

Abstract
X chromosome inactivation (XCI) is a key epigenetic gene expression regulatory process, which may play a role in women’s cancer. In particular tissues, some genes are known to escape XCI, yet patterns of XCI in ovarian cancer (OC) and their clinical associations are largely unknown. To examine XCI in OC, we integrated germline genotype with tumor copy number, gene expression and DNA methylation information from 99 OC patients. Approximately 10% of genes showed different XCI status (either escaping or being subject to XCI) compared with the studies of other tissues. Many of these genes are known oncogenes or tumor suppressors (e.g. DDX3X, TRAPPC2 and TCEANC). We also observed strong association between cis promoter DNA methylation and allele-specific expression imbalance (P = 2.0 × 10−10). Cluster analyses of the integrated data identified two molecular subgroups of OC patients representing those with regulated (N = 47) and dysregulated (N = 52) XCI. This XCI cluster membership was associated with expression of X inactive specific transcript (P = 0.002), a known driver of XCI, as well as age, grade, stage, tumor histology and extent of residual disease following surgical debulking. Patients with dysregulated XCI (N = 52) had shorter time to recurrence (HR = 2.34, P = 0.001) and overall survival time (HR = 1.87, P = 0.02) than those with regulated XCI, although results were attenuated after covariate adjustment. Similar findings were observed when restricted to high-grade serous tumors. We found evidence of a unique OC XCI profile, suggesting that XCI may play an important role in OC biology. Additional studies to examine somatic changes with paired tumor-normal tissue are needed.
Introduction
The biology of the X chromosome is complex, as one copy of the X chromosome in females must be transcriptionally silenced during embryonic development to ensure dosage of X-linked gene expression is similar to that in males (1–3). This X chromosome inactivation (XCI) is tissue specific and epigenetically initiated by X inactive specific transcript (XIST) (4), a long non-coding RNA that is transcribed on the inactive X (Xi) chromosome. XCI can be skewed, in that one particular copy of the chromosome shows preferential inactivation across cells in a given tissue (Fig. 1A). Additionally, 10–15% of genes escape this inactivation (‘escape genes’) and are expressed on both copies of the X chromosome (5–7).
Figure 1.
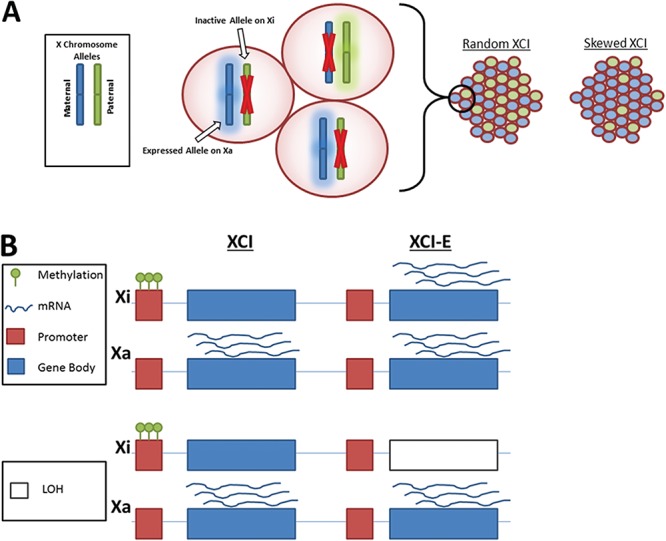
XCI is a complex process involving multiple levels of gene regulation. (A) Skewed XCI. In each female cell, the maternal or paternal X is inactivated. Females will yield a distribution of alleles. In random XCI, 50% of alleles are maternal, and 50% are paternal; in skewed XCI, the proportions vary from 50%. (B) For the Xi and Xa X chromosomes, promoter methylation and mRNA expression associated with genes subject to XCI (XCI) and escape genes (XCI-E), in regions with and without LOH.
Cancer of an ovary, fallopian tube or peritoneal tissue (collectively, ovarian cancer; OC) is diagnosed in over 230 000 women annually and is a leading cause of women’s cancer death (8–10). XCI appears to be important in female carcinogenesis, as ovarian and breast cancer cell lines can show loss of the Xi (11,12), Xi loss with active X (Xa) duplication and Xi reactivation (13–15); furthermore, there may be an interplay between Xi and BRCA1 (16). The X chromosome contains an excess of genes related to reproduction and hormones (e.g. the androgen receptor) (17), drug metabolism (18) (SLC6A8, SLC9A6 and SLC16A2), drug transport genes (19) (ABCB7 and ABCD1) and chemotherapy response (CTPS2 and DLG3) (20), along with an abundance of oncogenes and tumor suppressor genes (21), and tumor suppressor genes that escape XCI may contribute to the sex bias observed for some cancers (22). Additionally, XCI skewing has been shown to be more frequent in lymphocytes of ovarian and breast cancer patients compared with unaffected women (23–27) and increases with age (28). Finally, XCI in OC is thought to relate to the ‘two-hit hypothesis’ of tumorigenesis (29), as it is functionally equivalent to loss of heterozygosity (LOH), and X chromosome LOH has been reported in OC (30,31). Skewed XCI of tumor suppression genes, such as GPC3 (32,33) and MEF (34,35), may serve as the ‘first hit’ (21). The Cancer Genome Atlas (TCGA) previously investigated X chromosome copy number alterations and LOH in high-grade serious OCs and reported that X chromosome structural alterations were common and associated with worse survival (36).
We previously developed methods to estimate genes that escape from XCI in female samples without relying on male samples for comparison and identified 37 genes that differed in XCI escape status in ovarian tumor samples compared with the expected state (37). However, this prior investigation did not examine the association of XCI patterns with clinical factors and measured XCI patterns using allele-specific expression (ASE) estimated from RNA sequencing (RNA-seq) data without considering any epigenetic information such as DNA methylation. The process of XCI inherently involves multiple layers of gene regulation, as epigenetic mechanisms drive allele-specific transcription from either the maternally or paternally inherited alleles, which can be altered by structural variants (Fig. 1B). Furthermore, the estimation of ASE suffers from a large amount of missing data for each gene, limiting the ability to examine clinical associations at the gene level or across the X chromosome. Approaches that utilize genomic, epigenomic and transcriptomic data could improve the measurement of XCI patterns and at the same time enable examination of clinical relationships by ‘filling in the gaps’ of missing data. Therefore, a comprehensive multi-layered omics (‘multi-omics’) approach to examine XCI patterns in patient-derived ovarian tumor samples and association with clinical factors and outcomes is warranted. Here we leverage a novel multi-omics approach to evaluate XCI in nearly 100 women with OC and examine the clinical relevance of XCI patterns. We hypothesize that abnormal disruption of XCI promotes tumorigenesis through reactivation of oncogenes or deactivation of tumor suppressor genes and importantly that the disruption of XCI is associated with clinical features of OC.
Results
Summary of previously reported patterns of XCI escape across ovarian tumors
In analysis of tumors from a collection of 99 epithelial OC patients from the Mayo Clinic (Supplementary Material, Table S1) (38), we previously estimated ASE for X chromosome genes (37). As previously reported, a majority of tumors exhibited structural alteration on the X chromosome (N = 52; 53%) (37). Among 47 tumors with no structural alteration, 45 (96%) showed skewed XCI (P < 0.001, likelihood ratio test), meaning that a particular copy of the X chromosome is preferentially inactivated; a majority of these samples had highly skewed XCI (Supplementary Material, Fig. S1). For 372 genes, we observed at least 10 ASE reads not overlapping a structural variant in at least one tumor; per tumor, a median of 81 genes were evaluated. These results are generally consistent with prior studies of normal non-ovarian tissue types (37); across the tumor samples, 84.6% of genes had escape status concordant with prior studies from normal tissue (Table 1). Additionally, all tumors evaluated for XIST escape status (N = 16) showed evidence of inactivation.
Table 1.
Cross tabulation of numbers of genes (column %) by observed XCI status in ovarian tissue compared with the expected XCI status based on prior studies of normal tissue (based on Balaton et al.)
| Ovarian tumors | |||||
|---|---|---|---|---|---|
| Subject to XCI a | Variable escape from XCI b | Escape from XCI c | Total | ||
| Normal tissue | Subject to XCI | 172 (55.5%) | 0 (0.0%) | 13 (22.4%) | 185 (49.7%) |
| Mostly subject to XCI | 76 (24.5%) | 0 (0.0%) | 7 (12.1%) | 83 (22.3%) | |
| Variable escape from XCI | 12 (3.9%) | 0 (0.0%) | 1 (1.7%) | 13 (3.5%) | |
| Mostly variable escape from XCI | 7 (2.3%) | 0 (0.0%) | 1 (1.7%) | 8 (2.2%) | |
| Escape from XCI | 1 (0.3%) | 1 (25.0%) | 13 (22.4%) | 15 (4.0%) | |
| Mostly escape from XCI | 7 (2.3%) | 0 (0.0%) | 14 (24.1%) | 21 (5.6%) | |
| Discordant across tissues | 18 (5.8%) | 1 (25.0%) | 5 (5.8%) | 24 (6.5%) | |
| No call | 17 (5.5%) | 2 (50.0%) | 4 (5.2%) | 23 (6.2%) | |
| Total | 310 | 4 | 58 | 372 | |
A gene was classified as escaping XCI in a given tumor sample if PPE > 0.5 and subject to XCI if PPE < 0.5.
aGenes escaped XCI in > 80% of samples evaluated.
bGenes escaped XCI in < 20% of samples evaluated.
cGenes escaped XCI in between 20 and 80% of samples evaluated.
Genes were not able to be classified in any samples.
A median of 10% of genes (range: 0–0.39; IQR: 0.08–0.13) per tumor had a discrepant XCI status compared with expected status based on prior studies of normal tissue (39), where a gene was considered discrepant in a given tumor if the estimated escape status was the opposite of what was expected based on normal tissue (i.e. estimated as ‘escape’ when the expectation was ‘inactivated’ or estimated as ‘inactivated’ when the expectation was ‘escape’). Of the discrepant genes in a given tumor, 58% were expected to escape XCI but showed evidence of deactivation in ovarian tumors (posterior probability of escape <0.50). In particular, nine genes that escape XCI in other tissues and showed strong evidence of XCI in this study (posterior probability of escape <0.10 in at least one tumor) have been previously reported as tumor suppressors (KAL1, RBB7, TCEANC, TRAPPC2, DDX3X, USP9X and KDM5C) or markers of poor prognosis (CTPS2 and RPS4X). Additionally, five previously classified inactivated genes that showed strong evidence of escape in at least one tumor (posterior probability of escape >0.90) have been previously reported as oncogenes (CXorf36, SH3BGRL, ELF4, SLITRK4 and TAZ). This pattern is consistent with the two-hit model of carcinogenesis (Supplementary Material, Table S2).
Correlation between DNA methylation, posterior probability of escape and ASE
We observed a strong negative correlation between median promoter methylation and median posterior probability of escape (Spearman rho = −0.53; Fig. 2A), with near complete separation by expected escape status based on the prior literature (AUC = 0.99) (39). For genes that showed high probability of escape across ovarian tumors, methylation values were low (indicating expression of both copies; Q1–Q3 = 0.13–0.21), whereas for genes that showed low probability of escape across samples, methylation values were indicating inactivation of one copy (Q1–Q3 = 0.48–0.54).
Figure 2.
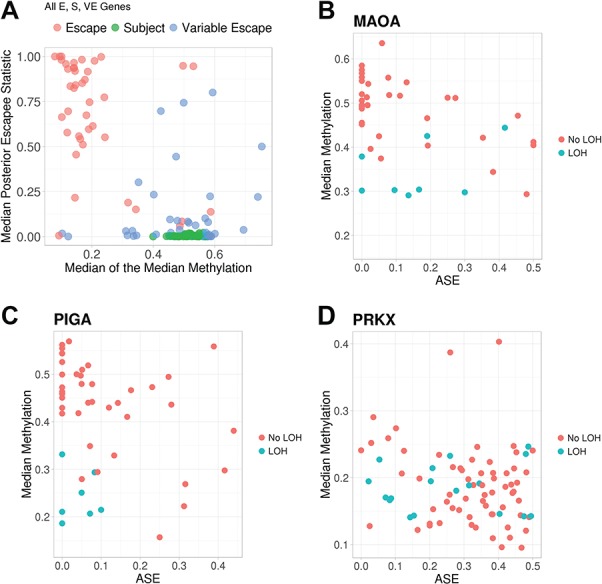
Correlation between the promoter methylation and the probability of escape from XCI (A) and ASE (B–D) of the corresponding gene. (A) For each gene, the median of the posterior probability of escape across 99 tumor samples is plotted against the median across tumor samples of the median DNA methylation across the CpGs in the promoter. Colors indicate the prior literature escape status of each gene by Balaton et al. (escape from XCI, subject to XCI or variable escape). For each tumor, patterns of median DNA methylation across CpGs in the promoter are plotted by ASE of select individual genes: (B) MAOA and (C) PIGA, which are subject to XCI and (D) PRKX, which escapes from XCI. Colors indicate presence of LOH in the gene region for that particular tumor.
In regions without LOH, we observed negative cis correlations between gene promoter methylation and corresponding ASE (Supplementary Material, Fig. S2; P = 2.1 × 10−10, linear mixed-effects model). A set of 129 genes had at least one corresponding methylation probe with a significant cis correlation (P < 0.1, Spearman correlation). Methylation values were lower in regions of LOH (median = 0.35) compared with regions without LOH (median = 0.48) (Supplementary Material, Fig. S3). Individual genes showed expected patterns between methylation, ASE and LOH. For example, inactivated genes MAOA and PIGA show methylation values near 0.5 and ASE values near zero for most tumors without LOH in the region, with methylation values decreasing as ASE values increase; for tumors with LOH in the region, methylation values are low (Fig. 2B and C). On the other hand, for escape gene PRKX, there is a negative correlation between methylation and ASE only in tumors without LOH in the region, and generally, methylation values are <0.5, and ASE values are >0 (Fig. 2D).
Clinical associations with XCI patterns at each gene
Because of the strong correlation between methylation and ASE when accounting for LOH, missing ASE information was imputed based on methylation and LOH data at 175 X chromosome genes (Supplemental Methods), resulting in an imputed ASE that represents integration of ASE, methylation and LOH data. The relationship between clinical factors and the imputed ASE was examined at each gene. In particular, ASE at 29 genes were associated with overall survival (P < 0.05; Supplementary Material, Table S3), including the previously reported discrepant genes TAZ, SLITRK4, CTPS2, KAL1, DOCK11, DMD and GEMIN8.
When considering the sum of discrepant genes in a given sample, patients with tumors showing greater than median XCI gene discrepancy were younger at age of diagnosis (median age 55 versus 60.5; P = 0.06); histology, grade and stage were not associated with the extent of XCI gene discrepancy (P > 0.50).
Patient sub-groups defined by chromosome-wide XCI patterns
Two patient clusters, or sub-groups, were identified based on ASE, promoter methylation and LOH (Supplementary Material, Fig. S4); consensus clustering was used to ensure that the two clusters were highly stable and reproducible (Supplementary Material, Fig. S5A and B). Cluster 1 indicates a subgroup of N = 47 patients with regulated XCI, where the process of XCI often acts as expected: ASE proportions near 0 for most genes expected to be inactivated and proportions near 0.5 for most genes expected to be escaping XCI. Alternatively, Cluster 2 represents a subgroup of N = 52 patients with dysregulated XCI, where the process of XCI is often not acting as expected: ASE proportions >0 for many genes expected to be inactivated and <0.5 for many genes expected to escape from XCI. Sequencing batch was not associated with ASE cluster (P = 0.78, Fisher’s exact test). Additionally, the clusters were not associated with molecular subtype (PASE = 0.88, Fisher’s exact test) (40), although the clusters were associated with the estimate of skewness based on genes subject to XCI based on prior studies of normal tissue (P = 0.003, Wilcoxon rank sum test).
We also sought to define patient sub-groups among those with high-grade serous tumors. Two patient clusters were identified (Supplementary Material, Fig. S6), representing a regulated XCI cluster (N = 23) and a dysregulated XCI cluster (N = 29).
Patient sub-groups and total XIST expression
We explored the biology associated with the XCI clusters. Because the XIST gene controls the process of XCI, patients with higher levels of XIST expression are likely exhibiting higher levels of inactivation than patients with lower XIST expression. Cluster 2 (the cluster of patients with dysregulated XCI patterns) had lower XIST expression after adjustment for sequencing batch (P = 0.002, Wilcoxon rank sum test; Supplementary Material, Fig. S7), confirming the hypothesis. Similarly, in High-Grade Serous (HGS) patients, Cluster 2 also had lower expression of XIST (P = 0.004, Wilcoxon rank sum test).
Genes defining the patient sub-groups
In discriminating ASE-derived tumor sub-groups, 53 genes/features comprised of two clusters were identified (Fig. 3; Supplementary Material, Table S4). These genes reside throughout the X chromosome, with no clear regional pattern (Supplementary Material, Fig. S8); annotation with DAVID (41) revealed that no biological gene sets were enriched in these genes (P > 0.01, Fisher’s exact test). Genes in the first feature cluster (N = 11 genes) were primarily those previously thought to undergo inactivation (39) (Supplementary Material, Table S5), and three of these showed aberrant escape in at least one ovarian tumor (DMD, WWC3 and DOCK11; Supplementary Material, Table S2) (37). Genes in the second feature cluster (N = 42 genes) were a mixture of inactivated (19 genes), escape genes (15 genes) and difficult to classify genes (8 genes) based on consensus calls across normal tissue types (39) (Supplementary Material, Table S5). Five of these were aberrantly escaping (GABRE, CXorf36, CXorf23, OPHN1 and BGN) and eight were aberrantly inactivated (TRAPPC2, ARSD, TCEANC, EIF2S3, NLGN4X, GPM6B, DDX3X and RPS4X) in at least one ovarian tumor (37) (Supplementary Material, Table S2).
Figure 3.
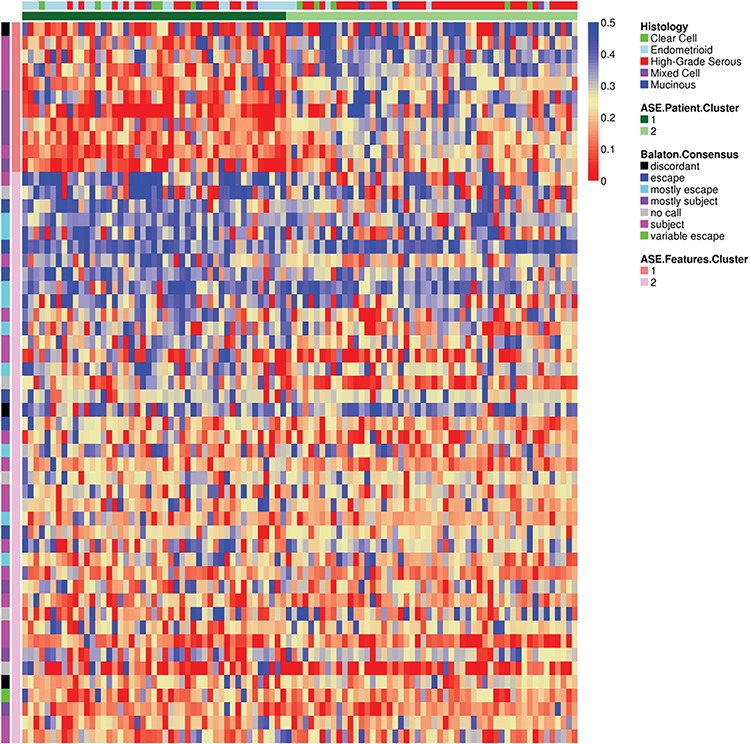
XCI patterns across ovarian tumors after feature extraction. Heatmap of the adjusted ASE for X chromosome genes after feature extraction. Rows are genes, and columns are samples. Color legend indicates the proportion of ASE reads. Previously published escapee patterns for each gene and the feature clusters are plotted on the left, and the characteristics of each sample and the patient clusters are plotted at the top.
Genes in the first feature cluster had lower ASE and posterior probabilities of escape than those in the second feature cluster, whereas those in the second feature cluster are more variable (Supplementary Material, Fig. S9). Additionally, the 11 genes in the first feature cluster have higher ASE for patients in the sub-group with dysregulated XCI [mediandysregulated = 0.25 (0.11–0.35), medianregulated = 0.09 (0.04–0.18); P = 2.57E-5, Wilcoxon rank sum test; Supplementary Material, Table S6]. The 42 genes in the second feature cluster include genes with slightly lower ASE for patients in the dysregulated subgroup [mediandysregulated = 0.20 (0.10–0.28), medianregulated = 0.26 (0.20–0.31); P = 0.038, Wilcoxon rank sum test; Supplementary Material, Table S6]. Furthermore, patients in the dysregulated sub-group have higher posterior probabilities of escape for the genes in the first feature cluster, but lower (and more variable) posterior probabilities of escape for the genes in the second feature cluster (Supplementary Material, Fig. S9).
Clinical associations with XCI patient subgroups
ASE cluster membership was associated with patient age at diagnosis, tumor histology, tumor grade, disease stage and results of surgical debulking (P < 0.05, linear regression and Fisher’s exact test; Table 2). Patients with tumors in the dysregulated XCI cluster were more likely to be older at diagnosis, high-grade serous histology, higher grade, higher stage and not optimally debulked. ASE clusters were not associated with BRCA mutation status (P = 0.81, Fisher’s exact test); six patients with BRCA1 mutations and four patients with BRCA2 mutations were equally split across the two clusters. ASE clusters were also associated with overall survival and time to disease recurrence (Table 3). Tumors with higher XCI dysregulation (i.e. higher ASE) had shorter overall survival (HR = 1.87, 95% CI = 1.12–3.12, Pscore = 0.015, Cox regression) and shorter time to recurrence (HR = 2.34, 95% CI = 1.41–3.88, Pscore = 0.0007, Cox regression; Fig. 4). However, these associations were attenuated after adjusting for debulking (HROS = 1.34; HRTTR = 1.75; Table 3). When restricted to 52 patients with high-grade serous disease, ASE clusters were not significantly associated with any clinical factors (Supplementary Material, Table S7); in particular, there were no associations with age at diagnosis, stage or surgical debulking status, as was observed across all histology. Although high-grade serous tumor XCI subgroups were not significantly associated with clinical outcomes (Supplementary Material, Table S8), the sample size was limited and patients in the dysregulated XCI subgroup trended toward having worse prognosis [HROS = 1.48 (0.79–2.79); HRTTR = 1.35 (0.74–2.50); Supplementary Material, Fig. S10]. Estimates of skewness based on genes subject to XCI in prior studies of normal tissue were not associated with clinical features, including grade, stage, histology, overall survival or time to recurrence (P > 0.17, linear regression).
Table 2.
Distribution of clinical features by XCI cluster membership
| Regulated cluster (N = 47) | Dysregulated cluster (N = 52) | P-value | ||
| Age at diagnosis, Mean (SD) | 59.1 (12.5) | 64.8 (11.6) | 0.0208b | |
| Grade, N(%) | 1 | 7 (14.9) | 4 (7.7) | 0.0116b |
| 2 | 13 (27.7) | 4 (7.7) | ||
| 3 | 27 (57.4) | 44 (84.6) | ||
| Stage, N(%) | I | 19 (40.4) | 6 (11.5) | 0.00121b |
| II | 5 (10.6) | 5 (9.6) | ||
| III | 17 (36.2) | 30 (57.7) | ||
| IV | 6 (12.8) | 11 (21.2) | ||
| Histotype, N(%) | High-grade serous | 15 (31.9) | 37 (71.2) | 7.1 × 10−5a |
| Endometrioid | 24 (51.1) | 6 (11.5) | ||
| Clear cell | 5 (10.6) | 5 (9.6) | ||
| Mucinous | 1 (2.1) | 2 (3.8) | ||
| Mixed | 2 (4.3) | 2 (3.8) | ||
| Surgical debulking outcome, N(%) | Sub-optimal (residual disease >1 cm) | 18 (38.3) | 39 (75.0) | 0.0002649a |
| Optimal (residual disease ≤1 cm) | 29 (61.7) | 13 (25.0) |
aFisher’s exact test.
bLinear model.
Table 3.
Clinical outcome associations with the dysregulated XCI cluster compared with the regulated XCI cluster for the clusters defined by ASE (Nregulated = 47, Ndysregulated = 52)
| Outcome | Covariates | HR (95% CI) | P-value a |
| Time to disease recurrence or death | None | 2.34 (1.41–3.88) | 0.001 |
| Debulking | 1.75 (1.04–2.94) | 0.03 | |
| Debulking, age, stage, grade | 1.36 (0.78–2.37) | 0.28 | |
| Survival time | None | 1.87 (1.12–3.12) | 0.017 |
| Debulking | 1.34 (0.79–2.26) | 0.27 | |
| Debulking, age, stage, grade | 1.01 (0.58–1.76) | 0.97 |
aCox regression.
Figure 4.
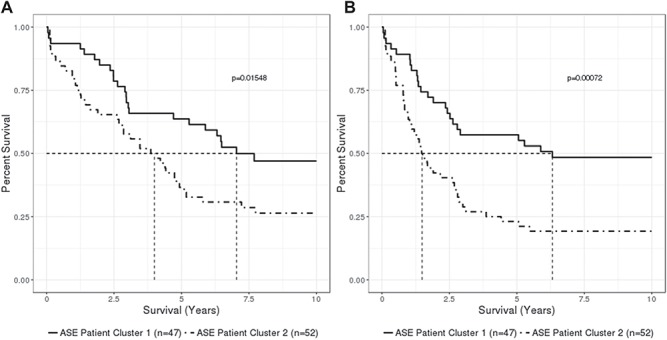
Kaplan–Meier curves of 10 year overall survival (A) and time to disease recurrence or death (B) by patient XCI subgroup defined by adjusted ASE. Percent survival is plotted against time in years. Dashed lines indicate median survival time. P-values indicate differences in curves from the log-rank test.
Discussion
In the most comprehensive study of XCI in ovarian tumors to date, we have identified molecular signatures of XCI based on multiple genomic data sources, including chromosome-wide XCI patterns and particular genes that escape XCI in ovarian tumors and have examined associations of XCI with clinical features. The probability of escape for each gene was inversely correlated with its DNA methylation status. Compared with the reported escape status in non-ovarian normal tissue types, the escape status of ~10% of genes differed. Although not associated with tumor characteristics, patients with higher numbers of discrepant genes tended to be younger at diagnosis. Notably, we observed that tumors with dysregulated XCI, defined as XCI patterns different from those expected in normal tissue, present at later age with higher grade and more advanced stage disease and are less likely to be optimally debulked in primary surgery. Tumors with dysregulated XCI patterns also have worse clinical outcomes, including shorter overall survival and time to disease recurrence. Similar relationships were observed when restricted to high-grade serous tumors.
When examining the level of inactivation at individual genes, reduced inactivation at a number of genes was associated with worse overall survival, including previously reported discrepant genes TAZ, SLITRK4, CTPS2, KAL1, DOCK11, DMD and GEMIN8 (37). In fact, overexpression of TAZ has been shown to promote the epithelial–mesenchymal transition and progression of OC, in addition to other cancer types (42,43). Additionally, increased expression of CTPS2 (along with DLG3) was associated with sensitivity to the platinum agents carboplatin and cisplatin, which are commonly used in conjunction with taxanes as first line therapy to treat OC (20); many OC patients who initially respond to these agents become resistant and recur.
Our analysis of XCI also revealed relevant biological insight. Among the two sets of genes differentiating the dysregulated and regulated XCI patient subgroups, genes in the first feature cluster were previously known to be inactivated in normal tissue types and also had low probabilities of escape in ovarian tumors; however, patients in the dysregulated XCI subgroup were more likely to have either complete or partial escape of XCI at these genes, including DMD, WWC3 and DOCK11. Genes in the second feature cluster included both inactivated and escape genes across normal tissue types and ovarian tumors, and patients in the dysregulated XCI subgroup had greater variability in probability of escape across these genes, including GABRE, CXorf36, CXorf23, OPHN1, BGN, TRAPPC2, ARSD, TCEANC, EIF2S3, NLGN4X, GPM6B, DDX3X and RPS4X. Genes with discrepant XCI status in ovarian tumors compared with previous reports tended to be enriched for tumor suppressors and oncogenes, suggesting the role that XCI may play in the two-hit hypothesis (29,30). In fact, the XCI patterns for some of these genes have previously been linked to cancer. For instance, DMD has a common fragile site that displays non-random inactivation in different cancers, including OC (44). DDX3X, a putative tumor suppressor, has been associated with escape from XCI that may contribute to male sex bias for some cancers (22,45). Although not previously associated with cancer, EIF2S3 has been reported as an escape gene that lies at an XCI boundary region (46). Additionally, many of these genes have been previously reported as relevant to OC. TCEANC may interact with BRCA1 in ovarian tumor suppression (47), and GPM6B has been proposed as a potential biomarker for early OC detection (48). Furthermore, low expression of RPS4X has been associated with poor prognosis in serous OC (49), and increased germline DNA methylation of GABRE has been associated with improved survival (50). Interestingly, TRAPPC2 and DDX3X have also been identified as possible drug targets (45,51). A number of these genes have been reported to be associated with other cancers [WWC3 (52,53), DOCK11 (54) and ARSD (55)]. In particular, NLGN4X expression was associated with survival in breast cancer (56). BGN expression was associated with proliferation in colon cancer (57) and poor prognosis in prostate cancer (58). Furthermore, OPHN1 (59) and CXorf36 (60) have been reported to be relevant for prostate cancer. Specifically, CXorf36 interacts with 8q24 in prostate cancer (60), and this region is also a risk region in breast cancer and OC (61).
Analyses of TCGA data, which include nine patients from the Mayo Clinic sample here, previously identified OC patient clusters based on DNA methylation and a subgroup of patients with dysregulated XCI, which seemed to be driven by LOH and other copy number alterations (36,62). TCGA patients in the dysregulated clusters tended to have full deletion of the inactive and duplication of the Xa (chromosome-wide copy-neutral LOH), partial reactivation or p or q arm deletion of the Xi. Similar to the patients in the dysregulated subgroup that we report, patients with these types of loss of XCI due to copy number alterations seemed to be associated with worse clinical prognosis. In our data, in regions of LOH we also observed that the Xi was more often lost, and the average promoter methylation was <50%. Prior to LOH adjustment, we also observed both ASE and methylation clustering patterns that were strongly influenced by LOH. Because copy number alterations and LOH are common in OC (62,63), including X chromosome LOH, we were interested in the role of XCI beyond structural alterations and took a number of steps in this study to eliminate this confounder. Compared with prior studies, our analyses have the advantages of accounting for potential confounding between the data types (such as methylation and LOH) and assessing consistency across data types (such as methylation and ASE).
Previous studies of XCI in OC cell lines (as well as breast cancer) have reported Barr body loss via deletion of the Xi and duplication of the Xa (11,12). In this study, we have found that the story is likely much more complicated in patient tissue, as we did not observe strong evidence of full reactivation of the entire chromosome. In contrast, our data are consistent with epigenetic erosion of XCI and local reactivation of genetic transcription on the Xi as seen in breast cancer cell lines (15). Abnormal deactivation of tumor suppressor genes and/or reactivation of oncogenes on the X chromosome may result in substantial dysregulation of cancer-related gene expression and contribute to tumor progression.
Strengths of this project are manifold, as we have leveraged types of X chromosome data that are typically ignored. Taking a multi-omic approach, we have used genotype, gene expression, DNA methylation and copy number data, as well as biological knowledge about how these data types interact, to create an innovative integrative analytical framework that has not previously been applied to studies of XCI. An additional strength is the use of a core set of OC patients with detailed clinical and follow-up data rather than cell lines or male tissue samples (64,65).
There are also a number of limitations; notably, statistical power is limited because of sample size, the need for RNA-seq reads to overlap heterozygous SNPs and low total expression for some genes. This limited our ability to adjust for multiple factors in clinical analyses, as well as restricting analyses to high-grade serous ovarian tumors, which is the most common histology with the worst clinical prognosis. To address this, we were able to impute this missing data using promoter methylation and copy number information from the corresponding regions, taking advantage of known biological relationships, further emphasizing the importance of an integrative approach to capture the biological complexity. Furthermore, no normal ovarian tissue samples were included in this study, and previous studies of XCI patterns in normal tissue did not include ovarian samples; therefore, differences described between tumors and expected normal patterns cannot be ruled out as tissue-related differences. Future studies on XCI patterns in normal ovarian tissues, as well as validation of our findings in ovarian tumors are needed.
In summary, the results of this study suggest that XCI may play an important role in ovarian tumor biology, and potentially clinical prognosis. Future work is warranted that directly incorporates copy number and total expression data, which assess XCI patterns in normal ovarian tissue to distinguish XCI patterns unique to ovarian tumors and that have larger sample sizes in independent data sets for validation. Functional studies and studies of somatic XCI patterns using paired tumor-normal data will also be critical to inform on the future clinical utility of these findings.
Materials and Methods
Study participants
Study participants included 99 patients enrolled into an Institutional Review Board (IRB)-approved protocol within 1 year of diagnosis of pathologically confirmed primary invasive epithelial ovarian, fallopian tube or primary peritoneal cancer at the Mayo Clinic between 2000 and 2009 (38). Tumor tissues were snap frozen immediately following surgery, were determined to have at least 70% tumor content and were a source of high-quality tumor DNA and RNA. Peripheral blood drawn prior to chemotherapy was used as a source of germline DNA. Follow-up clinical data were actively obtained through 2016 and revealed 66 recurrences and 62 deaths within 10 years of diagnosis.
Genomic data
Germline DNA was genotyped with Illumina Infinium Beadchips (Illumina, San Diego, CA, USA) (for 81 cases, Human610-Quad; for 18 cases, Omni 2.5 M-8) (61,66) and for a subset of 80 patients also genotyped on the Affymetrix Axiom Exome Array (Affymetrix, Santa Clara, CA, USA) (67), providing high-quality genotype data for 4543 rare variants on the X chromosome. Imputation was performed using the 1000 Genomes Project (68), resulting in a common set of Single Nucleotide Polymorphism (SNP) markers (438 944 X chromosome) measured on all samples (66).
Tumor RNA was sequenced on an Illumina HiSeq2000 with six samples per lane, following library preparation as previously described (69). A pilot study was performed for a subset of 15 patients whereby 500 ng RNA was used to generate polyA libraries using the Illumina TruSeq kit, and samples were run with 50 bp paired-end reads. For the remaining samples, 1 ug RNA was treated with riboZero, and libraries were made using the Illumina TruSeq Stranded Total RNA kit and samples were run with 100 bp paired-end reads. Both runs were found to be of consistent quality and coverage. Read alignment was performed with TopHat2 (70) and gene counts were generated with HTSeq (71). Median ASE was not associated with sequencing batch (P = 0.99).
Tumor DNA methylation was assayed using the Illumina Infinium 450 K DNA Methylation array, which included 4870 CpG probes in X chromosome gene promoters, which were considered in cis with the gene if within +/−200 KB of transcription start site. Quality control and normalization procedures have been previously described (38). CpG methylation was measured using beta values such that beta values near 0 indicate both alleles are active (‘escape’ from XCI), and CpGs with beta values near 0.5 indicate that one allele is silenced (‘subject’ to XCI).
Tumor copy number was estimated based on tumor DNA genotypes from Illumina OncoArray-500 k analysis. Large segmental (low resolution) instances of copy number aberrations and LOH were determined using OncoSNP, which accounts for intra-tumor heterogeneity and non-tumor content (72). Copy number was estimated with paired germline and tumor genotype data (‘tumor-normal’ mode), ‘intratumor’ and ‘stromal’ options, assuming >70% tumor content (72).
Gene-level XCI patterns
Estimation of ASE and identification of tumor XCI escape genes based on a two-stage analysis approach were described previously (37). Briefly, for each sample, we tested for preferential inactivation of one chromosome (i.e. ‘skewness’) using a composite likelihood ratio test based on a beta-binomial distribution (37). In the skewed samples (Bonferroni-adjusted P < 0.05), we determined genes that escape from XCI using a two-component Bayesian beta-binomial mixture model (37). XCI escape status was ascribed based on the posterior probability a gene is escaping XCI (PPE), where the gene was assigned ‘subject to XCI’ if the probability was <0.5 and the gene was assigned ‘escape’ if the probability was >0.5, unless otherwise noted (37). For each X chromosome gene, the tumor XCI status was compared with previously reported XCI status from multiple non-tumor tissues (39). For the purpose of calculating concordance between the ovarian tumor samples and the expected state based on the consensus call from normal tissue, a gene was called as either ‘subject’ or ‘escape’ based on a cut-point of 0.5 for PPE for a given sample. Across samples, a gene was called ‘subject to XCI’ if the proportion of samples called ‘subject’ was <0.2; a gene was called ‘escape from XCI’ if the proportion of samples called ‘escape’ was >0.8; and a gene was called ‘variable escape’ otherwise.
At the sample level, a gene was considered ‘discrepant’ compared with the consensus state from normal tissue if the XCI call was in the opposite direction (e.g. sample call was ‘subject’ but consensus call was ‘escape’ or ‘mostly escape’ or sample call was ‘escape’ but consensus call was ‘subject’ or ‘mostly subject’). For high-confidence discrepant calls, a cut-point of PPE < 0.1 or PPE > 0.9 was used.
We validated the XCI status derived from ASE data using methylation data (64). We evaluated the correlation of ASE and promoter methylation in the corresponding genes (+/−200 KB of transcription start site) using linear mixed models. For each gene, we also compared the average promoter methylation to the posterior probability of escape from XCI. We also visualized and compared the average promoter methylation across samples for estimated escape genes versus inactivated genes.
Missing ASE data were imputed using methylation and LOH data (Supplementary Material, Supplemental Methods). We assessed associations between imputed ASE at each gene and age at diagnosis, tumor histology, stage, grade, residual disease after surgery and percent tumor present in the sample using Fisher’s exact tests and linear regression. We tested associations with time to disease recurrence or death and overall survival time using Cox proportional hazards models.
Assessing global XCI patterns
In addition to the XCI escape patterns of individual genes, we also characterized patterns of the XCI process across the entire X chromosome to evaluate a global level of XCI disruption in ovarian tumors. We determined sub-groups, or ‘clusters’, of patients to characterize the level of disruption of the XCI process using multiple genomic data types (expression, methylation and LOH). To verify that ASE and methylation data provide consistent XCI signals, we examined cis correlations between ASE and methylation before and after LOH adjustment. Then missing ASE values were imputed using methylation and LOH data (Supplemental Methods). Patient clustering was performed with non-smooth non-negative matrix factorization (nsNMF) algorithm (73,74) in the imputed ASE data to identify sets of tumor samples with similar XCI patterns after LOH adjustment (Supplemental Methods). In order to ensure stability and reproducibility, consensus clustering with n = 100 runs was performed (75). The number of clusters was selected using consensus clustering and non-smooth NMF with a cophenetic correlation coefficient (74) assessment. As a sensitivity analysis, clustering was also restricted to high-grade serous samples. Clustering was performed using the R package ‘NMF’ (https://cran.r-project.org/web/packages/NMF/index.html).
Biological relevance and clinical associations of XCI patterns
In addition to sample-based clustering, we also performed feature extraction to determine which probes/genes had the greatest impact on the derived patient clusters (76). We determined sets of genes with similar XCI patterns and graphically inspected regional patterns. For the extracted feature sets, we summarized the previously estimated XCI escape status based on prior studies (39) and performed gene annotation and gene set analysis using the software DAVID (41) with X chromosome genes as the background. To gain insight into how the most influential genes were classifying patients, we compared the median estimated posterior probability of escape across all genes for each tumor across the patient clusters. We examined associations of the patient clusters with XIST total expression (as a surrogate of XCI activity), as well as molecular subtype (40) and level of skewness using Wilcoxon rank sum and Fisher’s exact tests.
We assessed associations between tumor-based XCI cluster and age at diagnosis, tumor histology, stage, grade, residual disease after surgery and percent tumor present in the sample using Fisher’s exact tests and linear regression. We tested associations with time to disease recurrence or death and overall survival time using Cox proportional hazards models. We also examined associations between each clinical feature and a high level of discrepant XCI genes [defined as having more than the median percentage (10%) of genes with discrepant XCI status compared with prior studies from normal tissue]. Analyses were repeated restricted to high-grade serous tumors and BRCA1 and BRCA2 pathogenic germline mutation carriers.
All analyses were conducted using the statistical software R 3.3.1.
Data access
Data have previously been submitted to public repositories (European Genotype Archive; EGAS00001002305), and additional molecular data will be submitted to the Gene Expression Omnibus repository. We assure that all gene and protein names used in this manuscript adhere to the approved nomenclature guidelines for humans.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors thank the following for their funding and support: The Walter and Evelyn Simmers Career Development Award for Ovarian Cancer Research, the Fraternal Order of the Eagles Cancer Research Fund, the National Institutes of Health National Cancer Institute and the National Institutes of Health Office of Women’s Health Research.
Conflict of Interest statement. All authors have reviewed the manuscript and have no conflict of interest to report. This manuscript has not been published, accepted for publication or under consideration by another journal.
Funding
National Institutes of Health, Office of Women’s Health (K12-HD065987); Fraternal Order of the Eagles Cancer Research Fund (FP00083572.02); Walter and Evelyn Simmers Career Development Award for Ovarian Cancer Research; National Institutes of Health, National Cancer Institute (4R00CA184415–02, P30-CA15083, R03-CA212127, R01-CA122443, P50-CA136393, R00CA184415).
References
- 1. Wise A.L., Gyi L. and Manolio T.A. (2013) eXclusion: toward integrating the X chromosome in genome-wide association analyses. Am. J. Hum. Genet., 92, 643–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Clayton D. (2008) Testing for association on the X chromosome. Biostatistics, 9, 593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ross M.T., Grafham D.V., Coffey A.J., Scherer S., McLay K., Muzny D., Platzer M., Howell G.R., Burrows C., Bird C.P. et al. (2005) The DNA sequence of the human X chromosome. Nature, 434, 325–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brown C.J., Ballabio A., Rupert J.L., Lafreniere R.G., Grompe M., Tonlorenzi R. and Willard H.F. (1991) A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature, 349, 38–44. [DOI] [PubMed] [Google Scholar]
- 5. Carrel L. and Willard H.F. (2005) X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature, 434, 400–404. [DOI] [PubMed] [Google Scholar]
- 6. Carrel L., Cottle A.A., Goglin K.C. and Willard H.F. (1999) A first-generation X-inactivation profile of the human X chromosome. Proc. Natl. Acad. Sci. U. S. A., 96, 14440–14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spatz A., Borg C. and Feunteun J. (2004) X-chromosome genetics and human cancer. Nat. Rev. Cancer, 4, 617–629. [DOI] [PubMed] [Google Scholar]
- 8. Ferlay J., Soerjomataram I., Dikshit R., Eser S., Mathers C., Rebelo M., Parkin D.M., Forman D. and Bray F. (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer, 136, E359–E386. [DOI] [PubMed] [Google Scholar]
- 9. McGuire V., Jesser C.A. and Whittemore A.S. (2002) Survival among U.S. women with invasive epithelial ovarian cancer. Gynecol. Oncol., 84, 399–403. [DOI] [PubMed] [Google Scholar]
- 10. Howlader N., Noone A., Krapcho M., Garshell J., Miller D., Altekruse S., Kosary C., Yu M., Ruhl J., Tatalovich Z. et al. (2014) SEER cancer statistics review. National Cancer Institute, Bethesda, MD,1975–2011. [Google Scholar]
- 11. Pageau G.J., Hall L.L., Ganesan S., Livingston D.M. and Lawrence J.B. (2007) The disappearing Barr body in breast and ovarian cancers. Nat. Rev. Cancer, 7, 628–633. [DOI] [PubMed] [Google Scholar]
- 12. Barr M.L. and Moore K.L. (1957) Chromosomes, sex chromatin, and cancer. Proc. Can. Cancer. Conf., 2, 3–16. [PubMed] [Google Scholar]
- 13. Benoit M.H., Hudson T.J., Maire G., Squire J.A., Arcand S.L., Provencher D., Mes-Masson A.M. and Tonin P.N. (2007) Global analysis of chromosome X gene expression in primary cultures of normal ovarian surface epithelial cells and epithelial ovarian cancer cell lines. Int. J. Oncol., 30, 5–17. [PubMed] [Google Scholar]
- 14. Chaligne R. and Heard E. (2014) X-chromosome inactivation in development and cancer. FEBS Lett., 588, 2514–2522. [DOI] [PubMed] [Google Scholar]
- 15. Chaligne R., Popova T., Mendoza-Parra M.A., Saleem M.A., Gentien D., Ban K., Piolot T., Leroy O., Mariani O., Gronemeyer H. et al. (2015) The inactive X chromosome is epigenetically unstable and transcriptionally labile in breast cancer. Genome Res., 25, 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Silver D.P., Dimitrov S.D., Feunteun J., Gelman R., Drapkin R., Lu S.D., Shestakova E., Velmurugan S., Denunzio N., Dragomir S. et al. (2007) Further evidence for BRCA1 communication with the inactive X chromosome. Cell, 128, 991–1002. [DOI] [PubMed] [Google Scholar]
- 17. Saifi G.M. and Chandra H.S. (1999) An apparent excess of sex- and reproduction-related genes on the human X chromosome. Proc. Biol. Sci., 266, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hediger M.A., Romero M.F., Peng J.B., Rolfs A., Takanaga H. and Bruford E.A. (2004) The ABCs of solute carriers: physiological, pathological and therapeutic implications of human membrane transport proteins. Pflugers Arch., 447, 465–468. [DOI] [PubMed] [Google Scholar]
- 19. Borst P. and Elferink R.O. (2002) Mammalian ABC transporters in health and disease. Annu. Rev. Biochem., 71, 537–592. [DOI] [PubMed] [Google Scholar]
- 20. Gamazon E.R., Im H.K., O’Donnell P.H., Ziliak D., Stark A.L., Cox N.J., Dolan M.E. and Huang R.S. (2011) Comprehensive evaluation of the contribution of X chromosome genes to platinum sensitivity. Mol. Cancer Ther., 10, 472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liao D.J., Du Q.Q., Yu B.W., Grignon D. and Sarkar F.H. (2003) Novel perspective: focusing on the X chromosome in reproductive cancers. Cancer Invest., 21, 641–658. [DOI] [PubMed] [Google Scholar]
- 22. Dunford A., Weinstock D.M., Savova V., Schumacher S.E., Cleary J.P., Yoda A., Sullivan T.J., Hess J.M., Gimelbrant A.A., Beroukhim R. et al. (2016) Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat. Genet. 2017Jan; 49: 10–16. doi: 10.1038/ng.3726 Epub 2016 Nov 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lose F., Duffy D.L., Kay G.F., Kedda M.A. and Spurdle A.B. (2008) Skewed X chromosome inactivation and breast and ovarian cancer status: evidence for X-linked modifiers of BRCA1. J. Natl. Cancer Inst., 100, 1519–1529. [DOI] [PubMed] [Google Scholar]
- 24. Buller R.E., Sood A.K., Lallas T., Buekers T. and Skilling J.S. (1999) Association between nonrandom X-chromosome inactivation and BRCA1 mutation in germline DNA of patients with ovarian cancer. J. Natl. Cancer Inst., 91, 339–346. [DOI] [PubMed] [Google Scholar]
- 25. Kristiansen M., Knudsen G.P., Maguire P., Margolin S., Pedersen J., Lindblom A. and Orstavik K.H. (2005) High incidence of skewed X chromosome inactivation in young patients with familial non-BRCA1/BRCA2 breast cancer. J. Med. Genet., 42, 877–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Struewing J.P., Pineda M.A., Sherman M.E., Lissowska J., Brinton L.A., Peplonska B., Bardin-Mikolajczak A. and Garcia-Closas M. (2006) Skewed X chromosome inactivation and early-onset breast cancer. J. Med. Genet., 43, 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Manoukian S., Verderio P., Tabano S., Colapietro P., Pizzamiglio S., Grati F.R., Calvello M., Peissel B., Burn J., Pensotti V. et al. (2013) X chromosome inactivation pattern in BRCA gene mutation carriers. Eur. J. Cancer, 49, 1136–1141. [DOI] [PubMed] [Google Scholar]
- 28. Sandovici I., Naumova A.K., Leppert M., Linares Y. and Sapienza C. (2004) A longitudinal study of X-inactivation ratio in human females. Hum. Genet., 115, 387–392. [DOI] [PubMed] [Google Scholar]
- 29. Knudson A.G., Jr. (1971) Mutation and cancer: statistical study of retinoblastoma. Proc. Natl. Acad. Sci. U. S. A., 68, 820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng P.C., Gosewehr J.A., Kim T.M., Velicescu M., Wan M., Zheng J., Felix J.C., Cofer K.F., Luo P., Biela B.H. et al. (1996) Potential role of the inactivated X chromosome in ovarian epithelial tumor development. J. Natl. Cancer Inst., 88, 510–518. [DOI] [PubMed] [Google Scholar]
- 31. Dodson M.K., Hartmann L.C., Cliby W.A., Delacey K.A., Keeney G.L., Ritland S.R., Su J.Q., Podratz K.C. and Jenkins R.B. (1993) Comparison of loss of heterozygosity patterns in invasive low-grade and high-grade epithelial ovarian carcinomas. Cancer Res., 53, 4456–4460. [PubMed] [Google Scholar]
- 32. Lin H.C., Huber R., Schlessinger D. and Morin P.J. (1999) Frequent silencing of the GPC3 gene in ovarian cancer cell lines. Cancer Res., 59, 807–810. [PubMed] [Google Scholar]
- 33. Choi C., Cho S.H., Horikawa I., Berchuck A., Wang N., Cedrone E., Jhung S.W., Lee J.B., Kerr J., Chenevixtrench G. et al. (1997) Loss of heterozygosity at chromosome segment Xq25-26.1 in advanced human ovarian carcinomas. Genes Chromosomes Cancer, 20, 234–242. [PubMed] [Google Scholar]
- 34. Seki Y., Suico M.A., Uto A., Hisatsune A., Shuto T., Isohama Y. and Kai H. (2002) The ETS transcription factor MEF is a candidate tumor suppressor gene on the X chromosome. Cancer Res., 62, 6579–6586. [PubMed] [Google Scholar]
- 35. Yao J.J., Liu Y., Lacorazza H.D., Soslow R.A., Scandura J.M., Nimer S.D. and Hedvat C.V. (2007) Tumor promoting properties of the ETS protein MEF in ovarian cancer. Oncogene, 26, 4032–4037. [DOI] [PubMed] [Google Scholar]
- 36. Kang J., Lee H.J., Kim J., Lee J.J. and Maeng L.S. (2015) Dysregulation of X chromosome inactivation in high grade ovarian serous adenocarcinoma. PLoS One, 10, e0118927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Larson N.B., Fogarty Z.C., Larson M.C., Kalli K.R., Lawrenson K., Gayther S., Fridley B.L., Goode E.L. and Winham S.J. (2017) An integrative approach to assess X-chromosome inactivation using allele-specific expression with applications to epithelial ovarian cancer. Genet. Epidemiol., 41, 898–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cicek M.S., Koestler D.C., Fridley B.L., Kalli K.R., Armasu S.M., Larson M.C., Wang C., Winham S.J., Vierkant R.A., Rider D.N. et al. (2013) Epigenome-wide ovarian cancer analysis identifies a methylation profile differentiating clear-cell histology with epigenetic silencing of the HERG K+ channel. Hum. Mol. Genet., 22, 3038–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Balaton B.P., Cotton A.M. and Brown C.J. (2015) Derivation of consensus inactivation status for X-linked genes from genome-wide studies. Biol. Sex Differ., 6, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Verhaak R.G., Tamayo P., Yang J.Y., Hubbard D., Zhang H., Creighton C.J., Fereday S., Lawrence M., Carter S.L., Mermel C.H. et al. (2013) Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Invest., 123, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang da W., Sherman B.T. and Lempicki R.A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc., 4, 44–57. [DOI] [PubMed] [Google Scholar]
- 42. Chen G., Xie J., Huang P. and Yang Z. (2016) Overexpression of TAZ promotes cell proliferation, migration and epithelial-mesenchymal transition in ovarian cancer. Oncol. Lett., 12, 1821–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moroishi T., Hansen C.G. and Guan K.L. (2015) The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer, 15, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McAvoy S., Ganapathiraju S.C., Ducharme-Smith A.L., Pritchett J.R., Kosari F., Perez D.S., Zhu Y., James C.D. and Smith D.I. (2007) Non-random inactivation of large common fragile site genes in different cancers. Cytogenet. Genome Res., 118, 260–269. [DOI] [PubMed] [Google Scholar]
- 45. Bol G.M., Xie M. and Raman V. (2015) DDX3, a potential target for cancer treatment. Mol. Cancer, 14, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Filippova G.N., Cheng M.K., Moore J.M., Truong J.P., Hu Y.J., Nguyen D.K., Tsuchiya K.D. and Disteche C.M. (2005) Boundaries between chromosomal domains of X inactivation and escape bind CTCF and lack CpG methylation during early development. Dev. Cell, 8, 31–42. [DOI] [PubMed] [Google Scholar]
- 47. Hill S.J., Rolland T., Adelmant G., Xia X., Owen M.S., Dricot A., Zack T.I., Sahni N., Jacob Y., Hao T. et al. (2014) Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes Dev., 28, 1957–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Urban P., Bilecova-Rabajdova M., Stefekova Z., Ostro A. and Marekova M. (2011) Overview of potential oncomarkers for detection of early stages of ovarian cancer. Klin. Onkol., 24, 106–111. [PubMed] [Google Scholar]
- 49. Tsofack S.P., Meunier L., Sanchez L., Madore J., Provencher D., Mes-Masson A.M. and Lebel M. (2013) Low expression of the X-linked ribosomal protein S4 in human serous epithelial ovarian cancer is associated with a poor prognosis. BMC Cancer, 13, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fridley B.L., Armasu S.M., Cicek M.S., Larson M.C., Wang C., Winham S.J., Kalli K.R., Koestler D.C., Rider D.N., Shridhar V. et al. (2014) Methylation of leukocyte DNA and ovarian cancer: relationships with disease status and outcome. BMC Med. Genomics, 7, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tokunaga M., Shiheido H., Tabata N., Sakuma-Yonemura Y., Takashima H., Horisawa K., Doi N. and Yanagawa H. (2013) MIP-2A is a novel target of an anilinoquinazoline derivative for inhibition of tumour cell proliferation. PLoS One, 8, e76774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Han Q., Lin X., Zhang X., Jiang G., Zhang Y., Miao Y., Rong X., Zheng X., Han Y., Han X. et al. (2017) WWC3 regulates the Wnt and Hippo pathways via Dishevelled proteins and large tumour suppressor 1, to suppress lung cancer invasion and metastasis. J. Pathol., 242, 435–447. [DOI] [PubMed] [Google Scholar]
- 53. Wang Y., Jiang M., Yao Y. and Cai Z. (2018) WWC3 inhibits glioma cell proliferation through suppressing the Wnt/β-catenin signaling pathway. DNA Cell Biol., 37, 31–37. [DOI] [PubMed] [Google Scholar]
- 54. Almstrup K., Leffers H., Lothe R.A., Skakkebaek N.E., Sonne S.B., Nielsen J.E., Rajpert-De Meyts E. and Skotheim R.I. (2007) Improved gene expression signature of testicular carcinoma in situ. Int. J. Androl., 30, 292–302discussion 303. [DOI] [PubMed] [Google Scholar]
- 55. Trojani A., Di Camillo B., Tedeschi A., Lodola M., Montesano S., Ricci F., Vismara E., Greco A., Veronese S., Orlacchio A. et al. (2011) Gene expression profiling identifies ARSD as a new marker of disease progression and the sphingolipid metabolism as a potential novel metabolism in chronic lymphocytic leukemia. Cancer Biomark., 11, 15–28. [DOI] [PubMed] [Google Scholar]
- 56. Henderson H.J., Karanam B., Samant R., Vig K., Singh S.R., Yates C. and Bedi D. (2017) Neuroligin 4X overexpression in human breast cancer is associated with poor relapse-free survival. PLoS One, 12, e0189662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xing X., Gu X. and Ma T. (2015) Knockdown of biglycan expression by RNA interference inhibits the proliferation and invasion of, and induces apoptosis in, the HCT116 colon cancer cell line. Mol. Med. Rep., 12, 7538–7544. [DOI] [PubMed] [Google Scholar]
- 58. Jacobsen F., Kraft J., Schroeder C., Hube-Magg C., Kluth M., Lang D.S., Simon R., Sauter G., Izbicki J.R., Clauditz T.S. et al. (2017) Up-regulation of biglycan is associated with poor prognosis and PTEN deletion in patients with prostate cancer. Neoplasia, 19, 707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goto K., Oue N., Hayashi T., Shinmei S., Sakamoto N., Sentani K., Teishima J., Matsubara A. and Yasui W. (2014) Oligophrenin-1 is associated with cell adhesion and migration in prostate cancer. Pathobiology, 81, 190–198. [DOI] [PubMed] [Google Scholar]
- 60. Du M., Yuan T., Schilter K.F., Dittmar R.L., Mackinnon A., Huang X., Tschannen M., Worthey E., Jacob H., Xia S. et al. (2015) Prostate cancer risk locus at 8q24 as a regulatory hub by physical interactions with multiple genomic loci across the genome. Hum. Mol. Genet., 24, 154–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Goode E.L., Chenevix-Trench G., Song H., Ramus S.J., Notaridou M., Lawrenson K., Widschwendter M., Vierkant R.A., Larson M.C., Kjaer S.K. et al. (2010) A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat. Genet., 42, 874–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature, 474, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Huang R.Y.J., Chen G.B., Matsumura N., Lai H.C., Mori S., Li J., Wong M.K., Konishi I., Thiery J.P. and Goh L. (2012) Histotype-specific copy-number alterations in ovarian cancer. BMC Med. Genomics, 5, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cotton A.M., Price E.M., Jones M.J., Balaton B.P., Kobor M.S. and Brown C.J. (2015) Landscape of DNA methylation on the X chromosome reflects CpG density, functional chromatin state and X-chromosome inactivation. Hum. Mol. Genet., 24, 1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tukiainen T., Villani A.C., Yen A., Rivas M.A., Marshall J.L., Satija R., Aguirre M., Gauthier L., Fleharty M., Kirby A. et al. (2017) Landscape of X chromosome inactivation across human tissues. Nature, 550, 244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kuchenbaecker K.B., Ramus S.J., Tyrer J., Lee A., Shen H.C., Beesley J., Lawrenson K., McGuffog L., Healey S., Lee J.M. et al. (2015) Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat. Genet., 47, 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Winham S.J., Pirie A., Chen Y.A., Larson M.C., Fogarty Z.C., Earp M.A., Anton-Culver H., Bandera E.V., Cramer D., Doherty J.A. et al. (2016) Investigation of exomic variants associated with overall survival in ovarian cancer. Cancer Epidemiol. Biomarkers Prev., 25, 446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E. and McVean G.A. (2010) A map of human genome variation from population-scale sequencing. Nature, 467, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Earp M.A., Raghavan R., Li Q., Dai J., Winham S.J., Cunningham J.M., Natanzon Y., Kalli K.R., Hou X., Weroha S.J. et al. (2017) Characterization of fusion genes in common and rare epithelial ovarian cancer histologic subtypes. Oncotarget, 8, 46891–46899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R. and Salzberg S.L. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol., 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Anders S., Pyl P.T. and Huber W. (2015) HTSeq––a Python framework to work with high-throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yau C., Mouradov D., Jorissen R.N., Colella S., Mirza G., Steers G., Harris A., Ragoussis J., Sieber O. and Holmes C.C. (2010) A statistical approach for detecting genomic aberrations in heterogeneous tumor samples from single nucleotide polymorphism genotyping data. Genome Biol., 11, R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pascual-Montano A., Carazo J.M., Kochi K., Lehmann D. and Pascual-Marqui R.D. (2006) Nonsmooth nonnegative matrix factorization (nsNMF). IEEE Trans. Pattern Anal. Mach. Intell., 28, 403–415. [DOI] [PubMed] [Google Scholar]
- 74. Brunet J.P., Tamayo P., Golub T.R. and Mesirov J.P. (2004) Metagenes and molecular pattern discovery using matrix factorization. Proc. Natl. Acad. Sci. U. S. A., 101, 4164–4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Monti S., Tamayo P., Mesirov J. and Golub T. (2003) Consensus clustering: a resampling-based method for class discovery and visualization of gene expression microarray data. Mach. Learn., 52, 91–118. [Google Scholar]
- 76. Carmona-Saez P., Pascual-Marqui R.D., Tirado F., Carazo J.M. and Pascual-Montano A. (2006) Biclustering of gene expression data by non-smooth non-negative matrix factorization. BMC Bioinformatics, 7, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.