

 Simple and direct determination of iron ion concentrations in water samples is achieved.
Simple and direct determination of iron ion concentrations in water samples is achieved.
Abstract
Iron is one of the most abundant elements in the environment and in the human body. As an essential nutrient, iron homeostasis is tightly regulated, and iron dysregulation is implicated in numerous pathologies, including neuro-degenerative diseases, atherosclerosis, and diabetes. Endogenous iron pool concentrations are directly linked to iron ion uptake from environmental sources such as drinking water, providing motivation for developing new technologies for assessing iron(ii) and iron(iii) levels in water. However, conventional methods for measuring aqueous iron pools remain laborious and costly and often require sophisticated equipment and/or additional processing steps to remove the iron ions from the original environmental source. We now report a simplified and accurate chemical platform for capturing and quantifying the iron present in aqueous samples through use of a post-synthetically modified porous aromatic framework (PAF). The ether/thioether-functionalized network polymer, PAF-1–ET, exhibits high selectivity for the uptake of iron(ii) and iron(iii) over other physiologically and environmentally relevant metal ions. Mössbauer spectroscopy, XANES, and EXAFS measurements provide evidence to support iron(iii) coordination to oxygen-based ligands within the material. The polymer is further successfully employed to adsorb and remove iron ions from groundwater, including field sources in West Bengal, India. Combined with an 8-hydroxyquinoline colorimetric indicator, PAF-1–ET enables the simple and direct determination of the iron(ii) and iron(iii) ion concentrations in these samples, providing a starting point for the design and use of molecularly-functionalized porous materials for potential dual detection and remediation applications.
Introduction
Iron is the fourth most abundant element in the earth's crust and the most abundant transition metal in the human body.1 It is required for sustaining a range of physiological processes such as electron transfer, oxygen transport, respiration, and gene expression,2–5 and iron deficiency leads to anemia.6 However, excess iron can increase production of reactive oxygen species, resulting in oxidative stress cascades that lead to lipid oxidation and DNA damage.2–4,7,8 Aberrant iron accumulation is implicated in aging and in several diseases, including cardiovascular diseases, neurodegenerative diseases, and cancer.6,9–14 In this context, chronic exposure to elevated iron levels in common drinking water is a potential contributor to abnormal iron accumulation. The World Health Organization (WHO) recommends an upper limit of 0.3 mg L–1 for iron ions in drinking water.15 Unfortunately, the reported iron ion concentrations in drinking water sources can vary over several orders of magnitude—for instance from 0.007 to 33.6 mg L–1 in West Bengal16 or from undetectable amounts to 950 mg L–1 based on Groundwater Ambient Monitoring and Assessment data from the San Francisco Bay (see ESI†). Because traditional methods for iron(ii) and iron(iii) detection require expensive instrumentation, such as inductively coupled plasma mass spectrometry or atomic absorption spectroscopy,17,18 it remains a challenge to rapidly and inexpensively screen drinking water for quantities of iron and other metal ion contaminants, particularly in developing countries and other lower-resource environments.11,16,17,19–24
To meet this challenge, we sought to develop a chemical strategy that would enable simultaneous detection and removal of both iron(ii) and iron(iii) ions from drinking water and other environmental and biomedical samples, with high selectivity over other metal ion contaminants. In particular, we envisioned a robust, molecularly-tailored solid-state adsorbent that would efficiently capture and remove iron ions from a complex water sample obtained in the field while also permitting a quantitative measure of the iron concentration. We chose to investigate porous aromatic frameworks (PAFs) for this dual remediation and quantitative detection function, owing to their high chemical and thermal stability—particularly in aqueous and biological samples—and because of their ability to be functionalized in a molecular fashion.25–37 The polymer PAF-1 is one such material that exhibits a high Brunauer–Emmett–Teller (BET) surface area38 of up to 5600 m2 g–1 and is readily functionalized through post-synthetic modification.38–43 Indeed, we recently reported a thioether-functionalized variant of this porous polymer, PAF-1–SMe, as a platform for selective copper ion capture from biofluid samples, ultimately as a diagnostic tool for Wilson's disease.42
Here, we present the synthesis of an iron-selective porous polymer via ether–thioether (ET) functionalization of PAF-1. The polymer PAF-1–ET (Fig. 1a) exhibits highly selective iron(ii) and iron(iii) ion uptake over competing metal ions in laboratory and field water samples. The combination of this polymer with 8-hydroxyquinoline indicator enables rapid and quantitative monitoring of iron levels with a simple colorimetric assay. We highlight the potential utility of this method for remediation and screening of synthetic groundwater as well as field samples of drinking water collected from West Bengal, India. This work provides a starting point for the development of new porous polymers for simplified, accurate, and rapid diagnostic and remediation applications without the need for bulky and expensive instrumentation.
Fig. 1. (a) Synthesis of PAF-1–ET. (b) Solid-state 13C NMR spectra of PAF-1–CH2Cl and PAF-1–ET illustrating new 13C chemical shifts arising from the –CH2OCH2CH2SCH3 group in PAF-1–ET (73, 48, 39, and 17 ppm) compared with a shift at 43 ppm for the –CH2Cl group in PAF-1–CH2Cl. (c) Nitrogen adsorption isotherms of PAF-1, PAF-1–CH2Cl, and PAF-1–ET measured at 77 K. Closed and open symbols represent adsorption and desorption results, respectively.

Experimental
General methods
All reactions were performed under a nitrogen or argon atmosphere and in dry solvents, unless otherwise stated. Tetrakis(4-bromophenyl)methane was purchased from TCI America as a pale yellow powder. All other starting materials and reagents were purchased from Sigma-Aldrich. Nitrogen adsorption isotherms were measured using a Micromeritics ASAP 2020 or 2420 instrument. Samples were transferred to a pre-weighed glass analysis tube that was capped with a Transeal and then evacuated on the ASAP until the outgas rate was less than 3 μbar min–1. Ultrahigh-purity grade (99.999%) nitrogen was used for gas adsorption measurements. Nitrogen isotherms were obtained using a 77 K liquid-N2 bath and were used to determine the surface areas and pore volumes using the Micromeritics software, assuming a value of 16.2 Å2 for the molecular cross-sectional area of N2. Infrared spectra were obtained on a PerkinElmer Spectrum 100 Optica FTIR spectrometer furnished with an attenuated total reflectance accessory. Thermal gravimetric analysis data were collected at a ramp rate of 5 °C min–1 under flowing nitrogen using a TA Instruments TGA Q5000. Scanning electron microscopy (SEM) samples were prepared by dispersing fine polymer powders into methanol and drop casting onto silicon chips. To dissipate charge, the samples were sputter coated with approximately 3 nm of Au (Denton Vacuum). Polymers were imaged at 5 keV and 12 μA by field emission SEM (JEOL FSM6430). Elemental analyses (C, H, N, S) were obtained from the Microanalytical Laboratory at the University of California, Berkeley. Elemental analysis for chlorine was performed at Galbraith Laboratories. UV-Vis spectroscopic measurements were performed in 100 mM HEPES buffer (pH = 6.7). Absorption spectra were recorded using a Varian Cary 50 spectrophotometer, and samples for absorption measurements were prepared in 1 × 0.5 cm quartz cuvettes (1.4 mL, Starna). Inductively coupled plasma-mass spectrometry (ICP-MS) was performed on samples that had been diluted into 2% nitric acid (made freshly from concentrated nitric acid [BDH Aristar Ultra] and Milli-Q water) containing 20 μg L–1 of a gallium internal standard (Inorganic Ventures, Christiansburg, VA). The samples were analyzed on a ThermoFisher iCAP-Qc ICP-MS in kinetic energy discrimination mode against a calibration curve of known metal concentrations (made from CMS-5, Inorganic Ventures, Christiansburg, VA). Low-temperature X-band EPR spectra were recorded using a Varian E109 EPR spectrometer equipped with a Model 102 microwave bridge. Sample temperature was maintained at 8 K by using an Air Products LTR liquid helium cryostat. The following spectrometer conditions were used: microwave frequency, 9.22 GHz; field modulation amplitude, 32 G at 100 kHz, and a microwave power of 20 mW.
Synthesis of PAF-1–ET
2-(Methylthio)ethan-1-ol (1.83 mL, 0.021 mol) and 3 equiv. of NaH (1.5 g, 0.063 mol) were mixed with toluene (100 mL) in a 250 mL Schlenk flask under N2. After 5 min, freshly-prepared PAF-1–CH2Cl (260 mg, see the ESI†) was added, and the mixture was stirred at 90 °C for 3 days. The resulting solid was collected, washed sequentially with 100 mL each of H2O, ethanol, CHCl3, and THF, and dried in a vacuum oven at 150 °C to produce PAF-1–ET as an off-white powder. Calc. for C32.5H34O2S2 (%): C 74.96, H 6.58, S 12.31, Cl 0.00; observed: C 74.89, H 5.08, S 5.50, Cl 1.97. Based on the sulfur elemental analysis, this preparation resulted in 45% substitution with 2-(methylthio)ethan-1-ol.§
Solid-state 13C NMR
NMR sample preparation
Samples of PAF-1–ET (35 mg) and PAF-1–CH2Cl (20 mg) were dried at 100 °C for 3 h before data collection. Iron(iii)-loaded PAF-1–ET was prepared by stirring PAF-1–ET (50 mg) at room temperature overnight in a solution of FeCl3 (10 mL, 100 mg L–1) dissolved in 100 mM HEPES buffer (pH = 6.7) with 2 equiv. of citric acid. The filtered iron(iii)-PAF-1–ET was washed with Milli-Q H2O, ethanol, dichloromethane, and THF (50 mL each) and dried at 100 °C for 3 h before data collection.
NMR experiments
All experiments were conducted at a 13C frequency of 75.5 MHz using a Tecmag Discovery spectrometer equipped with a 7.05 T magnet and a Chemagnetics 4 mm HX CP/MAS probe (magic-angle spinning rate of 10 kHz). Cross-polarization from 1H was used when acquiring spectra for PAF-1–CH2Cl and PAF-1–ET. The Hartmann–Hahn condition44 for cross-polarization experiments was obtained on solid adamantane, which is also a secondary 13C chemical shift reference (the methylene signal of adamantane was set to 38.48 ppm relative to TMS). The PAF-1–CH2Cl spectrum was collected using a CP contact time of 10 ms and a pulse delay of 4 s. A two-pulse phase modulation (TPPM) proton decoupling scheme was used, with a TPPM angle of 15 degrees and decoupling field strength of ∼60 kHz. The spectrum for PAF-1–ET was obtained using a contact time of 1 ms and a pulse delay of 4 s. Direct polarization (1 s pulse delay) was used to collect the spectrum for iron(iii)-loaded PAF-1–ET. Continuous wave proton decoupling was used for both the PAF-1–ET and iron(iii)–PAF-1–ET spectra, with a decoupling field strength of ∼60 kHz.
Metal ion adsorption in PAF-1–ET
Iron adsorption measurements
Samples of PAF-1–ET (2.0 mg) were added to conical tubes containing 5 mL of (NH4)2Fe(SO4)2·6H2O (dissolved in 100 mM HEPES buffer, pH = 6.7) with concentrations ranging from 10–3 to 275 mg L–1. Each mixture was capped under air and stored in a shaker at room temperature overnight. Each solution was subsequently filtered through a 0.45 μm membrane filter, and the filtrates were analyzed by ICP-MS to determine the residual iron content. Iron uptake (initial–residual iron concentration) data was fit using a Langmuir model given by: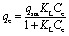 where qe is the adsorption capacity (mg g–1), Ce is the equilibrium iron ion concentration (mg L–1), qsat is the adsorption saturation capacity (mg g–1), and KL is the Langmuir constant (L mg–1), which is related to the binding affinity of the adsorption site. Langmuir fits are shown in Fig. 2a, and fit parameters are provided in Table S1.†
where qe is the adsorption capacity (mg g–1), Ce is the equilibrium iron ion concentration (mg L–1), qsat is the adsorption saturation capacity (mg g–1), and KL is the Langmuir constant (L mg–1), which is related to the binding affinity of the adsorption site. Langmuir fits are shown in Fig. 2a, and fit parameters are provided in Table S1.†
Fig. 2. (a) Iron(iii) adsorption isotherm for PAF-1–ET (blue squares) and PAF-1–CH2Cl (red circles) obtained for solutions of (NH4)2Fe(SO4)2 dissolved in 100 mM HEPES buffer solution (pH = 6.7, initial iron concentrations ranging from 10–3 to 275 mg L–1). Black lines represent fits obtained using a Langmuir model. (b) Adsorption by PAF-1–ET of several physiologically-relevant metal ions dissolved in 100 mM HEPES buffer solution at pH = 6.7 (initial concentrations of 0.3, 2, and 20 mg L–1). In the case of iron(iii), one equivalent of citric acid was also added to the solution to prevent precipitation of Fe(OH)3.

Metal ion adsorption selectivity studies
Samples of PAF-1–ET (2 mg) were added to conical tubes containing aqueous solutions (5 mL) of NaCl, KCl, MgCl2, CaCl2, MnCl2, (NH4)2Fe(SO4)2·6H2O, FeCl3, CoCl2, NiCl2, CuCl2, or ZnCl2 at initial concentrations of 0.3, 2, or 20 mg L–1 in 100 mM HEPES buffer (pH = 6.7). Iron(ii) samples were prepared and stored under anaerobic conditions until analysis by ICP-MS. In the case of iron(iii), one equivalent of citric acid was also added to the samples to prevent Fe(OH)3 precipitation. The slurries were stored in a shaker at room temperature overnight and then filtered through a 0.45 μm membrane. The filtrates were analyzed using ICP-MS, and the amount of metal ion adsorbed was calculated by subtracting the residual iron concentration from the initial iron concentration. The distribution coefficient, Kd, for each metal ion was determined by using PAF-1–ET (5 mg) in each metal solution (45 mL, 20 mg L–1 with an approximate flow rate of 0.5 mL min–1 as described in Section 3 of the ESI†).
Iron adsorption kinetics
An Erlenmeyer flask containing 2 mg of PAF-1–ET was charged with a solution of (NH4)2Fe(SO4)2·6H2O (10 mL, 10.2 ppm) in 100 mM HEPES buffer (pH = 6.7) and 1 equiv. of citric acid. The mixture was stirred at room temperature for 8 h. During this period, aliquots of the mixture were filtered at intervals through a 0.45 μm membrane. The filtrates were analyzed using ICP-MS to determine the iron ion concentration. The amount of iron adsorbed by PAF-1–ET was calculated by subtracting the residual from the initial iron concentration. The adsorption data were fit with the pseudo-second-order kinetic model (see Fig. S7†):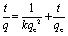 where k is the pseudo-second-order rate constant (g mg–1 min–1) and qe is the amount of iron adsorbed at equilibrium (mg g–1).
where k is the pseudo-second-order rate constant (g mg–1 min–1) and qe is the amount of iron adsorbed at equilibrium (mg g–1).
Mössbauer experiments
A sample of PAF-1–ET (∼50 mg) was added to an aqueous solution of 57FeCl3 (50 mg L–1, see the ESI†), and the mixture was stirred overnight at room temperature under N2. The resulting 57Fe(iii)-loaded PAF-1–ET was collected, washed with warm H2O (100 mL) and CHCl3 (100 mL), and then dried in a vacuum oven at 150 °C to yield a white powder. Mössbauer spectra were obtained between 5 and 300 K with a SEE Co. Mössbauer spectrometer equipped with a Co-57 source in Rh matrix. Reported isomer shifts are given relative to α-iron at 295 K. The spectral absorber was prepared in air by packing the sample into a 1.27 cm diameter Nylon washer before transferring to the spectrometer, where the absorber was always maintained under a He atmosphere. See the ESI† (Section 6) for full measurement details.
X-ray absorption measurements
X-ray absorption spectra were collected at the Stanford synchrotron radiation light source on beamline 9–3 with ring storage conditions of 3.0 GeV and 500 mA. The iron K-edge absorption spectra of the PAF samples, packed in 0.5 mm thick aluminum sample holders with Kapton film windows, were recorded at room temperature. Reference compounds were analyzed after dilution with boron nitride. The spectral data were collected in transmission mode for Fe, Fe2O3, and FeO and in fluorescence mode for PAF-1–ET with a 100-element Ge monolithic solid-state detector from Canberra. The incident radiation was monochromatized using a Si(220) double crystal monochromator, which was detuned to 50% of flux maximum at the iron K-edge to minimize the higher harmonics and reduce X-ray flux. A harmonic rejection mirror was used to further reduce the contamination from higher harmonics radiation. The incident and transmitted X-ray intensities were monitored with N2-filled ion chambers. An iron foil spectrum was concomitantly recorded for energy calibration where the first inflection point was assigned to 7111.2 eV. Even at the low X-ray flux density used, a slight photoreduction of PAF-1–ET was observed even after two scans at a given sample position. As a consequence, the spectral data were collected at multiple spots, and only the first two scans at each position were used for averaging the spectral data over multiple positions.
Data reduction was carried out with the SamView software obtained from SixPack software.¶ Athena software, Demeter version 0.9.25 45 was used for data averaging and removal of the pre-edge and post-edge background absorption. A five-domain cubic spline was used to remove low-frequency background in k-space. The resulting k-space data, as k3χ(k), was then Fourier transformed into r-space over a k range of 3.46 to 10.52 Å–1 and used for the extended X-ray absorption fine structure (EXAFS) refinement. The EXAFS fitting was carried out using the Artemis software45 with phase and amplitude functions obtained from FEFF, version 6.46 The average bond distance between the iron and scattering atom (R) and the mean square displacement of the bond distance (σ2) were allowed to vary, while N, the number of atoms in the shell, was systematically varied in integral steps. The value of E0, the energy of the zero value of the photoelectron wave vector k, was also varied but constrained to a common value for every shell in a given fit. The value for S02, the amplitude reduction factor, was extracted from the fit of the Fe foil data and was fixed at 0.95 during all other fits. The best fit parameters for EXAFS fits are given in Table S3.†
Results and discussion
Synthesis and characterization
Building upon the design of the thioether-functionalized polymer PAF-1–SMe for selective copper ion capture42 and the related PAF-1–SH for mercury ion adsorption,40 we initially synthesized a variety of PAF-1 derivative scaffolds. Subsequent testing for metal ion uptake from aqueous solutions showed that the variant with an extended ether–thioether pendant, referred to here as PAF-1–ET, is an effective material for selective capture of both iron(ii) and iron(iii). This work complements our recent efforts to develop activity-based fluorescent probes for iron(ii).47–52
The polymer PAF-1–ET was synthesized in three steps, starting with the synthesis of parent PAF-1 and PAF-1–CH2Cl (see the ESI†)40 followed by treatment of the latter with 2-(methylthio)ethan-1-ol to yield PAF-1–ET (Fig. 1a). Elemental analysis revealed a decrease in chlorine content from 13.60% in PAF-1–CH2Cl to 1.97 ± 1.5% in PAF-1–ET, and the sulfur content of the latter was found to be 5.50 ± 0.82%—corresponding to grafting of 45 ± 6.7% of the phenyl groups in PAF-1–ET or a loading of 1.7 mmol g–1. In the IR spectrum of PAF-1–ET, the absence of a peak at 1270 cm–1 (assigned to the C–H wagging mode of –CH2Cl in PAF-1–CH2Cl) further confirmed functionalization (Fig. S2†). Solid-state 1H–13C cross-polarization magic angle spinning NMR spectroscopy revealed new 13C chemical shifts at 73, 48, 39, and 17 ppm for PAF-1–ET, arising from the ether–thioether groups, and the absence of a shift at 43 ppm corresponding to the –CH2Cl groups of PAF-1–CH2Cl (Fig. 1b). Nitrogen adsorption isotherms obtained at 77 K revealed that PAF-1–ET retains permanent porosity with a high BET surface area of 1500 ± 420 m2 g–1 (Fig. 1c; error determined by measuring the N2 adsorption of four different PAF-1–ET samples). The expected drop in surface area upon PAF-1 functionalization may be due to the added mass and pore filling of the relatively bulky ether–thioether group.37,39,40,42,53–56 The average pore size distribution obtained from the adsorption isotherms was found to be <10 Å for PAF-1–ET, which is smaller than the average value of 12 Å for PAF-1 and supports incorporation of the ether–thioether groups (see Fig. S3†).
Selectivity and kinetics of iron uptake
Equilibrium iron(iii) isothermal adsorption data were collected for PAF-1–ET and PAF-1–CH2Cl over aqueous ion concentrations ranging from 1 μg L–1 to 275 mg L–1, and these data were fit using the Langmuir model29 to assess framework saturation capacities and binding affinities for iron(iii) (Fig. 2a and Table S1†). Notably, PAF-1–ET exhibited much higher iron(iii) uptake than PAF-1–CH2Cl over the entire concentration range and a binding affinity twice that of PAF-1–CH2Cl. At saturation, the maximum adsorption capacity (qsat) of PAF-1–ET was found to be 105(4) mg g–1, which corresponds to an uptake of 1.8 mmol of iron(iii) per gram of material—nearly three times the capacity of PAF-1–CH2Cl (37(2) mg g–1). Based on the maximum adsorption capacity of PAF-1–ET, the ET : iron ratio is at least 1.1. We also collected adsorption data using iron(iii) chloride, iron(iii) sulfate hydrate, or ammonium iron(iii) citrate to investigate the effect of the counterion on iron uptake in PAF-1–ET. For all salts, PAF-1–ET showed comparable iron uptake at low and high iron concentrations (see the ESI,† Section 4).
Importantly, PAF-1–ET also exhibited high selectivity for the adsorption of iron(ii) and iron(iii) ions over other biologically-relevant metal ions at initial concentrations of 0.3, 2, and 20 mg L–1 (Fig. 2b). For example, the distribution coefficient, Kd, for 10 mg L–1 iron(ii) in pH = 6.7 HEPES buffer was found to be 2.8(7) × 105 mL g–1, over an order of magnitude greater than the Kd values for 20 mg L–1 of Na+, K+, Mg2+, Ca2+, Cu2+, and Zn2+ (190, 180, 110, 280, 1700, and 150 mL g–1, respectively). Similarly, in a synthetic groundwater sample containing iron(iii) along with Mg2+, Ca2+, and Zn2+, the Kd value for iron(iii) was at least an order of magnitude greater than the Kd values for the competing cations (see ESI†). Given this exceptional performance, PAF-1–ET should be useful for iron capture from a variety of water samples.
The concentration dependence of iron(iii) uptake by PAF-1–ET was also evaluated by examining various pH = 6.7 aqueous solutions in HEPES buffer, and it was found that the adsorbed amount increases with increasing ion concentration in solution (Fig. 3a). In order to develop a colorimetric assay for detection of the adsorbed iron(iii), we evaluated the ability of 8-hydroxyquinoline to bind iron(iii) captured within the porous framework. Upon binding free iron(iii), 8-hydroxyquinoline undergoes a distinct change from colorless (315 nm absorption, ε = 1.95 × 103 M–1 cm–1) to blue-green (460 and 560 nm absorption, ε = 750 M–1 cm–1 at 460 nm). This absorption spectrum is known to be unique to an iron(iii) 8-hydroxyquinoline complex, differentiating iron(iii) from other metal complexes.57–60 Thus, successful binding of iron(iii) within PAF-1–ET by 8-hydroxyquinoline should permit a facile and quantitative determination of the quantity adsorbed. To confirm this capability, a 1 mM solution of 8-hydroxyquinoline in dimethyl sulfoxide was added to dried samples of PAF-1–ET that had been exposed to the aqueous iron samples in Fig. 3a. Gratifyingly, in the presence of these samples, the 8-hydroxyquinoline absorption spectra exhibited two new peaks at 460 and 560 nm (Fig. 3b), indicative of iron(iii) complex formation. The calculated amount of iron(iii) adsorbed by PAF-1–ET—based on the 460 nm absorbance peak for the highest sample concentration—correlated well with amounts determined directly via ICP-MS (Fig. 3c).
Fig. 3. (a) Iron(iii) adsorption by PAF-1–ET measured by ICP-MS in 100 mM HEPES (pH = 6.7) aqueous solutions using FeCl3 as the iron(iii) source. (b) Absorption spectra resulting from the addition of a 1 mM aqueous solution of 8-hydroxyquinoline to dried PAF-1–ET, obtained after exposure to the iron(iii) aqueous solutions shown in (a). The top red spectrum corresponds to the absorption of a complex of iron(iii) and 8-hydroxyquinoline, and the gray absorption spectra with increasing absorbance at 460 nm correspond to the solutions in (a) with increasing initial concentrations of iron(iii). (c) A close correlation was observed between the iron(iii) uptake measured by ICP-MS (gray squares) and the iron(iii) uptake obtained from the absorbance at 460 nm in the presence of the 8-hydroxyquinoline indicator (open circles).

Spectroscopic characterization
To obtain additional insight into the nature of the interactions between adsorbed iron(iii) and the framework functional groups, 57Fe Mössbauer spectra were collected between 5 and 300 K. Representative spectra at 100 and 5 K are shown in Fig. 4a and b (see also Fig. S8 and Table S2†). At all temperatures, the spectral fits indicated the predominant presence of paramagnetic, high-spin iron(iii) adsorbed within PAF-1–ET (red lines), with no evidence of long-range magnetic order. A small constrained component (green lines, 9% by area) was also present in all data, likely due to residual high-spin iron(ii) from sample preparation. The spectra were found to be very similar between 50 and 300 K, with a predominant bimodal distribution of quadrupole splittings, ΔEQ, between 0.6 and 1.0 mm s–1, centered about a unique high-spin iron(iii) isomer shift, δ, of 0.385(2) mm s–1 at 300 K (0.507(1) mm s–1 at 50 K, see Fig. S9†). These values are consistent with an iron(iii) ion residing in a pseudooctahedral coordination with a distribution of near-neighbor oxygen environments, a conclusion that is consistent with the extended X-ray absorption fine structure (EXAFS) data below. Interestingly, upon cooling to 20 K and below, some of the highly dispersed iron(iii) ions adsorbed in PAF-1–ET exhibit initial evidence for slow paramagnetic relaxation on the Mössbauer timescale (∼10–8 s), with predominant hyperfine fields of 45 T (17.6(6)% area), 46.6(1) T (50.4(6)% area), and 46.6(1) T (58.8(4)% area), respectively. The X-band EPR spectrum is compatible with the presence of high-spin iron(iii) (see Fig. S10†).
Fig. 4. Iron-57 Mössbauer spectra of PAF-1–ET obtained at (a) 100 and (b) 5 K. Red and green fits were assigned to iron(iii) (91%) and iron(ii) (9%), respectively. The total fit is shown in black. (c) The Fourier transform of the k3-weighted iron K-edge EXAFS data of iron(iii) adsorbed in PAF-1–ET (black dots); the best two-shell fit (red curve) was achieved with an immediate coordination environment of six O atoms at a distance of 2.00(1) Å and 12 C atoms at a distance of 3.06(4) Å; (inset) room-temperature XANES spectra obtained at the iron K-edge for PAF-1–ET (red curve) and Fe2O3, FeO, and Fe foil (gray, blue, and black curves, respectively).

Iron K-edge X-ray absorption spectroscopy was used to investigate the local coordination environment of the adsorbed iron. The X-ray absorption near-edge structure (XANES) spectrum of iron(iii) adsorbed in PAF-1–ET is shown in the inset of Fig. 4c, along with Fe2O3, FeO, and Fe foil for reference. The rising edge energy of the sample aligns well with that of Fe2O3, supporting the presence of iron(iii). Fig. 4c shows the k3-weighted EXAFS data for iron(iii)-adsorbed PAF-1–ET in r-space along with the best fit (see Table S3†). The horizontal axis represents the apparent distance R′, which is shorter than the actual distance by ∼0.5 Å due to a phase shift. For iron(iii)-adsorbed PAF-1–ET,61 the best two-shell fit was achieved with a coordination environment of six oxygen atoms at a distance of 2.00(1) Å and 12 carbon atoms at a distance of 3.06(4) Å.
We also obtained the 13C NMR spectrum of iron(iii)-loaded PAF-1–ET to compare with that of PAF-1–ET. The coordination of paramagnetic iron(iii) resulted in severe peak broadening in addition to an overall shift in the peaks observed for PAF-1–ET (Fig. S11†).62–64 The benzene ring resonances between 148–132 ppm for PAF-1–ET shifted to 147–140 ppm in the spectrum of iron(iii)-loaded PAF-1–ET, and all peaks corresponding to the ET functional groups of PAF-1–ET (73, 48, 39, and 17 ppm) shifted upfield in iron(iii)-loaded PAF-1–ET (to ∼34–5 ppm, see Fig. S11†). The more dramatic shift in the ether–thioether peaks provides additional evidence that this group is indeed bound to iron(iii).
Iron coordination in PAF-1–ET
To investigate possible iron coordination environments within PAF-1–ET, we used the program Materials Studio to generate a hypothetical portion of the PAF-1–ET structure, featuring one iron ion within a single diamond net. Based on the EXAFS data, it was assumed that a total of six oxygen atoms—and no sulfur atoms—coordinate to the iron. A monodentate sulfate anion was always included at one of the coordination sites for charge balance, and the remaining sites were coordinated by water and the ET oxygen atoms. The modeling revealed that up to three oxygen atoms from three different functional groups (ET : Fe = 2.9) can coordinate to the same iron ion at the low loading observed for genuine groundwater samples, provided that these groups are located at the 2-, 2′-, and 3-positions of two adjacent biphenyl groups (Fig. S12†). Importantly, the modeling also showed that the ET group flexibility may enable two groups to bind the same iron ion, regardless of their respective positions on a biphenyl unit (Fig. S12a–c†). Based on the maximum iron(iii) adsorption capacity data, for high iron loading (ET : Fe = 1.1), it is presumed that only a single ET group can bind to the iron. In this case, a coordinated water molecule might participate in a weak CH–π interaction with a benzene ring that stabilizes the iron ions (Fig. S13†), a hypothesis supported by the benzene ring shift in the 13C NMR spectrum of iron(iii)-loaded PAF-1–ET (Fig. S11†).
To investigate the importance of the ether–thioether orientation and pore environment in PAF-1–ET for iron(iii) uptake, we prepared a series of related porous polymers and evaluated their iron adsorption properties. The first of these polymers, PAF-1–TE, was synthesized from PAF-1–CH2Cl using 2-methoxyethane-1-thiol, yielding a material analogous to PAF-1–ET but featuring interchanged positions for the pendant oxygen and sulfur moieties. We also prepared ether-functionalized porous polymers PAF-1–OMe and PAF-1–Ethoxy (Fig. S14†) and a linear polysulfone polymer functionalized with the ether–thioether ligand, PSF–ET (Fig. S15†). When exposed to a solution of FeCl3 (20 mg L–1) dissolved in 100 mM HEPES buffer (pH = 6.7) with one equivalent of citric acid, each of the derivative polymers exhibited significantly lower iron(iii) uptake than PAF-1–ET (Fig. S16†). The much lower uptake of PAF-1–TE, –OMe, and –Ethoxy suggests that both the position of oxygen and the presence of sulfur are crucial for iron adsorption, while the low uptake of PSF–ET emphasizes the importance of a compact pore environment in tandem with the ET functionality. Accordingly, we hypothesize that presence of sulfur in the linker, along with its positioning, contributes an important pore packing effect to create the optimal pore environment in PAF-1–ET for iron adsorption, even if not involved in direct binding to the iron center.
Iron capture and detection in synthetic and environmental water samples
To verify the detection capability of PAF-1–ET when exposed to iron(iii) sources from different regions, synthetic groundwater was prepared according to the Gadgil65 procedure with iron(iii) concentrations of 1.8, 4.7, 6.7, and 37 mg L–1. The polymer was also used to treat genuine groundwater samples collected in West Bengal, India, reported to contain 14 mg L–1 of iron(iii) ions (Fig. 5a).16 Notably, PAF-1–ET adsorbed between ∼41 and 91% of the iron(iii) in the synthetic groundwater samples, from initial concentrations between 37 and 1.8 mg L–1, respectively. In the presence of PAF-1–ET, the concentration of iron(iii) in the genuine groundwater decreased with time as given by the expression y = Ae–t/t0 + C, where y is the detected amount of iron(iii), A is a scale factor, C is a constant, t0 is the decay time, and t is the elapsed time. The best fit shown in the inset to Fig. 5b corresponds to A = 9.2(3) mg L–1, C = 4.1(1) mg L–1, and t0 = 12(1) min. In other words, within 24 min PAF-1–ET captured 72% of the iron(iii) ions and was essentially saturated after ∼36 min, such that the iron(iii) concentration in the genuine groundwater reached a constant value of ∼3.92 mg L–1.
Fig. 5. (a) Adsorbed iron(iii) by PAF-1–ET in synthetic groundwater (initial iron concentrations were prepared as 1.8, 4.7, 6.7, and 37 mg L–1 using (NH4)2Fe(SO4)2 and groundwater collected in West Bengal, India16 (iron(iii) concentration of 14 mg L–1). For each measurement, 5 mL of each water sample were combined with 5 mL of citric acid solution (10–15 mg mL–1) and 2 mg of PAF-1–ET. (b) Absorption spectra after 8-hydroxyquinoline addition to dried PAF-1–ET samples following exposure to the corresponding groundwater samples in (a); (inset) time-dependent uptake of iron(iii) from the genuine groundwater sample fit to a single exponential decay y = Ae–t/t0 + C (black line), yielding parameters A = 9.2(3) mg L–1, C = 4.1(1) mg L–1, and t0 = 12(1) min. (c) Comparison of direct iron(iii) measurements by ICP-MS (black squares) and calculated iron(iii) concentrations determined from the absorbance at 460 nm in the presence of 8-hydroxyquinoline indicator (open circles).

The final concentration in the genuine groundwater sample is higher than that recommended for safe drinking water by the WHO (0.3 mg L–1), and the same is true for the synthetic groundwater solution with an initial concentration of 37 mg L–1. However, the saturation capacity of PAF-1–ET suggests that the framework is capable of reducing the iron(iii) content in both these solutions to levels lower than 0.3 mg L–1. It is likely that at higher iron(iii) concentrations, precipitation of Fe(OH)3 within the pores blocks some of the accessible coordination sites and reduces the effective capacity. Even still, PAF-1–ET functions exceptionally well in the removal of iron from solutions with relatively low initial concentrations—indeed, the framework was able to reduce the iron(iii) ion content to safe drinking levels for solutions with initial iron concentrations of 1.8, 4.7, and 6.7 mg L–1. For the effective treatment of water containing higher iron ion concentrations, it may be necessary to use larger quantities of citric acid to prevent Fe(OH)3 precipitation.
Analysis of the PAF-1–ET samples using an 8-hydroxyquinoline assay revealed an increase in absorbance at 460 and 560 nm with increasing iron(iii) concentration, as associated with the original water samples (Fig. 5b). The iron(iii) concentrations calculated from the absorption at 460 nm were again in good agreement with those determined from direct ICP-MS measurements (Fig. 5c). Finally, using the three-sigma method (3σ/k),66–68 the iron(iii) detection limit for the PAF-1–ET and 8-hydroxyquinoline assay was determined to be 150 μg L–1 (see Fig. S17 and the ESI for details†). Importantly, PAF-1–ET retains structural integrity and porosity following the addition of 8-hydroxyquinoline and can be cycled at least three times without a substantial loss of adsorption capacity (Fig. S18†).
Conclusions
We have demonstrated that the ether–thioether-functionalized porous aromatic framework PAF-1–ET is capable of selective and efficient iron ion uptake and removal from both synthetic water and environmental groundwater. In this material, captured iron(iii) is preferentially bound by oxygen in a pseudooctahedral coordination environment, as confirmed by Mössbauer and X-ray absorption spectroscopy. The introduction of oxygen functionality within the framework is thus responsible for a shift to iron ion selectivity from our previously reported copper-selective thioether-functionalized material, PAF-1–SMe.42 Finally, the combination of PAF-1–ET with 8-hydroxyquinoline as a colorimetric indicator provides an efficient and accurate tool for directly determining the iron ion concentrations from groundwater samples, with minimal processing and equipment needs.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We thank the NIH under award GM79465 for support of synthesis and biological studies in the laboratory of C. J. C. Gas adsorption, solid-state NMR measurements, and additional synthesis were supported by the Center for Gas Separations, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award DE-SC0001015. Mössbauer spectroscopy and scanning electron microscopy (SEM) measurements were supported by the Laboratory Directed Research and Development Program of Lawrence Berkeley National Laboratory under U.S. Department of Energy Contract DE-AC02-05CH11231. C. J. C. is an Investigator at the Howard Hughes Medical Institute and a CIFAR Senior Fellow. We thank the National Science Foundation (DGE 1752814) for providing graduate fellowship support for A. A. U. and M. K. T. We thank Kristen Colwell for collecting the SEM data, Dr C. Michael McGuirk for TGA analysis, and Dr Katie R. Meihaus for editorial assistance.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01441a
§Less than 100% loading of 2-(methylthio)-ethan-1-ol loading might be attributed to the reactivity of sodium hydride with benzyl chloride and the conversion of some of the –CH2Cl groups to other functionalities such as methyl, or the linkage of these groups to form polymeric 1,2-diphenylethane (see S. Bank and M. C. Prislopski, J. Chem. Soc. Chem. Commun., 1970, 1624; M. I. Watkins and G. A. Olah, J. Am. Chem. Soc., 1981, 103, 6566; and Y. Yuan and Y. Bian. Appl. Organomet. Chem., 2008, 22, 15).
References
- Weber K. A., Achenbach L. A., Coates J. D. Nat. Rev. Microbiol. 2006;4:752. doi: 10.1038/nrmicro1490. [DOI] [PubMed] [Google Scholar]
- Hentze M. W., Muckenthaler M. U., Andrews N. C. Cell. 2004;117:285. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- Dixon S. J., Stockwell B. R. Nat. Chem. Biol. 2014;10:9. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- Chang C. J. Nat. Chem. Biol. 2015;11:744. doi: 10.1038/nchembio.1913. [DOI] [PubMed] [Google Scholar]
- Spangler B., Morgan C. W., Fontaine S. D., Vander Wal M. N., Chang C. J., Wells J. A., Renslo A. R. Nat. Chem. Biol. 2016;12:680. doi: 10.1038/nchembio.2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew M. C. Liver Cancer. 2014;3:31. doi: 10.1159/000343856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theil E. C., Goss D. J. Chem. Rev. 2009;109:4568. doi: 10.1021/cr900052g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz T., Eaton J. W., Brunk U. T. Int. J. Biochem. Cell Biol. 2011;43:1686. doi: 10.1016/j.biocel.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Salonen J. T., Nyyssönen K., Korpela H., Tuomilehto J., Seppänen R., Salonen R. Circulation. 1992;86:803. doi: 10.1161/01.cir.86.3.803. [DOI] [PubMed] [Google Scholar]
- Martins S., Logan S. and Gilbert R. E., Iron Therapy for Improving Psychomotor Development and Cognitive Function in Children under the Age of Three with Iron Deficiency Anaemia, in Cochrane Database of Systematic Reviews; The Cochrane Collaboration, John Wiley & Sons, Ltd, Chichester, UK, 2001. [DOI] [PubMed] [Google Scholar]
- Carrier J., Aghdassi E., Cullen J., Allard J. P. J. Nutr. 2002;132:3146. doi: 10.1093/jn/131.10.3146. [DOI] [PubMed] [Google Scholar]
- Arezzini B., Lunghi B., Lungarella G., Gardi C. Int. J. Biochem. Cell Biol. 2003;35:486. doi: 10.1016/s1357-2725(02)00298-4. [DOI] [PubMed] [Google Scholar]
- Hoffbrand A. V., Cohen A., Hershko C. Blood. 2003;102:17. doi: 10.1182/blood-2002-06-1867. [DOI] [PubMed] [Google Scholar]
- Brewer G. J. Chem. Res. Toxicol. 2010;23:319. doi: 10.1021/tx900338d. [DOI] [PubMed] [Google Scholar]
- World Health Organization, World Health Organ. Guidel., 2003, vol. 2, p. 1. [Google Scholar]
- Amrose S. E., Bandaru S. R. S., Delaire C., van Genuch-ten C. M., Dutta A., DebSarkar A., Orr C., Roy J., Das A., Gadgil A. J. Sci. Total Environ. 2014;488–489:539. doi: 10.1016/j.scitotenv.2013.11.074. [DOI] [PubMed] [Google Scholar]
- Ikem A., Egiebor N. O., Nyavor K. Water, Air, Soil Pollut. 2003;149:51. [Google Scholar]
- Majzlan J., Alpers C. N., Koch C. B., McCleskey R. B., Myneni S. C. B., Neil J. M. Chem. Geol. 2011;284:296. [Google Scholar]
- Karabulut M., Marasinghe G. K., Ray C. S., Day D. E., Waddill G. D., Booth C. H., Allen P. G., Bucher J. J., Caulder D. L., Shuh D. K. J. Non-Cryst. Solids. 2002;306:182. [Google Scholar]
- Reimann C., Bjorvatn K., Frengstad B., Melaku Z., Tekle-Haimanot R., Siewers U. Sci. Total Environ. 2003;311:65. doi: 10.1016/S0048-9697(03)00137-2. [DOI] [PubMed] [Google Scholar]
- Berg M., Trang P. T. K., Stengel C., Buschmann J., Viet P. H., Van Dan N., Giger W., Stüben D. Chem. Geol. 2008;249:91. [Google Scholar]
- Buschmann J., Berg M., Stengel C., Winkel L., Sampson M. L., Trang P. T. K., Viet P. H. Environ. Int. 2008;34:756. doi: 10.1016/j.envint.2007.12.025. [DOI] [PubMed] [Google Scholar]
- Gyles P. A., Miller A. Environ. EarthEnviron. Earth Sci.Sci. 2011;62:1431. [Google Scholar]
- Waite T. D. Rev. Environ. Sci. Bio/Technol. 2002;1:9. [Google Scholar]
- Yaghi O. M., Li G., Li H. Nature. 1995;378:703. [Google Scholar]
- Jiang J.-X., Su F., Trewin A., Wood C. D., Campbell N. L., Niu H., Dickinson C., Ganin A. Y., Rosseinsky M. J., Khimyak Y. Z., Cooper A. I. Angew. Chem., Int. Ed. 2007;46:8574. doi: 10.1002/anie.200701595. [DOI] [PubMed] [Google Scholar]
- Adeyemo A. A., Adeoye I. O., Bello O. S. Toxicol. Environ. Chem. 2012;94:1846. [Google Scholar]
- Carboni M., Abney C. W., Liu S., Lin W. Chem. Sci. 2013;4:2396. [Google Scholar]
- Liu X., Demir N. K., Wu Z., Li K. J. Am. Chem. Soc. 2015;137:6999. doi: 10.1021/jacs.5b02276. [DOI] [PubMed] [Google Scholar]
- Wang C., Liu X., Demir N. K., Chen J. P., Li K. Chem. Soc. Rev. 2016;45:5107. doi: 10.1039/c6cs00362a. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Yuan S., Feng X., Li H., Zhou J., Wang B. J. Am. Chem. Soc. 2016;138:5785. doi: 10.1021/jacs.6b02553. [DOI] [PubMed] [Google Scholar]
- Ding S.-Y., Dong M., Wang Y.-W., Chen Y.-T., Wang H.-Z., Su C.-Y., Wang W. J. Am. Chem. Soc. 2016;13:3031. doi: 10.1021/jacs.5b10754. [DOI] [PubMed] [Google Scholar]
- Sapchenko S. A., Demakov P. A., Samsonenko D. G., Dybtsev D. N., Schröder M., Fedin V. P. Chem.–Eur. J. 2017;23:2286. doi: 10.1002/chem.201605895. [DOI] [PubMed] [Google Scholar]
- Li P.-Z., Su J., Liang J., Liu J., Zhang Y., Chen H., Zhao Y. Chem. Commun. 2017;53:3434. doi: 10.1039/c7cc01063j. [DOI] [PubMed] [Google Scholar]
- Bobbitt N. S., Mendonca M. L., Howarth A. J., Islamoglu T., Hupp J. T., Farha O. K., Snurr R. Q. Chem. Soc. Rev. 2017;46:3357. doi: 10.1039/c7cs00108h. [DOI] [PubMed] [Google Scholar]
- Sun D. T., Peng L., Reeder W. S., Moosavi S. M., Tiana D., Britt D. K., Oveisi E., Queen W. L. ACS Cent. Sci. 2018;4:349. doi: 10.1021/acscentsci.7b00605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y., Zhu G. ACS Cent. Sci. 2019;5:409. doi: 10.1021/acscentsci.9b00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben T., Ren H., Shengqian M., Cao D., Lan J., Jing X., Wang W., Xu J., Deng F., Simmons J. M., Qiu S., Zhu G. Angew. Chem., Int. Ed. 2009;48:9457. doi: 10.1002/anie.200904637. [DOI] [PubMed] [Google Scholar]
- Lu W., Yuan D., Sculley J., Zhao D., Krishna R., Zhou H.-C. J. Am. Chem. Soc. 2011;133:18126. doi: 10.1021/ja2087773. [DOI] [PubMed] [Google Scholar]
- Li B., Zhang Y., Ma D., Shi Z., Ma S. Nat. Commun. 2014;5:5537. doi: 10.1038/ncomms6537. [DOI] [PubMed] [Google Scholar]
- Van Humbeck J. F., McDonald T. M., Jing X., Wiers B. M., Zhu G., Long J. R. J. Am. Chem. Soc. 2014;136:2432. doi: 10.1021/ja4105478. [DOI] [PubMed] [Google Scholar]
- Lee S., Barin G., Ackerman C. M., Muchenditsi A., Xu J., Reimer J. A., Lutsenko S., Long J. R., Chang C. J. J. Am. Chem. Soc. 2016;138:7603. doi: 10.1021/jacs.6b02515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demir S., Brune N. K., Van Humbeck J. F., Mason J. A., Plakhova T. V., Wang S., Tian G., Minasian S. G., Tyliszczak T., Yaita T., Kobayashi T., Kalmykov S. N., Shiwaku H., Shuh D. K., Long J. R. ACS Cent. Sci. 2016;2:253. doi: 10.1021/acscentsci.6b00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H. W., Gaede H. C., Shore J., Reven L., Bowers C. R., Kritzenberger J., Pietrass T., Pines A., Tang P., Reimer J. A. J. Am. Chem. Soc. 1993;115:8491. [Google Scholar]
- Ravel B., Newville M. J. Synchrotron Radiat. 2005;12:537. doi: 10.1107/S0909049505012719. [DOI] [PubMed] [Google Scholar]
- Zabinsky S. I., Rehr J. J., Ankudinov A., Albers R. C., Eller M. J. Phys. Rev. B: Condens. Matter Mater. Phys. 1995;52:2995. doi: 10.1103/physrevb.52.2995. [DOI] [PubMed] [Google Scholar]
- Domaille D. W., Que E. L., Chang C. J. Nat. Chem. Biol. 2008;4:168. doi: 10.1038/nchembio.69. [DOI] [PubMed] [Google Scholar]
- Au-Yeung H. Y., Chan J., Chantarojsiri T., Chang C. J. J. Am. Chem. Soc. 2013;135:15165. doi: 10.1021/ja4072964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron A. T., Ramos-Torres K. M., Cotruvo J. A., Chang C. J. Acc. Chem. Res. 2015;48:2434. doi: 10.1021/acs.accounts.5b00221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron A. T., Loehr M. O., Bogena J., Chang C. J. J. Am. Chem. Soc. 2016;138:14338. doi: 10.1021/jacs.6b08016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron A. T., Heffern M. C., Lonergan Z. R., Wal M. N. V., Blank B. R., Spangler B., Zhang Y., Park H. M., Stahl A., Renslo A. R., Skaar E. P., Chang C. J. Proc. Natl. Acad. Sci. U. S. A. 2017;114:12669. doi: 10.1073/pnas.1708747114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron A. T., Reeves A. G., Chang C. J. Curr. Opin. Chem. Biol. 2018;43:113. doi: 10.1016/j.cbpa.2017.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Sculley J. P., Yuan D., Krishna R., Wei Z., Zhou H.-C. Angew. Chem., Int. Ed. 2012;51:7480. doi: 10.1002/anie.201202176. [DOI] [PubMed] [Google Scholar]
- Barin G., Peterson G. W., Crocellà V., Xu J., Colwell K. A., Nandy A., Reimer J. A., Bordiga S., Long J. R. Chem. Sci. 2017;8:4399. doi: 10.1039/c6sc05079d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Sun Q., Zhang Y., Abney C. W., Aguila B., Lin W., Ma S. ACS Appl. Mater. Interfaces. 2017;9:12511. doi: 10.1021/acsami.7b01711. [DOI] [PubMed] [Google Scholar]
- Kamcev J., Taylor M. K., Shin D.-M., Jarenwattananon N. N., Colwell K. A., Long J. R. Adv. Mater. 2019;31:1808027. doi: 10.1002/adma.201808027. [DOI] [PubMed] [Google Scholar]
- Serratrice G., Boukhalfa H., Béguin C., Baret P., Caris C., Pierre J.-L. Inorg. Chem. 1997;36:3898. [Google Scholar]
- Yang Y., Ding H., Hao S., Zhang Y., Kan Q. Appl. Organomet. Chem. 2011;25:262. [Google Scholar]
- Mladenova R., Ignatova M., Manolova N., Petrova T., Rashkov I. Eur. Polym. J. 2002;38:989. [Google Scholar]
- Goswami S., Aich K., Kumar Das A., Manna A., Das S. RSC Adv. 2013;3:2412. [Google Scholar]
- Teo B. K., EXAFS: Basic Principles and Data Analysis, Inorganic Chemistry Concepts, Springer-Verlag, Berlin Heidelberg, 1986. [Google Scholar]
- Grey C. P., Dupré N. Chem. Rev. 2004;104:4493. doi: 10.1021/cr020734p. [DOI] [PubMed] [Google Scholar]
- Aguiar P. M., Katz M. J., Leznoff D. B., Kroeker S. Phys. Chem. Chem. Phys. 2009;11:6925. doi: 10.1039/b907747b. [DOI] [PubMed] [Google Scholar]
- Pell A. J., Pintacuda G., Grey C. P. Prog. Nucl. Magn. Reson. Spectrosc. 2019;111:1. doi: 10.1016/j.pnmrs.2018.05.001. [DOI] [PubMed] [Google Scholar]
- Delaire C., van Genuchten C. M., Nelson K. L., Amrose S. E., Gadgil A. J. Environ. Sci. Technol. 2015;49:9945. doi: 10.1021/acs.est.5b01696. [DOI] [PubMed] [Google Scholar]
- Chen Y., Zhu C., Yang Z., Chen J., He Y., Jiao Y., He W., Qiu L., Cen J., Guo Z. Angew. Chem., Int. Ed. 2013;52:1688. doi: 10.1002/anie.201207701. [DOI] [PubMed] [Google Scholar]
- Ding X., Qu L., Yang R., Zhou Y., Li J. Luminescence. 2014;30:465. doi: 10.1002/bio.2761. [DOI] [PubMed] [Google Scholar]
- Zhu D., Xue L., Li G., Che Y., Jiang H. Org. Chem. Front. 2014;1:501. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.