

Abstract
During the 1970s, a Na+-independent, ouabain-insensitive, N-ethylmaleimide-stimulated K+-Cl− cotransport mechanism was identified in red blood cells for the first time and in a variety of cell types afterward. During and just after the mid-1990s, three closely related isoforms were shown to account for this mechanism. They were termed K+-Cl− cotransporter 1 (KCC1), KCC3, and KCC4 according to the nomenclature of Gillen et al. (1996) who had been the first research group to uncover the molecular identity of a KCC, that is, of KCC1 in rabbit kidney. Since then, KCC1 has been found to be the most widely distributed KCC isoform and considered to act as a housekeeping membrane protein. It has perhaps received less attention than the other isoforms for this reason, but as will be discussed in the following review, there is probably more to KCC1 than meets the eye. In particular, the so-called housekeeping gene also appears to play crucial and specific roles in normal as well as pathological hematopoietic and in cancer cells.
Keywords: Cation-Cl− cotransporter, K+-Cl− cotransporter, Red blood cells, Sickle cell anemia, Abnormal cell growth, Animal models, Submitted to Journal of Hematology and Oncology
Introduction
K+-Cl− cotransporter 1 (KCC1) is a membrane protein that mediates the symport of K+ and Cl− ions through the surface of most animal cells [1]. It is also referred to as SLC12A4 based on the Human Genome Organization (HUGO) nomenclature. It shares high levels of homology in amino acid sequence with three other KCC isoforms that are termed KCC2 (SLC12A5), KCC3 (SLC12A6), and KCC4 (SLC12A7). KCC1 also shares moderate levels of homology with three additional proteins that mediate the symport movement of Na+ and Cl− in the absence or presence of K+. Along with the four KCC isoforms, these additional proteins are all part of a larger family of proteins that are termed cation-Cl− cotransporters (CCC) in the literature [1–4].
The molecular identity of KCC1 was deciphered during the mid-1990s just after that of the Na+-dependent CCC. Of notice, however, pioneer work by three research groups had already led to the identification of a K+-Cl− cotransport mechanism during the seventies [5–7]. Subsequent to their discoveries, the KCC were eventually found to exhibit unique physiological roles and distribution patterns. KCC2 and KCC3 have received the most attention as they were ultimately linked to hereditary forms of neurological disorders in human [8–10]. KCC1 has received much less attention given that it was found to be ubiquitously distributed and assumed to act as a housekeeping gene [11–13].
There are yet many lines of evidence to suggest that KCC1 accomplishes dedicated physiological roles as well. In particular, this isoform has been shown to sustain normal erythropoiesis, sickle cell formation, cancer growth, and bone turnover [14–18]. The main goal of the following review will be to discuss the molecular features and tissue-specific functions of KCC1 from the hematological perspective for the greater part. As will be seen, the characterization of KCC1 has led the way to important findings and promising therapeutic avenues.
Main text
Identification of KCC1, a member of the cation-Cl− cotransporter family
Early functional characterization
In the seventies, a Na+-independent K+-Cl− cotransport mechanism was formally identified for the first time in red blood cells (RBC) by three research groups [5–7]. It was found to exhibit saturation kinetics and to be stimulated by cell swelling as well as N-ethylmaleimide, a thiol-reacting agent. It was also suspected of allowing reticulocytes (RTC) to decrease their cell volume while maturing into erythrocytes.
Soon after this discovery, several tissues and cell types were found to express a K+-Cl− cotransport mechanism that was more active under the hypotonic condition and that differed from another mechanism known as Na+-K+-Cl− cotransport. They included mouse ascites tumor cells [19], bovine aortic endothelial cells [20], salamander gallbladder, and proximal nephron [21, 22] as well as many other cell types or tissues.
Further studies eventually showed that the K+-Cl− cotransport mechanism was inhibited by the loop diuretic furosemide and the alkanoic acid DIOA [11, 23, 24]. They also shed light on the mechanisms by which this transport moiety is regulated in response to cell swelling. Such mechanisms were found to involve the cytoskeleton [25, 26] and signaling intermediates that cause the carrier to undergo dephosphorylation [27, 28].
Initial molecular characterization
A protein responsible for K+-Cl− cotransport was uncovered for the first time in rabbit kidney and rat brain during the mid-nineties, that is, almost 20 years after the initial functional characterizations in RBC. It was termed K+-Cl− cotransporter 1 (KCC1) by the research group who had made the discovery [11]. Another isoform (KCC2) was uncovered during the same time [29] and two other isoforms (KCC3 and KCC4) a few years later [30–32].
To clone KCC1, the strategy used was based on the observation that K+-Cl− and Na+-K+-Cl− cotransport shared various functional traits [33] and that the proteins responsible for either mechanism would thus share homology in residue sequences as well. Because the Na+-K+-Cl− cotransporters (NKCC) had already been cloned through previous work, they would then serve as queries to identify a putative KCC among orphan expressed sequence tags (EST) [2, 34, 35].
The strategy exploited led to the identification of EST that shared 20–50% homology with the NKCC sequences and encompassed the 3′ end of a candidate transporter [11]. A rabbit kidney medulla cDNA library was subsequently screened with one of the EST identified and found to include a large sequence that was comprised of a 3255-bp full-length open reading frame. This open reading frame was eventually predicted to encode a 12-transmembrane domain glycoprotein that shared moderate levels of homology in amino acid sequence with the NKCC (Fig. 1a).
Fig. 1.
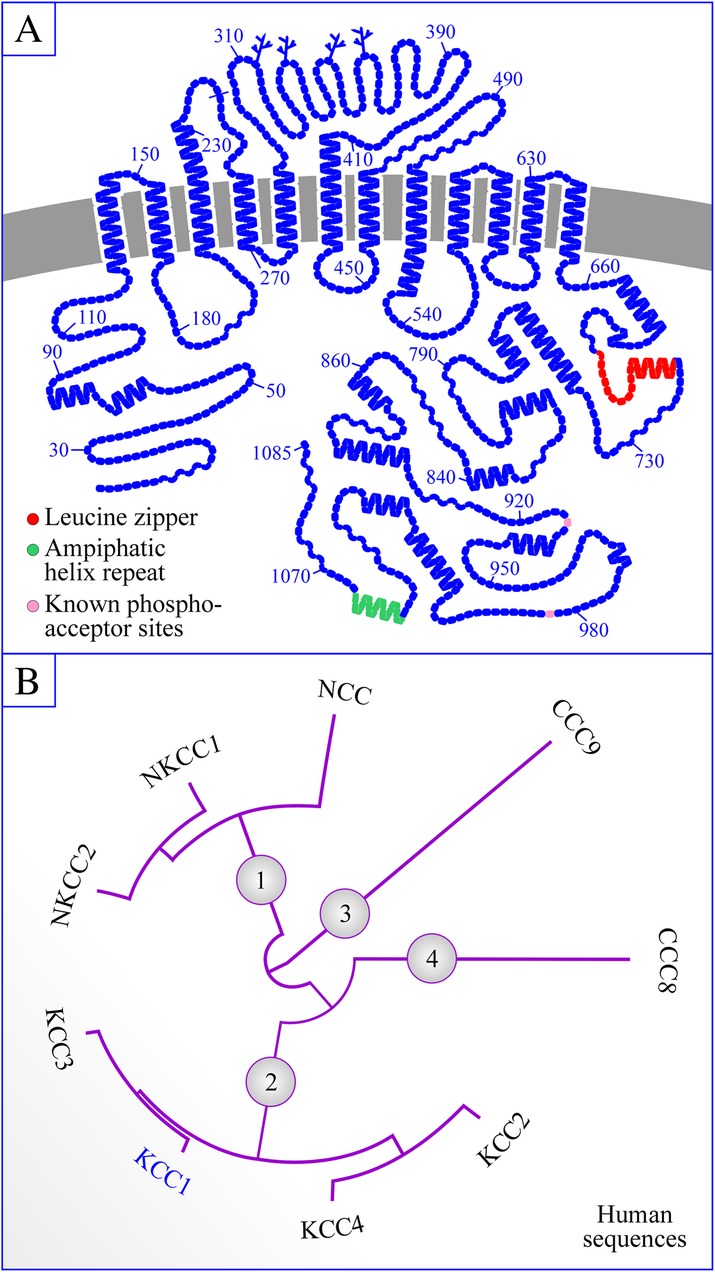
Structure of KCC1 and classification of the CCC family. a Structure. The topology model shown was drawn with the program PLOT by Biff Forbush (Yale University). Branched lines correspond to glycosylation sites, other symbols to residues and colors other than blue, to functional sites of potential importance. b Phylogenetic tree of the CCC family. The phylogram shown was obtained with the programs PhyML v3.1/3.0 aLRT and MUSCLE v3.8.31 [36, 37] using the most abundant human variants. GenBank accession numbers are provided in footnote 11
Studies in rabbit KCC1-expressing HEK-293 cells confirmed that the clone identified encoded a K+-Cl− cotransport mechanism [11]. In the presence of N-ethylmaleimide, for instance, the initial rate of furosemide-sensitive Rb+ efflux from these cells was 8-fold higher compared to controls cells. Along the same line, Rb+ efflux was stimulated further through cell swelling and Rb+ uptake by KCC1 was Cl−-dependent as well as Na+-independent.
Splice variants
KCC1 is expressed as four splice variants in the mouse as well as in human tissues. However, KCC1A is the only transcript to be fully conserved between the two species given that it is produced through the same initiation site in exon 1A and includes the same 24 exons. KCC1A is also the only transcript for which a role has been clearly defined. The other transcripts are formed through the alternative usage of three exons (called 1A, 1B, or 1C) in human and of two initiation sites along exon 1A in human and mouse. Some of the other transcripts also lack an exon in part or in full and one of the transcripts in mouse lacks most of the open reading frame.
Cation-Cl− cotransporter family
As it stands, nine CCC family members are known to exist. They fall into different phylogenetic branches as follows (and as shown in the cladogram of Fig. 1): KCC1 belongs to one branch along with the three other KCC, the Na+-dependent CCC belong to another branch and the remaining two CCC (called CCC8 and CCC9) belong to each an independent branch. Overall, KCC1 shares higher identity with KCC3 (~ 75%) than with any of the other CCC [1, 3].
Localization of KCC1 in animal species
Tissue distribution
Northern blot analyses of rat tissues initially revealed that KCC1 was ubiquitously expressed [11]. These observations are still supported today by the presence of KCC1 in numerous cell types and among the current EST sequences of multiple tissues from human, mouse, and rat. For this reason and due to its transport function, Kcc1 is often considered as a housekeeping gene that is involved in cell volume regulation and intracellular electrolyte balance [11–13].
As for the erythroid and lymphoid systems more specifically, KCC1 has been detected in bone marrow, lymph nodes, spleen, macrophages, pluripotent stem cells, RTC, mature RBC, megakaryocytes, circulating T cells, and monocytes according to several references and various online databanks2. It has also been detected in a variety of leukemic and lymphomatous cells, cancerous cell lines, and carcinomas as well as in myeloma cell1.
Cellular distribution
In non-epithelial cells, KCC1 acts mainly as a plasma membrane carrier system [11, 13]. Whether it could play a role in intracellular organelles has not been reported thus far. In epithelial cells, KCC1 also mainly acts at the cell surface but is confined to the basolateral membrane based on all accounts in the biomedical literature.
Function and regulation of KCC1 in animal species
Transport characteristics
There is a paucity of data regarding the transport characteristics of KCC1 per se. While it is mainly through transport assays in RBC that the functional signature of K+-Cl− cotransport was determined, it is now known that this transport function is accounted for by at least two KCC isoforms or splice variants in most cell types and tissues [15, 38–40]. Accordingly, K+-Cl− cotransport in native environments cannot be ascribed to the sole activity of KCC1.
While this limitation should be kept in mind, the studies in RBC showed that K+-Cl− cotransport was associated with the movement of one cation per one anion during each transport cycle [41] and that it was therefore outwardly directed. Of notice, however, the stoichiometry of ion transport by KCC1 per se has still not been confirmed experimentally and the number of ion binding sites for either of the Na+-independent family members has still not been determined [3, 4].
Through more recent work in heterologous expression systems, the affinity of KCC1 for the transported ions was found to be in the same range as that of KCC3 and KCC4 but its transport capacity to be relatively lower [11, 42, 43]. Interestingly, Bergeron et al. [43] further observed that K+ ions could be efficiently substituted for by NH4+ ions at the K+ translocation site of KCC1. For this reason, they came to the conclusion that this transporter could also be involved in acid uptake and had the potential to regulate both intracellular pH (pHi) and extracellular pH (pHe).
As for the effect of various agents on KCC1 activity per se, it was found to be as described in native cells types or tissues and similar among the isoforms. In essence, the pharmacological signature of KCC1 was characterized by the following traits: stimulation by N-ethylmaleimide, modest inhibition by bumetanide, DIDS, and barium, and stronger inhibition by furosemide and DIOA [11, 23, 24, 42, 43]. Importantly, several of these traits were observed in at least two heterologous expression systems.
Regulation
The N- and C-termini of KCC1 are both predicted to be cytosolically disposed, implying that they could interact with a number of signaling intermediates, cytoskeletal elements, and vesicle-associated membrane proteins. However, there are only a few consensus phosphoregulatory sites within these domains and most are for CK2 and PKC phosphorylation (Fig. 1a). In the C-terminus, there is also a leucine zipper domain and a paired amphipathic helix repeat that could sustain the assembly of KCC1 into CCC-based heterodimers [44] or its interaction with cytoskeletal elements [45].
Based on elegant studies by Rinehart et al. [46], cell swelling was found to activate KCC1 through dephosphorylation of residues T926 and T983 by a phosphatase type 1 (see Fig. 1a). In addition, the same residues were found to be phosphorylated under isotonic condition, i.e., when KCC1 is in its inactive state, through the WNK kinase/OSR1-dependent pathway. In subsequent studies, Frenette-Cotton et al. [47] have shown that additional Ser/Thr sites were probably at play given that cell swelling caused an overall increase in the phosphorylation state of another KCC isoform.
Otherwise, a number of studies have shown that K+-Cl− cotransport could be affected through changes in intracellular O2 pressure (pO2i) and Mg2+ concentration (Mg2+i) as well as through the involvement of cytoskeletal elements. The importance of these factors in KCC1 regulation will be outlined below while discussing the pathophysiology of RBC dehydration in sickle cell anemia, a disease where Hbα/α;β/β (HbA) is replaced by Hbα/α;S/S (HbS) through sickling mutations in both of the β chains.
Physiological roles of KCC1
Preamble
The physiological roles of KCC1 per se have not been studied as extensively as those of the other Na+-dependent CCC. There are probably at least three reasons as to why: (1) KCC1 might have been predicted to play the same role in many cell types by acting as a widely distributed housekeeping isoform. (2) KCC1 might have also been predicted to play a redundant role, especially in cell types where other Na+-dependent CCC with higher transport activity are expressed. (3) Over the years, KCC2 and KCC3 have generated most of the attention among the Na+-independent CCC because of their associations with neurological disorders [8–10].
Whether or not these reasons could have explained why KCC1 was subjected to more limited investigative efforts, they should be considered as unfounded as it stands. In particular, the characterization of mouse models inactivated for Kcc1 has now provided evidence to suggest that this gene could play important and specific roles in various cell types including those of the hematopoietic lineage. A review of the data available will be summarized hereafter.
Role of KCC1 in RBC
RBC is known to express KCC1, KCC3, and KCC4 [15, 38–40]. Two of the isoforms present also come as two splice variants each. However, there is evidence to suggest that K+-Cl− cotransport in this cell type could be accounted for by KCC3B predominantly. For instance, Pan et al. [39] have found that RTC and mature RBC from mouse and human expressed this variant protein at relatively comparable levels whereas RTC expressed KCC1 at much higher levels than mature RBC. Along the same line, Rust et al. [15] have found that Kcc3-null mouse RBC exhibited lower K+-Cl− cotransport than Kcc1-null mouse RBC.
For various reasons, however, it is not clear that KCC3 would play such a preponderant role in mature RBC. First, KCC1 and KCC3 were not detected by Pan et al. [39] through the same antibody. If KCC1 was actually much more abundant than KCC3 in RTC, it could then be as abundant as, or even more abundant than KCC3 in mature RBC. Second, while the genetic background used by Rust et al. [15] was not the same for all of the models characterized, it is known to affect K+-Cl− cotransport in RBC [48]. Third, KCC1 does appear to be involved in sickle cell anemia as will be discussed below.
The data of Pan et al. [39] are nonetheless consistent with the idea that KCC1 could play a role in erythroid maturation and are in keeping with those of two other research groups. Indeed, Pellegrino et al. [49] demonstrated that human or mouse HbA RTC only expressed full-length KCC1 transcripts during the early stages of cell differentiation, and Su et al. [12], that KCC1 expression was higher in enriched population of HbS RTC. Given that RTC and mature RBC are both endowed with K+-Cl− cotransport activity [12, 39, 50, 51], the data of Pan et al. also suggest that some of the carrier isoforms could exhibit exceptionally slow turnover rates beyond the RTC stage.
As alluded to already, it is now widely accepted that K+-Cl− cotransport at the RBC surface plays a central role in the pathophysiology of sickle cell anemia [14, 15]. In this inherited disorder, the abnormal hemoglobins expose a hydrophobic domain between the E and F helices when they are deoxygenated and polymerize with each other to form rigid precipitates that anchor themselves to the cytoskeleton [52, 53]. Sickling also comes with lower cell volumes due to an overactive K+-Cl− cotransport function [54, 55] that contributes to cell rigidity by increasing viscosity and polymer concentration.
Rust et al. [15] have demonstrated the importance of this mechanism by studying a mouse model of Kcc1 and Kcc3 inactivation in the SAD transgenic background of hyper sickling human HbS. In particular, they found that RBC in Kcc1−/−Kcc3−/−SAD mice was clearer and larger than in Kcc1+/+Kcc3+/+SAD mice. However, inactivation of either carrier in the SAD background revealed that KCC3 played a more important role than KCC1 in sickle cell formation. Given that mature RBC can also acquire the abnormal phenotype, excessive K+-Cl− cotransport could be contributed for by KCC1 beyond the RTC stage of erythroid differentiation, at least in the case of HbS cells.
Another group has demonstrated the role of KCC1 in sickle cell formation by studying a mouse model in which the transporter is constitutively activated through a phosphorylation-precluding mutation (M935K) in its C-terminus [14]. On its own, the Kcc1M935K/M935K mouse model resulted in semi-dominant RBC microcytosis, and when bred into the humanized heterozygote HHbα/α;β/S mouse model, in widespread sickling-induced tissue damage. As such, this group provided direct evidence that excessive K+-Cl− cotransport did contribute to sickle cell formation and that it could affect the erythropoietic lineage beyond the RTC stage of differentiation.
Despite the importance of previous findings, the mechanisms of increased K+-Cl− coefflux in sickle cell anemia are still largely elusive. A change in carrier abundance is probably partly at cause given expression levels of KCC1 and KCC3 are higher in HbS RTC than in HbA RTC and higher in mature HbS RBC that in mature HbA RBC [12, 49]. However, mature RBC does not have the capability of upregulating total protein expression, and they can also undergo sickling after only 2 h of hypoxia [56]. As such, dehydration of these cells by KCC1 would probably require unitary transport rates or capacity to increase as well.
One of the mechanisms that could account for the increase in K+-Cl− coefflux pertains to the dependence of this transport moiety on pO2i [57–61]. Even if K+-Cl− coefflux and pO2i are linearly interrelated in HbA cells and even if sickled cells form at low pO2i, there is evidence to suggest that hypoxia could still be a cause. Indeed, the relationship between K+-Cl− coefflux and pO2i in HbS cells is U-shaped instead of linear [57, 58, 61]. In the cytosol of sickled cells, additionally, the effect of hypoxia on O2i availability could be attenuated by an abnormal Bohr effect [62]. However, K+-Cl− coefflux in these cells is not much higher at very low pO2i than it is at 100% pO2i [63], and one would not expect the Bohr effect to counteract the effect of hypoxia completely.
How hypoxia could affect K+-Cl− coefflux is in itself unknown. Some investigators have argued that low pO2i could cause this carrier system to become more active in HbS cells by decreasing pHi [63–65]. However, other investigators have shown that the activity of both KCC1 and KCC3 decreased progressively below pHi levels of 7.0–7.1 and that the only isoform that could potentially increase its activity under such circumstances is KCC4 [43]. Thus far, however, the role of this other isoform in sickle cell anemia is controversial.
There is evidence to suggest that hypoxia might still explain why K+-Cl− coefflux is increased in HbS cells as it could do so through systemic rather than local effects. Indeed, placental growth factor (PIGF) has been found at high circulating levels in sickle cell anemia, probably as a result of HIF1α upregulation in ischemic tissues, and to increase KCC1 expression in an erythroid RTC type cell line [66]. Given, however, that PIGF is also upregulated in normal RTC by low pO2i [67], its synthesis by non-erythroid cells would have to be sufficiently important to bypass any inhibitory effects that low pO2i might exert on KCC in HbS cells (Fig. 2). It would not be predicted to affect mature RBC either if its effect was to alter total KCC1 expression primarily.
Fig. 2.
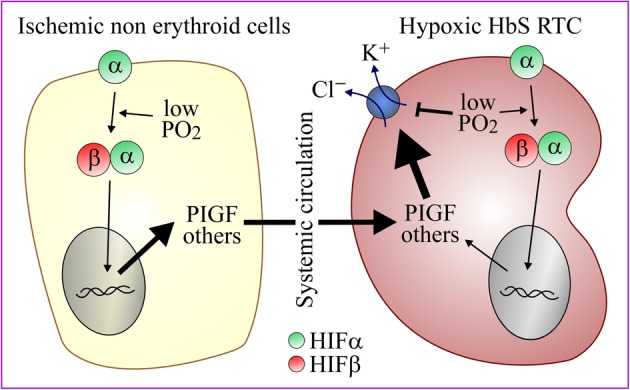
Regulation of K+-Cl− cotransport in HbS cells. During occlusive crises, PGIF are produced from ischemic non-erythroid tissues and taken up by RTC where it could increase KCC1 expression and overcome the potential inhibitory effect of low pO2i on K+-Cl− cotransport. Abbreviations: HbS, hemoglobin S; HIF, hypoxia-induced factor; PIGF, placental growth factor
The dependence of K+-Cl− coefflux on Mg2+i is another factor that could contribute to the transport phenotype of HbS cells. Indeed, while K+-Cl− coefflux is known to be stimulated at lower Mg2+i, the cytosol of sickled RBC is also known to be Mg2+-deficient [59, 68, 69]. Once again, however, the mechanisms and isoforms involved have not been deciphered. Some investigators have suggested that low Mg2+i could act by modulating the activity of signaling intermediates [63] and others by affecting the cytoskeleton (see below). Despite the unknowns, the sensitivity of K+-Cl− coefflux to Mg2+i is still of interest given that it has prompted clinical studies to determine the efficacy of Mg2+ supplementation in the treatment of sickling disorders [70].
A third mechanism could involve the disassembly of spectrin by HbS, i.e., of a cytoskeletal element that is normally formed of four subunits (α1/α1;β1/β1) and associates with the inner bilayer. This mechanism is suggested by two sets of deduced observations. The first one is that variety of intracellular inorganic cations have been found to inhibit K+-Cl− cotransport and that they could do so by shielding negative charges at the surface of the inner bilayer [45, 71]. The second one is that a rise in temperature has also been found to stimulate K+-Cl− cotransport [45, 72] and that it could do so by exposing negative charges from the same inner bilayer through spectrin disassembly [45].
As alluded to, regulatory enzymes are additional players that could contribute to upregulation of K+-Cl− cotransport in HbS cells. They include the WNK kinases that come as four isoforms and are known to inhibit KCC1 and KCC3 [46, 47, 73–76]. If these enzymes did play a role, their activity should thus be reduced in HbS cells. In this regard, interestingly, D368 in rat WNK1 and D293 in WNK3 have been shown to act as binding sites for Mg2+ and their replacement by Ala residues to abolish the kinase activity of these enzymes [77, 78]. The inner bilayer is also an important component of the WNK kinase-dependent signaling pathway [45, 77–80].
Other ion transport pathways could play a role in the dehydration of HbS cells [81–87]. They include the mechanosensitive ion channel PIEZO1 (also called Psickle) and the Gardos channel (also called KCNN4). In particular, both these pathways are sensitive to pO2i and are upregulated in sickled RBC [82, 88, 89]. In the past, inhibition of KCNN4 by clotrimazole and senicapoc has also been under clinical studies for the treatment of sickle cell anemia [87, 90, 91]. More recently, senicapoc has been renamed to PF-05416266 (Pfizer Inc., New York, NY, USA) and has become the object of a new trial for the same indication.
Cancer
At least three members of the KCC family have been shown to affect cancer cell proliferation, growth, and invasiveness. The mechanisms involved are still unknown but could implicate various effectors that are sensitive to changes in intracellular Cl− concentration (Cl−i), cell volume, or membrane potential. Alternatively, cancerous cell transformation could cause KCC activity to be affected secondarily through concomitant changes in pO2i, cell volume, signaling activity, cytoskeletal organization, and transcription efficiency.
As for KCC1 more specifically, it is suspected of facilitating growth and invasiveness for cervical and endometrial adenocarcinomas. In the presence of IGF, for instance, cell lines derived from such cancers have been found in some studies to exhibit increased ERK-dependent signaling and invasiveness, but not so if KCC1 activity was inhibited concomitantly through pharmacological agents or RNA interference [16, 92]. In these studies, KCC1 expression was also stimulated by IGF, but according to other accounts, it is typically low in many types of cancer cells, undetectable in lymphoma cells, and positively correlated with higher survival rates in renal cell carcinoma1.
We postulate that the HIF-dependent pathway could play an important role in regulating KCC1 expression at the surface of cancer cells. In particular, this pathway could be activated through somatic mutations in the VHL gene or through low pO2i levels as cancer cells proliferate into solid masses. The involvement of HIF under such circumstances could then explain why higher KCC1 expression is associated with a better prognosis in renal cell carcinomas as it would then point towards the presence of pathogenic modifications in the VHL gene [93, 94].
Bone
Kajiya et al. [18] have found that KCC1 was expressed in mouse osteoclasts based on reverse transcriptase (RT)-PCR measurements, immunohistochemical studies, and western blot analyses. Surprisingly, they found that KCC2 was also expressed in this cell type, albeit at much lower levels, and that KCC3 and KCC4 were undetectable. Transcript abundance inferred from the EST databanks is partly consistent with such findings1. In human and mouse bone, there are indeed 55 and 410 messages per million (MPM) for KCC1, respectively, and 0 MPM for KCC2 and KCC4. In human bone, however, there are also 97 MPM for KCC3, suggesting that this isoform is expressed in other cell types such as osteoblasts and osteocytes.
In the same study by Kajiya et al., KCC1-specific antisense oligonucleotides were also shown to suppress pit formation in calcified bone, and DIOA to increase Cl−i and H+i in osteoclasts. Although it was not clear in this work that KCC1 was expressed in the ruffled border of osteoclasts, Kajiya et al. suggested that at this location, KCC1 could provide an extrusion mechanism for Cl− during bone resorption to facilitate H+ secretion in forming pits (see Fig. 3). Interestingly, other studies have shown that the Cl− channel CIC7 also provided an extrusion mechanism for Cl− and that mice inactivated for the encoding gene exhibited severe osteopetrosis. Whether the same skeletal phenotype would be observed in Kcc1-null mouse does not appear to have been reported as of yet.
Fig. 3.
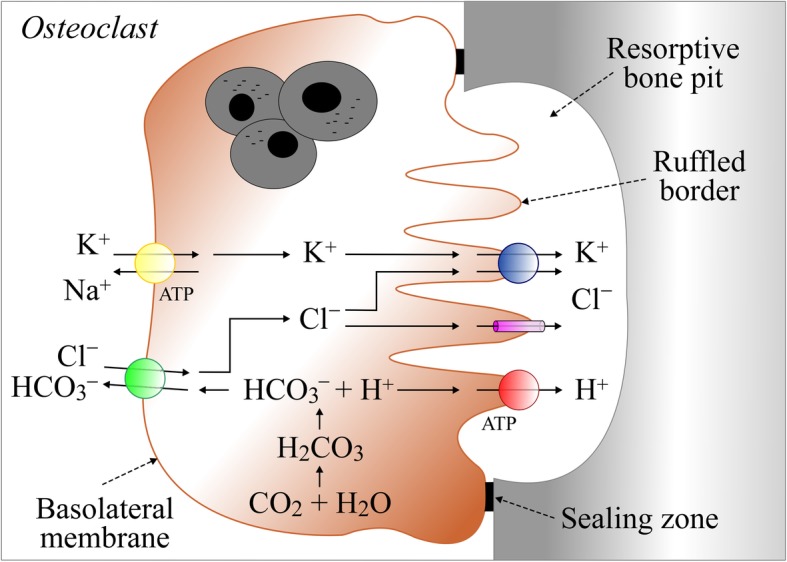
Role of KCC1 in osteoclasts. On the ruffled border, transport systems shown consist of KCC1, the Cl− channel CLC-7 [95], and the vacuolar H+-ATPase pump ATP6V1C1 [96]. On the basolateral membrane, they consist of the Na+/K+-ATPase pump ATPA1B1 [97] and the Cl−/HCO3− exchanger SLC4A2 [98]. On the ruffled border, the role of KCC1 could be to use the K+ gradient generated by the Na+ pump to provide an accessory route for Cl− secretion in resorptive pits [18]. If, alternatively, KCC1 was localized on the basolateral side, it could then serve two purposes. The first one would be to sustain Cl−/HCO3− exchange by providing the antiporter with a continued supply of Cl− ions. The presence of KCC1 at this location would thus allow secondarily for higher H+i and luminal H+ secretion. The second one would be to sustain Na+/K+-ATPase activity by providing the enzyme with a continued supplied of K+ ions. The presence of KCC1 at this location would thus allow secondarily for higher intracellular negativity and luminal Cl− secretion
NCC, another member of the CCC family, has been drawing attention in the field of osteoporosis for many years because its inactivation—through thiazide therapy or homozygous loss of function mutations—has been shown to increase bone mineral density [99]. While this effect has been generally attributed to the secondary role of NCC in Ca2+ handling by the gut and the kidney [100, 101]—NCC inactivation increases Ca2+ absorption in both epithelia—it could also be attributed to the presence of NCC in the bone. In particular, this CCC was shown to be much less abundant in differentiating than in proliferating human and fetal rat osteoblasts, and its inactivation to increase bone mineralization and expression of osteoblastic differentiation markers [102].
Taken together, the findings described in this section of the review highlight the potential importance of Cl−i or of cell surface Cl− transport on bone cell function. Higher Cl−i or lower Cl− efflux in osteoblasts (as would occur through decreased KCC1 activity) appears to be associated with decreased bone resorption, whereas higher Cl−i or lower Cl− efflux in osteoclasts (as would occur through increased NCC activity or decreased KCC3 activity) to be associated with decreased bone formation.
Conclusion
This review has allowed to show that KCC1 accomplishes specific physiological and pathophysiological roles in animal cells and that it does not act solely as a housekeeping K+-Cl− cotransport mechanism. As it stands, however, it is mainly in RBC maturation and sickling of RBC that such roles have been demonstrated more convincingly. There is still emerging evidence to suggest that KCC1 is also of functional relevance in cancer development and in bone resorption.
As mentioned, KCC1 is ubiquitously expressed and could thus play roles in many other cell types within the hematopoietic lineage. In this regard, KCC3 has been found to sustain hypochlorite synthesis by white blood cells through its Cl− cotransporter function in phagosomes [103, 104]. Along the same line, it is particularly intriguing that KCC1 is expressed in a variety of leukemic cells but that it is virtually absent from most types of lymphoma cells. It is thus tempting to postulate that the chromosomal locus of KCC1 (16q22.1), which is known to harbor cancer-associated genes such as CDH1 and CDH3, is altered in these cells through DNA rearrangements [105]1. Alternatively, low K+-Cl− cotransport activity could confer a survival benefit to a variety of lymphoma cells.
It is perhaps also intriguing that there are still no reports of human disorders that have been linked to pathogenic mutations in Kcc1. As suggested by the mouse models, the reason may be that this gene plays a redundant role and that its inactivation is thus tolerated under normal condition. If and when disease-causing mutations are identified, KCC1 will certainly find a place of honor among the other family members. The same will also be true if pharmacologic inactivation of this isoform in sickle cell anemia were to improve the morbidity and mortality that is associated with this prevalent disorder.
Acknowledgements
None.
Authors’ contributions
APG and PI contributed to the conception and design of the work. APG, SS, and PI helped in the acquisition, analysis, or interpretation of data for the work. All authors drafted the work and critically revised the work for intellectual content. All authors read and approved the final manuscript.
Funding
This work was funded by the Kidney Foundation of Canada and the Canadian Institute of Health Research.
Availability of data and materials
All data generated or analyzed during this study are included in this published article (see Footnotes).
Ethics approval and consent to participate
Not applicable.
Consent for publication
All persons designated as authors qualify for authorship and have approved the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Sequences used to generate the phylogram of Fig. 1b:
- CCC Accession numbers
- NKCC1 NP_001037.1
- NKCC2 NP_000329.2
- NCC NP_000330.2
- KCC1 NP_005063.1
- KCC2 NP_001128243.1
- KCC3 NP_598408.1
- KCC4 NP_006589.2
- CCC8 NP_064631.2
- CCC9 NP_078904.3
Web links exploited:
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Garneau A. P., Marcoux A. A., Slimani S., Tremblay L. E., Frenette‐Cotton R., Mac‐Way F., Isenring P. Physiological roles and molecular mechanisms of K + ‐Cl − cotransport in the mammalian kidney and cardiovascular system: where are we? The Journal of Physiology. 2019;597(6):1451–1465. doi: 10.1113/JP276807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu JC, Lytle C, Zhu TT, Payne JA, Benz E, Jr, Forbush B., 3rd Molecular cloning and functional expression of the bumetanide-sensitive Na-K-Cl cotransporter. Proc Natl Acad Sci U S A. 1994;91(6):2201–2205. doi: 10.1073/pnas.91.6.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garneau AP, Marcoux AA, Frenette-Cotton R, Mac-Way F, Lavoie JL, Isenring P. Molecular insights into the normal operation, regulation, and multisystemic roles of K(+)-Cl(-) cotransporter 3 (KCC3) Am J Physiol Cell Physiol. 2017;313(5):C516–CC32. doi: 10.1152/ajpcell.00106.2017. [DOI] [PubMed] [Google Scholar]
- 4.Marcoux AA, Garneau AP, Frenette-Cotton R, Slimani S, Mac-Way F, Isenring P. Molecular features and physiological roles of K(+)-Cl(-) cotransporter 4 (KCC4) Biochim Biophys Acta Gen Subj. 2017;1861(12):3154–3166. doi: 10.1016/j.bbagen.2017.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Lauf PK, Theg BE. A chloride dependent K+ flux induced by N-ethylmaleimide in genetically low K+ sheep and goat erythrocytes. Biochem Biophys Res Commun. 1980;92(4):1422–1428. doi: 10.1016/0006-291X(80)90445-3. [DOI] [PubMed] [Google Scholar]
- 6.Dunham PB, Stewart GW, Ellory JC. Chloride-activated passive potassium transport in human erythrocytes. Proc Natl Acad Sci U S A. 1980;77(3):1711–1715. doi: 10.1073/pnas.77.3.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kregenow FM. The response of duck erythrocytes to nonhemolytic hypotonic media. Evidence for a volume-controlling mechanism. J Gen Physiol. 1971;58(4):372–395. doi: 10.1085/jgp.58.4.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ. Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron. 2001;30(2):515–524. doi: 10.1016/S0896-6273(01)00297-5. [DOI] [PubMed] [Google Scholar]
- 9.Casaubon LK, Melanson M, Lopes-Cendes I, Marineau C, Andermann E, Andermann F, et al. The gene responsible for a severe form of peripheral neuropathy and agenesis of the corpus callosum maps to chromosome 15q. Am J Hum Genet. 1996;58(1):28–34. [PMC free article] [PubMed] [Google Scholar]
- 10.Kahle KT, Merner ND, Friedel P, Silayeva L, Liang B, Khanna A, et al. Genetically encoded impairment of neuronal KCC2 cotransporter function in human idiopathic generalized epilepsy. EMBO Rep. 2014;15(7):766–774. doi: 10.15252/embr.201438840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gillen CM, Brill S, Payne JA, Forbush B., 3rd Molecular cloning and functional expression of the K-Cl cotransporter from rabbit, rat, and human. A new member of the cation-chloride cotransporter family. J Biol Chem. 1996;271(27):16237–16244. doi: 10.1074/jbc.271.27.16237. [DOI] [PubMed] [Google Scholar]
- 12.Su W, Shmukler BE, Chernova MN, Stuart-Tilley AK, de Franceschi L, Brugnara C, et al. Mouse K-Cl cotransporter KCC1: cloning, mapping, pathological expression, and functional regulation. Am J Physiol. 1999;277(5):C899–C912. doi: 10.1152/ajpcell.1999.277.5.C899. [DOI] [PubMed] [Google Scholar]
- 13.Cell Physiology Source Book - Essentials of Membrane Biophysics. 4th ed: Elsevier; 2011. 248 p.
- 14.Brown FC, Conway AJ, Cerruti L, Collinge JE, McLean C, Wiley JS, et al. Activation of the erythroid K-Cl cotransporter Kcc1 enhances sickle cell disease pathology in a humanized mouse model. Blood. 2015;126(26):2863–2870. doi: 10.1182/blood-2014-10-609362. [DOI] [PubMed] [Google Scholar]
- 15.Rust MB, Alper SL, Rudhard Y, Shmukler BE, Vicente R, Brugnara C, et al. Disruption of erythroid K-Cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J Clin Invest. 2007;117(6):1708–1717. doi: 10.1172/JCI30630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang S, Wu X, Jiang T, Lu Y, Ma L, Liang M, et al. The up-regulation of KCC1 gene expression in cervical cancer cells by IGF-II through the ERK1/2MAPK and PI3K/AKT pathways and its significance. Eur J Gynaecol Oncol. 2009;30(1):29–34. [PubMed] [Google Scholar]
- 17.Kajiya H, Okamoto F, Ohgi K, Nakao A, Fukushima H, Okabe K. Characteristics of ClC7 Cl- channels and their inhibition in mutant (G215R) associated with autosomal dominant osteopetrosis type II in native osteoclasts and hClcn7 gene-expressing cells. Pflugers Arch. 2009;458(6):1049–1059. doi: 10.1007/s00424-009-0689-4. [DOI] [PubMed] [Google Scholar]
- 18.Kajiya H, Okamoto F, Li JP, Nakao A, Okabe K. Expression of mouse osteoclast K-Cl Co-transporter-1 and its role during bone resorption. J Bone Miner Res. 2006;21(7):984–992. doi: 10.1359/jbmr.060407. [DOI] [PubMed] [Google Scholar]
- 19.Aull F. Potassium chloride cotransport in steady-state ascites tumor cells. Does bumetanide inhibit? Biochim Biophys Acta. 1981;643(2):339–345. doi: 10.1016/0005-2736(81)90079-1. [DOI] [PubMed] [Google Scholar]
- 20.Perry PB, O'Neill WC. Swelling-activated K fluxes in vascular endothelial cells: volume regulation via K-Cl cotransport and K channels. Am J Physiol. 1993;265(3 Pt 1):C763–C769. doi: 10.1152/ajpcell.1993.265.3.C763. [DOI] [PubMed] [Google Scholar]
- 21.Larson M, Spring KR. Volume regulation by Necturus gallbladder: basolateral KCl exit. J Membr Biol. 1984;81(3):219–232. doi: 10.1007/BF01868715. [DOI] [PubMed] [Google Scholar]
- 22.Anagnostopoulos T, Edelman A, Planelles G, Teulon J, Thomas SR. Transport of chlorine in the proximal tubule. Its effects on water-electrolyte absorption. J Physiol (Paris). 1984;79(3):132–138. [PubMed] [Google Scholar]
- 23.Garay RP, Nazaret C, Hannaert PA, Cragoe EJ., Jr Demonstration of a [K+,Cl-]-cotransport system in human red cells by its sensitivity to [(dihydroindenyl)oxy]alkanoic acids: regulation of cell swelling and distinction from the bumetanide-sensitive [Na+,K+,Cl-]-cotransport system. Mol Pharmacol. 1988;33(6):696–701. [PubMed] [Google Scholar]
- 24.Gusev GP, Agalakova NI, Lapin AV. Kinetics of K-Cl cotransport in frog erythrocyte membrane: effect of external sodium. J Membr Biol. 1999;172(3):203–213. doi: 10.1007/s002329900597. [DOI] [PubMed] [Google Scholar]
- 25.Sachs JR, Martin DW. The role of ATP in swelling-stimulated K-Cl cotransport in human red cell ghosts. Phosphorylation-dephosphorylation events are not in the signal transduction pathway. J Gen Physiol. 1993;102(3):551–573. doi: 10.1085/jgp.102.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelley SJ, Dunham PB. Mechanism of swelling activation of K-Cl cotransport in inside-out vesicles of LK sheep erythrocyte membranes. Am J Physiol. 1996;270(4 Pt 1):C1122–C1130. doi: 10.1152/ajpcell.1996.270.4.C1122. [DOI] [PubMed] [Google Scholar]
- 27.Jennings ML. al-Rohil N. Kinetics of activation and inactivation of swelling-stimulated K+/Cl- transport. The volume-sensitive parameter is the rate constant for inactivation. J Gen Physiol. 1990;95(6):1021–1040. doi: 10.1085/jgp.95.6.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jennings ML, Schulz RK. Okadaic acid inhibition of KCl cotransport. Evidence that protein dephosphorylation is necessary for activation of transport by either cell swelling or N-ethylmaleimide. J Gen Physiol. 1991;97(4):799–817. doi: 10.1085/jgp.97.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Payne JA, Stevenson TJ, Donaldson LF. Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem. 1996;271(27):16245–16252. doi: 10.1074/jbc.271.27.16245. [DOI] [PubMed] [Google Scholar]
- 30.Hiki K, D'Andrea RJ, Furze J, Crawford J, Woollatt E, Sutherland GR, et al. Cloning, characterization, and chromosomal location of a novel human K+-Cl- cotransporter. J Biol Chem. 1999;274(15):10661–10667. doi: 10.1074/jbc.274.15.10661. [DOI] [PubMed] [Google Scholar]
- 31.Mount DB, Mercado A, Song L, Xu J, George AL, Jr, Delpire E, et al. Cloning and characterization of KCC3 and KCC4, new members of the cation-chloride cotransporter gene family. J Biol Chem. 1999;274(23):16355–16362. doi: 10.1074/jbc.274.23.16355. [DOI] [PubMed] [Google Scholar]
- 32.Race JE, Makhlouf FN, Logue PJ, Wilson FH, Dunham PB, Holtzman EJ. Molecular cloning and functional characterization of KCC3, a new K-Cl cotransporter. Am J Physiol. 1999;277(6 Pt 1):C1210–C1219. doi: 10.1152/ajpcell.1999.277.6.C1210. [DOI] [PubMed] [Google Scholar]
- 33.Payne JA, Xu JC, Haas M, Lytle CY, Ward D, Forbush B., 3rd Primary structure, functional expression, and chromosomal localization of the bumetanide-sensitive Na-K-Cl cotransporter in human colon. J Biol Chem. 1995;270(30):17977–17985. doi: 10.1074/jbc.270.30.17977. [DOI] [PubMed] [Google Scholar]
- 34.Igarashi P, Vanden Heuvel GB, Payne JA, Forbush B., 3rd Cloning, embryonic expression, and alternative splicing of a murine kidney-specific Na-K-Cl cotransporter. Am J Physiol. 1995;269(3 Pt 2):F405–F418. doi: 10.1152/ajprenal.1995.269.3.F405. [DOI] [PubMed] [Google Scholar]
- 35.Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR. Molecular cloning and chromosome localization of a putative basolateral Na(+)-K(+)-2Cl- cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem. 1994;269(41):25677–25683. [PubMed] [Google Scholar]
- 36.Dereeper A, Audic S, Claverie JM, Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010;10:8. doi: 10.1186/1471-2148-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.-F., Guindon S., Lefort V., Lescot M., Claverie J.-M., Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Research. 2008;36(Web Server):W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crable SC, Hammond SM, Papes R, Rettig RK, Zhou GP, Gallagher PG, et al. Multiple isoforms of the KC1 cotransporter are expressed in sickle and normal erythroid cells. Exp Hematol. 2005;33(6):624–631. doi: 10.1016/j.exphem.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 39.Pan D, Kalfa TA, Wang D, Risinger M, Crable S, Ottlinger A, et al. K-Cl cotransporter gene expression during human and murine erythroid differentiation. J Biol Chem. 2011;286(35):30492–30503. doi: 10.1074/jbc.M110.206516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lauf PK, Zhang J, Delpire E, Fyffe RE, Mount DB, Adragna NC. K-Cl co-transport: immunocytochemical and functional evidence for more than one KCC isoform in high K and low K sheep erythrocytes. Comp Biochem Physiol A Mol Integr Physiol. 2001;130(3):499–509. doi: 10.1016/S1095-6433(01)00421-4. [DOI] [PubMed] [Google Scholar]
- 41.Jennings ML, Adame MF. Direct estimate of 1:1 stoichiometry of K(+)-Cl(-) cotransport in rabbit erythrocytes. Am J Physiol Cell Physiol. 2001;281(3):C825–C832. doi: 10.1152/ajpcell.2001.281.3.C825. [DOI] [PubMed] [Google Scholar]
- 42.Mercado A, Song L, Vazquez N, Mount DB, Gamba G. Functional comparison of the K+-Cl- cotransporters KCC1 and KCC4. J Biol Chem. 2000;275(39):30326–30334. doi: 10.1074/jbc.M003112200. [DOI] [PubMed] [Google Scholar]
- 43.Bergeron MJ, Gagnon E, Wallendorff B, Lapointe JY, Isenring P. Ammonium transport and pH regulation by K(+)-Cl(-) cotransporters. Am J Physiol Renal Physiol. 2003;285(1):F68–F78. doi: 10.1152/ajprenal.00032.2003. [DOI] [PubMed] [Google Scholar]
- 44.Simard CF, Bergeron MJ, Frenette-Cotton R, Carpentier GA, Pelchat ME, Caron L, et al. Homooligomeric and heterooligomeric associations between K+-Cl- cotransporter isoforms and between K+-Cl- and Na+-K+-Cl- cotransporters. J Biol Chem. 2007;282(25):18083–18093. doi: 10.1074/jbc.M607811200. [DOI] [PubMed] [Google Scholar]
- 45.Sachs JR, Martin DW. Role of polyamine structure in inhibition of K+-Cl- cotransport in human red cell ghosts. J Physiol. 1999;520(Pt 3):723–735. doi: 10.1111/j.1469-7793.1999.00723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rinehart J, Maksimova YD, Tanis JE, Stone KL, Hodson CA, Zhang J, et al. Sites of regulated phosphorylation that control K-Cl cotransporter activity. Cell. 2009;138(3):525–536. doi: 10.1016/j.cell.2009.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frenette-Cotton R, Marcoux AA, Garneau AP, Noel M, Isenring P. Phosphoregulation of K(+) -Cl(-) cotransporters during cell swelling: novel insights. J Cell Physiol. 2018;233(1):396–408. doi: 10.1002/jcp.25899. [DOI] [PubMed] [Google Scholar]
- 48.Armsby CC, Stuart-Tilley AK, Alper SL, Brugnara C. Resistance to osmotic lysis in BXD-31 mouse erythrocytes: association with upregulated K-Cl cotransport. Am J Physiol. 1996;270(3 Pt 1):C866–C877. doi: 10.1152/ajpcell.1996.270.3.C866. [DOI] [PubMed] [Google Scholar]
- 49.Pellegrino CM, Rybicki AC, Musto S, Nagel RL, Schwartz RS. Molecular identification and expression of erythroid K:Cl cotransporter in human and mouse erythroleukemic cells. Blood Cells Mol Dis. 1998;24(1):31–40. doi: 10.1006/bcmd.1998.0168. [DOI] [PubMed] [Google Scholar]
- 50.Canessa M, Romero JR, Lawrence C, Nagel RL, Fabry ME. Rate of activation and deactivation of K:Cl cotransport by changes in cell volume in hemoglobin SS, CC and AA red cells. J Membr Biol. 1994;142(3):349–362. doi: 10.1007/BF00233441. [DOI] [PubMed] [Google Scholar]
- 51.Parshina EY, Yusipovich AI, Platonova AA, Grygorczyk R, Maksimov GV, Orlov SN. Thermal inactivation of volume-sensitive K(+),Cl(-) cotransport and plasma membrane relief changes in human erythrocytes. Pflugers Arch. 2013;465(7):977–983. doi: 10.1007/s00424-013-1221-4. [DOI] [PubMed] [Google Scholar]
- 52.Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. doi: 10.1038/nrdp.2018.10. [DOI] [PubMed] [Google Scholar]
- 53.Yuditskaya S, Suffredini AF, Kato GJ. The proteome of sickle cell disease: insights from exploratory proteomic profiling. Expert Rev Proteomics. 2010;7(6):833–848. doi: 10.1586/epr.10.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brugnara C. Sickle cell dehydration: Pathophysiology and therapeutic applications. Clin Hemorheol Microcirc. 2018;68(2-3):187–204. doi: 10.3233/CH-189007. [DOI] [PubMed] [Google Scholar]
- 55.Brugnara C, Van Ha T, Tosteson DC. Role of chloride in potassium transport through a K-Cl cotransport system in human red blood cells. Am J Physiol. 1989;256(5 Pt 1):C994–1003. doi: 10.1152/ajpcell.1989.256.5.C994. [DOI] [PubMed] [Google Scholar]
- 56.Darrow MC, Zhang Y, Cinquin BP, Smith EA, Boudreau R, Rochat RH, et al. Visualizing red blood cell sickling and the effects of inhibition of sphingosine kinase 1 using soft X-ray tomography. J Cell Sci. 2016;129(18):3511–3517. doi: 10.1242/jcs.189225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gibson JS, Speake PF, Ellory JC. Differential oxygen sensitivity of the K+-Cl- cotransporter in normal and sickle human red blood cells. J Physiol. 1998;511(Pt 1):225–234. doi: 10.1111/j.1469-7793.1998.225bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gibson JS, Khan A, Speake PF, Ellory JC. O2 dependence of K+ transport in sickle cells: the effect of different cell populations and the substituted benzaldehyde 12C79. FASEB J. 2001;15(3):823–832. doi: 10.1096/fj.00-0177com. [DOI] [PubMed] [Google Scholar]
- 59.Muzyamba MC, Campbell EH, Gibson JS. Effect of intracellular magnesium and oxygen tension on K+-Cl- cotransport in normal and sickle human red cells. Cell Physiol Biochem. 2006;17(3-4):121–128. doi: 10.1159/000092073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gibson JS, Speake PF, Muzyamba MC, Husain F, Luckas MC, Ellory JC. K(+) transport in red blood cells from human umbilical cord. Biochim Biophys Acta. 2001;1512(2):231–238. doi: 10.1016/S0005-2736(01)00323-6. [DOI] [PubMed] [Google Scholar]
- 61.Gibson JS, Muzyamba MC, Ball SE, Ellory JC. K+ transport in HbSC-containing human red blood cells. Journal of Physiology. 2001;535:27. [Google Scholar]
- 62.Ueda Y, Nagel RL, Bookchin RM. An increased Bohr effect in sickle cell anemia. Blood. 1979;53(3):472–480. [PubMed] [Google Scholar]
- 63.Hannemann A, Weiss E, Rees DC, Dalibalta S, Ellory JC, Gibson JS. The Properties of Red Blood Cells from Patients Heterozygous for HbS and HbC (HbSC Genotype) Anemia. 2011;2011:248527. doi: 10.1155/2011/248527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ellory JC, Hall AC, Ody SA. Factors affecting the activation and inactivation of KCl cotransport in 'young' human red cells. Biomed Biochim Acta. 1990;49(2-3):S64–S69. [PubMed] [Google Scholar]
- 65.Brugnara C, Bunn HF, Tosteson DC. Regulation of erythrocyte cation and water content in sickle cell anemia. Science. 1986;232(4748):388–390. doi: 10.1126/science.3961486. [DOI] [PubMed] [Google Scholar]
- 66.Gonsalves CS, Crable S, Chandra S, Li W, Kalra VK, Joiner CH. Angiogenic growth factors augment K-Cl cotransporter expression in erythroid cells via hypoxia-inducible factor-1alpha. Am J Hematol. 2014;89(3):273–281. doi: 10.1002/ajh.23631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonsalves CS, Li C, Mpollo MS, Pullarkat V, Malik P, Tahara SM, et al. Erythropoietin-mediated expression of placenta growth factor is regulated via activation of hypoxia-inducible factor-1alpha and post-transcriptionally by miR-214 in sickle cell disease. Biochem J. 2015;468(3):409–423. doi: 10.1042/BJ20141138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ortiz OE, Lew VL, Bookchin RM. Deoxygenation permeabilizes sickle cell anaemia red cells to magnesium and reverses its gradient in the dense cells. J Physiol. 1990;427:211–226. doi: 10.1113/jphysiol.1990.sp018168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willcocks JP, Mulquiney PJ, Ellory JC, Veech RL, Radda GK, Clarke K. Simultaneous determination of low free Mg2+ and pH in human sickle cells using 31P NMR spectroscopy. J Biol Chem. 2002;277(51):49911–49920. doi: 10.1074/jbc.M207551200. [DOI] [PubMed] [Google Scholar]
- 70.De Franceschi L, Bachir D, Galacteros F, Tchernia G, Cynober T, Alper S, et al. Oral magnesium supplements reduce erythrocyte dehydration in patients with sickle cell disease. J Clin Invest. 1997;100(7):1847–1852. doi: 10.1172/JCI119713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sachs JR. Soluble polycations and cationic amphiphiles inhibit volume-sensitive K-Cl cotransport in human red cell ghosts. Am J Physiol. 1994;266(4 Pt 1):C997–1005. doi: 10.1152/ajpcell.1994.266.4.C997. [DOI] [PubMed] [Google Scholar]
- 72.Orlov SN, Kolosova IA, Cragoe EJ, Gurlo TG, Mongin AA, Aksentsev SL, et al. Kinetics and peculiarities of thermal inactivation of volume-induced Na+/H+ exchange, Na+,K+,2Cl- cotransport and K+,Cl- cotransport in rat erythrocytes. Biochim Biophys Acta. 1993;1151(2):186–192. doi: 10.1016/0005-2736(93)90103-7. [DOI] [PubMed] [Google Scholar]
- 73.de Los HP, Alessi DR, Gourlay R, Campbell DG, Deak M, Macartney TJ, et al. The WNK-regulated SPAK/OSR1 kinases directly phosphorylate and inhibit the K+-Cl- co-transporters. Biochem J. 2014;458(3):559–573. doi: 10.1042/BJ20131478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kahle KT, Flores B, Bharucha-Goebel D, Zhang J, Donkervoort S, Hegde M, et al. Peripheral motor neuropathy is associated with defective kinase regulation of the KCC3 cotransporter. Sci Signal. 2016;9(439):ra77. doi: 10.1126/scisignal.aae0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Melo Z, de los Heros P, Cruz-Rangel S, Vazquez N, Bobadilla NA, Pasantes-Morales H, et al. N-terminal serine dephosphorylation is required for KCC3 cotransporter full activation by cell swelling. J Biol Chem. 2013;288(44):31468–31476. doi: 10.1074/jbc.M113.475574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mercado A, de Los HP, Melo Z, Chavez-Canales M, Murillo-de-Ozores AR, Moreno E, et al. With no lysine L-WNK1 isoforms are negative regulators of the K+-Cl- cotransporters. Am J Physiol Cell Physiol. 2016;311(1):C54–C66. doi: 10.1152/ajpcell.00193.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem. 2000;275(22):16795–16801. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- 78.Rinehart J, Kahle KT, de Los HP, Vazquez N, Meade P, Wilson FH, et al. WNK3 kinase is a positive regulator of NKCC2 and NCC, renal cation-Cl- cotransporters required for normal blood pressure homeostasis. Proc Natl Acad Sci U S A. 2005;102(46):16777–16782. doi: 10.1073/pnas.0508303102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sasaki E, Susa K, Mori T, Isobe K, Araki Y, Inoue Y, et al. KLHL3 Knockout Mice Reveal the Physiological Role of KLHL3 and the Pathophysiology of Pseudohypoaldosteronism Type II Caused by Mutant KLHL3. Mol Cell Biol. 2017;37(7):e00508–16. [DOI] [PMC free article] [PubMed]
- 80.Glover M, Ware JS, Henry A, Wolley M, Walsh R, Wain LV, et al. Detection of mutations in KLHL3 and CUL3 in families with FHHt (familial hyperkalaemic hypertension or Gordon's syndrome) Clin Sci (Lond). 2014;126(10):721–726. doi: 10.1042/CS20130326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ranney HM. Psickle, the temporary leaky link between sickling and cellular dehydration. J Clin Invest. 1997;99(11):2559–2560. doi: 10.1172/JCI119441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lew VL, Ortiz OE, Bookchin RM. Stochastic nature and red cell population distribution of the sickling-induced Ca2+ permeability. J Clin Invest. 1997;99(11):2727–2735. doi: 10.1172/JCI119462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma YL, Rees DC, Gibson JS, Ellory JC. The conductance of red blood cells from sickle cell patients: ion selectivity and inhibitors. J Physiol. 2012;590(9):2095–2105. doi: 10.1113/jphysiol.2012.229609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Demolombe S, Duprat F, Honore E, Patel A. Slower Piezo1 inactivation in dehydrated hereditary stomatocytosis (xerocytosis) Biophys J. 2013;105(4):833–834. doi: 10.1016/j.bpj.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cahalan SM, Lukacs V, Ranade SS, Chien S, Bandell M, Patapoutian A. Piezo1 links mechanical forces to red blood cell volume. Elife. 2015;4. [DOI] [PMC free article] [PubMed]
- 86.Maher AD, Kuchel PW. The Gardos channel: a review of the Ca2+-activated K+ channel in human erythrocytes. Int J Biochem Cell Biol. 2003;35(8):1182–1197. doi: 10.1016/S1357-2725(02)00310-2. [DOI] [PubMed] [Google Scholar]
- 87.Ataga KI, Smith WR, De Castro LM, Swerdlow P, Saunthararajah Y, Castro O, et al. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood. 2008;111(8):3991–3997. doi: 10.1182/blood-2007-08-110098. [DOI] [PubMed] [Google Scholar]
- 88.Mohandas N, Rossi ME, Clark MR. Association between morphologic distortion of sickle cells and deoxygenation-induced cation permeability increase. Blood. 1986;68(2):450–454. [PubMed] [Google Scholar]
- 89.Gardos G. The function of calcium in the potassium permeability of human erythrocytes. Biochim Biophys Acta. 1958;30(3):653–654. doi: 10.1016/0006-3002(58)90124-0. [DOI] [PubMed] [Google Scholar]
- 90.Castro OL, Gordeuk VR, Gladwin MT, Steinberg MH. Senicapoc trial results support the existence of different sub-phenotypes of sickle cell disease with possible drug-induced phenotypic shifts. Br J Haematol. 2011;155(5):636–638. doi: 10.1111/j.1365-2141.2011.08758.x. [DOI] [PubMed] [Google Scholar]
- 91.McNaughton-Smith GA, Burns JF, Stocker JW, Rigdon GC, Creech C, Arrington S, et al. Novel inhibitors of the Gardos channel for the treatment of sickle cell disease. J Med Chem. 2008;51(4):976–982. doi: 10.1021/jm070663s. [DOI] [PubMed] [Google Scholar]
- 92.Shang C, Lu YM, Meng LR. KCC1 gene advances cell invasion ability by regulating ERK signaling pathway in endometrial cancer HEC-1B cell line. Int J Gynecol Cancer. 2011;21(5):795–799. doi: 10.1097/IGC.0b013e318216a169. [DOI] [PubMed] [Google Scholar]
- 93.Yao M, Yoshida M, Kishida T, Nakaigawa N, Baba M, Kobayashi K, et al. VHL tumor suppressor gene alterations associated with good prognosis in sporadic clear-cell renal carcinoma. J Natl Cancer Inst. 2002;94(20):1569–1575. doi: 10.1093/jnci/94.20.1569. [DOI] [PubMed] [Google Scholar]
- 94.Patard JJ, Fergelot P, Karakiewicz PI, Klatte T, Trinh QD, Rioux-Leclercq N, et al. Low CAIX expression and absence of VHL gene mutation are associated with tumor aggressiveness and poor survival of clear cell renal cell carcinoma. Int J Cancer. 2008;123(2):395–400. doi: 10.1002/ijc.23496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kornak U, Kasper D, Bosl MR, Kaiser E, Schweizer M, Schulz A, et al. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104(2):205–215. doi: 10.1016/S0092-8674(01)00206-9. [DOI] [PubMed] [Google Scholar]
- 96.Feng S, Deng L, Chen W, Shao J, Xu G, Li YP. Atp6v1c1 is an essential component of the osteoclast proton pump and in F-actin ring formation in osteoclasts. Biochem J. 2009;417(1):195–203. doi: 10.1042/BJ20081073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baron R, Neff L, Roy C, Boisvert A, Caplan M. Evidence for a high and specific concentration of (Na+,K+)ATPase in the plasma membrane of the osteoclast. Cell. 1986;46(2):311–320. doi: 10.1016/0092-8674(86)90748-8. [DOI] [PubMed] [Google Scholar]
- 98.Wu J, Glimcher LH, Aliprantis AO. HCO3-/Cl- anion exchanger SLC4A2 is required for proper osteoclast differentiation and function. Proc Natl Acad Sci U S A. 2008;105(44):16934–16939. doi: 10.1073/pnas.0808763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cheng L, Zhang K, Zhang Z. Effectiveness of thiazides on serum and urinary calcium levels and bone mineral density in patients with osteoporosis: a systematic review and meta-analysis. Drug Des Devel Ther. 2018;12:3929–3935. doi: 10.2147/DDDT.S179568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hsu YJ, Yang SS, Cheng CJ, Liu ST, Huang SM, Chau T, et al. Thiazide-sensitive Na+ -Cl- cotransporter (NCC) gene inactivation results in increased duodenal Ca2+ absorption, enhanced osteoblast differentiation and elevated bone mineral density. J Bone Miner Res. 2015;30(1):116–127. doi: 10.1002/jbmr.2306. [DOI] [PubMed] [Google Scholar]
- 101.Alexander RT, Dimke H. Effect of diuretics on renal tubular transport of calcium and magnesium. Am J Physiol Renal Physiol. 2017;312(6):F998–F1015. doi: 10.1152/ajprenal.00032.2017. [DOI] [PubMed] [Google Scholar]
- 102.Dvorak MM, De Joussineau C, Carter DH, Pisitkun T, Knepper MA, Gamba G, et al. Thiazide diuretics directly induce osteoblast differentiation and mineralized nodule formation by interacting with a sodium chloride co-transporter in bone. J Am Soc Nephrol. 2007;18(9):2509–2516. doi: 10.1681/ASN.2007030348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun YT, Shieh CC, Delpire E, Shen MR. K(+)-Cl(-) cotransport mediates the bactericidal activity of neutrophils by regulating NADPH oxidase activation. J Physiol. 2012;590(14):3231–3243. doi: 10.1113/jphysiol.2011.225300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Foote JR, Behe P, Frampton M, Levine AP, Segal AW. An exploration of charge compensating ion channels across the phagocytic vacuole of neutrophils. Front Pharmacol. 2017;8:94. doi: 10.3389/fphar.2017.00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Paredes J, Figueiredo J, Albergaria A, Oliveira P, Carvalho J, Ribeiro AS, et al. Epithelial E- and P-cadherins: role and clinical significance in cancer. Biochim Biophys Acta. 2012;1826(2):297–311. doi: 10.1016/j.bbcan.2012.05.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article (see Footnotes).