

Abstract
Background
The CRISPR/Cas9 system has opened new perspectives to study the molecular basis of cerebral cavernous malformations (CCMs) in personalized disease models. However, precise genome editing in endothelial and other hard‐to‐transfect cells remains challenging.
Methods
In a proof‐of‐principle study, we first isolated blood outgrowth endothelial cells (BOECs) from a CCM1 mutation carrier with multiple CCMs. In a CRISPR/Cas9 gene correction approach, a high‐fidelity Cas9 variant was then transfected into patient‐derived BOECs using a ribonucleoprotein complex and a single‐strand DNA oligonucleotide. In addition, patient‐specific CCM1 knockout clones were expanded after CRISPR/Cas9 gene inactivation.
Results
Deep sequencing demonstrated correction of the mutant allele in nearly 33% of all cells whereas no CRISPR/Cas9‐induced mutations in predicted off‐target loci were identified. Corrected BOECs could be cultured in cell mixtures but demonstrated impaired clonal survival. In contrast, CCM1‐deficient BOECs displayed increased resistance to stress‐induced apoptotic cell death and could be clonally expanded to high passages. When cultured together, CCM1‐deficient BOECs largely replaced corrected as well as heterozygous BOECs.
Conclusion
We here demonstrate that a non‐viral CRISPR/Cas9 approach can not only be used for gene knockout but also for precise gene correction in hard‐to‐transfect endothelial cells (ECs). Comparing patient‐derived isogenic CCM1+/+, CCM1+/−, and CCM1−/− ECs, we show that the inactivation of the second allele results in clonal evolution of ECs lacking CCM1 which likely reflects the initiation phase of CCM genesis.
Keywords: blood outgrowth endothelial cells, CCM1, cerebral cavernous malformation, CRISPR/Cas9, mutation correction
1. INTRODUCTION
Cerebral cavernous malformations (CCM) are angiographically occult clusters of enlarged and tightly packed blood vessels of the venous‐capillary bed. Due to disorganized tight and adherens junctions between the lining endothelial cells (ECs), CCMs tend to leak into the neighboring brain tissue. Such intracranial hemorrhages from CCMs can lead to focal neurological deficits, epileptic seizures, and recurrent headaches (Draheim, Fisher, Boggon, & Calderwood, 2014; Spiegler, Rath, Paperlein, & Felbor, 2018). A hereditary etiology has first been assumed by H. Kufs in 1928 who described CCMs in a father–daughter duo (Kufs, 1928). Several decades later, three CCM disease genes have been identified: CCM1 (KRIT1; OMIM: *604214) (Laberge‐le Couteulx et al., 1999; Sahoo et al., 1999), CCM2 (Malcavernin; OSM; *607929) (Denier et al., 2004; Liquori et al., 2003), and CCM3 (PDCD10, TFAR15; *609118) (Bergametti et al., 2005). The familial form (OMIM 116860, 603284, 603285) is inherited in an autosomal‐dominant manner and usually presents with multiple CCMs that are thought to occur after a second somatic mutation within CCM1, CCM2, or CCM3 in the endothelial compartment (Akers, Johnson, Steinberg, Zabramski, & Marchuk, 2009; Gault, Shenkar, Recksiek, & Awad, 2005; McDonald et al., 2014).
No targeted CCM therapies are available yet and our understanding of the basic mechanisms that initiate CCM formation and growth is still incomplete. Recent in vitro and in vivo studies suggested that clonal expansion of CCM3‐deficient ECs might be a key feature of CCM3 biology (Detter, Snellings, & Marchuk, 2018; Schwefel et al., 2019). However, this novel model of clonal CCM evolution has neither been directly validated for CCM1 nor in a patient‐specific context. Blood outgrowth endothelial cells (BOECs) are a perfect tool to answer this question in a personalized disease model. BOECs are fully differentiated, true ECs that can be established from peripheral or cord blood samples by culturing mononuclear cells in an endothelial‐supportive medium on collagen‐coated plates (Chong, Ng, & Chan, 2016; Hebbel, 2017; Lin, Weisdorf, Solovey, & Hebbel, 2000). BOECs originate from circulating endothelial colony‐forming cells (ECFCs), demonstrate enhanced proliferative properties, and high phenotypic stability (Hirschi, Ingram, & Yoder, 2008; Martin‐Ramirez, Hofman, Biggelaar, Hebbel, & Voorberg, 2012). Of note, CRISPR/Cas9‐mediated gene disruption in human ECs has first been demonstrated in outgrowth ECs derived from cord blood ECFCs. Abrahimi and co‐workers used lentiviral vectors for CRISPR/Cas9 delivery and efficiently inactivated the human CIITA gene which encodes for an MHC class II transactivator (Abrahimi et al., 2015).
Using a non‐viral and plasmid‐free CRISPR/Cas9 approach, we here demonstrate that not only CCM1 gene ablation but also precise gene correction is feasible in hard‐to‐transfect primary ECs. In a patient‐specific cell culture model of Knudson's two‐hit hypothesis, we show for the first time that it is the acquisition of a compound heterozygous second CCM1 mutation which leads to a clonogenic survival advantage.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
The patient participated in this study with written informed consent according to the German Gene Diagnostic Act and approval of the local ethics committee (University Medicine Greifswald, Germany; No.: BB 047/14a).
2.2. Generation of blood outgrowth endothelial cells
Blood outgrowth ECs were established from 30 ml peripheral blood as described (Martin‐Ramirez et al., 2012). In brief, mononuclear cells were separated from whole blood using Ficoll‐Paque PLUS (GE Healthcare, Little Chalfont, UK) and maintained in EGM‐2 medium (Lonza, Basel, Switzerland) supplemented with 18% fetal calf serum (FCS, Thermo Fisher Scientific, Waltham, Massachusetts, USA) on collagen I‐coated plates (Ibidi, Martinsried, Germany).
2.3. In vitro studies
Immunofluorescent staining was performed with cells that had been fixed on 96‐well plates, permeabilized, and washed several times. The following antibodies were used: monoclonal mouse anti‐human CD31 (BBA7, R&D Systems, Minneapolis, MN), polyclonal rabbit anti‐SM22α (ab14106, Abcam, Cambridge, UK), monoclonal mouse anti‐human CD146 (MAB932, R&D Systems), polyclonal rabbit anti‐human CD34 (HPA036723, Sigma‐Aldrich, St. Louis, MO), polyclonal rabbit anti‐KLF4 (PA5‐27441, Thermo Fisher Scientific), secondary goat anti‐mouse IgG antibody, Alexa Fluor 488 (A‐11029, Thermo Fisher Scientific), and goat anti‐rabbit IgG antibody, Alexa Fluor 555 (A‐21429, Thermo Fisher Scientific). Cell nuclei were stained with DAPI (D9542, Sigma‐Aldrich) and samples were overlayed with mounting medium (50001, Ibidi) prior to image acquisition with an EVOS FL microscope (Thermo Fisher Scientific). F‐actin was stained with phalloidin (Abcam). Spheroid sprouting was performed as described (Spiegler et al., 2016).
2.4. CRISPR/Cas9‐mediated gene editing
For gene correction, the Alt‐R® CRISPR‐Cas9 crRNA 5'‐AUCUCCUCACAUGGAAACUA‐3' (Integrated DNA Technologies (IDT), Coralville, Iowa, US) targeting the patient‐specific variant c.2012delA located in exon 18 of the CCM1 gene (LRG_650t1) was determined using IDT's Custom design tool (https://eu.idtdna.com/site/order/designtool/index/CRISPR_CUSTOM). For cotransfection, a single‐strand oligodeoxynucleotide template (5'‐GGAGTGAATATAAAAGGACTTCATCTCCTCAATATGGAAACTAAGGTAGATTTTCAGCTCTTT‐3', Ultramer® DNA Oligo, IDT) containing a synonymous single nucleotide change was designed with Edit‐R HDR Donor Designer (http://dharmacon.horizondiscovery.com/gene-editing/crispr-cas9/edit-r-hdr-donor-designer-oligo/#). One micromolar duplexes of crRNA and tracrRNA (IDT) were complexed with 1 µM S.p. Cas9 HiFi protein (IDT) in Opti‐MEM I reduced serum medium (Thermo Fisher Scientific) to a final concentration of 60 nM. Reverse transfection of crRNA:tracrRNA:Cas9 ribonucleoprotein (RNP) complexes into BOECs was accomplished using Lipofectamine RNAiMAX (4,8 µl/well of a 24‐well plate; Thermo Fisher Scientific). The HDR template was added to a final concentration of 10 nM. To achieve a complete CCM1 knockout, the crRNA 5'‐UCUCCUCAACAUGGAAACUA‐3' (IDT) targeting the corresponding wild type allele of the patient's variant was transfected as described above.
2.5. Transfection readout
After cells reached approximately 90% confluency, DNA was harvested by resuspending the cells in QuickExtract solution (Epicentre, Madison, WI) and heating samples to 65°C and 95°C for 15 min each. A PCR for the CCM1 target site (forward primer 5'‐CAACCAGGTCAGCAAACTATAGCTTATAGCC‐3', reverse primer 5'‐TCTCCAACCCAGAAAAACGCTCTCACTAGAATC‐3') was performed with 5 ng genomic DNA using OneTaq DNA Polymerase (New England Biolabs, Ipswich, Massachusetts, USA). Purification of the amplicons with Agencourt® AMPure® XP beads (Beckman Coulter, Pasadena, USA) was followed by library preparation with Nextera XT Kit according to the manufacturer's protocol (Illumina®, San Diego, California, USA). Deep sequencing was performed with 2 x 150 cycles on a MiSeq instrument (Illumina®). The data were analyzed using SeqNext software (JSI Medical Systems, Ettenheim, Germany). Only variants with quality scores ≥30 and combined read frequencies ≥1% were called.
2.6. Subculturing of transfected BOECs
Transfected cell mixtures were subcultured until grown to confluence on a T25 flask. Clonal expansion was accomplished by seeding 0.5 cells/well of a 96‐well plate.
To analyze the proportion of cells with different genotypes in the cell mixtures over time, 10% of CCM1‐deficient cells harboring c.2012delA and c.2014_2021del alleles were added to the corrected cell mixture, grown, and passaged on 6‐well plates. After 16 and 34 days at roughly 90% confluency, the cells were splitted 1:2 with one half used for DNA isolation and amplicon deep sequencing as described above.
2.7. Potential off‐target analysis
Possible off‐target sites were predicted by the CCTop – CRISPR/Cas9 target online predictor and IDT's CRISPR‐Cas9 Design checker. The six most likely candidate sequences were selected based on the position of mismatches and their genomic location. Amplicons were generated from DNA of cell mixtures after transfection using the following primer combinations: RIMS2 (forward primer 5'‐GAGTTCACACATCCACCTCTG‐3', reverse primer 5'‐GGACAGATGTTTATTGAGCAGC‐3'), WDFY3 (forward primer 5'‐TGGCTGTTAGAGGAAGTGGA‐3', reverse primer 5'‐ACAAAGGTAGAGTTGCTGATGG‐3'), VASN (forward primer 5'‐CTCTGCAGAGTTTTCCCAGG‐3', reverse primer 5'‐AGAATGGTGGGACTTGGAGC‐3'), MAP4K4 (forward primer 5'‐GACTTCTCCAGGCATGTGAG‐3', reverse primer 5'‐CACCTGGAGAAAAGCAGTGA‐3'), RP11‐179A10.1 (forward primer 5'‐AGCTGAAAACTGCACCGATC‐3', reverse primer 5'‐GTCAGGGCACATCAGGGAT‐3'), and ATP13A4 (forward primer 5'‐CAGCCCGGATAAGGACTGTA‐3', reverse primer 5'‐AGACCCCATCAACTGAGGAT‐3'). Amplicons were either prepared for deep sequencing as described above or digested using T7 Endonuclease I (New England Biolabs) and analyzed on a Bioanalyzer instrument using Agilent's DNA 1000 kit (Agilent, Santa Clara, CA). For Sanger sequencing, DNA amplicons were purified with ExoSAP‐IT (Affymetrix, Santa Clara, CA), marked with BigDye Terminator Cycle Sequencing v3.1 kit (Thermo Fisher Scientific) and sequenced on an ABI 3130XL (Applied Biosystems, Foster City, CA).
2.8. Caspase‐3 activity
Fluorometric analysis of Caspase‐3 activity was performed using Caspase‐3 DEVD‐R110 Assay Kit (Biotium; Fremont, CA) according to the manufacturer's instructions after induction of apoptosis with 1 µM staurosporine for 2 hr.
3. RESULTS
3.1. Establishment and characterization of patient‐derived BOECs
The 31‐year‐old index case (III:2, Figure 1a) became symptomatic at the age of 27 with recurrent headaches and epileptic seizures. Two cavernous malformations were identified by magnetic resonance imaging (MRI) in his right and left frontal lobe. The larger one was resected after symptomatic bleeding. CCMs have also been detected in his symptomatic father (II:2), brother (III:1), and paternal aunt (II:1). Molecular genetic analyses of all three CCM genes revealed the heterozygous one base pair deletion c.2012delA in CCM1 (Figure 1b) which has not been described in the literature so far. The deletion leads to a frameshift and therefore disrupts part of the C‐terminal FERM domain [p.(Asn671Thrfs*36)] which is important for the interaction of CCM1 with binding partners like HEG1 and RAP1 (Figure 1c) (Fisher & Boggon, 2014; Spiegler et al., 2014). Apart from this novel CCM1 frameshift variant, another seven nonsense and 14 frameshift mutations within exon 18 of CCM1 have been reported previously in CCM families and are listed as disease‐causing in the human gene mutation database (HGMD Professional 2018.4). Consequently, the identified CCM1 variant was classified as pathogenic for CCM according to the ACMG guidelines for variant interpretation (Richards et al., 2015).
Figure 1.

Generation and characterization of patient‐derived BOECs. (a) Pedigree of the CCM index case (III:2; arrow). (b) Sequence of the heterozygous frameshift variant c.2012delA in the CCM1 gene of III:2. (c) Schematic domain structure for CCM1 and localization of the pathogenic c.2012delA; p.(Asn671Thrfs*36) mutation in its FERM domain. ANK = ankyrin repeat domain; FERM = band Four.1 Ezrin Radixin Moesin; NPxY/F = Asn‐Pro‐X‐Tyr/Phe motif. (d) Brightfield and (e) spheroid sprouting of patient‐derived BOECs. Strong expression of CD31/PECAM‐1 (green) and very few cells expressing SM22α (red, *)(f, g) as well as immunopositivity for CD146 (green, h, i) and CD34 (green, j, k) in BOECs established from a healthy donor (f, h, j) and the index patient III:2 (g, i, k) confirmed their endothelial phenotype. Scale bars indicate 100 µm (f, g) or 400 µm (d, h‐k)
BOECs were established from peripheral blood of the index proband within 21 days. Bright‐field microscopy demonstrated the typical cobblestone morphology (Figure 1d) and immunofluorescence imaging verified strong expression of the endothelial marker proteins CD31/PECAM‐1 (Figure 1g), CD146 (Figure 1i) and CD34 (Figure 1k) with no differences compared to BOECs from a healthy control (Figure 1f,h,j). In contrast, only very few cells were positively stained for the mesenchymal marker protein SM22α (Figure 1g). We also tested the ability of the patient‐derived cells to form endothelial sprouts which is a distinctive feature between hematopoietic and endothelial progenitor lineages (Medina et al., 2017) (Figure 1e).
BOECs from healthy controls and the germline mutation carrier could be passaged approximately 18 times consistent with previous reports (Groeneveld et al., 2015; Lin et al., 2000).
3.2. Efficient gene correction with CRISPR/Cas9
Using a crRNA:tracrRNA:Cas9 RNP approach, we have recently demonstrated that highly efficient gene disruption can be achieved in human ECs (Schwefel et al., 2019). However, nonviral CRISPR/Cas9‐mediated gene corrections that are even more challenging in hard‐to‐transfect primary ECs have not yet been reported. Thus, we used the patient‐derived BOECs in a proof‐of‐concept study for precise CCM1 gene correction. A crRNA:tracrRNA:Cas9 RNP complex that targeted the c.2012delA CCM1 allele was cotransfected with a single‐strand oligodeoxynucleotide (ssODN) as donor template into the patient‐derived BOECs (Figure 2a,b). A silent variant was introduced into the ssODN (c.2013C>T; p.Asn671=) to enable tracking of the homology‐directed repair (HDR) efficiency. Ten days after transfection, amplicon deep sequencing identified the precisely corrected CCM1 allele (c.[2012=;2013C>T]) with a read frequency of 5.1% in DNA isolated from the cell mixture (Figure 2c). Given that the BOECs had been heterozygous for the c.2012delA allele prior to CRISPR/Cas9 genome editing, HDR‐mediated gene correction occurred in 10% of all cells. Notably, a second CRISPR/Cas9‐induced CCM1 variant which restores the open reading frame was found in 17% of all reads which would correspond to 34% of all cells (Figure 2c). This variant, c.[2012delA;2020dupA], probably originated from a one base pair duplication within a stretch of three adenosines near the PAM site on the c.2012delA allele. Remarkably, an increase in the reference allele frequency (c.2012=) of 11.2% was observed suggesting nonhomologous end joining (NHEJ)‐driven correction without the usage of HDR template (Figure 2c) in about 22% of all cells. Consistent with the high efficiency of the CRISPR/Cas9‐mediated CCM1 gene correction, the read frequency of the c.2012delA allele was reduced to 7.2%. To demonstrate the specificity of our genome editing approach, six potential off‐target loci were screened for CRISPR/Cas9‐induced variants. These loci were selected from bioinformatic predictions based on the number and position of mismatches in the crRNA binding site. No off‐target mutations were identified by amplicon deep sequencing and T7 endonuclease cleavage assay (Figure 2d, Table 1). Of note, these results demonstrate for the first time that a CRISPR/Cas9 RNP approach can be used for efficient in vitro gene correction in human ECs.
Figure 2.
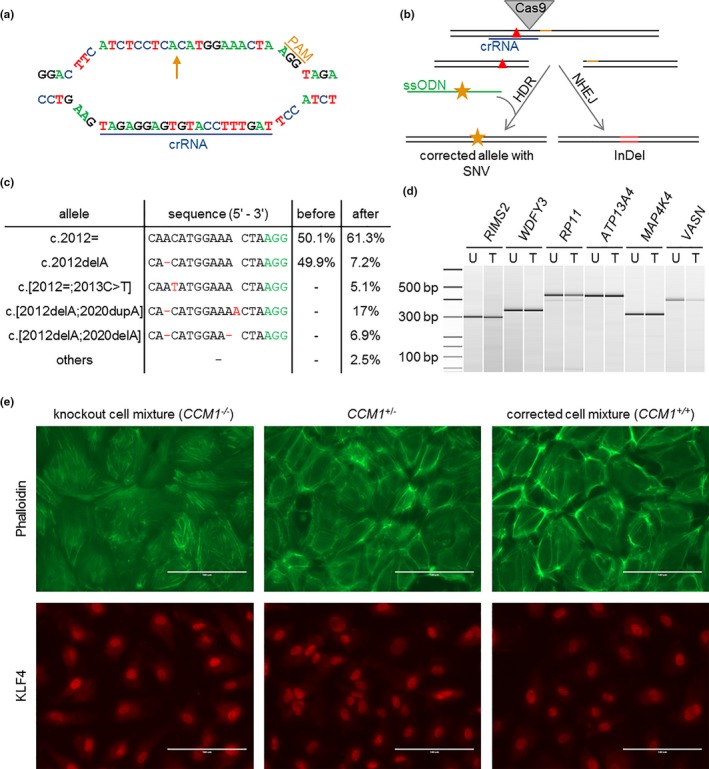
Correction of the patient‐specific pathogenic CCM1 variant and comparison of CCM1+/+, CCM1+/− and CCM1−/− BOECs by immunofluorescence. (a) crRNA and PAM sequence at the CCM1 allele c.2012delA (arrow). (b) Strategy of HDR‐mediated correction of a pathogenic CCM1 variant (red triangle) with CRISPR/Cas9 genome editing. ssODN = single‐strand oligodeoxynucleotide, orange star = silent SNV. (c) Amplicon deep sequencing results after CRISPR/Cas9 genome editing. The variant read frequencies before and after CRISPR/Cas9 transfection are given next to the sequence alignments. (d) T7 endonuclease I cleavage of PCR products from untreated (U) and treated (T) cells spanning possible off‐target sites. Ladder bands are given in base pairs (bp). (e) Characterization of the knockout cell mixture (left), the unmodified, patient‐derived BOECs (middle), and the corrected cell mixture (right). Visualization of the cytoskeleton using phalloidin reveals a regular F‐actin organization without the formation of stress fibers in the patients‐derived and corrected BOEC mixtures while CCM1‐/‐ cells demonstrated stress fiber bundles (upper panel). The strongest expression of the transcription factor KLF4 can be found in the knockout cell mixture when compared to unmodified and corrected BOECs (lower panel)
Table 1.
Selected possible off‐target sites of the crRNA for CCM1 c.2012delA. Mismatches (MM) are highlighted in red, the PAM sequence is marked in green
| Gene | Sequence | # MM | Position |
|---|---|---|---|
| RIMS2 |

|
3 | Exonic |
| WDFY3 |
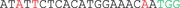
|
3 | Intronic |
| MAP4K4 |

|
4 | Intronic |
| RP11−179A10.1 |

|
4 | Exonic |
| ATP13A4 |

|
4 | Intronic |
| VASN |
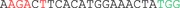
|
4 | Intronic |
3.3. Characterization of BOECs after CRISPR/Cas9‐mediated CCM1 gene correction and disruption
CCM1 inactivation induces profound endothelial dysfunction such as upregulation of the transcription factor KLF4 and increased actin stress fiber assembly (Cuttano et al., 2016; Glading, Han, Stockton, & Ginsberg, 2007; Zhou et al., 2015). To address these changes in our personalized disease model, we also used the CRISPR/Cas9 system to induce chronic CCM1 inactivation by targeting the CCM1 wild type allele in patient‐derived BOECs. Increased stress fiber formation was observed after complete CCM1 inactivation (CCM1−/−). In contrast, BOECs with the heterozygous germline variant (CCM1+/−) and also the crRNA:tracrRNA:Cas9:HDR‐treated cell mixture (CCM1+/+) demonstrated regular cortical actin assembly (Figure 2e, upper panel). As expected, the highest KLF4 expression was found in CCM1‐/‐ BOECs. When compared to CCM1+/− BOECs, we observed a slightly reduced KLF4 level in CCM1+/+ BOECs which might indicate a mild phenotype for CCM1+/− BOECs on a molecular level (Figure 2e, lower panel).
Twenty‐six clonal BOEC colonies were established by limiting dilution after CRISPR/Cas9‐mediated gene correction. Heterozygosity for the corrected CCM1 allele c.[2012=;2013C>T] was found in two colonies (Figure 3a). The heterozygous in‐frame variant c.[2012delA;2020dupA] (Figure 3b) was identified in 13 clones and two colonies demonstrated the reference sequence without the silent SNV on both CCM1 alleles. The remaining nine colonies harbored the pathogenic c.2012delA allele in heterozygous state (Figure 3c). This genotype ratio correlates well with the observed allele frequencies in our amplicon deep sequencing analysis of the cell mixtures and indicates that 65.4% of all cells (17/26 colonies) underwent targeted gene repair which would completely or largely recover protein function.
Figure 3.

CCM1‐/‐ mutant BOECs acquire clonal dominance in co‐culture. (a–d) Sanger sequencing analyses of DNA samples from clonally expanded BOEC colonies after CRISPR/Cas9 genome editing demonstrated (a) the HDR‐corrected sequence with the silent variant c.2013C>T, p.(Asn671=), (b) the in‐frame variant c.[2012delA;2020dupA], p.(Asn671_Glu673delinsThrTrpLys), (c) the patient‐specific one base pair deletion c.2012delA, p.(Asn671Thrfs*36) in heterozygous states and (d) the compound heterozygous knockout alleles c.[2012delA];[2014_2021del], p.[Asn671Thrfs*36];[Met672*]. Nucleotide calls and amino acid changes (single letter code) are given above the electropherograms. Predicted consequences of the nucleotide changes are depicted at the bottom of each subpanel. E = exon, AA = amino acid. (e) CCM1‐/‐ clones display significantly reduced caspase‐3 activity under induction of apoptosis with 1 µM staurosporine. Data are presented as mean and SEM. Two‐way ANOVA was used for statistical analysis: **p < 0.01. (f) Amplicon deep sequencing results for cell mixtures over time. The corrected cell mixture shows a constant allele ratio for 34 days of culture (left) whereas the addition of 10% compound heterozygous CCM1‐deficient cells (c.[2012delA];[2014_2021del] corresponds to c.[2014_2021del] in 5% of all alleles, red color) to the corrected cell mixture leads to a strong shift toward knockout alleles already after 16 days (right)
3.4. Survival advantage of patient‐derived BOECs after complete CCM1 inactivation
When we aimed to expand and characterize clonal CCM1−/− and CCM1+/+ BOECs, we observed dramatic differences between both conditions. While CCM1‐deficient clones proliferate well to very high passages (>30), corrected clones could not be further expanded after limiting dilution cloning. Since BOECs are primary cells with a limited life span, we hypothesized that CCM1−/− cells have a clonal survival advantage and show impaired induction of apoptotic cell death like human CCM3−/− ECs (Schwefel et al., 2019). Therefore, caspase‐3 activity was measured under basal culture conditions and staurosporine stress. Noteworthy, we observed a significant resistance of CCM1−/− BOECs to staurosporine‐induced apoptosis (Figure 3e).
Their clonal survival advantage was also evident when we added 10% CCM1−/− ECs (c.[2012delA];[2014_2021del], p.[Asn671Thrfs*36];[Met672*], Figure 3d) to the corrected BOEC mixture (90%). While the frequency of corrected CCM1 alleles was stable over various passages when no CCM1−/− ECs were added (Figure 3f, left columns), a significant shift was observed after addition of the CCM1−/− clone. The CCM1+/+ and CCM1+/− cells were overgrown by CCM1−/− BOECs and the corrected c.[2012=;2013C>T] allele was undetectable by amplicon deep sequencing already 16 days after starting the coculture experiment (Figure 3f, right columns).
4. DISCUSSION
Using patient‐derived endothelial progenitor cells (EPCs) in a personalized CCM disease model, we here demonstrate that a no‐nviral and plasmid‐free CRISPR/Cas9 approach allows not only efficient gene knockout but also precise CCM1 gene correction in human ECs. In accordance with Knudson's two‐hit hypothesis (Knudson, 1971), the introduction of a somatic CCM1 mutation into the second allele of a CCM1 mutation carrier resulted in a clear phenotype while heterozygosity for the germline variant alone did not. Within a short period of time, CCM1−/− EPCs dominated the cell culture (Figure 3f) which likely reflects the early phase of lesion genesis as recently visualized in an inducible Ccm3 mouse model (Detter et al., 2018). Consequently, our work suggests that the concept of clonal evolution seen after CCM3 inactivation in mice (Detter et al., 2018) and immortalized human umbilical vein ECs (Schwefel et al., 2019) also applies to CCM1.
The delivery of the CRISPR/Cas9 components into primary endothelial and other hard‐to‐transfect cells has been a major obstacle for cardiovascular research. Plasmids, lenti‐, adeno‐ or adeno‐associated viral vectors have most often been used to achieve sufficient gene knockout rates in ECs (Abrahimi et al., 2015; Cullere, Plovie, Bennett, MacRae, & Mayadas, 2015; Gong et al., 2017; Miao et al., 2018; Wu et al., 2017). Just recently, we have demonstrated that gene knockouts in human ECs can be established with a crRNA:tracrRNA:Cas9 RNP approach (Schwefel et al., 2019). However, effective CRISPR/Cas9‐driven gene knock‐in or precise single nucleotide corrections have been hampered by low efficiency of homology‐directed repair (HDR) strategies when compared to non‐homologous end joining (NHEJ)‐mediated gene knockout approaches (Mali et al., 2013). Even with the use of the small molecule L755507 that can enhance HDR efficiency, CRISPR/Cas9‐mediated knock‐in rates in primary ECs have been reported to be lower than 3% (Yu et al., 2015). To the best of our knowledge, precise editing of single nucleotide variants (SNV) or small frameshift mutations in primary ECs or endothelial progenitor cells have not yet been reported.
It therefore seems remarkable for CCM but also for cardiovascular research in general that we were able to precisely correct 10% of all patient‐derived CCM1+/− ECs by homology‐directed repair in a crRNA:tracrRNA:Cas9 RNP approach. It should be noted that crRNA:tracrRNA:Cas9 RNPs have several advantages when compared to viral or plasmid delivery systems. In particular, the risk of off‐target effects is minimized due to the transient expression of RNPs (Kim, Kim, Cho, Kim, & Kim, 2014). The absence of CRISPR/Cas9‐induced variants on the patient's wild type CCM1 allele and of mutations in predicted off‐target loci highlights the specificity of our non‐viral and plasmid‐free approach.
Given that a Knudsonian two‐step inactivation of CCM1 in ECs initiates CCM formation in germline mutation carriers (Akers et al., 2009; Gault et al., 2009, 2005; McDonald et al., 2014; Pagenstecher, Stahl, Sure, & Felbor, 2009), a clonal survival advantage of CCM1−/− ECs as demonstrated in our study is a major limitation for any therapeutic somatic gene correction approach. While the allele ratio was stable in corrected cell mixtures over several passages, co‐culture experiments indicated that CCM1−/− ECs rapidly superseded CCM1+/− and corrected CCM1+/+ ECs. Furthermore, CCM1−/− ECs could be clonally expanded by limiting dilution and subcultured for various passages whereas CCM1+/+ and CCM1+/− cells could not be cloned or the clones became senescent after only a few passages as it would have been expected for primary ECs, respectively. Although EPCs are important components of the neurovascular unit (Malinovskaya et al., 2016) and contribute to endothelial regeneration, vascular stability and de novo formation of functional blood vessels (Banno & Yoder, 2018; Critser & Yoder, 2010), our results therefore suggest that the therapeutic potential of somatic gene correction in CCM is limited.
CONFLICT OF INTEREST
The authors have reported no conflicts of interest.
ACKNOWLEDGMENTS
The authors thank the patient and his family for their participation. This work was funded by grants from the Research Network Molecular Medicine of the University Medicine Greifswald to SS (FVMM, Grant No FOVB‐2017‐03, FOVB‐2018‐06). MR was supported by the German Research Foundation (DFG RA2876/2‐1).
Spiegler S, Rath M, Much CD, Sendtner BS, Felbor U. Precise CCM1 gene correction and inactivation in patient‐derived endothelial cells: Modeling Knudson's two‐hit hypothesis in vitro. Mol Genet Genomic Med. 2019;7:e755 10.1002/mgg3.755
Funding information
This work was funded by grants from the Research Network Molecular Medicine of the University Medicine Greifswald to SS (FVMM, Grant No FOVB‐2017–03, FOVB‐2018–06). MR was supported by the German Research Foundation (DFG RA2876/2‐1).
REFERENCES
- Abrahimi, P. , Chang, W. G. , Kluger, M. S. , Qyang, Y. , Tellides, G. , Saltzman, W. M. , & Pober, J. S. (2015). Efficient gene disruption in cultured primary human endothelial cells by CRISPR/Cas9. Circulation Research, 117(2), 121–128. 10.1161/CIRCRESAHA.117.306290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akers, A. L. , Johnson, E. , Steinberg, G. K. , Zabramski, J. M. , & Marchuk, D. A. (2009). Biallelic somatic and germline mutations in cerebral cavernous malformations (CCMs): Evidence for a two‐hit mechanism of CCM pathogenesis. Human Molecular Genetics, 18(5), 919–930. 10.1093/hmg/ddn430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banno, K. , & Yoder, M. C. (2018). Tissue regeneration using endothelial colony‐forming cells: Promising cells for vascular repair. Pediatric Research, 83(1–2), 283–290. 10.1038/pr.2017.231 [DOI] [PubMed] [Google Scholar]
- Bergametti, F. , Denier, C. , Labauge, P. , Arnoult, M. , Boetto, S. , Clanet, M. , … Tournier‐Lasserve, E. (2005). Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. American Journal of Human Genetics, 76(1), 42–51. 10.1086/426952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong, M. S. , Ng, W. K. , & Chan, J. K. (2016). Concise review: Endothelial progenitor cells in regenerative medicine: applications and challenges. Stem Cells Translational Medicine, 5(4), 530–538. 10.5966/sctm.2015-0227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couteulx, S.‐L. , Jung, H. H. , Labauge, P. , Houtteville, J.‐P. , Lescoat, C. , Cecillon, M. , … Tournier‐Lasserve, E. (1999). Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nature Genetics, 23(2), 189–193. 10.1038/13815 [DOI] [PubMed] [Google Scholar]
- Critser, P. J. , & Yoder, M. C. (2010). Endothelial colony‐forming cell role in neoangiogenesis and tissue repair. Current Opinion in Organ Transplantation, 15(1), 68–72. 10.1097/MOT.0b013e32833454b5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullere, X. , Plovie, E. , Bennett, P. M. , MacRae, C. A. , & Mayadas, T. N. (2015). The cerebral cavernous malformation proteins CCM2L and CCM2 prevent the activation of the MAP kinase MEKK3. Proceedings of the National Academy of Sciences of the United States of America, 112(46), 14284–14289. 10.1073/pnas.1510495112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttano, R. , Rudini, N. , Bravi, L. , Corada, M. , Giampietro, C. , Papa, E. , … Dejana, E. (2016). KLF4 is a key determinant in the development and progression of cerebral cavernous malformations. EMBO Molecular Medicine, 8(1), 6–24. 10.15252/emmm.201505433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denier, C. , Goutagny, S. , Labauge, P. , Krivosic, V. , Arnoult, M. , Cousin, A. , … Tournier‐Lasserve, E. (2004). Mutations within the MGC4607 gene cause cerebral cavernous malformations. American Journal of Human Genetics, 74(2), 326–337. 10.1086/381718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detter, M. R. , Snellings, D. A. , & Marchuk, D. A. (2018). Cerebral cavernous malformations develop through clonal expansion of mutant endothelial cells. Circulation Research, 123(10), 1143–1151. 10.1161/CIRCRESAHA.118.313970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draheim, K. M. , Fisher, O. S. , Boggon, T. J. , & Calderwood, D. A. (2014). Cerebral cavernous malformation proteins at a glance. Journal of Cell Science, 127(Pt 4), 701–707. 10.1242/jcs.138388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, O. S. , & Boggon, T. J. (2014). Signaling pathways and the cerebral cavernous malformations proteins: Lessons from structural biology. Cellular and Molecular Life Sciences, 71(10), 1881–1892. 10.1007/s00018-013-1532-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault, J. , Awad, I. A. , Recksiek, P. , Shenkar, R. , Breeze, R. , Handler, M. , & Kleinschmidt‐DeMasters, B. K. (2009). Cerebral cavernous malformations: Somatic mutations in vascular endothelial cells. Neurosurgery, 65(1), 138–144. 10.1227/01.NEU.0000348049.81121.C1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault, J. , Shenkar, R. , Recksiek, P. , & Awad, I. A. (2005). Biallelic somatic and germ line CCM1 truncating mutations in a cerebral cavernous malformation lesion. Stroke, 36(4), 872–874. 10.1161/01.STR.0000157586.20479.fd [DOI] [PubMed] [Google Scholar]
- Glading, A. , Han, J. , Stockton, R. A. , & Ginsberg, M. H. (2007). KRIT‐1/CCM1 is a Rap1 effector that regulates endothelial cell cell junctions. Journal of Cell Biology, 179(2), 247–254. 10.1083/jcb.200705175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, H. , Liu, M. , Klomp, J. , Merrill, B. J. , Rehman, J. , & Malik, A. B. (2017). Method for dual viral vector mediated CRISPR‐Cas9 gene disruption in primary human endothelial cells. Scientific Reports, 7, 42127 10.1038/srep42127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeneveld, D. J. , van Bekkum, T. , Dirven, R. J. , Wang, J. W. , Voorberg, J. , Reitsma, P. H. , & Eikenboom, J. (2015). Angiogenic characteristics of blood outgrowth endothelial cells from patients with von Willebrand disease. Journal of Thrombosis and Haemostasis, 13(10), 1854–1866. 10.1111/jth.13112 [DOI] [PubMed] [Google Scholar]
- Hebbel, R. P. (2017). Blood endothelial cells: Utility from ambiguity. Journal of Clinical Investigation, 127(5), 1613–1615. 10.1172/JCI93649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschi, K. K. , Ingram, D. A. , & Yoder, M. C. (2008). Assessing identity, phenotype, and fate of endothelial progenitor cells. Arteriosclerosis, Thrombosis, and Vascular Biology, 28(9), 1584–1595. 10.1161/ATVBAHA.107.155960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. , Kim, D. , Cho, S. W. , Kim, J. , & Kim, J. S. (2014). Highly efficient RNA‐guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Research, 24(6), 1012–1019. 10.1101/gr.171322.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudson, A. G. Jr (1971). Mutation and cancer: Statistical study of retinoblastoma. Proceedings of the National Academy of Sciences of the United States of America, 68(4), 820–823. 10.1073/pnas.68.4.820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufs, H. (1928). Über die heredofamiläre Angiomatose des Gehirns und der Retina, ihre Beziehungen zueinander und zur Angiomatose der Haut. Zeitschrift für die gesamte Neurologie und Psychiatrie, 113, 651– 686. 10.1007/BF02884519 [DOI] [Google Scholar]
- Lin, Y. , Weisdorf, D. J. , Solovey, A. , & Hebbel, R. P. (2000). Origins of circulating endothelial cells and endothelial outgrowth from blood. Journal of Clinical Investigation, 105(1), 71–77. 10.1172/JCI8071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori, C. L. , Berg, M. J. , Siegel, A. M. , Huang, E. , Zawistowski, J. S. , Stoffer, T’. P. , … Marchuk, D. A. (2003). Mutations in a gene encoding a novel protein containing a phosphotyrosine‐binding domain cause type 2 cerebral cavernous malformations. American Journal of Human Genetics, 73(6), 1459–1464. 10.1086/380314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali, P. , Yang, L. , Esvelt, K. M. , Aach, J. , Guell, M. , DiCarlo, J. E. , … Church, G. M. (2013). RNA‐guided human genome engineering via Cas9. Science, 339(6121), 823–826. 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinovskaya, N. A. , Komleva, Y. K. , Salmin, V. V. , Morgun, A. V. , Shuvaev, A. N. , Panina, Y. A. , … Salmina, A. B. (2016). Endothelial progenitor cells physiology and metabolic plasticity in brain angiogenesis and blood‐brain barrier modeling. Frontiers in Physiology, 7, 599 10.3389/fphys.2016.00599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Ramirez, J. , Hofman, M. , van den Biggelaar, M. , Hebbel, R. P. , & Voorberg, J. (2012). Establishment of outgrowth endothelial cells from peripheral blood. Nature Protocols, 7(9), 1709–1715. 10.1038/nprot.2012.093 [DOI] [PubMed] [Google Scholar]
- McDonald, D. A. , Shi, C. , Shenkar, R. , Gallione, C. J. , Akers, A. L. , Li, S. , … Marchuk, D. A. (2014). Lesions from patients with sporadic cerebral cavernous malformations harbor somatic mutations in the CCM genes: Evidence for a common biochemical pathway for CCM pathogenesis. Human Molecular Genetics, 23(16), 4357–4370. 10.1093/hmg/ddu153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina, R. J. , Barber, C. L. , Sabatier, F. , Dignat‐George, F. , Melero‐Martin, J. M. , Khosrotehrani, K. , … Stitt, A. W. (2017). Endothelial progenitors: A consensus statement on nomenclature. Stem Cells Translational Medicine, 6(5), 1316–1320. 10.1002/sctm.16-0360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, Y. , Ajami, N. E. , Huang, T.‐S. , Lin, F.‐M. , Lou, C.‐H. , Wang, Y.‐T. , … Chen, Z. (2018). Enhancer‐associated long non‐coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nature Communications, 9(1), 292 10.1038/s41467-017-02113-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagenstecher, A. , Stahl, S. , Sure, U. , & Felbor, U. (2009). A two‐hit mechanism causes cerebral cavernous malformations: Complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Human Molecular Genetics, 18(5), 911–918. 10.1093/hmg/ddn420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo, T. , Johnson, E. W. , Thomas, J. W. , Kuehl, P. M. , Jones, T. L. , Dokken, C. G. , … Marchuk, D. A. (1999). Mutations in the gene encoding KRIT1, a Krev‐1/rap1a binding protein, cause cerebral cavernous malformations (CCM1). Human Molecular Genetics, 8(12), 2325–2333. 10.1093/hmg/8.12.2325 [DOI] [PubMed] [Google Scholar]
- Schwefel, K. , Spiegler, S. , Ameling, S. , Much, C. D. , Pilz, R. A. , Otto, O. , … Rath, M. ( 2019). Biallelic CCM3 mutations cause a clonogenic survival advantage and endothelial cell stiffening. Journal of Cellular and Molecular Medicine, 23( 3), 1771 – 1783. 10.1111/jcmm.14075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegler, S. , Kirchmaier, B. , Rath, M. , Korenke, G. C. , Tetzlaff, F. , van de Vorst, M. , … Felbor, U. (2016). FAM222B is not a likely novel candidate gene for cerebral cavernous malformations. Molecular Syndromology, 7(3), 144–152. 10.1159/000446884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegler, S. , Najm, J. , Liu, J. , Gkalympoudis, S. , Schröder, W. , Borck, G. , … Felbor, U. (2014). High mutation detection rates in cerebral cavernous malformation upon stringent inclusion criteria: One‐third of probands are minors. Molecular Genetics & Genomic Medicine, 2(2), 176–185. 10.1002/mgg3.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegler, S. , Rath, M. , Paperlein, C. , & Felbor, U. (2018). Cerebral cavernous malformations: An update on prevalence, molecular genetic analyses, and genetic counselling. Molecular Syndromology, 9(2), 60–69. 10.1159/000486292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, W. , Duan, Y. , Ma, G. , Zhou, G. , Park‐Windhol, C. , D'Amore, P. A. , & Lei, H. (2017). AAV‐CRISPR/Cas9‐mediated depletion of VEGFR2 blocks angiogenesis in vitro. Investigative Ophthalmology & Visual Science, 58(14), 6082–6090. 10.1167/iovs.17-21902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, C. , Liu, Y. , Ma, T. , Liu, K. , Xu, S. , Zhang, Y. U. , … Qi, L. S. (2015). Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell, 16(2), 142–147. 10.1016/j.stem.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z. , Rawnsley, D. R. , Goddard, L. M. , Pan, W. , Cao, X.‐J. , Jakus, Z. , … Kahn, M. L. (2015). The cerebral cavernous malformation pathway controls cardiac development via regulation of endocardial MEKK3 signaling and KLF expression. Developmental Cell, 32(2), 168–180. 10.1016/j.devcel.2014.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]