in the model yeast Saccharomyces cerevisiae. Here, epigenetic control regulates the expression of FLO genes that define cellular phenotypes, but loss-of-function mutations in IRA1/IRA2 (GTPase-activating factors) can also activate FLO gene expression (Halme et al., 2004). Thus, genetic and epigenetic mechanisms act in concert to promote heterogeneity in both C. albicans and S. cerevisiae. The interplay between these mechanisms is striking in C. albicans as EFG1 is both a regulator of epigenetic white-opaque switching and the target of a mutational mechanism that increases phenotypic variation. Genetic and epigenetic regulation therefore combine to produce four (or more) phenotypic states that are readily distinguishable at the colony level (Figure 7).
Figure 7. Genetic and epigenetic mechanisms act together to generate phenotypic heterogeneity in C. albicans.
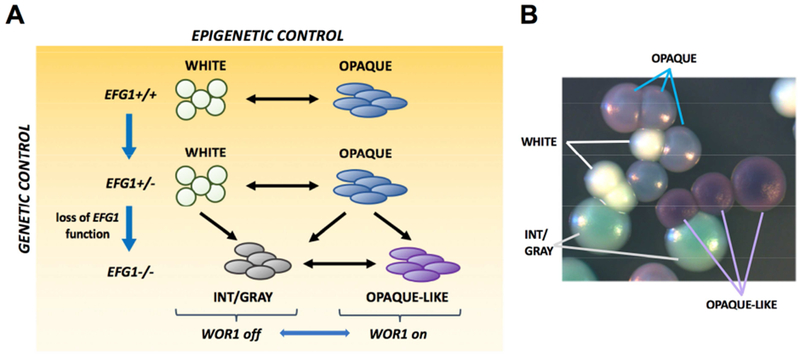
(A) Epigenetic switching occurs between white and opaque states. EFG1 is essential for the white state and mutational loss of this gene causes white cells to adopt the gray state. efg1 null cells can still undergo epigenetic switching between gray and opaque-like states. Both opaque and opaque-like states require WOR1 expression. C. albicans therefore uses both genetic and epigenetic mechanisms to adopt different cell states, with EFG1 playing a central role in both mechanisms.
(B) Four alternative cell states are distinguishable on CHROMagar medium.
See also Figure S7.
The genetic regulation of phenotypic diversity in C. albicans also shows parallels to phase variation in bacterial pathogens. Here, phenotypic change can arise via instability in hypermutable sequences (Bidmos and Bayliss, 2014; van der Woude and Baumler, 2004). This is exemplified in Neisseria gonorrhoeae, where changes in the number of polynucleotide repeats creates/removes premature stop mutations thereby regulating the expression of key virulence factors (Rotman and Seifert, 2014). We therefore note that mutational processes generate phenotypic heterogeneity in pathogens from different kingdoms. It is also expected that additional hemizygous loci will be identified whose inactivation can trigger switch-like behavior in C. albicans.
C. albicans Cell States Determine Fitness and Virulence in the Host
Loss of EFG1 function resulted in increased fitness of C. albicans cells in the GI, consistent with previous studies (Hirakawa et al., 2015; Pande et al., 2013; Pierce et al., 2013; Pierce and Kumamoto, 2012). Of greater import, clinical isolates that were naturally EFG1 hemizygous underwent frequent white-to-gray switching during GI passage, with gray cells subsequently taking over the population. These results establish that loss of EFG1 readily occurs in EFG1+/− strains passaged in the GI and that the resulting efg1 null (gray) cells then dominate this niche.
In a systemic infection, white cells were more virulent than gray cells, consistent with the critical role of EFG1 in pathogenesis (Lo et al., 1997; Park et al., 2005). In contrast, however, co-infection experiments revealed that gray cells outcompeted white cells in most internal organs, indicating that virulence (mortality) and fitness (relative fungal burden) may be partially uncoupled. This extends prior observations in which kidney fungal burdens did not correlate with virulence in strains with a decreased ability to undergo filamentation (Bendel et al., 2003; Felk et al., 2002; Saville et al., 2003). Additional studies will determine if differences in filamentation, metabolic activity, immune responses or toxin secretion (Moyes et al., 2016) underlie these differences in fitness and pathogenesis.
While most of our analyses examined MTL heterozygous strains, we also examined MTL homozygous strains which adopted white/opaque states when EFG1+ and gray/opaque-like states when efg1 null. Here, white cells were again the most virulent, whereas gray cells outcompeted white cells in the GI tract. While both opaque and opaque-like cells were unstable in the GI, there was a complex interplay between different cell types in the systemic model; white and gray cells both outcompeted opaque-like cells, and yet gray cells often switched to the opaque-like state in the kidney. These results further highlight that phenotypic transitions between alternative cell states frequently occur in vivo with consequences for infection of host niches.
Concluding Remarks
Our observation that clinical C. albicans isolates are a mixture of different genotypes (0, 1 or 2 functional EFG1 alleles) suggests that each of these genotypes is under positive selection in nature. EFG1 hemizygous cells exhibit phenotypic differences with EFG1+/+ cells (Glazier et al., 2017; Zavrel et al., 2012) so that hemizygous cells could provide a heterozygote advantage in certain environments. As evidenced here, the efg1 null state also provides a fitness advantage in several infection types, yet previous work has established that EFG1 function is critical for infection of other host niches (Harriott et al., 2010; Stoldt et al., 1997; Yano et al., 2016). This suggests that different EFG1 genotypes are maintained in C. albicans populations due to the multiple selection pressures encountered by the species. Regardless of how selection is precisely shaping natural populations, we establish that genetic and epigenetic mechanisms act in tandem to increase phenotypic heterogeneity in C. albicans and that different cell state transitions provide an effective strategy for regulating interactions with the mammalian host.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Richard J. Bennett (Richard_Bennett@brown.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
BALB/c mice (female, 15-20 g, 8-9 weeks old) were purchased from Charles River Laboratories. All animals were housed in a centralized and AAALAC-accredited research animal facility that is fully staffed with trained husbandry, technical and veterinary personnel. For antibiotic treatment, mice were given 1,500 U/ml of penicillin, 2 mg/ml of streptomycin and 5% glucose for taste in the drinking water. Mice were randomly grouped to perform the experiments. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals as defined by the National Institutes of Health (PHS Assurance #A3284-01). Animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Brown University.
Yeast
Candida albicans SC5314 was obtained from A. Johnson (UCSF), a set of 20 clinical isolates (Hirakawa et al., 2015) were provided by D. Soll (U. Iowa), and 43 serial isolates (Ford et al., 2015) were provided by C. Cuomo (Broad Institute) and T. Patterson (UTHSA). Clinical isolates BJ1097, HJ039, and HJ071 (Tao et al., 2014) were provided by G. Huang (Fudan U.). Additional strains used in this study were derived from C. albicans isolates SC5314, P75063, 1619, P37037, BJ1097, P76055, P37005, GC75, P57072 backgrounds. Construction of C. albicans strains is described below.
METHOD DETAILS
Media and reagents
Yeast extract peptone dextrose (YPD) and synthetic complete dextrose (SCD) media were prepared as previously described (Guthrie and Fink, 1991). YPS medium was prepared as for YPD but using 2% sucrose instead of 2% dextrose. YPD containing 200 μg/mL nourseothricin (Werner BioAgents, Jena, Germany) was used to select nourseothricin-resistant (NATR) strains. CHROMagar Candida medium (CHROMagar Candida, Paris, France) was used to differentiate C. albicans phenotypes.
Plasmids and strains
Oligonucleotides and strains described in this paper are listed in Tables S2 and S3, respectively. To delete EFG1 in SC5314, PCR using oligos 2251/2252 and 2253/2254 was used to amplify the 5’ and 3’ flanks of the EFG1 gene. PCR was also used to amplify HIS1 or LEU2 markers from plasmids pSN52 and pSN40, respectively, as described (Noble and Johnson, 2005). Products were combined by fusion PCR using oligos 2251/2254 and the HIS1-containing product transformed into SC5314-derived strains SN87 (Noble and Johnson, 2005) and RBY1132 (Bennett and Johnson, 2006) to generate efg1Δ/EFG1 strains in MTLa/α and MTLa/a backgrounds (CAY6970, 6973, 6976 and 7064). Correct integration of the EFG1 deletion cassette was verified by PCR across the 5’ and 3’ disruption junctions using oligos 2284/690 and 2285/691. To generate efg1Δ/Δ null mutants, the LEU2-containing PCR fusion product was transformed into efg1Δ/EFG1 strains (CAY6970) and junction checks performed by PCR with oligos 2284/692 and 2285/693 to generate efg1Δ/Δ strains. Absence of the EFG1 ORF was confirmed by PCR with oligos 2291/2292. For construction of heterozygous efg1Δ/EFG1 strains, monitoring of which EFG1 allele was deleted was performed by PCR of the remaining EFG1 ORF (oligos 3497/3499) and digestion with KpnI which cuts once in EFG1 allele A (CR_07890W_A) at position 1180, but does not cut in EFG1 allele B (CR_07890W_B) (Skrzypek et al., 2017).
To delete EFG1 in clinical isolates of C. albicans, a pSFS-EFG1 knockout plasmid (pRB721) was constructed by PCR amplification of the 5’ and 3’ flanks of EFG1 with oligos 4104/4105 and 4106/4107, and cloning into plasmid pSFS2A (Reuss et al., 2004) using ApaI/XhoI and SacI/SacII, respectively. pRB721 was linearized with ApaI/SacI before transformation. One copy of EFG1 was deleted in isolates P76055, P37005, GC75 and P57072 to generate CAY8673, CAY8724, CAY8718 and CAY8721, respectively. PCR with oligos 2284/11 and 2286/12 was used to check the 5’ and 3’ junctions of transformants.
To generate a strain expressing the Efg1(G252D) mutant, a plasmid containing SC5314 EFG1 (allele B) and its promoter region was constructed by PCR amplification of this region with oligos 1838/1839. The PCR product was cloned into pSFS2A (Reuss et al., 2004) using ApaI and KpnI to produce plasmid pRB360. This plasmid was mutagenized by the QuickChange method using oligos 4290/4291. The mutated plasmid was confirmed to contain the G252D mutation by Sanger sequencing. The resulting plasmid (pRB823) was digested with HpaI and transformed into efg1Δ/Δ strain CAY6977 resulting in efg1Δ/Δ+EFG1(G252D) strain CAY8646. Integration into the EFG1 locus was confirmed by PCR using oligos 1840/2933.
To construct Efg1-GFP and Wor1-GFP strains, PCR was performed on plasmid pADH76 (a gift from Aaron Hernday, UCMerced) with oligos 3821/3685 and 3819/3687, respectively. The resulting PCR products were transformed into CAY6970 to generate efg1Δ/EFG1-GFP (CAY8128) and efg1Δ/EFG1 WOR1/WOR1-GFP (CAY8131) strains. Correct integration was checked by PCR with oligos 11/828 and 11/25 for EFG1-GFP and WOR1-GFP, respectively. Efg1-GFP and Wor1-GFP reporters were also generated into a wildtype MTLa/a background. Plasmids pMBL162 and pMBL165 were digested with SphI and AatII as described (Lohse and Johnson, 2010) and introduced into RBY1136 to generate EFG1-GFP (CAY7292) and WOR1-GFP (CAY7286).
The same strategy described above was used to tag the intact EFG1 in clinical isolates with a GFP marker. PCR was used to amplify GFP using oligos 3821/3685 and the product transformed into P37037, P75063, 1619 and BJ1097 to create EFG1null/EFG1-GFP (CAY9155, 9159, 9165, 9195). Integration was checked by PCR with oligos 11/828 and fluorescence microscopy.
C-terminal epitope tagging of Efg1 was performed by PCR amplification of a 13 × MYC-SAT1 construct (Hernday et al., 2010) with oligos 3685/3821 to target the construct to the endogenous EFG1 locus. Epitope tagging was performed in wildtype and efg1Δ/EFG1 strains so that the remaining allele of EFG1 was engineered to express Efg1 with a C-terminal 13 × myc tag generating CAY8162 and CAY8940, respectively. This strain was able to form the white phenotypic state indicating that C-terminal tagging of the gene did not interfere with Efg1 function.
To generate NATR strains for in vivo competition assays, plasmid pDis3 was introduced into a neutral locus in the genome (Gerami-Nejad et al., 2013). This plasmid was linearized with NgoMIV and transformed into efg1Δ/EFG1 and efg1Δ/efg1Δ strains to create NATR versions of CAY7768, CAY7868 and CAY7869. PCR with primers 3055/3056 was used to check correct integration.
To re-integrate HIS1 gene into his1 auxotroph strains, PCR with oligos 4114/4115 was used to amplify the HIS1 gene from SC5314 and transformed into target strains. Integration was checked by PCR with oligos 2609/2610 in strain CAY8282 and CAY9962.
One allele of ADE2 was deleted from strain SN87 by PCR amplification of the HIS1 gene from pSN52 with oligos 4864/4865 that have flanking homology to the ADE2 gene and integration checked by PCR with oligos 690/397 and 691/398 in strain CAY9628.
Scanning electron microscopy
C. albicans cells were grown on SCD medium at 20°C for 72 h. Cells were suspended in water to an OD600 of 1 and incubated on poly-L-lysine coated coverslips. Cells were then fixed in 2.5% glutaraldehyde in 0.1 M Na-Cacodylate buffer, pH 7.4, overnight at 4°C. Cells were washed with 0.1 M Na-Cacodylate buffer, pH 7.4, and post-fixed in 0.1% aqueous osmium tetroxide in 0.1 M Na-Cacodylate buffer, pH 7.4, at 20°C for 90 min. Cells were dehydrated using a graded ethanol series starting with 30% ethanol and increasing in 15% increments. The prepared coverslips were dried in a Ladd critical point drier and coated in gold in an Emitech K-550 sputter coater (2 cycles, 20 mA at the 45 nm position for 3 min). Cells were imaged using a Hitachi 2700 Scanning Electron Microscope with Quartz PCI software.
Phenotypic assays
To obtain cells in different phenotypic states cells were passaged for single colonies on YPD, SCD or CHROMagar Candida media at 22°C. Stochastic transitions between states were sufficient to obtain the phenotypic states described in this work. Quantitative analysis of phenotypes was performed by plating pure populations of cells from one phenotypic state onto CHROMagar Candida or YPD for single colonies. Transition events to a different phenotypic state were subsequently quantified (both sectors and whole colonies) after growth at 22°C for 7 days, analogous to previous studies (Miller and Johnson, 2002).
Analysis of filamentous growth
For analysis of filamentation phenotypes, cells were cultured in SCD overnight at 22°C and individual colonies plated onto YPD or Spider and incubated at 37°C for 5 days. Colony morphologies were imaged using an Infinity 2 digital camera with Infinity analyzer software (Lumenera Corporation). For embedded phenotypes, cells were prepared as described above, diluted in molten YPS and plates allowed to set. Embedded cells were incubated at 22°C and 37°C, and morphologies examined after 5 days using a Zeiss Axio Observer Z1.
RNA-Seq analysis
Cells were grown in SCD overnight, diluted in fresh SCD to an OD600 of 0.1 and harvested in exponential growth once the OD600 reached 1.0. The purity of cultures in different cell states was confirmed by plating cells to CHROMagar Candida. The MasterPure Yeast RNA Purification Kit (Epicentre) was used to purify RNA. RNA was treated with Turbo DNaseI (Ambion) and polyA RNA isolated and used to construct strand-specific libraries using the dUTP second strand marking method as previously described (Parkhomchuk et al., 2009). Samples were pooled and sequenced on the Illumina HiSeq 2500 (Beckman) to generate 101 base reads.
To measure gene expression, reads were aligned to the C. albicans SC5314 A21 genome and assigned to individual A21 genome features (http://candidagenome.org/download/gff) using STAR (version 2.5)(Dobin et al., 2013). Transcript abundance measurements were calculated using HTseq (version 0.9.1)(Anders et al., 2015) as transcripts per million (TPM). Differentially expressed genes were identified using EdgeR (Bioconductor version 3.5)(Robinson et al., 2010). Gene Ontology (GO) analysis was performed using the GO term finder incorporated into the Candida Genome Database (http://www.candidagenome.org/cgi-bin/GO/goTermFinder)(Inglis et al., 2012) with corrections for multiple hypothesis testing. All of the RNA-seq data associated with this project has been uploaded to GEO (Gene Expression Omnibus) and has a provisional accession number (GSE105399).
Quantitative RT-PCR Analysis
RNA for quantitative RT-PCR assays were prepared as for whole genome RNA-seq. cDNA was prepared using SuperScript IV reverse transcriptase (Thermo Fisher) using the manufacturer’s protocol. Quantitative PCR was performed using SYBR green master mix (Thermo Fisher) run on a LightCycler 96 instrument (Roche). RNA expression was determined relative to actin controls. Oligos used for PCR were 819/828 for EFG1, 25/26 for WOR1, and 3429/3430 for ACT1.
Fluorescence microscopy
To image Efg1-GFP and Wor1-GFP expressing strains, cells were grown in exponential phase in SCD medium and examined by Differential Interference Contrast (DIC) and GFP microscopy using an Axio Observer Z1 Microscope (Zeiss).
Western blotting
Cells were incubated on SCD plates and grown overnight at 22°C. Cells were removed from colonies on plates and lysates prepared by boiling for 5 minutes in Laemmli buffer. Samples were electrophoresed on a 4-15% TGX Strain-Free gel (Bio-Rad), then transferred to PVDF membrane (Immobilon-P, Millipore). The membrane was blocked with 5% skim milk powder in TBS-T at room temperature, followed by incubation with a 1:20,000 mouse anti-MYC antibody (Millipore #05-724) in blocking buffer for 1 h. The membrane was washed and incubated with a 1:20,000 dilution of donkey anti-mouse HRP antibody (Jackson ImmunoResearch #715-036-151) in blocking buffer for 1 h. After washing with TBS-T without milk, membranes were treated using Supersignal West Pico Chemiluminescent Substrate (Thermo Fisher) and the ECL signal detected using a ChemiDoc MP imaging system (Bio-Rad). To control for gel loading, images showing total protein in the whole cell lysate are shown prior to transfer.
Flow cytometry of C. albicans cells
To monitor the expression of EFG1, the functional EFG1 allele was tagged by a GFP marker in C. albicans. Cells were grown in SCD medium at 22°C overnight, washed in PBS buffer for 3 times, and resuspended in PBS. Flow cytometry analysis was performed on a BD FACSAria III. 100,000 cells were examined for each isolate and data was analyzed by FlowJo software.
Analysis of C. albicans genome sequences
Whole genome sequences of the 21 clinical isolates (NCBI BioProject ID: PRJNA193498) and 43 clinical isolates from the OPC patients (NCBI BioProject ID: PRJNA257929) were obtained from previous studies (Ford et al., 2015; Hirakawa et al., 2015) and aligned to the SC5314 reference genome v21 (http://www.candidagenome.org). SNP variants were called using the Genome Analysis Toolkit (GATK) (McKenna et al., 2010). Polymorphic features of high fidelity were filtered using the GATK VariantFiltration tool with pre-specified hard filters (QD<2.0, MQ<40.0, FS>60.0, MQRankSum<-12.5, ReadPosRankSum<-8.0). Subsets of variant calls were manually checked in the Integrative Genomics Viewer (IGV). EFG1 mutations in clinical isolates P75063, P37037, 1619 and 4018 were confirmed here by Sanger sequencing, while that in P94015 was also sequenced (Hirakawa et al., 2015). An additional heterozygous EFG1 mutation (E282K) was found in strain 1649 which may also impact EFG1 function but was not included in the current analysis. We similarly examined SC5314 and the 63 clinical isolates for potential ORF-disrupting mutations in established white-opaque network transcription factors including AHR1, CZF1, SSN6, WOR1, WOR2, WOR3, and WOR4 (Hernday et al., 2016; Lohse et al., 2013; Lohse and Johnson, 2016; Zordan et al., 2007). Recently, Ropars et al. sequenced the genomes of 182 C. albicans isolates from diverse sources (Ropars et al., 2018). Four of the 182 strains contained disruptive mutations in EFG1: CEC2021, heterozygous mutation disrupting the start codon, CEC4497, heterozygous nonsense mutation A128*, CEC2022, heterozygous nonsense mutation Q130*, and CEC4486, homozygous V230L mutation within the conserved DNA binding domain.
Analysis of de novo mutations in EFG1
To identify EFG1 mutations in SC5314-derived strains, PCR was performed with oligos 4476/2910 on gray cells derived from efg1Δ/EFG1 white cells (CAY6970/7064). For gray cells found to contain the EFG1 amplicon, DNA fragments were gel purified using a GeneJET Gel Extraction Kit (Thermo Fisher Scientific) and Sanger sequencing performed. For clinical isolates, PCR was performed on gray cells originating from EFG1-GFP tagged strains (EFG1null/EFG1-GFP) with oligos 4476/1473 or 4476/2381 to amplify the intact EFG1 allele. Amplicons were purified using a DNA Clean & Concentrator kit (Zymo Research) and analyzed by Sanger sequencing. Mutations were determined by BLAST at the Candida Genome Database (http://www.candidagenome.org/cgi-bin/compute/blast_clade.pl).
SNP-RFLP analysis
SNP-RFLP was performed as described previously (Forche et al., 2009). Primers and restriction enzymes are listed in Table S4. SNP markers were amplified by PCR in a total volume of 25 μL. Half volume of the PCR reaction was separated and subsequently cut by restriction enzyme overnight. Genotypes were analyzed by running an agarose gel with digested products along with undigested PCR fragments.
Virulence experiments
BALB/c mice (Charles River Laboratories, MA, USA, 15-20 g, 8-9 weeks old) were used for virulence assays. White and gray cells from an SC5314 efg1/EFG1 strain (CAY7064 and CAY7065 cells, respectively) were grown in 3 mL of SCD overnight at 22°C. The overnight culture was diluted 10-fold in SCD and diluted cultures grown for 4 h at 22°C. Cells were harvested and sterile phosphate-buffered saline (PBS) used to wash the cells three times. Mice were infected via tail-vein injection using 27G1/2 needles (Becton Dickinson) with an inoculum of 6.0 × 105 colony forming units (CFUs) in 0.2 ml in PBS. Mouse weight, activity, and behavior were observed twice a day and euthanized when morbid (defined as weight loss >15%, hunched posture, reduced activity). CFUs and C. albicans cell phenotypes were determined after euthanasia by plating of cells from kidneys onto CHROMagar.
Murine competition assays
BALB/c mice (Charles River Laboratories, MA, 15-20 g, 8-9 weeks old) were used to perform competition assays in systemic and commensal infection models. For the systemic model, cells were grown and prepared as for virulence assays, above. The inoculum was a 50:50 mixture of one NAT-resistant (NATR) isolate and one NAT-sensitive (NATS) isolate. A total inoculum of ~6.0 × 105 CFUs was used to infect mice via the tail-vein and four mice were used for each competition experiment. The activity, behavior and weight of the mice were monitored twice daily and mice were euthanized when morbid (defined as weight loss >15%, hunched posture, reduced activity) or 7 days post-infection. Cells were recovered from the kidneys and plated on YPD, YPD+NAT and CHROMagar Candida media. Genotype data (‘geno’) was determined by examining the numbers of NATR and NATS colonies on YPD+NAT and YPD media. The phenotypes (‘pheno’) of recovered cells were examined following growth on CHROMagar to determine the phenotypic composition of the fungal population.
For the commensal GI model, an antibiotics-treated mouse model was used (Pande et al., 2013). Unless stated otherwise, animals were housed together with free access to food and water. Mice were given standard rodent chow (FormuLab 5001; PMI Nutrition International), and their water was supplemented with antibiotics (1,500 U/ml of penicillin, 2 mg/ml of streptomycin) and 5% glucose for taste. Antibiotic treatment was initiated 4 days prior to infection, and mice remained on antibiotics for the duration of the infection. Cells were prepared as for virulence assays (see above) and washed with sterile water three times. Mice were infected with a total of ~108 CFUs in 0.5 ml of H2O via oral gavage using plastic feeding tubes (Instech Laboratories). The inoculum was composed of a 50:50, 90:10, or 99:1 mixture of two isolates (one NATR and one NATS). Three mice were infected for each competition experiment. Mouse fecal pellets were collected daily and homogenized using a PBS solution supplemented with antibiotics (500 μg/ml penicillin, 500 μg/ml ampicillin, 250 μg/ml streptomycin, 225 μg/ml kanamycin, 125 μg/ml chloramphenicol, and 25 μg/ml doxycycline). Cells were plated to YPD, YPD+NAT and CHROMagar Candida media to determine the genotype and phenotype of the fungal population, as described for the systemic infection.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of individual experiments can be found in the manuscript text and figure legends. Additional details of data analysis are provided below.
Statistical analysis
Phenotypic switching assays were performed three times in independent experiments. For RNA-seq analysis, differentially expressed genes were defined using the approach of quantile-adjusted conditional maximum likelihood followed by an exact test. In this study, we show differentially expressed genes with at least a 2-fold change in abundance and q < 0.05. For animal experiments, each group contained 2-5 mice both for GI infections and for competition assays in the systemic model. For virulence assays, 20 mice and 9 mice were used for infections with a/α and a/a cells, respectively. The actual number of mice used in each experiment is provided in the corresponding figure legend. The log-rank Mantel-Cox test was used for pairwise comparison of survival curves. Student’s t-test was used to analyze statistical significance in mouse GI and systemic infection models.
DATA AND SOFTWARE AVAILABILITY
All of the RNA-seq data associated with this project has been uploaded to GEO (Gene Expression Omnibus) and has a provisional accession number (GSE105399).
Supplementary Material
Table S1. RNA-sequencing data for C. albicans cells in different cell states and genetic backgrounds. Related to Figure 3. TPM values, differentially expressed genes and GO term analysis are listed in separate sheets. * indicates cells derived from efg1Δ/EFG1 white state.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-MYC antibody | Millipore | CAT# 05-724 |
| Donkey anti-mouse HRP antibody | Jackson ImmunoResearch | CAT# 715-036-151 |
| Supersignal West Pico chemiluminescent substrate | Thermo Fisher | CAT# PI34580 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nourseothricin | Werner BioAgents | CAT# 5.010.000 |
| Chloramphenicol | Fisher | CAT# BP904 |
| Streptomycin sulfate | MP Biomedicals | CAT# 0219454180 |
| Penicillin-G potassium | Fisher Bioreagents | CAT# BP914100 |
| Sucrose | Sigma-Aldrich | CAT# S0389 |
| Dextrose | Fisher Scientific | CAT# D16-3 |
| 2.5% glutaraldehyde in 0.1 M Na-Cacodylate buffer, pH 7.4 | Electron Microscopy Sciences | CAT# 15960 |
| 0.1 M Na-Cacodylate buffer, pH 7.4 | Electron Microscopy Sciences | CAT# 11652 |
| Osmium tetroxide | Electron Microscopy Sciences | CAT# 19150 |
| Ethanol | Pharmco-Aaper | CAT# 111000200 |
| Ampicillin sodium salt | Fisher Bioreagents | CAT# BP1760 |
| Kanamycin sulfate | Fisher Bioreagents | CAT# BP906-5 |
| Doxycycline hyclate | Sigma-Aldrich | CAT# D9891 |
| Critical Commercial Assays | ||
| MasterPure Yeast RNA Purification Kit | Epicentre | CAT# MPY03100 |
| DNA Clean & Concentrator kit | Zymo Research | CAT# 11-305C |
| CHROMagar Candida medium | Fisher Scientific | CAT# NC9514149 |
| Turbo DNase | Fisher Scientific | CAT# am2238 |
| SuperScript IV Reverse Transcriptase | Thermo Fisher | CAT# 18090050 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | CAT# 172-5121 |
| 4-15% TGX Strain-Free gel | Bio-Rad | CAT# 4568084 |
| Deposited Data | ||
| Raw data files for RNA sequencing | NCBI Gene Expression Omnibus | GSE105399 |
| Experimental Models: Organisms/Strains | ||
| Mouse: BALB/c | Charles River Laboratories | CAT# 028 |
| Candida albicans: full strain list is presented in Table S3 | This paper | N/A |
| Oligonucleotides | ||
| Full list of primers is presented in Table S2 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: pRB360 | This paper | N/A |
| Plasmid: PRB823 | This paper | N/A |
| Plasmid: pRB721 | This paper | N/A |
| Plasmid: pADH76 | Aaron Hernday, UCMerced | N/A |
| Plasmid: pMBL162 | (Lohse and Johnson, 2010) | N/A |
| Plasmid: pMBL165 | (Lohse and Johnson, 2010) | N/A |
| Plasmid: pSFS-13 × MYC-SAT1 | (Hernday et al., 2010) | N/A |
| Plasmid: pDis3 | (Gerami-Nejad et al., 2013) | N/A |
| Software and Algorithms | ||
| EdgeR (version 3.5) | (Robinson et al., 2010) | https://www.bioconductor.org/packages/release/bioc/html/edgeR.html |
| HTseq (version 0.9.1) | (Anders et al., 2015) | https://htseq.readthedocs.io/en/release_0.10.0/ |
| STAR (version 2.5) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| GO term finder | (Inglis et al., 2012) | http://www.candidagenome.org/cgi-bin/GO/goTermFinder |
| Genome Analysis Toolkit | Broad Institute | https://software.broadinstitute.org/gatk/ |
| Integrative Genomics Viewer | Broad Institute | http://software.broadinstitute.org/software/igv/ |
| BLAST | Candida Genome Database | http://www.candidagenome.org/cgi-bin/compute/blast_clade.pl |
| FlowJo (version 10.2) | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo |
Highlights.
Hemizygosity of the EFG1 TF licenses a phenotypic transition in C. albicans cells
Numerous clinical isolates are EFG1 hemizygous and thus can undergo this transition
This ‘phenotypic switch’ corresponds to mutational loss of EFG1 function
TF loss frequently occurs during murine infections and drives host adaptation
Acknowledgements
We thank J. Berman, A. Hernday, A. Johnson and M. Lohse for plasmids, C. Cuomo, G. Huang, A. Johnson, and T. Patterson for strains, I. Escobar and J. Howells for assistance with strain construction, and C. Cuomo for genome analysis advice. This work was supported by NIH grants AI081704/AI141893 and by a Burroughs Wellcome Fund PATH award to R.J.B.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Ackermann M (2015). A functional perspective on phenotypic heterogeneity in microorganisms. Nat. Rev. Microbiol 13, 497–508. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendel CM, Hess DJ, Garni RM, Henry-Stanley M, and Wells CL (2003). Comparative virulence of Candida albicans yeast and filamentous forms in orally and intravenously inoculated mice. Critical Care Med. 31, 501–507. [DOI] [PubMed] [Google Scholar]
- Bennett RJ, Forche A, and Berman J (2014). Rapid mechanisms for generating genome diversity: whole ploidy shifts, aneuploidy, and loss of heterozygosity. Cold Spring Harb. Perspect. Med 4, a019604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett RJ, and Johnson AD (2006). The role of nutrient regulation and the Gpa2 protein in the mating pheromone response of C. albicans. Mol. Microbiol 62, 100–119. [DOI] [PubMed] [Google Scholar]
- Bidmos FA, and Bayliss CD (2014). Genomic and global approaches to unravelling how hypermutable sequences influence bacterial pathogenesis. Pathogens 3, 164–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, and White TC (2012). Hidden killers: human fungal infections. Sci. Transl. Med 4, 165rv113. [DOI] [PubMed] [Google Scholar]
- Calo S, Billmyre RB, and Heitman J (2013). Generators of phenotypic diversity in the evolution of pathogenic microorganisms. PLoS Pathog. 9, e1003181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste AT, Turner V, Ischer F, Morschhauser J, Forche A, Selmecki A, Berman J, Bille J, and Sanglard D (2006). A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at Chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics 172, 2139–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deitsch KW, Lukehart SA, and Stringer JR (2009). Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat. Rev. Microbiol 7, 493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedt T, Krishnamurthy S, Bockmuhl DP, Tebarth B, Stempel C, Russell CL, Brown AJ, and Ernst JF (2004). APSES proteins regulate morphogenesis and metabolism in Candida albicans. Mol. Biol. Cell 15, 3167–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene IV, Farrer RA, Hirakawa MP, Agwamba K, Cuomo CA, and Bennett RJ (2018). Global analysis of mutations driving microevolution of a heterozygous diploid fungal pathogen. Proc. Natl. Acad. Sci. USA 115, E8688–E8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felk A, Kretschmar M, Albrecht A, Schaller M, Beinhauer S, Nichterlein T, Sanglard D, Korting HC, Schafer W, and Hube B (2002). Candida albicans hyphal formation and the expression of the Efg1-regulated proteinases Sap4 to Sap6 are required for the invasion of parenchymal organs. Infect. Immun 70, 3689–3700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forche A, Abbey D, Pisithkul T, Weinzierl MA, Ringstrom T, Bruck D, Petersen K, and Berman J (2011). Stress alters rates and types of loss of heterozygosity in Candida albicans. MBio 2, e00129–00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forche A, Magee PT, Magee BB, and May G (2004). Genome-wide single-nucleotide polymorphism map for Candida albicans. Eukaryot. Cell 3, 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forche A, Steinbach M, and Berman J (2009). Efficient and rapid identification of Candida albicans allelic status using SNP-RFLP. FEMS Yeast Res. 9, 1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CB, Funt JM, Abbey D, Issi L, Guiducci C, Martinez DA, Delorey T, Li BY, White TC, Cuomo C, et al. (2015). The evolution of drug resistance in clinical isolates of Candida albicans. eLife 4, e00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fungtammasan A, Walsh E, Chiaromonte F, Eckert KA, and Makova KD (2012). A genome-wide analysis of common fragile sites: what features determine chromosomal instability in the human genome? Genome Res. 22, 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerami-Nejad M, Zacchi LF, McClellan M, Matter K, and Berman J (2013). Shuttle vectors for facile gap repair cloning and integration into a neutral locus in Candida albicans. Microbiology 159, 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazier VE, Murante T, Murante D, Koselny K, Liu Y, Kim D, Koo H, and Krysan DJ (2017). Genetic analysis of the Candida albicans biofilm transcription factor network using simple and complex haploinsufficiency. PLoS Genet. 13, e1006948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, and Fink GR (1991). Guide to Yeast Genetics and Molecular Biology (San Diego: Academic Press; ). [Google Scholar]
- Halme A, Bumgarner S, Styles C, and Fink GR (2004). Genetic and epigenetic regulation of the FLO gene family generates cell-surface variation in yeast. Cell 116, 405–415. [DOI] [PubMed] [Google Scholar]
- Harriott MM, Lilly EA, Rodriguez TE, Fidel PL Jr., and Noverr MC (2010). Candida albicans forms biofilms on the vaginal mucosa. Microbiology 156, 3635–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman J, Carter DA, Dyer PS, and Soll DR (2014). Sexual reproduction of human fungal pathogens. Cold Spring Harbor Perspect. Med. 4, a019281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernday AD, Lohse MB, Fordyce PM, Nobile CJ, DeRisi JL, and Johnson AD (2013). Structure of the transcriptional network controlling white-opaque switching in Candida albicans. Mol. Microbiol 90, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernday AD, Lohse MB, Nobile CJ, Noiman L, Laksana CN, and Johnson AD (2016). Ssn6 defines a new level of regulation of white-opaque switching in Candida albicans and is required for the stochasticity of the switch. MBio 7, e01565–01515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernday AD, Noble SM, Mitrovich QM, and Johnson AD (2010). Genetics and molecular biology in Candida albicans. Methods Enzymol. 470, 737–758. [DOI] [PubMed] [Google Scholar]
- Hirakawa MP, Martinez DA, Sakthikumar S, Anderson MZ, Berlin A, Gujja S, Zeng Q, Zisson E, Wang JM, Greenberg JM, et al. (2015). Genetic and phenotypic intra-species variation in Candida albicans. Genome Res. 25, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Wang H, Chou S, Nie X, Chen J, and Liu H (2006). Bistable expression of WOR1, a master regulator of white-opaque switching in Candida albicans. Proc. Natl. Acad. Sci. USA 103, 12813–12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis DO, Arnaud MB, Binkley J, Shah P, Skrzypek MS, Wymore F, Binkley G, Miyasato SR, Simison M, and Sherlock G (2012). The Candida genome database incorporates multiple Candida species: multispecies search and analysis tools with curated gene and protein information for Candida albicans and Candida glabrata. Nucleic Acids Res. 40, D667–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvaal CA, Srikantha T, and Soll DR (1997). Misexpression of the white-phase-specific gene WH11 in the opaque phase of Candida albicans affects switching and virulence. Infect. Immun 65, 4468–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon-Chung KJ, and Chang YC (2012). Aneuploidy and drug resistance in pathogenic fungi. PLoS Pathog. 8, e1003022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan CY, Newport G, Murillo LA, Jones T, Scherer S, Davis RW, and Agabian N (2002). Metabolic specialization associated with phenotypic switching in Candida albicans. Proc. Natl. Acad. Sci. USA 99, 14907–14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo HJ, Kohler JR, DiDomenico B, Loebenberg D, Cacciapuoti A, and Fink GR (1997). Nonfilamentous C. albicans mutants are avirulent. Cell 90, 939–949. [DOI] [PubMed] [Google Scholar]
- Lohse MB, Hernday AD, Fordyce PM, Noiman L, Sorrells TR, Hanson-Smith V, Nobile CJ, DeRisi JL, and Johnson AD (2013). Identification and characterization of a previously undescribed family of sequence-specific DNA-binding domains. Proc. Natl. Acad. Sci. USA 110, 7660–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MB, and Johnson AD (2010). Temporal anatomy of an epigenetic switch in cell programming: the white-opaque transition of C. albicans. Mol. Microbiol 78, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MB, and Johnson AD (2016). Identification and characterization of Wor4, a new transcriptional regulator of white-opaque switching. G3 (Bethesda) 6, 721–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez Castel A, Cleary JD, and Pearson CE (2010). Repeat instability as the basis for human diseases and as a potential target for therapy. Nature Rev. Mol. Cell Biol. 11, 165–170. [DOI] [PubMed] [Google Scholar]
- Martins BM, and Locke JC (2015). Microbial individuality: how single-cell heterogeneity enables population level strategies. Curr. Opin. Microbiol 24, 104–112. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MG, and Johnson AD (2002). White-opaque switching in Candida albicans is controlled by mating-type locus homeodomain proteins and allows efficient mating. Cell 110, 293–302. [DOI] [PubMed] [Google Scholar]
- Moyes DL, Wilson D, Richardson JP, Mogavero S, Tang SX, Wernecke J, Hofs S, Gratacap RL, Robbins J, Runglall M, et al. (2016). Candidalysin is a fungal peptide toxin critical for mucosal infection. Nature 532, 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble SM, French S, Kohn LA, Chen V, and Johnson AD (2010). Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat. Genet 42, 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble SM, Gianetti BA, and Witchley JN (2017). Candida albicans cell-type switching and functional plasticity in the mammalian host. Nat. Rev. Microbiol 15, 96–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble SM, and Johnson AD (2005). Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot. Cell 4, 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman TM, Lord ND, Paulsson J, and Losick R (2015). Stochastic switching of cell fate in microbes. Annu. Rev. Microbiol 69, 381–403. [DOI] [PubMed] [Google Scholar]
- Odds FC, and Bernaerts R (1994). CHROMagar Candida, a new differential isolation medium for presumptive identification of clinically important Candida species. J. Clin. Microbiol 32, 1923–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande K, Chen C, and Noble SM (2013). Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat. Genet 45, 1088–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Myers CL, Sheppard DC, Phan QT, Sanchez AA, J EE, and Filler SG (2005). Role of the fungal Ras-protein kinase A pathway in governing epithelial cell interactions during oropharyngeal candidiasis. Cell. Microbiol 7, 499–510. [DOI] [PubMed] [Google Scholar]
- Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, and Soldatov A (2009). Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 37, e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaller MA, and Diekema DJ (2007). Epidemiology of invasive candidiasis: a persistent public health problem. Clin. Microbiol. Rev 20, 133–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JV, Dignard D, Whiteway M, and Kumamoto CA (2013). Normal adaptation of Candida albicans to the murine gastrointestinal tract requires Efg1p-dependent regulation of metabolic and host defense genes. Eukaryot. Cell 12, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce JV, and Kumamoto CA (2012). Variation in Candida albicans EFG1 expression enables host-dependent changes in colonizing fungal populations. MBio 3, e00117–00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramage G, VandeWalle K, Lopez-Ribot JL, and Wickes BL (2002). The filamentation pathway controlled by the Efg1 regulator protein is required for normal biofilm formation and development in Candida albicans. FEMS Microbiol. Lett. 214, 95–100. [DOI] [PubMed] [Google Scholar]
- Reuss O, Vik A, Kolter R, and Morschhauser J (2004). The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341, 119–127. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropars J, Maufrais C, Diogo D, Marcet-Houben M, Perin A, Sertour N, Mosca K, Permal E, Laval G, Bouchier C, et al. (2018). Gene flow contributes to diversification of the major fungal pathogen Candida albicans. Nature Comm. 9, 2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotman E, and Seifert HS (2014). The genetics of Neisseria species. Annu. Rev. Genet 48, 405–431. [DOI] [PubMed] [Google Scholar]
- Saville SP, Lazzell AL, Monteagudo C, and Lopez-Ribot JL (2003). Engineered control of cell morphology in vivo reveals distinct roles for yeast and filamentous forms of Candida albicans during infection. Eukaryot. Cell 2, 1053–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrzypek MS, Binkley J, Binkley G, Miyasato SR, Simison M, and Sherlock G (2017). The Candida Genome Database (CGD): incorporation of Assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucleic Acids Res. 45, D592–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutsky B, Staebell M, Anderson J, Risen L, Pfaller M, and Soll DR (1987). “White-opaque transition”: a second high-frequency switching system in Candida albicans. J. Bacteriol 169, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonneborn A, Tebarth B, and Ernst JF (1999). Control of white-opaque phenotypic switching in Candida albicans by the Efg1p morphogenetic regulator. Infect. Immun 67, 4655–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikantha T, Borneman AR, Daniels KJ, Pujol C, Wu W, Seringhaus MR, Gerstein M, Yi S, Snyder M, and Soll DR (2006). TOS9 regulates white-opaque switching in Candida albicans. Eukaryot. Cell 5, 1674–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoldt VR, Sonneborn A, Leuker CE, and Ernst JF (1997). Efg1p, an essential regulator of morphogenesis of the human pathogen Candida albicans, is a member of a conserved class of bHLH proteins regulating morphogenetic processes in fungi. EMBO J 16, 1982–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudbery PE (2011). Growth of Candida albicans hyphae. Nat. Rev. Microbiol 9, 737–748. [DOI] [PubMed] [Google Scholar]
- Tao L, Du H, Guan G, Dai Y, Nobile CJ, Liang W, Cao C, Zhang Q, Zhong J, and Huang G (2014). Discovery of a “white-gray-opaque” tristable phenotypic switching system in Candida albicans: roles of non-genetic diversity in host adaptation. PLoS Biol. 12, e1001830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Woude MW, and Baumler AJ (2004). Phase and antigenic variation in bacteria. Clin Microbiol Rev 17, 581–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TC (1997). The presence of an R467K amino acid substitution and loss of allelic variation correlate with an azole-resistant lanosterol 14alpha demethylase in Candida albicans. Antimicrob. Agents Chemother. 41, 1488–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, and Edmond MB (2004). Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin. Infect. Dis 39, 309–317. [DOI] [PubMed] [Google Scholar]
- Yano J, Yu A, Fidel PL Jr., and Noverr MC (2016). Transcription factors Efg1 and Bcr1 regulate biofilm formation and virulence during Candida albicans-associated denture stomatitis. PLoS One 11, e0159692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavrel M, Majer O, Kuchler K, and Rupp S (2012). Transcription factor Efg1 shows a haploinsufficiency phenotype in modulating the cell wall architecture and immunogenicity of Candida albicans. Eukaryot. Cell 11, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan RE, Galgoczy DJ, and Johnson AD (2006). Epigenetic properties of white-opaque switching in Candida albicans are based on a self-sustaining transcriptional feedback loop. Proc. Natl. Acad. Sci. USA 103, 12807–12812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zordan RE, Miller MG, Galgoczy DJ, Tuch BB, and Johnson AD (2007). Interlocking transcriptional feedback loops control white-opaque switching in Candida albicans. PLoS Biol. 5, e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. RNA-sequencing data for C. albicans cells in different cell states and genetic backgrounds. Related to Figure 3. TPM values, differentially expressed genes and GO term analysis are listed in separate sheets. * indicates cells derived from efg1Δ/EFG1 white state.
Data Availability Statement
All of the RNA-seq data associated with this project has been uploaded to GEO (Gene Expression Omnibus) and has a provisional accession number (GSE105399).