

Abstract
Background
Pathogenic variants in SCN1A cause variable epilepsy disorders with different disease severities. We here investigate whether common variation in the promoter region of the unaffected SCN1A allele could reduce normal expression, leading to a decreased residual function of Nav1.1, and therefore to more severe clinical outcomes in patients affected by pathogenic SCN1A variants.
Methods
Five different SCN1A promoter‐haplotypes were functionally assessed in SH‐SY5Y cells using Firefly and Renilla luciferase assays. The SCN1A promoter region was analyzed in a cohort of 143 participants with SCN1A pathogenic variants. Differences in clinical features and outcomes between participants with and without common variants in the SCN1A promoter‐region of their unaffected allele were investigated.
Results
All non‐wildtype haplotypes showed a significant reduction in luciferase expression, compared to the wildtype promoter‐region (65%–80%, p = 0.039–0.0023). No statistically significant differences in clinical outcomes were observed between patients with and without common promoter variants. However, patients with a wildtype promoter‐haplotype on their unaffected SCN1A allele showed a nonsignificant trend for milder phenotypes.
Conclusion
The nonsignificant observed trends in our study warrant replication studies in larger cohorts to explore the potential modifying role of these common SCN1A promoter‐haplotypes.
Keywords: Dravet, GEFS+, promoter, SCN1A, variable expression
1. INTRODUCTION
Dravet syndrome is one of the most well‐known genetic epilepsy syndromes. The main characteristics of the disease are early onset intractable epileptic seizures and a delayed psychomotor development that results in mild to severe intellectual disability (ID). Furthermore, many patients experience walking difficulties and/or behavioral problems (Brunklaus, Ellis, Reavey, Forbes, & Zuberi, 2012; Dravet, 1978, 2011; Gitiaux et al., 2016; Rilstone, Coelho, Minassian, & Andrade, 2012). Mutations in the SCN1A gene (OMIM 182389) are the cause of disease and detected in the majority of Dravet syndrome patients (Parihar & Ganesh, 2013). SCN1A encodes for the α‐subunit of a neuronal sodium channel, Nav1.1. The main disease mechanism in SCN1A‐related Dravet syndrome is haploinsufficiency, caused by complete or partial loss of function of the channel, which leads to disturbances in neuronal excitability (Catterall, Kalume, & Oakley, 2010; Escayg & Goldin, 2010).
Pathogenic variants in SCN1A are also found in patients with much milder phenotypes, such as Genetic Epilepsy Febrile Seizures Plus (GEFS+) syndrome or febrile seizures only (Escayg et al., 2000). The association of SCN1A with multiple phenotypes may be partly explained by the varying effects of different pathogenic variants: variants that cause a complete loss of function (LoF) of the channel are virtually always associated with severe phenotypes, whereas variants that cause milder disturbances are usually found in milder phenotypes (Meng et al., 2015). However, this does not fully explain the variability that is observed in SCN1A related phenotypes: varying phenotypes have been associated with the exact same variant, even within families, and Dravet syndrome patients with similar LoF variants may show very different clinical outcomes (Akiyama, Kobayashi, Yoshinaga, & Ohtsuka, 2010; Depienne et al., 2010; Guerrini et al., 2010; Harkin et al., 2007; Jansen et al., 2006; Mahoney et al., 2009; Passamonti et al., 2015; Pineda‐Trujillo et al., 2005; Suls et al., 2010). Several modifying factors have already been proven or suggested to have an influence on these outcomes, such as mosaicism for the pathogenic SCN1A variants, the presence of variants in modifier genes and environmental factors such as anti‐epileptic treatment (Ceulemans, 2011; Depienne et al., 2010; Gennaro et al., 2006; Guerrini et al., 1998; Lange, Gunning, et al., 2018; Lange, Koudijs, et al., 2018; Marini, Mei, Helen Cross, & Guerrini, 2006).
Another factor that could potentially contribute to phenotypic variability is additional variation in the SCN1A gene itself. Genome‐wide association studies (GWAS) have shown a significant association between SCN1A and genetic generalized, focal and unclassified epilepsies in general, and hippocampal sclerosis and febrile seizures (Anney et al., 2014; Kasperavičiute et al., 2013). This observation suggests that common, low risk variation may affect normal function and/or expression of SCN1A. SCN1A has at least three major promoters that are simultaneously active in various brain regions including the cerebellum, cerebral cortex, putamen, hippocampus, and thalamus (Dong et al., 2014; Long et al., 2008; Nakayama et al., 2010). Promoter 1 (P1a) alone yielded transcription activity in a neuronal cell culture assay, though the activity was greatly enhanced when 5’ untranslated exons (UE) were added (Long et al., 2008). A total of five 5’ UEs of SCN1A are currently known, all of them carrying multiple putative transcription factor binding sites (Long et al., 2008; Martin, Tang, Ta, & Escayg, 2007). Adding to the complexity of SCN1A transcription, the 5’ untranslated region including the three promoters are located in a span of 75 Kb upstream of the first coding exon (Long et al., 2008; Martin et al., 2007) (Figure 1). This region has not been studied extensively in Dravet syndrome patients, but may harbor mutations that could either be the cause of their epilepsy, or include variants that could modify the phenotype caused by another major mutation in the coding region of the gene. So far, two reports have been published that suggested that pathogenic mutations in the regulatory 5’ region of SCN1A were likely the cause of disease in two Dravet syndrome patients, as no SCN1A coding mutations could be detected. Interestingly, the novel promoter mutations were found to reduce transcription in vitro, increasing the likelihood of their causality (Gao et al., 2017; Nakayama et al., 2010). These findings stress the importance of the SCN1A promoter‐regions for correct functioning of the Nav1.1 channel. It has previously been suggested that part of the 20%–30% of Dravet syndrome patients in whom no coding variants in SCN1A could be detected, harbor mutations in its regulatory regions (Djémié et al., 2016). However, in most diagnostic centers the promoter regions are not routinely sequenced when analyzing SCN1A, so its exact role remains unclear.
Figure 1.
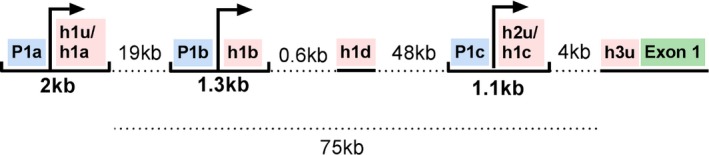
Overview SCN1A 5' UTR. SCN1A has a complex 5’ UTR. Three major promoter regions (blue) and five 5’ UE (pink) are currently known. The half‐tick up lines indicate a promoter region with a subsequent 5’ UE which together carry consensus regions for multiple transcription factor binding sites and initiator elements. Transcription start sites are indicated with an arrow. Dashed lines indicate the distance to next element. Underlined elements indicate the remaining two 5’ UEs and the first coding exon of SCN1A (green). Figure established based on previous work (Dong et al., 2014; Long et al., 2008; Nakayama et al., 2010)
We hypothesize that not only pathogenic mutations, but also common variation on the promoter regions of SCN1A can interfere with normal expression. Although the effects of common variation are likely milder than those of a true pathogenic mutation in the promoter regions, a clinical effect might be detectable when common variation in the promoter regions coexists with a pathogenic mutation in the coding region of SCN1A on the other allele. A small decrease in expression of SCN1A could lead to a decreased residual function of Nav1.1 in patients that are already haploinsufficient, and therefore lead to more severe clinical outcomes. Previously, no significant differences in expression were observed for a group of common variants in the first SCN1A promoter region (Gao et al., 2017). We have cloned a new set of haplotypes and used a slightly altered promoter region that includes the first 5’ UE in the functional expression analysis. In this study, we analyze the first SCN1A promoter‐region of 143 participants affected by pathogenic SCN1A variants, to investigate whether common variation in this region can affect phenotypic outcomes.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
The study was approved by the Ethical Committee of the University Medical Center Utrecht. Informed consent was obtained from participants or their legal caretakers according to the Declaration of Helsinki.
2.1.1. Participants and clinical data
Participants
A cohort of 143 participants with SCN1A pathogenic variants was evaluated, of which most have previously been described (Lange, Gunning, et al., 2018; Lange, Koudijs, et al., 2018). Only participants with pathogenic variants (class V) or likely pathogenic variants (class IV) in SCN1A were included, according to the American College of Medical Genetics and Genomics criteria (Richards et al., 2015). All variants had been detected and classified in genetic diagnostic laboratories. Patients who had previously been shown to be mosaic (n = 4) for their pathogenic SCN1A variant were excluded from analyses, as mosaicism may greatly influence outcomes (Lange, Koudijs, et al., 2018). Our cohort comprised patients with Dravet syndrome, GEFS+, febrile seizures, and also four participants who had been seizure‐free their entire lives, but did have a child with Dravet syndrome that carried the same pathogenic SCN1A variant. Dravet syndrome was diagnosed based on previously published criteria (Verbeek et al., 2013) and in line with recently published recommendations (Wirrell et al., 2017). Our main statistical analyses of clinical outcomes were performed on patients with Dravet syndrome only. Non‐Dravet syndrome patients remained included in the molecular analyses to separately investigate whether different promoter haplotypes could explain the inter‐familial phenotypic variability of Dravet syndrome patients and their more mildly affected family members.
Clinical data
Detailed clinical data were collected from medical records for all participants, and a semi‐structured telephone interview was conducted when possible (n = 130). A classification of the developmental outcome was made, rated in a consensus meeting by a child neurologist, neuropsychologist, and clinical geneticist. Developmental outcome was rated on a five‐point scale based on available data on IQ and developmental level (1 = no ID (IQ or developmental quotient (DQ) >85), 2 = borderline ID (IQ or DQ 70–85), 3 = mild ID (IQ or DQ 50–70), 4 = moderate ID (IQ or DQ 30–50), 5 = severe or profound ID (IQ or DQ <30)). When no (recent) IQ or DQ was available, the assessment was made based on school functioning, communication and adaptive behavior. Furthermore, approximated IQ/DQ scores after 5 years of disease were calculated, to obtain a cognitive outcome measurement unaffected by the influence of the different ages at assessment of the participants. For this, all IQ‐ and developmental assessment scores of each patient, conducted at different ages, were interpolated by linear regression as previously described (Lange, Gunning, et al., 2018). When the first official assessment was made later than 5 years after seizure onset we used the age at which a developmental delay was first observed (by either parents or clinicians) as the first moment of decline, and IQ/DQ scores up until that age were estimated to be average (=100).
2.1.2. Molecular analyses
Functional characterization of common SCN1A promoter variants
The SCN1A (Homo sapiens chromosome 2, GRCh38.p12: 166148836 to 166151403, NC_000002.12) promoter 1 region including h1u was PCR amplified from human control DNA using primers with a 15bp extension arm used for cloning. Five different haplotypes of 2568bp were selected and ligated in Psicheck‐2 plasmids using In‐Fusion cloning (clontech). The Psicheck‐2 plasmid enables dual‐reporter luciferase read‐out as it carries both the renilla (Renilla reniformis derived) and firefly (Photinus pyralis derived) luciferase genes. Firefly luciferase is expressed by the HSV‐TK promoter and can therefore be used as a control, while Renilla luciferase is controlled by the SCN1A promoter. As both genes are present on the same plasmid, unlike single reporter luciferase systems that require the transfection of two plasmids, the normalization and therefore read‐out is more accurate. Plasmids were subsequently sequenced to confirm the haplotypes. SH‐SY5Y cells were seeded in 24‐well cell culture plates until 80% confluency was reached. Psicheck‐2 plasmids carrying the SCN1A promoter haplotypes were transfected using polyethylenimine. After 48 hr, cells were lysed and the lysate transferred to a white opaque 96‐well plate in which the luciferase recording took place. Firefly and Renilla luciferase activities were detected using the dual luciferase reporter assay system (Promega) in the Varioskan FLASH luminometer (Thermo Fisher Scientific). Read‐out was performed twice as a technical replicate, and averaged values were taken as final mean. Luciferase experiments were replicated eight times. Differences in expression between haplotypes were not normally distributed and therefore analyzed using the Mann‐Whitney U‐test. For primer sequences see Supplementary Data S1.
Reconstruction of SCN1A promoter‐haplotypes of the unaffected allele in participants
SCN1A was re‐analysed in all participants as previously described (Lange, Koudijs, et al., 2018). In short, all SCN1A exons were captured by single molecule molecular inversion probes (smMIPs) and sequenced on a NextSeq500 (Illumina, San Diego, CA). The resulting data were analyzed using commercial software (SeqNext module of Sequence Pilot; JSI medical systems, Ettenheim, Germany). Reads with the same single‐molecule tag were assembled into one consensus read, to correct for PCR and sequencing artefacts. SCN1A pseudogene reads were removed from alignment and analysis. The used smMIP design included the 5’ promoter region to capture three common promoter‐variants (−1964 (rs2212657), −1036 (rs4319946) and −52 (rs16851666)). The promoter‐haplotypes of the unaffected SCN1A allele of each patient was reconstructed based on the genotypes on these positions when possible. Direct assignment of genotypes to the unaffected allele was only possible in the case of homozygous genotypes, when the same genotype is present on both alleles. In the case of heterozygous genotypes, assignment of genotypes to the affected and unaffected alleles was only possible if the following condition was met: the participant had an affected family member with a homozygous genotype at the same position, with whom they shared the same inherited pathogenic SCN1A variant. If so, the genotype present on the shared, affected allele is known and the genotype of the unaffected allele can be deducted. When the genotypes of the unaffected allele on all three positions could be reconstructed, one of the five described haplotypes could be matched and assigned. The frequency of the SCN1A promoter haplotypes in the non‐Finnish European population, which best resembles our cohort, was estimated by extracting these haplotypes from the 1,000 genomes Phase3 phased haplotype dataset (https://mathgen.stats.ox.ac.uk/impute/1000GP_Phase3.html), based on the −1964, −1036 and −52 SNPs.
Association of promoter variants with common epilepsies
A recent genome‐wide association study (GWAS) of the epilepsies identified a strong association with SNPs in SCN1A (The International League Against Epilepsy Consortium on Complex Epilepsies, 2018). We analyzed the association of the three common promoter variants with epilepsy using data from the latest epilepsy GWAS. We tested for independent associations of our promoter variants with epilepsy by performing a linear regression on each variant while conditioning on the most significant SCN1A‐SNP (rs6432877) from the GWAS. Conversely, we then tested to see if the GWAS association with SCN1A could be explained by our promoter SNPs by conditioning in the opposite direction.
Statistical analyses of clinical outcomes
Differences in clinical features and outcomes between Dravet syndrome patients with and without common variants in the SCN1A promoter‐region on their unaffected allele were investigated. Ordinal regression, corrected for age, was used to investigate cognitive outcome scores; the Mann‐Whitney U test was used to investigate age at seizure onset, age at first notice of developmental delay, age at first afebrile seizure and interpolated IQ/DQ scores after 5 years of disease. A similar analysis was performed for Dravet syndrome patients with nonmosaic truncating pathogenic variants only, to limit the influence of different pathogenic SCN1A variants themselves on the results. All reported tests were performed two‐tailed with an alpha‐level for significance of p < 0.05. We furthermore separately investigated whether family members, that carry the same pathogenic SCN1A variant but show varying disease severities, may have different promoter‐haplotypes that could explain their different outcomes.
3. RESULTS
3.1. Functional characterization of common SCN1A promoter variants
Five different SCN1A promoter‐haplotypes were defined, based on eight SNPs in the −2,271 to 297 region (Figure 2). Haplotype 1 lacks all eight SNPs and was therefore regarded wild‐type, from which the relative SCN1A expression levels were estimated for haplotypes 2–5. All non‐wildtype haplotypes (2–5) showed a significant reduction in luciferase expression, compared to the wildtype promoter‐region (Figure 2). The relative SCN1A expression decreased for 73% by haplotype 2, 64% by haplotype 3, 75% by haplotype 4, or haplotype 5).
Figure 2.

Functional effect of common promoter variants in SCN1A. Top: Simplified SCN1A 5’ UTR, adapted from Figure 1. Middle: Promoter haplotypes tested in this study. Haplotype 1 depicts a promoter + h1u region without common variants. Haplotype 2, 3, 4 and 5 carry multiple common variants spread over the promoter region: −1964 (rs2212657, MAF 0.43), −1663 (rs151217464, MAF 0.01), −1449 (rs7606233, MAF 0.50), −1036 (rs4319946, MAF 0.49), −688 (rs16851669, MAF 0.50), −244 (rs80169419, MAF 0.07), −52 (rs16851666, MAF 0.50) and 34 (rs757291646, MAF < 0.01). Bottom: Luciferase expression analysis of SCN1A promoter haplotypes as depicted above. Empty vector (SV40) expression was set to 100%. Haplotype 1, without common variants was used as control haplotype of which the expression reduction of Haplotype 2, 3, 4 and 5 was measured
3.2. Reconstruction of SCN1A promoter‐haplotypes of the unaffected allele in participants
SmMIP‐sequencing results were obtained for all participants. In all patients their known SCN1A pathogenic variant could be identified, except for variants undetectable by whole exome sequencing (e.g., deletions of the complete SCN1A gene), meaning no samples swaps had occurred. In 46 patients the promoter variant genotype of their unaffected SCN1A allele could be reconstructed. All other patients had heterozygous genotypes at at least one of the three promoter variant locations, and had no included family members who could be used for haplotype phasing.
3.3. Epilepsy‐GWAS associations of the common promoter variants
The strongest association with common epilepsy, which broadly comprises generalized and focal epilepsies, has been mapped to the SCN1A region in a recent GWAS of common epilepsy, with the most significantly associated SNP being rs6432877. The −52 and −1,036 promoter variants showed borderline genome wide significant with the “all epilepsy” phenotype, whereas the association of the −1964 variant was much weaker (p = 6,20E−8, 1,00E−7 and 0.68 respectively). In order to test whether the promoter variants were correlated to the SCN1A GWAS signal, we conditioned on the top GWAS SCN1A‐SNP (rs6432877) and observed that the associations were no longer significant indicating that these promoter variants SNPs are in variable Linkage Disequilibrium with the top GWAS SNP (r 2 = 0.21, 0.63 and 0.12 respectively). Conversely, we also tested to see if the signal from the top GWAS SCN1A‐SNP could be explained by one of the promoter variants by conditioning on each in turn. The strength of the GWAS signal diminished marginally when conditioning on the −52 and −1036 variants but was not affected by conditioning on the −1964 variant (p cond = 3.99E−08, 1.16E−05, and 1.12E−13, respectively), indicating that the GWAS signal was not entirely dependent on the promoter variants.
3.4. Clinical outcomes
Forty of the 46 participants with reconstructed promoter‐haplotypes had been diagnosed with Dravet syndrome; the others had either GEFS+ syndrome or febrile seizures, and one participant had never experienced any seizures. Regarding the 40 Dravet syndrome patients: in nine patients a wildtype promoter‐region was detected (haplotype 1); none of the patients carried haplotype 2; haplotype 3 was identified in only one patient; haplotype 4 was present in 12, and haplotype 5 was found in 18 participants. The estimated population frequency of the fivehaplotypes, based on the 1000genomes Phase3 phased haplotype dataset, was 0.232 for the haplotype 1, 0 for haplotype 2, 0.0025 for haplotype 3, 0.3899 for haplotype 4, and 0.375 for haplotype 5, which roughly resembles the distribution of haplotypes in our patients. An overview of the clinical outcomes of the 40 Dravet syndrome patients is shown in Table 1. No statistically significant differences were seen between patients with and without the common promoter variants (Table 1, Figures 3, 4, 5, 6, 7). However, patients with a wildtype promoter‐haplotype on their unaffected SCN1A allele showed a nonsignificant trend for milder phenotypes, when compared to patients that carried a variant promoter haplotype: on average, seizure onsets occurred at an older age (6.1 vs. 5.1 months, p = 0.746), as did developmental delays (median 36–47 months vs. median 24–35 months, p = 0.265). Furthermore, cognitive capacities declined slower (IQ after 5 years of disease 73 vs. 65.9, p = 0.566). More favorable cognitive outcome scores were also observed, although this is likely to be at least partly due to the wildtype‐patients being younger than the other group. Similar outcomes were seen for Dravet syndrome patients with truncating variants only (Table 2): although this group consisted of only 19 patients, leading to a lower detection power, a similar nonsignificant trend for milder phenotypes was observed in patients with wildtype promoters.
Table 1.
Clinical outcomes of patients with different promoter‐haplotypes (all nonmosaic Dravet syndrome patients)
| Promoter‐haplotype unaffected allelea | 1 | 3 | 4 | 5 | Any variant (haplotype 3, 4 or 5) | p‐value (test)e |
|---|---|---|---|---|---|---|
| Number of patients | 9 | 1 | 12 | 18 | 31 | |
| Age (years, mean/median) | 12/7 | 14/14 | 14/13 | 16/14 | 15/13 | |
| Cognitive outcomeb (median) | 3 | 5 | 4 | 4 | 4 | 0.859 (Ordinal regression corrected for age) |
| Age at seizure onset (months, mean) | 6.1 (missing: 1) | 5.0 | 5.7 | 5.2 | 5.4 | 0.746 (MWU test) |
| Age at first notice of developmental delayc (median) | 3 (missing: 1) | 1 | 2 (missing: 1) | 2 | 2 (missing: 1) | 0.265 (MWU test) |
| Age at first afebrile seizured (median) | 0 (missing: 1) | 0 | 0 (missing: 1) | 0 (missing: 2) | 0 (missing: 3) | 0.837 (MWU test) |
| Interpolated IQ/DQ score after 5 years of disease (mean) | 73.0 (missing: 3) | 33.0 | 64.5 (missing: 5) | 68.9 (missing: 4) | 65.9 (missing: 9) | 0.566 (MWU test) |
1: Wildtype (no variants). 2: variant at −52. 3: variant at −1964. 4: variant at −52 and −1964. 5: variant at −1964 and −1036.
Based on available data on IQ and developmental level, adjusted for age at assessment (1 = no ID (IQ or developmental quotient (DQ) >85), 2 = borderline ID (IQ or DQ 70–85), 3 = mild ID (IQ or DQ 50–70), 4 = moderate ID (IQ or DQ 30–50), 5 = severe or profound ID (IQ or DQ <30)). When no (recent) IQ or DQ was available, the assessment was made based on school functioning, communication and adaptive behavior.
By parents or physicians. 0 = <12 months, 1 = 12–23 months, 2 = 24–35 months, 3 = 36–47 months, 4 = >48 months, 5 = no developmental delay.
0 = <12 months, 1 = 12–23 months, 2 = 24–47 months, 3 = >48 months, 4 = never had afebrile seizures.
p‐values are based on statistical analyses of differences between group 1 (wiltype) and all other haplotypes combined (any variant). All reported tests were performed two‐tailed with an alpha‐level for significance of p < 0.05. MWU‐test = Mann Whitney U‐test.
Figure 3.
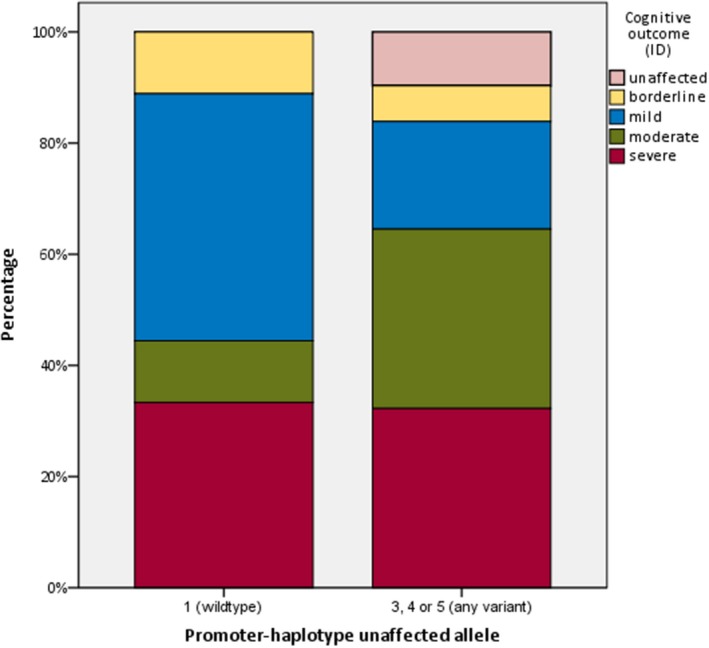
distribution of different cognitive outcome scores between patients with and without variants in the promoter‐region of their unaffected SCN1A allele
Figure 4.
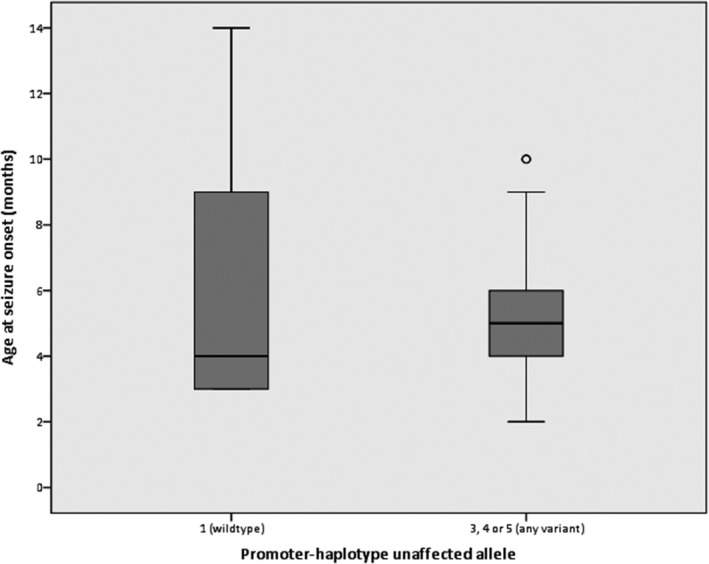
distribution of age at seizure onset between patients with and without variants in the promoter‐region of their unaffected SCN1A allele
Figure 5.
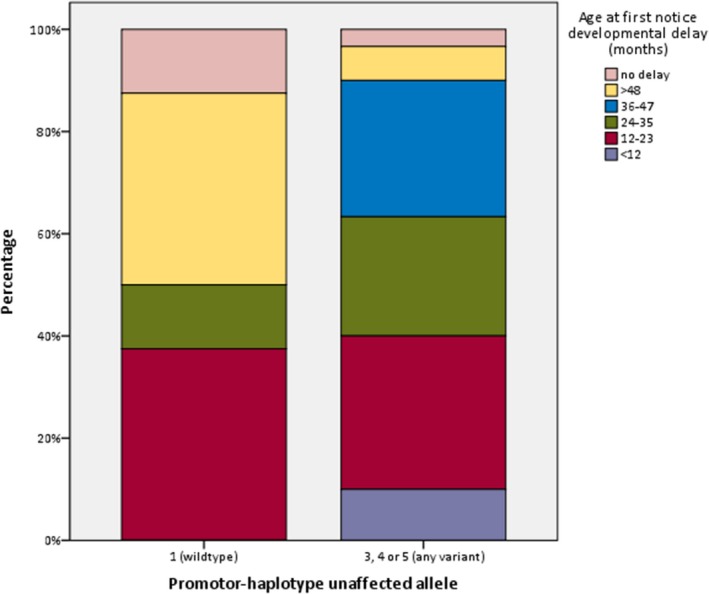
distribution of onset of developmental delay between patients with and without variants in the promoter‐region of their unaffected SCN1A allele
Figure 6.
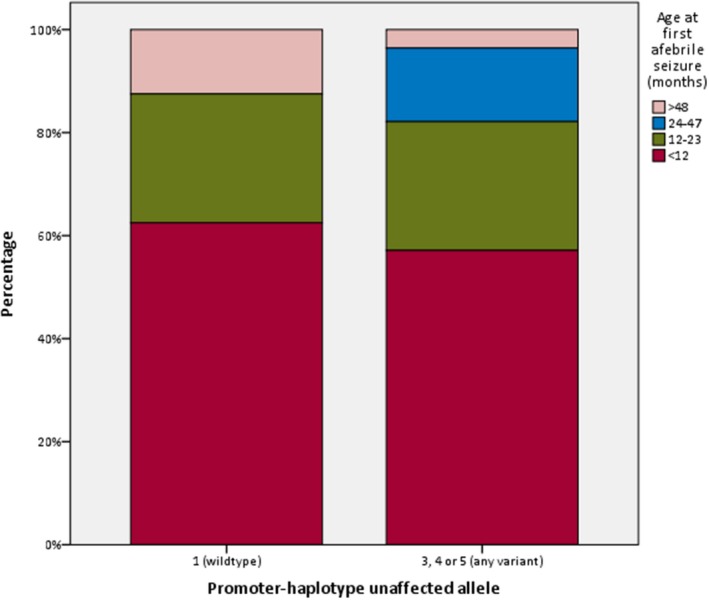
distribution of age at first afebrile seizure between patients with and without variants in the promoter‐region of their unaffected SCN1A allele
Figure 7.
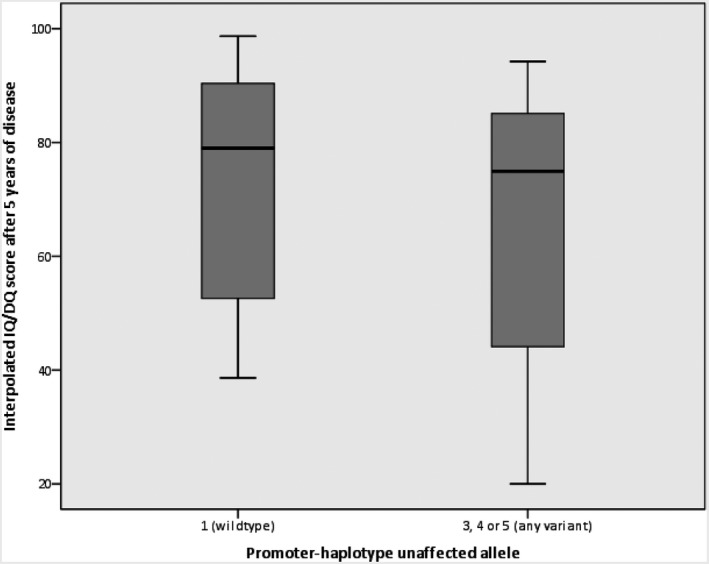
distribution of IQ/DQ scores after five years of disease between patients with and without variants in the promoter‐region of their unaffected SCN1A allele
Table 2.
Clinical outcomes of patients with different promoter‐haplotypes (non‐mosaic Dravet syndrome patients with truncating SCN1A variants)
| Promoter‐haplotype unaffected allelea | 1 | 3 | 4 | 5 | Any variant (haplotype 3, 4 or 5) | p‐value (test)e |
|---|---|---|---|---|---|---|
| Number of patients | 5 | 1 | 5 | 8 | 14 | |
| Age (years, mean/median) | 14/7 | 14/14 | 11/13 | 22/24 | 17.3/14.5 | |
| Cognitive outcomeb (median) | 3 | 5 | 4 | 4.5 | 4 | 0.547 (Multiple regression corrected for age) |
| Age at seizure onset (months, mean) | 6.6 | 5.0 | 6.0 | 5.25 | 5.5 | 0.823 (MWU test) |
| Age at first notice of developmental delayc (median) | 4 | 1 | 2 | 2.5 | 2 | 0.298 (MWU test) |
| Age at first afebrile seizured (median) | 0 | 0 | 0 | 0 (missing: 2) | 0 (missing: 2) | 0.712 (MWU test) |
| Interpolated IQ/DQ score after 5 years of disease (mean) | 75.1 (missing: 2) | 33.0 | 65.7 (missing: 3) | 74.9 (missing: 1) | 68.9 (missing: 4) | 0.973 (MWU test) |
1: Wildtype (no variants). 2: variant at −52. 3: variant at −1964. 4: variant at −52 and −1964. 5: variant at −1964 and −1036.
Based on available data on IQ and developmental level, adjusted for age at assessment (1 = no ID (IQ or developmental quotient (DQ) >85), 2 = borderline ID (IQ or DQ 70–85), 3 = mild ID (IQ or DQ 50–70), 4 = moderate ID (IQ or DQ 30–50), 5 = severe or profound ID (IQ or DQ <30). When no (recent) IQ or DQ was available, the assessment was made based on school functioning, communication, and adaptive behavior.
By parents or physicians. 0 = <12 months, 1 = 12–23 months, 2 = 24–35 months, 3 = 36–47 months, 4 = >48 months, 5 = no developmental delay.
0 = <12 months, 1 = 12–23 months, 2 = 24–47 months, 3 = >48 months, 4 = never had afebrile seizures.
p‐values are based on statistical analyses of differences between group 1 (wiltype) and all other haplotypes combined (any variant). All reported tests were performed two‐tailed with an alpha‐level for significance of p < 0.05. MWU‐test = Mann Whitney U‐test.
3.5. Anecdotal family studies
Among the complete group of 46 participants with reconstructed promoter‐haplotypes were eight participants, belonging to four different families that showed a clear intra‐familial variability (Figure 8): family 1 consists of a severely affected 10‐year‐old proband with Dravet syndrome, and a father with mild epilepsy and normal cognitive functioning. Family 2 consists of two brothers with Dravet syndrome, one of whom is more severely affected than the other. Family 3 consists of a proband with a phenotype on the border of Dravet syndrome and GEFS+, with regression over the years. His father has never had any seizures. Family 4 consists of two brothers of whom the oldest has severe Dravet syndrome and the youngest has a much milder phenotype. In family 2, 3 and 4, each of the milder participants carried haplotype 5, and each of the more severely affected participants carried haplotype 4. Since only very small, insignificant differences in luciferase expression between haplotype 4 and 5 were observed, the different promoter‐haplotypes are unlikely to explain the clinical differences between these patients. However, in family 1, the severely affected patient carried haplotype 5, whereas the milder patient had a wildtype promoter, for which we did observe a large difference in luciferase expression.
Figure 8.
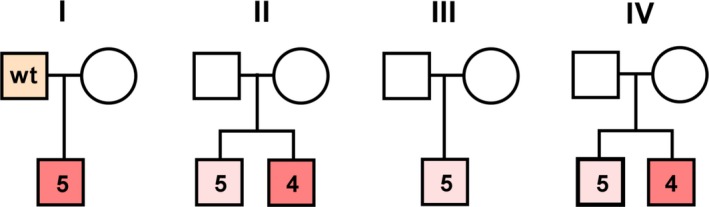
Family tree I‐IV. Orange box indicates mild epilepsy phenotype with normal cognitive functioning. Light red box indicates a mild DS, or borderline DS/GEFS+ phenotype. Dark red boxes indicate severe DS. Numbers in the boxes correspond to promoter haplotype number or wildtype haplotype from Figure 2
4. DISCUSSION
Our experiments showed that the presence of common variants in the promoter‐region of SCN1A cause a significant decrease in luciferase activity, compared to the wildtype promoter. This indicates that SCN1A expression and function may be negatively influenced by such variants, likely due to disturbance of RNA polymerase II and/or transcription factor binding. Although this reduced expression cannot cause epilepsy independently, since a large part of the healthy populations carries these common variants as well, it may modulate the effect of other variants that are present. Our results can only in part be compared to those of Gao et al. (Huang et al., 2014), who found no differences in expression between the most common promoter‐haplotypes. These different results may be attributed to three factors. First, we are measuring a different group of variants, which results in different expression levels. Second, we have cloned a slightly altered promoter region that is shorter on the 5’ side, but extended on the 3’ side to include the complete h1u. H1u, the first 5’ UE contains transcription factor binding sites such as EBF and the Initiator element that is required to form the transcription complex. Third, we use the Promega dual‐assay luciferase plasmid, which has both the renilla and firefly luciferase gene incorporated. In single‐assay luciferase assays, using two plasmids, normalization of luciferase data could be less sensitive. In general, the luciferase reporter assay is currently the fastest tool to measure gene expression at the transcriptional level. Nevertheless, it should be noted that in vitro assays can never fully mimic an in vivo state, especially in complex structures such as the brain. While for this study a neuronal cell line was used to perform the expression studies, this can be improved by introducing the luciferase constructs in the brain of an animal model. In this way, the interactions between cell types in the brain are included, approaching the in vivo state more accurately. Also, SCN1A has at least three promoters which are consecutively active and five 5’ UE's currently known, adding up to the difficulty of interpreting SCN1A expression. Nevertheless, we found that a combination of common and rare variants in the SCN1A promoter 1region, reduced expression on transcription level. The reduced luciferase expression was in line with our hypothesis that a set of SCN1A variants may affect expression of the gene and thus lead to more severe phenotypes, when present on the unaffected SCN1A allele of a Dravet syndrome patient. However, the clinical consequences of these different haplotypes were less convincing: no statistically significant differences were seen between patients with and without the common promoter variants, although we did observe a minor trend of more severe outcomes on multiple clinical variables in patients with common promoter variants. There may be several reasons for this. First, it is likely that common variants in the promoter region only have small phenotypic effects, since they otherwise would have been subject to negative selection. This limited effect was also illustrated by Gao et al., (2017); although a pathogenic point mutation in the SCN1A promoter‐region led to an in vitro decrease of expression and mild epilepsy in a proband, the same variant was found in the asymptomatic mother of the patient. This indicates that promoter‐variants by themselves may only have a limited influence on phenotypes. To detect such small effects, large sample sizes are prerequisite. Our study sample is likely too small to reliably detect any phenotypic consequences. Second, other (stronger) modifiers may simultaneously modulate the effect of promoter‐variants. Although we excluded patients with mosaic pathogenic variants, we cannot eliminate the influence of variants in modifier genes and environmental factors on outcomes. If these other factors are strong influencers, they may override any effects the promoter‐variants have.
Besides a small sample size, our study has several other limitations. While the luciferase plasmids were fully sequenced, the SCN1A patient promoter haplotypes were reconstructed based on three common SNPs. No sequencing of the complete promoter‐region was performed in the participants. The patients’ haplotypes may therefore not fully correspond to the haplotypes tested during the luciferase experiments. Theoretically, patients may harbor additional promoter‐variants that could either rescue or aggravate impaired expression. This could have large effects on outcomes in a sample size as small as ours. Furthermore, different primary pathogenic SCN1A variants may influence outcomes; however, a trend for milder phenotypes in patients with wildtype promoters was seen for the group of patients with truncating mutations only as well, which indicates that this effect is limited.
We also analyzed four families of which multiple members were affected by the same pathogenic SCN1A variant, but showed different phenotypes nonetheless; in these cases the effect of the primary mutation on the resulting phenotype is expected to be equal. Since in three of the four families both members had variant‐haplotypes, our hypothesis could not explain their phenotypic differences. This is however not surprising, since in two of these families both members were affected by different clinical syndromes; as stated before, the modifying effect of promoter‐variants is likely not strong enough to cause this independently. In only one family, consisting of two brothers with Dravet syndrome, the milder brother carried a wildtype promoter on his unaffected allele, whereas the more severe brother carried a variant‐promoter. According to our hypothesis, this might explain their phenotypic differences; however, as mentioned previously, we cannot exclude other influencers and definitive conclusions are not possible based on only one family.
In conclusion, we found that common variants in the SCN1A promoter reduce transcription in neuronal cell culture, which may indicate that promoter haplotypes can act as a disease modifier in epilepsy. We however only found a small, nonsignificant effect of the SCN1A promoter on clinical outcomes of Dravet syndrome patients. These results are inconclusive due to a limited detection power; however, the observed trends in our cohort warrant replication studies in larger cohorts to explore the potential modifying role of these common SCN1A promoter‐haplotypes. The inclusion of large numbers of Dravet syndrome patients, ideally all with similar primary LoF variants, is essential to detect the likely small effect these haplotypes might have on phenotypes. Sequencing of all three complete SCN1A promoter‐regions, preferably including the 5’‐UEs, would be required to obtain conclusive results.
CONFLICT OF INTEREST
None declared.
Supporting information
ACKNOWLEDGMENTS
We thank Dr. Vamshidhar R. Vangoor for leading the luciferase read‐out experiments. This study was supported by the “Stichting Vrienden WKZ” (project 1614054) on behalf of Stichting Panta Rhei, and the Dutch Epilepsy Foundation (project 2017‐01). MMC has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska‐Curie grant agreement No 751761.
de Lange IM, Weuring W, van ‘t Slot R, et al. Influence of common SCN1A promoter variants on the severity of SCN1A‐related phenotypes. Mol Genet Genomic Med. 2019;7:e727 10.1002/mgg3.727
Contributor Information
Iris M. de Lange, Email: i.m.delange-2@umcutrecht.nl.
Wout Weuring, Email: w.j.weuring-2@umcutrecht.nl.
REFERENCES
- Akiyama, M. , Kobayashi, K. , Yoshinaga, H. , & Ohtsuka, Y. (2010). A long‐term follow‐up study of Dravet syndrome up to adulthood. Epilepsia, 51(6), 1043–1052. 10.1111/j.1528-1167.2009.02466.x [DOI] [PubMed] [Google Scholar]
- Anney, R. J. L. , Avbersek, A. , Balding, D. , Baum, L. , Becker, F. , Berkovic, S. F. , … Zimprich, F. (2014). Genetic determinants of common epilepsies: A meta‐analysis of genome‐wide association studies. The Lancet Neurology, 13(9), 893–903. 10.1016/S1474-4422(14)70171-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunklaus, A. , Ellis, R. , Reavey, E. , Forbes, G. , & Zuberi, S. (2012). Prognostic, clinical and demographic features in SCN1A mutation‐positive Dravet syndrome. Brain, 135(8), 2329–2336. 10.1093/brain/aws151 [DOI] [PubMed] [Google Scholar]
- Catterall, W. A. , Kalume, F. , & Oakley, J. C. (2010). NaV1.1 channels and epilepsy. The Journal of Physiology, 588(11), 1849–1859. 10.1113/jphysiol.2010.187484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceulemans, B. (2011). Overall management of patients with Dravet syndrome. Developmental Medicine and Child Neurology, 53(Suppl. 2), 19–23. 10.1111/j.1469-8749.2011.03968.x [DOI] [PubMed] [Google Scholar]
- de Lange, I. M. , Gunning, B. , Sonsma, A. C. M. , van Gemert, L. , van Kempen, M. , Verbeek, N. E. , … Brilstra, E. H. (2018). Influence of contraindicated medication use on cognitive outcome in Dravet syndrome and age at first afebrile seizure as a clinical predictor in SCN1A‐related seizure phenotypes. Epilepsia, 59(6), 1154–1165. 10.1111/epi.14191 [DOI] [PubMed] [Google Scholar]
- de Lange, I. M. , Koudijs, M. J. , van ’t Slot, R. , Gunning, B. , Sonsma, A. C. M. , van Gemert, L. J. J. M. , … Koeleman, B. P. C. (2018). Mosaicism of de novo pathogenic SCN1A variants in epilepsy is a frequent phenomenon that correlates with variable phenotypes. Epilepsia, 59, 690–703. 10.1111/epi.14021 [DOI] [PubMed] [Google Scholar]
- Depienne, C. , Trouillard, O. , Gourfinkel‐An, I. , Saint‐Martin, C. , Bouteiller, D. , Graber, D. , … LeGuern, E. (2010). Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. Journal of Medical Genetics, 47(6), 404–410. 10.1136/jmg.2009.074328 [DOI] [PubMed] [Google Scholar]
- Djémié, T. , Weckhuysen, S. , von Spiczak, S. , Carvill, G. L. , Jaehn, J. , Anttonen, A. K. , … Suls, A. (2016). Pitfalls in genetic testing: The story of missed SCN1A mutations. Molecular Genetics & Genomic Medicine, 4(4), 457–464. 10.1002/mgg3.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, Z. F. , Tang, L. J. , Deng, G. F. , Zeng, T. , Liu, S. J. , Wan, R. P. , … Long, Y. S. (2014). Transcription of the human sodium channel SCN1A gene is repressed by a scaffolding protein RACK1. Molecular Neurobiology, 50(2), 438–448. 10.1007/s12035-014-8633-9 [DOI] [PubMed] [Google Scholar]
- Dravet, C. (1978). Les epilepsies graves de l’enfant. Vie Med, 8(2), 543–548. [Google Scholar]
- Dravet, C. (2011). The core Dravet syndrome phenotype. Epilepsia, 52(suppl. 2), 3–9. 10.1111/j.1528-1167.2011.02994.x [DOI] [PubMed] [Google Scholar]
- Escayg, A. , & Goldin, A. (2010). Sodium channel SCN1A and epilepsy: Mutations and mechanisms. Epilepsia, 51(9), 1650–1658. 10.1111/j.1528-1167.2010.02640.x.Sodium [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escayg, A. , MacDonald, B. T. , Meisler, M. H. , Baulac, S. , Huberfeld, G. , An‐Gourfinkel, I. , … Malafosse, A. (2000). Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nature Genetics, 24, 343–345. 10.1038/nature4441023a [DOI] [PubMed] [Google Scholar]
- Gao, Q. W. , Hua, L. D. , Wang, J. , Fan, C. X. , Deng, W. Y. , Li, B. , … Shi, Y. W. (2017). A point mutation in SCN1A 5′ genomic region decreases the promoter activity and is associated with mild epilepsy and seizure aggravation induced by antiepileptic drug. Molecular Neurobiology, 54(4), 2428–2434. 10.1007/s12035-016-9800-y [DOI] [PubMed] [Google Scholar]
- Gennaro, E. , Santorelli, F. M. , Bertini, E. , Buti, D. , Gaggero, R. , Gobbi, G. , … Zara, F. (2006). Somatic and germline mosaicisms in Severe Myoclonic Epilepsy of Infancy. Biochemical and Biophysical Research Communications, 341(2), 489–493. 10.1016/j.bbrc.2005.12.209 [DOI] [PubMed] [Google Scholar]
- Gitiaux, C. , Chemaly, N. , Quijano‐Roy, S. , Barnerias, C. , Desguerre, I. , Hully, M. , … Nabbout, R. (2016). Motor neuropathy contributes to crouching in patients with Dravet syndrome. Neurology, 87(3), 277–281. 10.1212/WNL.0000000000002859 [DOI] [PubMed] [Google Scholar]
- Guerrini, R. , Cellini, E. , Mei, D. , Metitieri, T. , Petrelli, C. , Pucatti, D. , … Zamponi, N. (2010). Variable epilepsy phenotypes associated with a familial intragenic deletion of the SCN1A gene. Epilepsia, 51(12), 2474–2477. 10.1111/j.1528-1167.2010.02790.x [DOI] [PubMed] [Google Scholar]
- Guerrini, R. , Dravet, C. , Genton, P. , Belmonte, A. , Kaminska, A. , & Dulac, O. (1998). Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia, 39(5), 508–512. 10.1111/j.1528-1157.1998.tb01413.x [DOI] [PubMed] [Google Scholar]
- Harkin, L. A. , McMahon, J. M. , Iona, X. , Dibbens, L. , Pelekanos, J. T. , Zuberi, S. M. , … Wirrell, E. (2007). The spectrum of SCN1A‐related infantile epileptic encephalopathies. Brain, 130(3), 843–852. 10.1093/brain/awm002 [DOI] [PubMed] [Google Scholar]
- Huang, A. Y. , Xu, X. , Ye, A. Y. , Wu, Q. , Yan, L. , Zhao, B. , … Wei, L. (2014). Postzygotic single‐nucleotide mosaicisms in whole‐genome sequences of clinically unremarkable individuals. Cell Research, 24(11), 1311–1327. 10.1038/cr.2014.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, F. , Sadleir, L. , Harkin, L. , Vadlamudi, L. , McMahon, J. , Mulley, J. , … Berkovic, S. (2006). Severe myoclonic epilepsy of infancy (Dravet syndrome): Recognition and diagnosis in adults. Neurology, 67(12), 2224–2226. [DOI] [PubMed] [Google Scholar]
- Kasperavičiute, D. , Catarino, C. B. , Matarin, M. , Leu, C. , Novy, J. , Tostevin, A. , … Sisodiya, S. M. (2013). Epilepsy, hippocampal sclerosis and febrile seizures linked by common genetic variation around SCN1A. Brain, 136(10), 3140–3150. 10.1093/brain/awt233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, Y. S. , Zhao, Q. H. , Su, T. , Cai, Y. L. , Zeng, Y. , Shi, Y. W. , … Liao, W. P. (2008). Identification of the promoter region and the 5’‐untranslated exons of the human voltage‐gated sodium channel Nav1.1 gene (SCN1A) and enhancement of gene expression by the 5’‐untranslated exons. Journal of Neuroscience Research, 86(15), 3375–3381. 10.1002/jnr.21790 [DOI] [PubMed] [Google Scholar]
- Mahoney, K. , Moore, S. J. , Buckley, D. , Alam, M. , Parfrey, P. , Penney, S. , … Young, T. L. (2009). Variable neurologic phenotype in a GEFS+ family with a novel mutation in SCN1A. Seizure, 18(7), 492–497. 10.1016/j.seizure.2009.04.009 [DOI] [PubMed] [Google Scholar]
- Marini, C. , Mei, D. , Helen Cross, J. , & Guerrini, R. (2006). Mosaic SCN1A mutation in familial severe myoclonic epilepsy of infancy. Epilepsia, 47(10), 1737–1740. 10.1111/j.1528-1167.2006.00675.x [DOI] [PubMed] [Google Scholar]
- Martin, M. S. , Tang, B. , Ta, N. , & Escayg, A. (2007). Characterization of 5′ untranslated regions of the voltage‐gated sodium channels SCN1A, SCN2A, and SCN3A and identification of cis‐conserved noncoding sequences. Genomics, 10.1016/j.ygeno.2007.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, H. , Xu, H. Q. , Yu, L. , Lin, G. W. , He, N. , Su, T. , … Liao, W. P. (2015). The SCN1A mutation database: Updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Human Mutation, 36(6), 573–580. 10.1002/humu.22782 [DOI] [PubMed] [Google Scholar]
- Nakayama, T. , Ogiwara, I. , Ito, K. , Kaneda, M. , Mazaki, E. , Osaka, H. , … Yamakawa, K. (2010). Deletions of SCN1A 5′ genomic region with promoter activity in Dravet syndrome. Human Mutation, 31(7), 820–829. 10.1002/humu.21275 [DOI] [PubMed] [Google Scholar]
- Parihar, R. , & Ganesh, S. (2013). The SCN1A gene variants and epileptic encephalopathies. Journal of Human Genetics, 58(9), 573–580. 10.1038/jhg.2013.77 [DOI] [PubMed] [Google Scholar]
- Passamonti, C. , Petrelli, C. , Mei, D. , Foschi, N. , Guerrini, R. , Provinciali, L. , & Zamponi, N. (2015). A novel inherited SCN1A mutation associated with different neuropsychological phenotypes: Is there a common core deficit? Epilepsy and Behavior, 43, 89–92. 10.1016/j.yebeh.2014.11.009 [DOI] [PubMed] [Google Scholar]
- Pineda‐Trujillo, N. , Carrizosa, J. , Cornejo, W. , Arias, W. , Franco, C. , Cabrera, D. , … Ruíz‐Linares, A. (2005). A novel SCN1A mutation associated with severe GEFS+ in a large South American pedigree. Seizure, 14(2), 123–128. 10.1016/j.seizure.2004.12.007 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Azi, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30.Standards [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rilstone, J. J. , Coelho, F. M. , Minassian, B. A. , & Andrade, D. M. (2012). Dravet syndrome: Seizure control and gait in adults with different SCN1A mutations. Epilepsia, 53(8), 1421–1428. 10.1111/j.1528-1167.2012.03583.x [DOI] [PubMed] [Google Scholar]
- Suls, A. , Velizarova, R. , Yordanova, I. , Deprez, L. , Van Dyck, T. , Wauters, J. , … De Jonghe, P. (2010). Four generations of epilepsy caused by an inherited microdeletion of the SCN1A gene. Neurology, 75(1), 72–76. 10.1212/WNL.0b013e3181e62088 [DOI] [PubMed] [Google Scholar]
- The International League Against Epilepsy Consortium on Complex Epilepsies (2018). Genome‐wide mega‐analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nature Communications, 9(1), 5269 10.1038/s41467-018-07524-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek, N. E. , Van Der Maas, N. A. T. , Jansen, F. E. , Van Kempen, M. J. A. , Lindhout, D. , & Brilstra, E. H. (2013). Prevalence of SCN1A‐related Dravet syndrome among children reported with seizures following vaccination: A population‐based ten‐year cohort study. PLoS ONE, 8(6), e65758 10.1371/journal.pone.0065758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirrell, E. C. , Laux, L. , Donner, E. , Jette, N. , Knupp, K. , Meskis, M. A. , … Berg, A. T. (2017). Optimizing the diagnosis and management of Dravet syndrome: Recommendations from a North American consensus panel. Pediatric Neurology, 68, 18–34.e3. 10.1016/j.pediatrneurol.2017.01.025 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials