

Abstract
Zinc phenoxide complexes L1ZnOAr 1–4 (L1=Me2NC2H4NC(Me)CHC(Me)O) and L2ZnOAr 5–8 (L2=Me2NC3H6NC(Me)CHC(Me)O) with donor‐functionalized β‐ketoiminate ligands (L1/2) and OAr substituents (Ar=Ph 1, 5; 2,6‐Me2‐C6H3 2, 6; 3,5‐Me2‐C6H3 3, 7; 4‐Bu‐C6H4 4, 8) with tuneable electronic and steric properties were synthesized and characterized. 1–8 adopt binuclear structures in the solid state except for 5, while they are monomeric in CDCl3 solution. 1–8 are active catalysts for the ring opening polymerization (ROP) of lactide (LA) in CH2Cl2 at ambient temperature and the catalytic activity is controlled by the electronic and steric properties of the OAr substituent, yielding polymers with high average molecular weight (M n) and moderately controlled molecular weight distribution (MWDs). 1 and 5 showed a living polymerization character and kinetic studies on the ROP of L–LA with 1 and 5 proved first order dependencies on the monomer concentration. Homonuclear decoupled 1H‐NMR analyses of polylactic acid (PLA) formed with rac‐LA proved isotactic enrichment of the PLA microstructure.
Keywords: zinc, ketoiminate, lactide, polymerization reactions, substituent effects
1. Introduction
Polylactide (PLA) is of technological interest since it represents environmentally friendly bio‐polymer resources, which are promising alternatives to non‐renewable petroleum‐based polymeric materials.1 Due to its remarkable biodegradable, biocompatible, nontoxic, and permeable properties, PLA is used in protein encapsulation, microsphere advancement, drug delivery, and other biomedical applications.2 Several methods have been reported for the synthesis of PLA, however, the most effective method is the ring opening polymerization (ROP) of cyclic esters catalysed by metal catalysts3 as well as organic catalysts.4 For biomedical applications, nontoxic metal catalysts are required for the polymerization of cyclic esters, since complete removal of catalyst residues from the polymer is often impossible. Zinc is a very promising metal candidate for such applications, mainly due to its high activity, minimal toxicity, and low price.5 In the last decade, a number of effective zinc‐based catalysts containing a variety of ligands with different steric and electronic properties have been established in the ROP of cyclic esters.6 The ancillary ligands play a key role in the catalytic activity, and fine‐tuning of steric and electronic effects of the ligand was used to manipulate and to control the polymerization process.
Tridentate NNO‐chelating β‐ketoiminate ligands containing a modest π‐electron rich framework are known to effectively stabilize the metal centre. By varying the amino moieties of the tridentate β‐ketoiminate ligand, its steric and electronic properties can be tuned, hence allowing to control the coordination behaviour towards the metal centre7 and therefore also to control the polymerization process.7c, 8 In 2009, Lin et. al reported on Zn and Mg alkoxide complexes containing a tridentate NNO‐chelating β‐ketoiminate ligand, which showed remarkable catalytic activities and stereoselectivities in the ROP of lactide (LA).9c The reactivity and stereoselectivity was found to be drastically influenced by the substituents of the β‐ketoiminate ligand as well as by the (initiating) substituent bound to the metal centre. Later on, the same group showed that the catalytic activity of the complexes increased upon introduction of electron‐donating substituents at the phenyl rings, whereas the catalytic activity was found to decrease with introducing electron‐withdrawing substituents at the β‐ketoiminate ligands.9a, 9b Recently Huang and Yang et al. reported on magnesium and zinc complexes containing tridentate NNO‐chelating pyrazoline‐β‐ketoiminate ligands with different electron‐donating substituents. These complexes showed high activities towards ROP of LA and produced controlled molecular weight polymers with narrow molecular weight distribution (MWD).10 They also found that the metal substituents as well as the electron‐donating substituents of the pyrrolidine‐β‐ketoiminate ligands play an important role in the activity and stereoselectivity of the polymerization process. Recently, we reported the ROP of LA by using homoleptic (L2Zn) and heteroleptic (LZnOAr) β‐ketoiminate zinc complexes and demonstrated that the heteroleptic complexes LZnOAr containing a supplementary phenoxide or Lewis base coordinated to the Zn atom are highly active catalysts.11 In addition, a large number of β‐ketoiminate metal complexes were reported, which showed excellent reactivity to the ROP of cyclic esters under milder reaction condition in a controlled mechanism.12
Since β‐ketoiminate zinc phenoxide complexes were found to exhibit remarkable activities for the polymerization of lactide, we became interested to evaluate the distinctive role of the phenoxide ligand in more detail. We herein report on the synthesis and characterization of eight heteroleptic zinc complexes of the general type L1/2ZnOAr, in which four different phenoxide substituents (OAr) with varying electronic and steric properties and two tridentate NNO‐chelating β‐ketoiminate ligands with different sidearm donor moieties were used. The structure of the complexes was determined in solution (NMR) and in the solid state (XRD) and their catalytic activity towards the ROP of lactide was investigated in detail.
2. Results and Discussion
2.1. Synthesis and Characterization
Two β‐ketoimine ligands L1H and L2H (L1=Me2NC2H4NC(Me)CHC(Me)O, L2=Me2NC3H6NC(Me)CHC(Me)O) with different hemilabile sidearm donor groups were prepared according to literature procedures.13 The heteroleptic complexes L1ZnEt and L2ZnEt were first synthesized by alkane elimination reaction of L1/2H with one equivalent of diethylzinc (1 M in n‐hexane) at −30 °C in n‐hexane14 and then reacted with one equivalent of the corresponding phenol ArOH (Ar=Ph, 2,6‐Me2‐C6H3, 3,5‐Me2‐C6H3, 4‐n‐Bu‐C6H4) in hexane at 25 °C, yielding the expected heteroleptic phenoxide complexes L1ZnOAr 1–4 and L2ZnOAr 5–8 in high yields (Scheme 1). The reactions occurred with elimination of ethane and the reaction products were purified by re‐crystallization from saturated solutions in THF at 0 °C. Complexes 1–8 were characterized by heteronuclear NMR (1H, 13C) and IR spectroscopy and their purity was proven by elemental analysis (C, H, N).
Scheme 1.
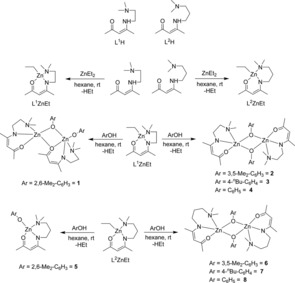
Synthesis of β‐ketoiminate zinc phenoxide complexes 1–8.
The 1H NMR spectra of 1–8 show the expected resonances of the corresponding β‐ketoiminate ligand (L1, L2) as well as the phenoxide substituent in the expected equimolar ratios. The proton resonances of 1–8 are slightly shifted to lower field in comparison to the starting ligands. The proton signal of the CγH group is shifted from 4.69 ppm (ligand) to 4.72–5.04 ppm for complexes 1–4, and from 4.7 ppm (ligand) to 4.73−4.8 for complexes 5–8. In addition, the resonances of the CH 2N(CH3)2 protons of 5–8 in CDCl3 solution at 25 °C appear as broad signals rather than as triplets, pointing to a hindered rotation (Figures S9, S12, S15 and S18). In contrast, the 1H NMR spectra of 5–8 in toluene‐d8 at 40 °C show the expected triplets (Figures S10, S13, S16 and S19). The 13C NMR spectra of 1–8 also show the expected resonances of the β‐ketoiminate ligand and the phenoxide groups. The ipso‐phenyl carbon atoms of the phenoxide groups of 1–8 are significantly shifted to higher field compared to the corresponding phenol. Binding the phenoxide substituent to the Lewis acidic zinc centre results in a partial positive charge at the oxygen atom and, resulting from the stronger polarization of the O−C(Ar) bond, a partial positive charge at the ipso‐phenyl carbon atom. FT‐IR studies show the characteristic peaks of the corresponding functional groups in each complex.
To clarify whether the complexes are monomeric or dimeric in solution, diffusion‐ordered spectroscopy (DOSY) NMR experiments in CDCl3 were performed for complexes 1, 3–6 and 8, respectively, to calculate their hydrodynamic radii (R H).15 The calculated R H values of 3.33 (1), 3.95 (3), 4.21 (4), 3.22 (5, 6), and 3.57 Å (8) are very adjacent to half of the hydrodynamic radii of 6.51 (1), 7.97 (3), 6.34 (4), 6.75 (6) and 6.36 Å (8), respectively, which were calculated from the solid state structure (see Supporting Information). According to these studies, complexes 1–8 adopt mononuclear structures in CDCl3 solution at ambient temperature.
2.2. Single Crystal X‐Ray Diffraction Studies
Single crystals of 1–7 were obtained from saturated solutions in THF upon storage at 0 °C for 24 hours, whereas crystals of 8 were grown from a saturated solution in toluene at 0 °C within 2 days. The molecular structures of 1, 2 (as representative example for the dimer structures observed for 2–4 and 6–8), and 5 are depicted in Figures 1–3, whereas the structures of 3, 4, and 6–8 are given in the electronic supplement (Figures S44‐S48). 1, 3, 4 and 7 crystallize in the monoclinic space group P21 /c, 5 and 8 in the monoclinic space group P21, 2 in the triclinic space group P , and 6 in the tetragonal space group I41 /a, respectively.16
Figure 1.
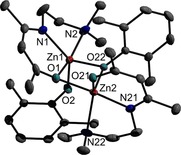
ORTEP representation of solid‐state structure of compound 1. H atoms have been omitted for clarity and thermal ellipsoids are shown with 50 % probability level. Selected bond lengths [Å] and angles [°]: Zn(1)‐O(2) 1.9219(14), Zn(1)‐O(22) 1.9974(14), Zn(1)‐N(1) 2.0235(17), Zn(1)‐O(1) 2.1328(14), Zn(1)‐N(2) 2.2949(18), Zn(2)‐O(21) 1.9683(15), Zn(2)‐O(22) 2.0180(13), Zn(2)‐N(21) 2.0468(18), Zn(2)‐O(1) 2.1181(14), Zn(2)‐N(22), 2.1553(18), O(21)‐Zn(2)‐O(1) 98.71(6), O(22)‐Zn(2)‐O(1) 77.74(5), N(21)‐Zn(2)‐O(1) 171.16(6), O(21)‐Zn(2)‐N(22) 128.03(7), O(22)‐Zn(2)‐N(22) 114.44(6), N(21)‐Zn(2)‐N(22) 79.46(7), O(1)‐Zn(2)‐N(22) 94.47(7).
Figure 3.
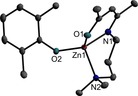
ORTEP representation of solid‐state structure of compound 5. H atoms have been omitted for clarity and thermal ellipsoids are shown with 50 % probability level. Selected bond lengths [Å] and angles [°]: Zn(1)‐O(2) 1.8917(13), Zn(1)‐O(1) 1.9443(13), Zn(1)‐N(1) 1.9830(14), Zn(1)‐N(2) 2.0899, O(2)‐Zn(1)‐O(1) 117.58(6), O(2)‐Zn(1)‐N(1) 128.80(6), O(1)‐Zn(1)‐N(1) 97.17(5), O(2)‐Zn(1)‐N(2) 100.03(5), O(1)‐Zn(1)‐N(2) 114.21(5), N(1)‐Zn(1)‐N(2) 97.99(6).
Complexes 2–4, 6, and 7 adopt phenoxide‐bridged binuclear structures in the solid state with a central Zn2O2 four‐membered ring. The molecules are centrosymmetric, hence the central Zn2O2 rings are perfectly flat and located on corresponding special positions. The oxygen atoms of the phenoxide ligands bridge the Zn atoms, which are further threefold‐coordinated by the NNO‐ligand, resulting in a coordination number of five for each Zn atom. To access the coordination sphere, τ was calculated by literature method.17 The τ values show a preference for a trigonal‐bipyramidal arrangement of the Zn atom in complexes 6 (τ=0.88), 7 (τ=0.84) and 8 (τ=0.83), which contain the C3H6 sidearm donor. In contrast, a square pyramidal arrangement of the Zn centre is observed for complexes 2 (τ=0.09), 3 (τ=0.03) and 4 (τ=0.1), respectively, containing the C2H4 sidearm donor functionalized β‐ketoiminate substituent.
The Zn−O bond lengths within the four‐membered Zn2O2 ring differ by about 0.03 Å except for 6 (Table 1). Complex 8 shows the same molecular connectivity, but the molecule is not centrosymmetric. The τ value for the Zn centre agrees well with the observations for the centrosymmetric molecules. As was observed for 6 and 7, the difference between the Zn−O bond lengths within the Zn2O2 ring is more pronounced, and the Zn−O bond to the oxygen atom of the ligand is shorter. The solid‐state structures of 1 (Figure 1) and 5 (Figure 3), which carry two methyl groups in ortho‐position of the phenolate substituent, differ from the other complexes, for which the structure of 2 is exemplarily shown (Figure 2). This is most obvious for complex 5, which adopts a mononuclear structure in the solid state. The Zn atom in 5 is coordinated by two oxygen and two nitrogen atoms of the phenoxide and the NNO‐ligand, resulting in a distorted tetrahedral coordination geometry (bond angles range from 97.17(5)° to 128.80(6)°). In contrast, the binuclear complex 1 also forms a central four‐membered Zn2O2 ring, but only one of the bridging O atoms originates from a phenoxide group, whereas the second one belongs to the NNO‐ligand. The second phenoxide group is end‐on coordinated to the Zn atom. Despite the C2H4 sidearm donor chain, the τ value suggests a tendency toward a trigonal‐bipyramidal coordination. However, the coordination sphere of the Zn atom with the end‐of coordinated phenoxide ligand is strongly distorted (τ=∼0.5).
Table 1.
Selected Zn‐OPh bond lengths [Å] and Zn‐OPh‐Zn and OPh‐Zn‐OPh bond angles [°] of 1–8.
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Zn−OPh | 1.9219(14)[a] | 2.0081(7) | 2.0044(9) | 2.0077(8) |
| 1.9974(14)[b] | 2.0358(7) | 2.0370(9) | 2.0395(8) | |
| 2.0180(13)[b] | ||||
| Zn−OLig | 1.9683(15)[a] | 2.0158(8) | 2.0499(9) | 2.0339(8) |
| 2.1328(14)[b] | ||||
| 2.1181(14)[b] | ||||
| OPh−Zn−OPh | – | 80.76(3) | 80.03(4) | 80.48(3) |
| Zn−OPh−Zn | 104.71(6) | 99.24(3) | 99.97(4) | 99.52(3) |
| 5 | 6 | 7 | 8 | |
| Zn−OPh | 1.8917(13) | 2.0034(7) | 2.0022(5) | 2.0268(13) |
| 2.1420(6) | 2.1598(5) | 2.1012(13) | ||
| 1.9999(13) | ||||
| 2.1385(13) | ||||
| Zn−OLig | 1.9443(13) | 1.9589(8) | 1.9681(6) | 1.9789(12) |
| 1.9645(12) | ||||
| OPh−Zn−OPh | – | 79.53(3) | 77.80(2) | 78.05(5) |
| 77.76(4) | ||||
| Zn−OPh–Zn | – | 100.47(3) | 102.20(2) | 98.43(6) |
[a] End‐on coordinated O atoms. [b] Bridging O atoms. Symmetry‐equivalent bond lengths of the centrosymmetric compounds are omitted.
Figure 2.
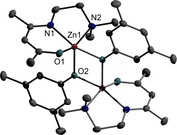
ORTEP representation of solid‐state structure of compound 2. H atoms have been omitted for clarity and thermal ellipsoids are shown with 50 % probability level. i 2‐x, 2‐y, 1‐z. Selected bond lengths [Å] and angles [°]: Zn(1)‐O(2) 2.0081(7), Zn(1)‐O(1) 2.0158(8), Zn(1)‐O(2)i 2.0358(7), Zn(1)‐N(1) 2.0389(10), Zn(1)‐O(1′) 2.115(8), Zn(1)‐N(1′) 2.177(9), Zn(1)‐N(2) 2.2126(8), Zn(1)‐Zn(1)i 3.0804(4), O(2)‐Zn(1)‐O(1) 104.08(4), O(2)‐Zn(1)‐O(2)i 80.76(3), O(1)‐Zn(1)‐O(2)i 90.26(3), O(2)‐Zn(1)‐N(1) 117.44(3), O(1)‐Zn(1)‐N(1) 88.91(4), O(2)i‐Zn(1)‐N(1) 161.38(3), O(2)‐Zn(1)‐O(1′) 92.2(2), O(2)i‐Zn(1)‐O(1′) 103.6(3), O(2)‐Zn(1)‐N(1′) 165.2(2), O(2)i‐Zn(1)‐N(1′) 113.9(2), O(1′)‐Zn(1)‐N(1′) 82.9(3), O(2)‐Zn(1)‐N(2) 100.31(3), O(1)‐Zn(1)‐N(2) 155.59(4), O(2)i‐Zn(1)‐N(2) 92.60(3), N(1)‐Zn(1)‐N(2) 80.76(3), O(1′)‐Zn(1)‐N(2) 160.9(2), N(1′)‐Zn(1)‐N(2) 81.3(2).
Very likely, the different structures of 1 and 5 are caused by steric hindrance due to the ortho‐methyl groups of the phenoxide. In 1 both oxygen atoms in the ring symmetrically bridge the Zn atoms. The Zn−O bonds to the OAr group are roughly 0.1 Å shorter compared to the Zn−O bond of the NNO‐ligand. In 5, the Zn−O bonds are shorter compared to the binuclear complexes, resulting from the lower coordination number of the Zn atom (4 vs. 5). The Zn−O bond length of the phenoxide ligand (Zn1−O2) is approximately equal to the longer bond length within the Zn2O2 ring except for 2, which shows a slightly shorter Zn−O bond lengths. The Zn−O bond lengths within the Zn2O2 rings in 6 and 7 differ by about 0.15 Å and the Zn−O bond to the oxygen atom of the NNO‐ligand is significantly shorter.
2.3. Polymerization Studies
We systematically investigated the catalytic activity of the heteroleptic zinc complexes 1–8 in the ROP of lactide in dichloromethane at ambient temperature (25 °C) under argon atmosphere. We first explored the polymerization activity towards L–Lactide (L–LA) using a 200 : 1 [M]0/[C]0 molar ratio. The polymerization results are summarized in Table 2, proving that 1–8 exhibit excellent activities for the ROP of L–LA. Complete conversion was achieved within a very short period of time and the isolated yields of the PLA were typically higher than 95 %. The polymerization process proceeds with moderate molecular weight control and the molecular weight distributions were found to range from 1.2 to 1.5. The catalytic activity of 1–8 was compared with respect to the TOF values (Table 2), which were calculated based on one zinc atom, which is reliable due to the mononuclear nature of the catalyst in solution.
Table 2.
Polymerization data of L–LA using catalyst 1–8 with [M]o/[C]o ratio=200 : 1 in CH2Cl2 at 25 °C under argon atmosphere.
| Catalyst | Time[a] [s] | Yield[b] [%] | M n (°bs)[c] [kg/mol] | M n (the°)[d] [kg/mol] | M n/M n | TOF[e] [h−1] |
|---|---|---|---|---|---|---|
| 1 | 110 | 98 | 27.3 | 28.8 | 1.4 | 3200 |
| 2 | 140 | 99 | 47.3 | 28.8 | 1.2 | 2550 |
| 3 | 540 | 98 | 45.4 | 28.8 | 1.3 | 650 |
| 4 | 1300 | 99 | 56.3 | 28.8 | 1.3 | 270 |
| 5 | 70 | 99 | 36.1 | 28.8 | 1.2 | 10200 |
| 6 | 85 | 99 | 49.1 | 28.8 | 1.5 | 4200 |
| 7 | 260 | 97 | 45.2 | 28.8 | 1.3 | 1350 |
| 8 | 320 | 96 | 52.6 | 28.8 | 1.5 | 1080 |
[a] Time required for complete conversion. [b] Isolated yield of the polymer after quenching. [c] Measured by GPC at 40 °C in THF, relative to polystyrene standards. [d] M n (The°)at 100 % conversion=[M]0/[C]0×mol. Wt. of monomer. [e] The rate is expressed in terms of the turnover frequency (TOF), mol of LA consumed/(mol catalyst per h)) calculated with respect to number of Zn atom in the catalyst (monomeric structure in solution). The given values represent average values obtained from two different runs.
The analysis of the resulting TOF values revealed that the L1‐substituted complexes (1–4) containing the C2‐spacer are less active than the L2‐substituted ones (5–8). The L2‐substituted complexes, which contain the propyl (C3) spacer, form six‐membered ZnC4N rings upon coordination of the amine sidearm donor (NMe2 group) to the electrophilic Zn atom, whereas the L1‐substituted complexes form five‐membered ZnC3N rings. The higher catalytic activity of 5–8 most likely originates from the formation of highly puckered six‐membered ZnC4N rings, resulting in a less effective shielding of the Zn atom in 5–8 and hence favouring the coordination of the nucleophilic substrate (lactide) to the electrophilic Zn centre. Comparable findings have been previously reported.5d
The TOF values as summarized in Table 2 demonstrate the strong influence of the substitution pattern of the phenoxide substituents on the catalytic activity of the L1‐substituted (1>2>3>4) and L2‐substituted complexes (5>6>7>8). The catalytic activity in both groups follows the same trend and was found to increase upon introduction of an electron donating group R at the phenyl ring as was previously reported.9a, 9b, 10 Moreover, complexes 1 and 5 containing two methyl substituents in ortho‐position of the phenoxide ligand are more active than complexes 2 and 6, which contain two methyl groups in meta‐position. The ortho‐substitution provides higher electron density at the oxygen atom due to the +I effect of the Me groups, resulting in a higher nucleophilicity of the phenoxide ligand. Moreover, the higher steric demand of the ortho‐substituted phenoxide may favour the ligand dissociation. Both effects result in higher activities of complexes 1 and 5 compared to 2 and 6 (Table 2).
To study the electronic structures of 1 to 8 in detail, gas phase structures and natural bond orbital (NBO) analyses were calculated using density functional theory (DFT) with the Gaussian program package18 at the B3LYP LANL2DZ level of theory.19 While the charge of the phenoxide oxygen atom in both type of complexes (1 0.98, 2 0.96, 3 0.96, 4 0.95; 5 0.97, 6 0.95, 7 0.95, 8 0.95) is almost identical (Table S4), compounds 1 and 5 show the highest values. These findings are in accordance with the largest electron‐donating property of the 2,6‐Me2‐C6H3 substituent, resulting in the highest nucleophilicity for the 2,6‐Me2‐C6H3‐substituted complexes 1 and 5, respectively, and also perfectly agree with the observed activity trend in catalysis.
The activity of complex 5 in ROP of lactide is remarkable and a very high TOF value (>10.000 h−1) was observed. Furthermore, the M n°bs values observed for complexes 1 and 5 agree with the calculated values (M n the° values), whereas those of the other complexes showed significantly higher M n°bs values. Since we observed distinct initiation phases in the polymerization studies using complexes 2–4 and 6–8, the high M n°bs values most likely result from rather slow rates of initiation (k i) compared to the propagating rate of polymerization (k p), resulting in polymers with higher than expected molecular weights as was previously reported in ROP polymerization processes.5d
The polymerization reactions using complexes 1–8 were also performed with racemic‐lactide (rac‐LA) in a molar ratio [M]o/[C]o=200 in CH2Cl2 at ambient temperature under argon atmosphere (Table 3). As was expected, the catalytic activity of complexes 1–8 follow the same trend as was observed with L–LA. To gain more information of the polymer microstructure, homonuclear decoupled 1H NMR spectra of the PLA obtained with catalysts 1–8 were recorded. The methine proton signal of the homonuclear‐decoupled 1H NMR spectra of the polymer were assigned according to Hillmyer and co‐workers,20 demonstrating isotactic enhancement in the polymer microstructure. This finding is supported by the fact that the iii tetrads, which is typically used as an indication for the formation of isotactically enriched PLA, exhibits a major peak in the spectra (Figures S35 and S36).21 The P i values were found to range from 0.6 to 0.7, proving that these catalysts showed a moderate polymerization selectivity.
Table 3.
Polymerization data of rac‐LA using complexes 1–8 with [M]o/[C]o ratio=200 : 1 in CH2Cl2 at 25 °C under argon atmosphere.
| Catalyst | Time[a] [s] | Yield[b] [%] | M n (°bs)[c] [kg/mol] | M n (the°)[d] [kg/mol] | M w/M n | Pi [e] |
|---|---|---|---|---|---|---|
| 1 | 110 | 93 | 32.0 | 28.8 | 1.4 | 0.70 |
| 2 | 140 | 90 | 36.1 | 28.8 | 1.2 | 0.66 |
| 3 | 540 | 95 | 42.8 | 28.8 | 1.4 | 0.63 |
| 4 | 1300 | 97 | 56.5 | 28.8 | 1.3 | 0.62 |
| 5 | 70 | 95 | 28.9 | 28.8 | 1.3 | 0.68 |
| 6 | 85 | 95 | 38.2 | 28.8 | 1.5 | 0.64 |
| 7 | 260 | 97 | 48.3 | 28.8 | 1.4 | 0.62 |
| 8 | 320 | 95 | 51.8 | 28.8 | 1.4 | 0.60 |
[a] Time required for complete conversion. [b] Isolated yield of the polymer after quenching. [c] Measured by GPC at 40 °C in THF, relative to polystyrene standards. [d] M n (The°)at 100 % conversion=[M]0/[C]0 x mol. Wt. of monomer. [e] Calculated from homonuclear decoupled 1H NMR spectra.
We furthermore systematically investigated polymerization reactions with increasing L–LA monomer to catalyst molar ratios for 1 and 5 as the most active catalysts at ambient temperature (25 °C) in CH2Cl2 under argon atmosphere. The results are summarized in Table 4 (5) and table S3 (1). The polymerization processes were completed within a few minutes with high [M]0/[C]0 molar ratios (400–1000) and a moderate correlation between M n°bs and the M n the° was observed. M n was found to linearly increase with increasing monomer‐to‐catalyst molar ratio (Figure 4), demonstrating that the polymerization reaction shows certain “living” character. Furthermore, the “living” character was confirmed by a polymerization resumption experiment, in which an additional amount of L–LA monomer ([M]0/[C]0=400) was added after the polymerization of the first amount of L–LA monomer ([M]0/[C]0=400) was finished. The polymerization of the second amount of L–LA monomer was completed within a few minutes (Table 4, Entry 6 and Table S1, Entry 5).
Table 4.
Polymerization data of L–LA using catalyst 5 with varying [M]o/[C]o molar ratios in CH2Cl2 at 25 °C under argon atmosphere.
| Entry | [M]g/[C] | Time[a] [s] | Yield[b] [%] | M n (°bs)[c] [kg/mol] | M w/M n | TOF[e] [h−1] |
|---|---|---|---|---|---|---|
| 1 | 200 : 1 | 70 | 99 | 36.1 | 1.2 | 10200 |
| 2 | 400 : 1 | 170 | 99 | 59.8 | 1.5 | 8400 |
| 3 | 600 : 1 | 280 | 98 | 94.9 | 1.5 | 7560 |
| 4 | 800 : 1 | 365 | 98 | 125.0 | 1.6 | 7730 |
| 5 | 1000 : 1 | 420 | 97 | 162.0 | 1.7 | 8300 |
| 6 | (400+400):1 | 175+195 | 95 | 120.6 | 1.5 | 7400 |
[a] Time required for complete conversion. [b] Isolated yield of the polymer after quenching. [c] Measured by GPC at 40 °C in THF, relative to polystyrene standards. [d] M n (The°)at 100 % conversion=[M]0/[C]0×mol. Wt. of monomer. [e] The rate is expressed in terms of the turnover frequency (TOF), mol of LA consumed/(mol catalyst per h)) calculated with respect to number of Zn atom in the catalyst (monomeric structure in solution). The given values represent average values obtained from two different runs.
Figure 4.
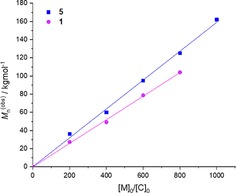
Plot of M n (°bs)c (kg/mol) vs. [M]0/[C]0 for L–LA using complexes 1 and 5 in CH2Cl2 at 25 °C.
2.4. Polymerization Kinetics
Complexes 1 and 5 are the most active catalysts, hence polymerization kinetics of rac‐LA with a monomer‐to‐catalyst ratio [M]0/[C]0 of 1600 (1) and 800 (5) were performed at 25 °C in CDCl3‐solution in a J‐Young NMR tube. The reaction order with respect to the monomer concentration was determined by monitoring the time‐dependent conversion of rac‐lactide by 1H NMR spectroscopy. The linear dependency of ln[M]0/[M]t vs time (Figure 5) proves first‐order kinetics of the monomer concentration on the polymerization rate. Rate constants (k p) of the polymerization reactions were determined from the slope of the lines (1: 10.88 Lmol−1s−1; 5: 11.31 Lmol−1s−1), assuming that every zinc atom propagates one chain.
Figure 5.
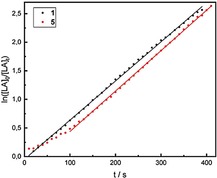
Semi‐logarithmic plots of rac‐LA conversion initiated by 1 and 5 vs. time: [rac‐LA]o/[C]o=800 per zinc atom at 25 °C in CDCl3.
To gain a deeper understanding of the reaction mechanism, low molecular weight L‐PLA was synthesized by polymerizing L–LA with catalyst 1 and 5 with molar ratios of 50 : 1 in CH2Cl2 at 25 °C. The solvent was removed under vacuum and the residue was dissolved in a minimum amount of CH2Cl2 and precipitated by pouring into cold ethanol. The resultant polymer was completely dried under vacuum and systematically characterized using NMR and IR spectroscopy as well as MALDI‐TOF mass spectrometry. The 1H NMR spectra (Figure S41 in the ESI) of the low molecular weight polymer proved that the polymer chain is end‐capped with phenol groups, which initiated the polymerization reaction according to the well‐known coordination‐insertion mechanism (CIM). These results were confirmed by MALDI‐TOF mass spectroscopy analyses of the oligomer (Figure S47).
Three series could be identified in the MALDI spectra: one series is capped by the phenolate as end‐group (23 % of all chains), one series by the NNO ligand (14 %) and one series by the ligand plus zinc ion (35 %). Hence, the anionic ligand is also capable of opening the lactide rings. The polymer treatment after polymerization consists of precipitation of the dissolved polymer in cold methanol. Hereby, the zinc ion seems to hold to the ligand at the chain end. Accordingly, many chains are started by the phenolate, but some are also started by the anionic NNO donor ligand. The anionic ligand at the end of the chain is also typical for the coordination‐insertion mechanism.22 The presence of hydroxyl group in the end chain of the polymer was further confirmed by IR due to the presence of the small band ν(H−O) at 3528 cm−1 (Figure S42 in the ESI). These findings prove that the polymerization process proceeds through the well‐known “coordination‐insertion” (CIM) pathway.9b, 12e, 12h, 22
In 2016 Williams et al. published on the syntheses and catalytic activities of both a binuclear and a mononuclear zinc complex.6h The mononuclear complex was found to be slower than the binuclear complex in terms of catalytic activity. Complexes 1 and 5 reported herein almost reach the k p value of 24 mol L−1 s−1 of the mononuclear Williams complex. However, the TOF value of 60.000 h−1 for the binuclear Williams complex is six times higher than that of the complex 5 (TOF approx. 10.000 h−1). Nevertheless, the complexes presented here stand out from the two reference complexes due to their simple ligand design, their easy synthetic accessibility and their high activity in the ring opening polymerization of lactide.
3. Conclusions
A series of eight heteroleptic zinc phenoxide complexes of the type L1/2ZnOR containing NNO‐tridentate β‐ketoiminate ligands with four different phenoxide substituents with various electronic properties were synthesized in high yield and completely characterized. Complexes 1–8 form dimers in the solid state except for complex 5, which adopts a mononuclear structure, whereas diffusion‐ordered spectroscopy (DOSY) NMR analysis in CDCl3 proved that the complexes are monomeric in solution. 1–8 are active catalysts in the ROP of LA in CH2Cl2 at ambient temperature (25 °C), producing high molecular weight polymers with moderately controlled PDI with a high conversion (>95 %). The catalytic activity increases upon introduction of electron‐donating groups at the phenoxide substituent in ortho‐position. Complex 5, which is the most active catalyst, showed a moderate correlation between the observed molecular weight (M n°bs) and the theoretical molecular weight (M n the°). Analyses of low molecular weight polymers by 1H NMR spectroscopy proved that the polymerization process follows the coordination‐insertion mechanism (CIM). Furthermore, polymerization studies with complexes 1 and 5 showed continuously increasing M n values with increasing monomer to catalyst molar ratio. The polydispersities are small, pointing to a living polymerization character. Homonuclear decoupled 1H NMR studies showed slight isotactic enrichment of the PLA obtained from rac‐LA, and kinetic studies with the most active catalysts 1 and 5 demonstrate that the polymerization reactions have first order dependencies on the monomer concentration. In summary, both catalysts reach enormous polymerisation rate constants, which demonstrate that zinc ketoiminates are a versatile class of ROP catalysts.
Experimental Section
General Experimental Details
All reactions were performed under a dry argon atmosphere with rigorous exclusion of moisture and air using standard Schlenk and glovebox techniques. Argon gas was purified by passing through preheated Cu2O pellets and molecular sieve columns. ZnEt2 (1 M in hexane), amines, and calcium hydride were purchased from Sigma‐Aldrich and used as received, while the phenols were purified prior to use by sublimation. rac‐LA was purchased from Sigma‐Aldrich and sublimed twice under argon atmosphere and stored under a dry argon atmosphere in a glovebox. β‐ketoimines (L1H, L2H) and the heteroleptic complexes L1/2ZnEt were prepared according to literature procedures.13, 14 Toluene and n‐hexane were dried using a MBraun Solvent Purification System, degassed, and stored in Schlenk flasks under argon atmosphere. THF was dried by heating under reflux for 12 h over sodium and benzophenone and freshly distilled prior to use. Deuterated solvents were dried over activated molecular sieves (4 Å) and degassed prior to use. Karl Fischer titration of the dry solvents showed values below 2 ppm.
1H and 13C NMR spectra were recorded at 297 K in CDCl3 or toluene‐d8 solution using a Bruker Avance 300 spectrometer with a QNP probe head (1H: 300 MHz, 13C: 75 MHz) or Bruker Avance 500 (1H: 400 MHz, 13C: 125 MHz). 1H and 13C NMR spectra were referenced to internal CDCl3 (1H=7.26 ppm, 13C=77.16 ppm) and toluene‐d8 (1H=7.00 ppm, 13C=128.33 ppm), respectively. IR spectra were recorded in a glovebox under argon atmosphere using an ALPHA−T FT‐IR spectrometer equipped with a single reflection ATR sampling module. Elemental analyses were performed with a Perkin Elmer Series 11 Analyzer at the elemental analysis laboratory of the University of Duisburg‐Essen. ESI‐MS spectra of the samples were recorded using Waters Q‐Tof micro mass spectrometer. Melting points were determined in sealed glass capillaries and are not corrected.
The number‐average molecular weight (M n), molecular weight distribution (MWD) and dispersity (M w/M n) of all polylactide polymers were determined by Gel Permeation Chromatography (GPC) on a 1260 Infinity instrument (Polymer Standard Service, Mainz) equipped with 3 SDV columns (pore sizes 106, 105, 103 Å) and a SECcurity differential refractometer. The PLA was measured using HPLC grade THF as eluent at a flow rate of 1.0 mL min−1 at 40 °C (column oven TCC6000). A calibration setup was obtained using 12 narrow molecular weight polystyrene standards (Polymer Standard Service, Mainz) and the WinGPC UniChrom software. Number average molecular weights (M n) and MWDs (M w/M n) of polymers were measured relative to polystyrene standards.
The average molecular masses and the mass distributions of the obtained polylactide samples (kinetic measurements) were determined by GPC in THF as the mobile phase at a flow rate of 1 mL min−1. The utilised GPCmax VE‐2001 from Viscotek was a combination of an HPLC pump, two Malvern Viscothek T columns (porous styrene divinylbenzene copolymer) with a maximum pore size of 500 and 5000 Å, a refractive index detector (VE‐3580), and a viscometer (Viscotek 270 Dual Detector). Universal calibration was applied to evaluate the chromatographic results.
The end group analysis was performed by MALDI‐TOF on a Bruker ultrafleXtreme equipped with a 337 nm smartbeam laser in the reflective mode. THF solutions of trans‐2‐[3‐(4‐tert‐butylphenyl)‐2‐methyl‐2‐propenylidene]malononitrile (DCTB) (5 μL of a 20 mg/mL solution), sodium trifluoroacetate (0.1 μL of a 10 mg/mL solution), and analyte (5 μL of a 10 mg/mL) were mixed and a droplet thereof applied on the sample target. Protein 1 calibration standard is the name of the protein mixture used for calibration. For spectra 4000 laser shots with 24 % laser power were collected. The laser repetition rate was 1000 Hz. The homopolymer analysis was performed using Polymerix software (Sierra Analytics).
General Synthesis of 1–8
A solution of the respective phenol in 5 ml of hexane was added at ambient temperature dropwise to a stirred solution of the heteroleptic complex L1/2ZnEt in 5 ml of n‐hexane. After the gas evolution has stopped, the resulting solution was stirred for additional 3 h at ambient temperature (25 °C). The solvent was evaporated under reduced pressure to yield a white solid, which was re‐crystallized from a saturated solution in THF at 0 °C, isolated by filtration and dried under vacuum.
1. 2,6‐dimethylphenol (139 mg, 1.14 mmol), L1ZnEt (300 mg, 1.14 mmol). Yield: 330 mg (93 %). Mp. 170 °C. Anal. Calcd for C34H52N4O4Zn2: C, 57.39; H, 7.37; N, 7.87. Found: C, 56.92; H, 7.40; N, 7.43. 1H NMR (300 MHz, CDCl3, 25 °C): δ 1.81 (s, 3H, CβCH 3), 1.91 (s, 3H, CH 3CN), 2.12 (s, 6H, o–CCH 3), 2.21 (s, 6H, N(CH 3)2), 2.68 (t, 3 J HH=6.3 Hz, 2H, CH 2N(CH3)2), 3.29 (t, 3 J HH=6.0 Hz, 2H, CβNCH 2), 4.72 (s, 1H, CH), 6.43 (d, 3 J HH=7.2 Hz, 1H, p‐H), 6.84 (d, 3 J HH=7.2 Hz, 2H, m‐H); 13C NMR (75 MHz, CDCl3, 25 °C): δ 17.6 (Ar−CH3), 22.2 (CH3‐CN), 27.8 (β‐CH3), 44.8 (CH2N), 45.5 (N(CH 3)2), 58.3 (CH2N(CH3)2), 97.6 (CH), 115.3 (Arp−C), 127.0 (Aro−C), 128.0 (Arm‐C), 160.1 (Ari−C), 172.8 (CN), 183.0 (CO). ATR−IR: ν 3059, 2998, 2966,2943, 2910, 2889, 2839, 2792, 1589, 1499, 1458, 1398, 1339, 1261, 1225, 1089, 1024, 933, 847, 799, 781, 743, 688, 640, 581, 550, 519, 436, 406 cm–1.
2. 3,5‐dimethylphenol (139 mg, 1.14 mmol), L1ZnEt (300 mg, 1.14 mmol). Yield: 324 mg (91 %). Mp. 235 °C. Anal. Calcd for C34H52N4O4Zn2: C, 57.39; H, 7.37; N, 7.87. Found: C, 56.90; H, 7.39; N, 7.67. 1H NMR (300 MHz, CDCl3, 25 °C): δ 1.93 (s, 6H, CβCH 3, CH 3CNC), 2.17 (s, 6H, N(CH 3)2), 2.28 (s, 6H, CH 2N(CH3)2), 2.40 (t, 3 J HH=6.0 Hz, 2H, o–CCH 3), 3.15 (t, 3 J HH=6.0 Hz, 2H, C βNCH 2), 5.05 (s, 1H, CH), 6.2 (s, 1H, p‐H), 6.45 (s, 2H, m–H); 13C NMR (75 MHz, CDCl3, 25 °C): δ 21.6 (Ar−CH3), 22.0 (CH3−CN), 27.8 (β‐CH3), 44.4 (CH2N), 46.1 (N(CH 3)2), 58.6 (CH2N(CH3)2), 97.3 (CH), 117.1 (Aro−C), 117.6 (Arp−C), 138.1 (Arm−C), 164.3 (Ari−C), 172.3 (CN), 183.2 (CO). ATR–IR: ν 3068, 3002, 2963, 2911, 2843, 2799, 2657,1585, 1508, 1464, 1404, 1398, 1320, 1259, 1218, 1164, 1102, 1023, 936, 843, 819, 799, 784, 748, 689, 615, 598, 573, 532, 404 cm–1.
3. 4‐n‐butylphenol (171 mg, 1.14 mmol), L1ZnEt (300 mg, 1.14 mmol). Yield: 340 mg (88 %). Mp. 167 °C. Anal. Calcd for C38H60N4O4Zn2: C, 59.45; H, 7.88; N, 7.30. Found: C, 59.86; H, 7.74; N, 7.44. 1H NMR (300 MHz, CDCl3, 25 °C): δ 0.91 (t, 3 J HH=7.5 Hz, 3H, CH2CH 3), 1.26–1.39 (m, 2H, CH 2CH3), 1.48‐1.58 (m, 2H, CH2CH 2), 1.91 (s, 3H, CβCH 3), 1.92 (s, 3H CH 3CN), 2.27 (s, 6H, N(CH 3)2), 2.43 (t, 3 J HH=6.0 Hz, 2H, CH 2Ar), 2.47 (t, 3 J HH=7.8 Hz, 2H, CH 2N(CH3)2), 3.13 (t, 3 J HH=6.0 Hz, 2H, C βNCH 2), 5.01 (s, 1H, CH), 6.70 (d, 3 J HH=8.1 Hz, 2H, o‐H), 6.86 (d, 3 J HH=7.8 Hz, 2H, m‐H); 13C NMR (75 MHz, CDCl3, 25 °C): δ 14.2 (CH2 CH3), 22.1 (CH3‐CN), 22.6 (CH3 CH2), 27.8 (β‐CH3), 34.2 (CH3 CH2), 35.0 (Ar−CH2), 44.4 (CH2N), 46.0 (N(CH 3)2), 59.5 (CH2N(CH3)2), 96.2 (CH), 118.6 (Aro−C), 128.8 (Arm−C), 129.9 (Arm−C), 162.3 (Ari−C), 172.4 (CN), 184.0 (CO). ATR−IR: ν 3009, 2949, 2923, 2847, 2792, 2554, 1601, 1578, 1462, 1401, 1344, 1266, 1171, 1109, 1050, 1026, 960, 935, 849, 832, 788, 751, 722, 673, 642, 541, 495, 4041 cm–1.
4. Phenol (107 mg, 1.14 mmol), L1ZnEt (300 mg, 1.14 mmol). Yield: 306 mg (93 %). Mp. 227 °C. Anal. Calcd for C30H44N4O4Zn2: C, 54.97; H, 6.77; N, 8.55. Found: C, 54.40; H, 6.73; N, 8.35. 1H NMR (300 MHz, CDCl3, 25 °C): δ 1.91 (s, 3H, CβCH 3), 1.93 (s, 3H CH 3CN), 2.28 (s, 6H, N(CH 3)2), 2.40 (t, 3 J HH=6.0 Hz, 2H, CH 2N(CH3)2), 3.13 (t, 3 J HH=6.0 Hz, 2H, C βNCH 2), 5.01 (s, 1H, CH), 6.58 (t, 3 J HH=7.2 Hz, 1H, p−H), 6.81 (d, 3 J HH=4.8 Hz, 2H, o‐H), 7.03‐7.08 (m, 2H, m‐H); 13C NMR (75 MHz, CDCl3, 25 °C): δ 22.1 (CH3‐CN), 27.8 (β‐CH3), 44.4 (CH2N), 46.0 (N(CH 3)2), 59.5 (CH2N(CH3)2), 96.3 (CH), 119.2 (Aro−C), 119.4 (Arm–C), 129.1 (Arm−C), 162.3 (Ari−C), 172.4 (CN), 184.0 (CO). ATR−IR: ν 3067, 3041, 3019, 2991, 2961, 2903, 2870, 2835, 2797, 2545, 1589, 1484, 1460, 1401, 1342, 1261, 1220, 1162, 1024, 993, 935, 890, 824, 758, 697, 643, 559, 489, 4028, 408 cm−1.
5. 2,6‐dimethylphenol (132 mg, 1.08 mmol), L2ZnEt (300 mg, 1.08 mmol). Yield: 345 mg (93 %). Mp. 109 °C. Anal. Calcd for C18H28N2O2Zn: C, 58.46; H, 7.63; N 7.58. Found: C, 57.83; H, 7.71; N, 2.19. 1H NMR (300 MHz, CDCl3, 25 °C): δ 1.89 (br, 8H, CβCH 3, CH 3CN, CH2CH2), 2.21 (s, 6H, o‐CCH 3), 2.51 (s, 6H, N(CH 3)2), 2.80 (br, 2H, CH 2N(CH3)2), 3.60 (t, 3 J HH=5.7 Hz, 2H, CβNCH 2), 4.74 (s, 1H, CH), 6.42 (t, 3 J HH=7.2 Hz, 1H, p‐H), 6.89 (d, 3 J HH=7.5 Hz, 2H, m‐H); 1H NMR (300 MHz, toluene‐d8, 40 °C): δ 1.20‐1.27 (m, 2H, CH2CH2), 1.42 (s, 3H, CβCH 3,), 1.88 (s, 3H, CH 3CN,), 1.99 (s, 6H, o‐CCH 3), 2.15 (t, 3 J HH=5.1 Hz, 2H, CH 2N(CH3)2), 2.43 (s, 6H, N(CH 3)2), 3.08 (t, 3 J HH=5.4 Hz, 2H, CβNCH 2), 4.60 (s, 1H, CH), 6.45 (t, 3 J HH=7.2 Hz, 1H, p‐H), 7.10 (d, 3 J HH=7.5 Hz, 2H, m–H); 13C NMR (75 MHz, CDCl3, 25 °C): δ 17.7 (Ar‐CH3), 20.5(CH3‐CN), 27.2(CH2CH2), 27.7 (β‐CH3), 48.4 (CH2N), 50.1 (N(CH 3)2), 61.8 (CH2N(CH3)2), 97.1 (CH), 114.3 (Arp−C), 126.4 (Aro−C), 128.0 (Arm−C), 163.1 (Ari−C), 171.3 (CN), 182.5 (CO). ATR−IR: ν 3061, 3035, 3000, 2959, 2923, 2890, 2851, 2808, 1587, 1502, 1459, 1424, 1293, 1274, 1091, 1012, 969, 933, 866, 847, 748, 700, 675, 642, 578, 523, 480, 443, 386 cm−1.
6. 3,5‐dimethylphenol (132 mg, 1.08 mmol), L2ZnEt (300 mg, 1.08 mmol). Yield: 319 mg (86 %). Mp. 107 °C. Anal. Calcd for C36H56N4O4Zn2: C, 58.46; H, 7.63; N, 7.58. Found C, 58.40; H, 7.50; N, 8.25. 1H NMR (300 MHz, CDCl3, 25 °C): δ 1.78‐1.85 (m, 2H, CH 2CH2), 1.90 (s, 3H, CβCH 3,), 1.95 (s, 3H, CH 3CN), 2.18 (s, 6H, o−CCH 3), 2.46 (s, 6H, N(CH 3)2), 2.69 (br, 2H, CH 2N(CH3)2), 3.52 (t, 3 J HH=5.4 Hz, 2H, C βNCH 2), 4.80 (s, 1H, CH), 6.22 (s, 1H, p‐H), 6.35 (s, 2H, m‐H); 1H NMR (300 MHz, toluene‐d8, 40 °C): δ 1.42–1.50 (m, 2H, CH 2CH2), 1.53 (s, 3H, CβCH 3,), 2.00 (t, 3 J HH=5.7 2H, CH 2N(CH3)2), 2.11 (s, 6H, o–CH 3), 2.12 (s, 3H, CH 3CN), 2.26 (s, 6H, N(CH 3)2), 2.85 (t, 3 J HH=6.6 Hz, 2H, C βNCH 2), 4.85 (s, 1H, CH), 6.39 (s, 1H, p‐H), 6.85 (s, 2H, m‐H); 13C NMR (75 MHz, CDCl3, 25 °C): δ 20.9 (Ar‐CH3), 21.5 (CH3−CN), 27.0 (CH2CH2), 27.7 (β‐CH3), 47.2 (CH2N), 50.1 (N(CH 3)2), 61.6 (CH2N(CH3)2), 97.2 (CH), 116.9 (Aro−C), 116.9 (Arp−C), 138.7 (Arm−C), 166.6 (Ari−C), 172.1 (CN), 182.9 (CO). ATR−IR: ν 3041, 2935, 2905, 2859, 2834, 2788, 1578, 1508, 1464, 1408, 1307, 1150, 1013, 974, 941, 878, 821, 802, 777, 704, 671, 641, 600, 531, 515, 481, 456,440, 403 cm–1.
7. 4‐n‐butylphenol (162 mg, 1.08 mmol), L2ZnEt (300 mg, 1.08 mmol). Yield: 320 mg (80 %). Mp. 96 °C. Anal. Calcd for C40H64N4O4Zn2: C, 60.38; H, 8.11; N, 7.04. Found: C, 59.83; H, 8.17; N, 7.35. 1H NMR (300 MHz, CDCl3, 25 °C): δ 0.90 (t, 3 J HH=7.2 Hz, 3H, CH2CH 3), 1.27‐1.39 (m, 2H, CH 2CH3), 1.48‐1.58 (m, 2H, CH2CH 2), 1.74‐1.82 (m, 2H, CH2CH 2), 1.88 (s, 3H, CβCH 3), 1.95 (s, 3H CH 3CN), 2.42 (s, 6H, N(CH 3)2), 2.58 (t, 3 J HH=8.1 Hz, 2H, CH 2Ar), 2.64 (br, 2H, CH 2N(CH3)2), 3.46 (br, 2H, C βNCH 2), 4.79 (s, 1H, CH), 6.62 (d, 3 J HH=7.8 Hz, 2H, o‐H), 6.87 (d, 3 J HH=8.1 Hz, 2H, m‐H); 1H NMR (300 MHz, toluene‐d8, 40 °C): δ 0.86 (t, 3 J HH=7.2 Hz, 3H, CH2CH 3), 1.2–1.36 (m, 2H, CH 2CH3), 1.42‐1.48 (m, 2H, CH2CH 2), 1.53 (s, 3H, CβCH 3), 1.53‐1.58 (m, 2H, CH2CH 2), 2.03 (t, 3 J HH=5.7 Hz, 2H, CH 2Ar), 2.08 (s, 3H CH 3CN), 2.09 (s, 6H, N(CH 3)2), 2.49 (t, 3 J HH=7.8 Hz, 2H, CH 2N(CH3)2), 2.89 (t, 3 J HH=6.6 Hz, 2H, C βNCH 2), 4.80 (s, 1H, CH), 6.95 (d, 3 J HH=2.7 Hz, 2H, o‐H), 7.08 (d, 3 J HH=1.8 Hz, 2H, m–H); 13C NMR (75 MHz, CDCl3, 25 °C): δ 14.1 (CH2 CH3), 21.0 (CH3‐CN), 22.5 (CH3 CH2), 27.0 (CH3 CH2), 27.8 (β‐CH3), 34.3 (CH3 CH2), 34.9 (Ar‐CH2), 47.1 (CH2N), 50.0 (N(CH 3)2), 61.4 (CH2N(CH3)2), 97.1 (CH), 118.5 (Aro−C), 129.1 (Arm−C), 129.1 (Arm−C), 164.3 (Ari−C), 172.2 (CN), 182.9 (CO). ATR–IR: ν 3067, 2985, 2957, 2924, 2836, 2792, 2521, 1583, 1496, 1464, 1410, 1375, 1344, 1246, 1166, 1107, 1073, 1059,1012, 974, 900, 838, 780, 738, 669, 640, 575, 521, 479, 4033, 387 cm–1.
8. Phenol (102 mg, 1.08 mmol), L2ZnEt (300 mg, 1.08 mmol). Yield: 285 mg (83 %). Mp. 79 °C. Anal. Calcd for C32H48N4O4Zn2: C, 56.23; H, 7.08; N, 8.20. Found: C, 56.40; H, 6.73; N, 8.25. 1H NMR (300 MHz, CDCl3, 25 °C): δ 1.80‐1.87 (m, 2H, CH2CH 2), 1.90 (s, 3H, CβCH 3), 1.95 (s, 3H CH 3CN), 2.48 (s, 6H, N(CH 3)2), 2.72 (br, 2H, CH 2N(CH3)2), 3.55 (t, 3 J HH=5.4 Hz, 2H, C βNCH 2), 4.78 (s, 1H, CH), 6.61 (d, 3 J HH=6.6 Hz, 1H, p–H), 6.73 (d, 3 J HH=7.8 Hz, 2H, o‐H), 7.10 (t, 3 J HH=7.2 Hz, 2H, m‐H); 1H NMR (300 MHz, toluene‐d8, 40 °C): δ 1.38–1.42 (m, 2H, CH2CH 2), 1.50 (s, 3H, CβCH 3), 2.00 (t, 3 J HH=5.4 Hz, 2H, CH 2N(CH3)2), 2.07 (s, 3H CH 3CN), 2.08 (s, 6H, N(CH 3)2), 2.82 (t, 3 J HH=6.1 Hz, 2H, C βNCH 2), 4.79 (s, 1H, CH), 6.68‐6.73 (m, 1H, p‐H), 7.15 (d, 3 J HH=6.0 Hz, 2H, o‐H), 7.17 (d, 3 J HH=2.4 Hz, 2H, m‐H); 13C NMR (75 MHz, CDCl3, 25 °C): δ 20.9 (CH3‐CN), 27.0 (CH2CH2), 27.8 (β‐CH3), 47.3 (CH2N), 50.2 (N(CH 3)2), 61.8 (CH2N(CH3)2), 97.4 (CH), 121.1 (Aro−C), 126.4 (Arm−C), 129.4 (Arm−C), 161.1 (Ari−C), 172.3 (CN), 183.0 (CO). ATR–IR: ν 3067,3048, 2982, 2947, 2911, 2879, 2841, 2798, 2532, 1584, 1510, 1462, 1411, 1353, 1340, 1248, 1159, 1107, 1068, 1011, 992, 975, 939, 897, 824, 767, 750, 703, 641, 549, 480, 4030, 387 cm–1.
Single Crystal X‐ray Analysis
The crystals were mounted on nylon loops in inert oil. Data were collected on a Bruker AXS D8 Kappa diffractometer with APEX2 detector (monochromated Mo−Kα radiation, λ=0.71073 Å) at 100(2) K. The structures of 1–8 were solved by Direct Methods (SHELXS‐97)23 and refined anisotropically by full‐matrix least‐squares on F2 (SHELXL‐2014).24, 25 Absorption corrections were performed semi‐empirically from equivalent reflections on basis of multi‐scans (Bruker AXS APEX2). Hydrogen atoms were refined using a riding model or rigid methyl groups. In 1 the side chain was disordered over two sites. These were refined with fixed occupancies of 2/3 and 1/3, respectively. In 2 most parts of the ligand were disordered. The respective C−C bond lengths were restrained to be equal (SADI). RIGU and SIMU restraints were applied to the ADP of the atoms of the disordered parts. For C5’ an additional ISOR restraints was necessary (Table S1 and S2).
General Procedure for the Bulk Polymerization of Lactide
Polymerization reactions were performed in 200 : 1 molar ratios of the monomers and 1–8. A solution of 17.34 μmol of 1–8 and 0.5 g of lactide (rac‐LA or L‐LA) in 5 ml of CH2Cl2 was stirred at ambient temperature in a Schlenk‐flask equipped with a magnetic bar under argon atmosphere. After the desired time, the reaction was terminated by exposing to air and the volatiles were removed under vacuum. The resulting residues were dissolved into a minimum amount of CH2Cl2 and poured into cold methanol. The polymer precipitated immediately and was isolated by filtration. The filtered product was dried under vacuum until constant weight was observed. The conversion rate of the monomer was determined by 1H NMR analysis. An analogous procedure was applied for polymerization reactions using higher monomer to catalyst molar ratios. All polymerisation reactions have been performed twice.
General Procedure for the Polymerization Kinetics
The reaction kinetics were also determined for 1 and 5, using a solution of rac‐lactide (0.0504 g, 0.350 mmol) and the desired catalyst in 0.7 mL of CDCl3 (0.5 molL−1) in a Young NMR tube. The conversion rate of LA was monitored by 1H NMR spectroscopy at 26 °C. The ln{[M]0/[M]t} ratio was calculated by integration of the peak corresponding to the methyl proton for the polymer and monomer. Apparent rate constants were obtained from the slopes of the best fit line. All polymerisation reactions have been performed twice.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support by Evonik Industries (P.S., A.T., A.H.G.) and the University of Duisburg Essen (St.S.) is acknowledged. A.H.G. is grateful for financial support from the DFG through the Emmy‐Noether Program (No. 376920678). P.M.S. thanks the Hanns‐Seidel‐Foundation (fellowship) for funding (Bundesministerium für Bildung und Forschung, BMBF). S.H.‐P. thanks Total Corbion PLA for lactide donations.
S. Ghosh, P. M. Schäfer, D. Dittrich, C. Scheiper, P. Steiniger, G. Fink, A. N. Ksiazkiewicz, A. Tjaberings, C. Wölper, A. H. Gröschel, A. Pich, S. Herres-Pawlis, S. Schulz, ChemistryOpen 2019, 8, 951.
Dedicated to Prof. G. Meyer on the occasion of his 70th birthday
Contributor Information
Prof. Dr. Sonja Herres‐Pawlis, Email: sonja.herres-pawlis@ac.rwth-aachen.de
Prof. Dr. Stephan Schulz, Email: stephan.schulz@uni-due.de.
References
- 1.
- 1a. Bakewell C., White A. J., Long N. J., Williams C. K., Angew. Chem. Int. Ed. 2014, 53, 9226–9230; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9380–9384; [Google Scholar]
- 1b. Rhim J. W., Park H. M., Ha C. S., Prog. Polym. Sci. 2013, 38, 1629–1652; [Google Scholar]
- 1c. Chen G. Q., Patel M. K., Chem. Rev. 2012, 112, 2082–2099; [DOI] [PubMed] [Google Scholar]
- 1d. Thomas C. M., Lutz J. F., Angew. Chem. Int. Ed. 2011, 50, 9244–9246; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9412–9414; [Google Scholar]
- 1e. Inkinen S., Hakkarainen M., Albertsson A. C., Sodergard A., Biomacromolecules 2011, 12, 523–532; [DOI] [PubMed] [Google Scholar]
- 1f. Arbaoui A., Redshaw C., Polym. Chem. 2010, 1, 801–826; [Google Scholar]
- 1g. Thomas C. M., Chem. Soc. Rev. 2010, 39, 165–173; [DOI] [PubMed] [Google Scholar]
- 1h. Woodruff M. A., Hutmacher D. W., Prog. Polym. Sci. 2010, 35, 1217–1256; [Google Scholar]
- 1i. Ragauskas A. J., Williams C. K., Davison B. H., Britovsek G., Cairney J., Eckert C. A., Frederick W. J. J., Hallett J. P., Leak D. J., Liotta C. L., Mielenz J. R., Murphy R., Templer R., Tschaplinski T., Science 2006, 311, 484–489. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Wu W., Wang W., Li J., Prog. Polym. Sci. 2015, 46, 55–85; [Google Scholar]
- 2b. Lih E., Oh S. H., Joung Y. K., Lee J. H., Han D. K., Prog. Polym. Sci. 2015, 46, 28–61; [Google Scholar]
- 2c. Nofar M., Park C. B., Prog. Polym. Sci. 2014, 39, 1721–1741; [Google Scholar]
- 2d. Raquez J.-M., Habibi Y., Murariu M., Dubois P., Prog. Polym. Sci. 2013, 38, 1504–1542; [Google Scholar]
- 2e. Armentano I., Bitinis N., Fortunati E., Mattioli S., Rescignano N., Verdejo R., Lopez-Manchado M. A., Kenny J. M., Prog. Polym. Sci. 2013, 38, 1720–1747. [Google Scholar]
- 3.
- 3a. Kremer A. B., Mehrkhodavandi P., Coord. Chem. Rev. 2019, 380, 35–57; [Google Scholar]
- 3b. Li X., Zhang Q., Li Z., Xu S., Zhao C., Chen C., Zhi X., Wang H., Zhua N., Guo K., Polym. Chem. 2016, 7, 1368–1374; [Google Scholar]
- 3c. Kireenko M. M., Kuchuk E. A., Zaitsev K. V., Tafeenko V. A., Oprunenko Y. F., Churakov A. V., Lermontova E. K., Zaitseva G. S., Karlov S. S., Dalton Trans. 2015, 44, 11963–11976; [DOI] [PubMed] [Google Scholar]
- 3d. Horeglad P., Cybularczyk M., Trzaskowski B., Zukowska G. Z., Dranka M., Zachara J., Organometallics 2015, 34, 3480–3496; [Google Scholar]
- 3e. Silvino A. C., Rodrigues A. L. C., Resende J. A. L. C., Inorg. Chem. Commun. 2015, 55, 39–42; [Google Scholar]
- 3f. Quan S. M., Diaconescu P. L., Chem. Commun. 2015, 51, 9643–9646; [DOI] [PubMed] [Google Scholar]
- 3g. Peng K.-F., Chen Y., Chen C.-T., Dalton Trans. 2015, 44, 9610–9619; [DOI] [PubMed] [Google Scholar]
- 3h. Ghosh S., Chakraborty D., Ramkumar V., J. Polym. Sci., Part A: Polym. Chem. 2015, 53, 1474–1491; [Google Scholar]
- 3i. Yang Y., Wang H., Ma H., Inorg. Chem. 2015, 54, 5839–5854; [DOI] [PubMed] [Google Scholar]
- 3j. Ghosh S., Gowda R. R., Jagana R., Chakraborty D., Dalton Trans. 2015, 44, 10410–10422; [DOI] [PubMed] [Google Scholar]
- 3k. Wongmahasirikun P., Promon P., Sangtrirutnugul P., Kongsaeree P., Phomphrai K., Dalton Trans. 2015, 44, 12357–12364; [DOI] [PubMed] [Google Scholar]
- 3l. Yamada S., Takasu A., Takayama S., Kawamura K., Polym. Chem. 2014, 5, 5283–5288; [Google Scholar]
- 3m. Monica F. D., Luciano E., Roviello G., Grassi A., Milione S., Capacchione C., Macromolecules 2014, 47, 2830–2841; [Google Scholar]
- 3n. Whitehorne T. J. J., Schaper F., Inorg. Chem. 2013, 52, 13612–13622; [DOI] [PubMed] [Google Scholar]
- 3o. Dijkstra P. J., Du H., Feijen J., Polym. Chem. 2011, 2, 520–527; [Google Scholar]
- 3p. Kricheldorf H. R., Chem. Rev. 2009, 109, 5579–5594; [DOI] [PubMed] [Google Scholar]
- 3q. Davidson M. G., O′Hara C. T., Jones M. D., Keir C. G., Mahon M. F., Kociok-Köhn G., Inorg. Chem. 2007, 46, 7686–7688; [DOI] [PubMed] [Google Scholar]
- 3r. Chmura A. J., Chuck C. J., Davidson M. G., Jones M. D., Lunn M. D., Bull S. D., Mahon M. F., Angew. Chem. Int. Ed. 2007, 46, 2280–2283; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 2330–2333; [Google Scholar]
- 3s. Wu J., Yu T. L., Chen C. T., Lin C. C., Coord. Chem. Rev. 2006, 250, 602–626; [Google Scholar]
- 3t. Ray L., Katiyar V., Raihan M. J., Nanavati H., Shaikh M. M., Ghosh P., Eur. J. Inorg. Chem. 2006, 3724–3730; [Google Scholar]
- 3u. Cabaret O. D., Vaca B. M., Bourissou D., Chem. Rev. 2004, 104, 6147–6176. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Zhi X., Liu J., Li Z., Wang H., Wang X., Cui S., Chen C., Zhao C., Li X., Guo K., Polym. Chem. 2016, 7, 339–349; [Google Scholar]
- 4b. Kazakov O. I., Kiesewetter M. K., Macromolecules 2015, 48, 6121–6126; [Google Scholar]
- 4c. Saito T., Aizawa Y., Tajima K., Isonob T., Satoh T., Polym. Chem. 2015, 6, 4374–4384; [Google Scholar]
- 4d. Zhu J.-B., Chen E. Y.-X., J. Am. Chem. Soc. 2015, 137, 12506–12509; [DOI] [PubMed] [Google Scholar]
- 4e. Spink S. S., Kazakov O. I., Kiesewetter E. T., Kiesewetter M. K., Macromolecules 2015, 48, 6127–6131; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4f. Coulembier O., Winter J. D., Josse T., Mespouille L., Gerbaux P., Dubois P., Polym. Chem. 2014, 5, 2103–2108; [Google Scholar]
- 4g. Zhao J., Pahovnik D., Gnanou Y., Hadjichristidis N., Polym. Chem. 2014, 5, 3750–3753. [Google Scholar]
- 5.
- 5a. Wheaton C. A., Hayes P. G., Ireland B. J., Dalton Trans. 2009, 38, 4832–4846; [DOI] [PubMed] [Google Scholar]
- 5b. Williams C. K., Chem. Soc. Rev. 2007, 36, 1573–1580; [DOI] [PubMed] [Google Scholar]
- 5c. Chisholm M. H., Gallucci J., Phomphrai K., Inorg. Chem. 2002, 41, 2785–2794; [DOI] [PubMed] [Google Scholar]
- 5d. Chamberlain B. M., Cheng M., Moore D. R., Ovitt T. M., Lobkovsky E. B., Coates G. W., J. Am. Chem. Soc. 2001, 123, 3229–3238. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a.P. M. Schäfer, P. McKeown, M. Fuchs, R. D. Rittinghaus, A. Hermann, J. Henkel, S. Seidel, C. Roitzheim, A. N. Ksiazkiewicz, A. Hoffmann, A. Pich, M. D. Jones, S. Herres-Pawlis, Dalton Trans 2019, 48, 6071–6084; [DOI] [PubMed]
- 6b. Rittinghaus R. D., Schäfer P. M., Albrecht P., Conrads C., Hoffmann A., Ksiazkiewicz A. N., Bienemann O., Pich A., Herres-Pawlis S., ChemSusChem 2019, 1, 1–6; [DOI] [PubMed] [Google Scholar]
- 6c. Schäfer P. M., Fuchs M., Ohligschläger A., Rittinghaus R., McKeown P., Akin E., Schmidt M., Hoffmann A., Liauw M. A., Jones M. D., Herres-Pawlis S., ChemSusChem 2017, 10, 3547–3556; [DOI] [PubMed] [Google Scholar]
- 6d. Jedrzkiewicz D., Adamus G., Kwiecień M., John L., Ejfler J. , Inorg. Chem. 2017, 56, 1349–1365; [DOI] [PubMed] [Google Scholar]
- 6e. Ghosh S., Spannenberg A., Mejía E., Helv. Chim. Acta 2017, 100, −1700176; [Google Scholar]
- 6f. Wang H., Yang Y., Ma H., Inorg. Chem. 2016, 55, 7356–7372; [DOI] [PubMed] [Google Scholar]
- 6g. Ebrahimi T., Mamleeva E., Yu I., Hatzikiriakos S. G., Mehrkhodavandi P., Inorg. Chem. 2016, 55, 9445–9453; [DOI] [PubMed] [Google Scholar]
- 6h. Thevenon A., Romain C., Bennington M. S., White A. J. P., Davidson H. J., Brooker S., Williams C. K., Angew. Chem. Int. Ed. 2016, 55, 8680–8685; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8822–8827; [Google Scholar]
- 6i. Wang J.-H., Tsai C.-Y., Su J.-K., Huang B.-H., Lin C.-C., Ko B.-T., Dalton Trans. 2015, 44, 12401–12410; [DOI] [PubMed] [Google Scholar]
- 6j. Mou Z., Liu B., Wang M., Xie H., Li P., Li L., Li S., Cui D., Chem. Commun. 2014, 50, 11411–11414; [DOI] [PubMed] [Google Scholar]
- 6k. Wang H., Ma H., Chem. Commun. 2013, 49, 8686–8688; [DOI] [PubMed] [Google Scholar]
- 6l. Dai Z., Zhang J., Gao Y., Tang N., Huang Y., Wu J., Catal. Sci. Technol. 2013, 3, 3268–3277; [Google Scholar]
- 6m. Piedra-Arroni E., Ladavière C., Amgoune A., Bourissou D., J. Am. Chem. Soc. 2013, 135, 13306–13309; [DOI] [PubMed] [Google Scholar]
- 6n. Vieira I. dos Santos, Herres-Pawlis S., Eur. J. Inorg. Chem. 2012, 765–774; [Google Scholar]
- 6o. Chen M.-T., Chen C.-T., Dalton Trans. 2011, 40, 12886–12894; [DOI] [PubMed] [Google Scholar]
- 6p. Haak R. M., Decortes A., Escudero-Adán E. C., Belmonte M. Martínez, Martin E., Benet-Buchholz J. A., Kleij W., Inorg. Chem. 2011, 50, 7934–7936; [DOI] [PubMed] [Google Scholar]
- 6q. Wang L., Ma H., Dalton Trans. 2010, 39, 7897–7910; [DOI] [PubMed] [Google Scholar]
- 6r. Darensbourg D. J., Karroonnirun O., Inorg. Chem. 2010, 49, 2360–2371; [DOI] [PubMed] [Google Scholar]
- 6s. Silvernail C. M., Yao L. J., Hill L. M. R., Hillmyer M. A., Tolman W. B., Inorg. Chem. 2007, 46, 6565–6574; [DOI] [PubMed] [Google Scholar]
- 6t. Chen H.-Y., Tang H.-Y., Lin Cu.-C., Macromolecules 2006, 39, 3745–3752; [Google Scholar]
- 6u. Chen H.-Y., Huang B.-H., Lin C.-C., Macromolecules 2005, 38, 5400–5405; [Google Scholar]
- 6v.T. R. Jensen, L. E. Breyfogle, M. A. Hillmyer, W. B. Tolman, Chem. Commun 2004, 2504–2505; [DOI] [PubMed]
- 6w. Cheng M., Attygalle A. B., Lobkovsky E. B., Coates G. W., J. Am. Chem. Soc. 1999, 121, 11583–11584; [Google Scholar]
- 6x. Fliedel C., Vila-Viçosa D., Calhorda M. J., Dagorne S., Avilés T., ChemCatChem 2014, 6, 1357–1367; [Google Scholar]
- 6y. Fliedel C., Rosa V., Alves F. M., Martins A. M., Avilés T., Dagorne S., Dalton Trans. 2015, 44, 12376–12387. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Beavers C. M., Talbo G. H., Richards A. F., J. Organomet. Chem. 2011, 696, 2507–2511; [Google Scholar]
- 7b. Laurence B. M., Lappert M. F., Severn J. R., Chem. Rev. 2002, 102, 3031–3066; [DOI] [PubMed] [Google Scholar]
- 7c. Yu R. C., Hung C. H., Huang J. H., Lee H. Y., Chen J. T., Inorg. Chem. 2002, 41, 6450–6455. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Tang H.-Y., Chen H.-Y., Huang J.-H., Lin C.-C., Macromolecules 2007, 40, 8855–8860; [Google Scholar]
- 8b. Lee W.-Y., Hsieh H.-H., Hsieh C.-C., Lee H. M., Lee G.-H., Huang J.-H., Wu T.-C., Chuang S.-H., J. Organomet. Chem. 2007, 692, 1131–1137. [Google Scholar]
- 9.
- 9a. Chuang H.-J., Chen H.-L., Ye J.-L., Chen Z.-Y., Huang P.-L., Liao T.-T., Tsai T.-E., Lin C.-C., J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 696–707; [Google Scholar]
- 9b. Chuang H.-J., Chen H.-L., Huang B.-H., Tsai T.-E., Huang P.-L., Liao T.-T., Lin C.-C., J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 1185–1196; [Google Scholar]
- 9c. Huang Y., Hung W.-C., Liao M.-Y., Tsai T.-E., Peng Y.-L., Lin C.-C., J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 2318–2329. [Google Scholar]
- 10. Huang Y., Kou X., Duan Y.-L., Ding F.-F., Yin Y.-F., Wang W., Yang Y., Dalton Trans. 2018, 47, 8121–8133. [DOI] [PubMed] [Google Scholar]
- 11. Steiniger P., Schafer P. M., Wolper C., Henkel J., Ksiazkiewicz A. N., Pich A., Herres-Pawlis S., Schulz S., Eur. J. Inorg. Chem. 2018, 4014–4021. [Google Scholar]
- 12.
- 12a. Raghavendra B., Shashank P. V. S., Pandey M. K., Reddy N. D., Organometallics, 2018, 37, 1656–1664; [Google Scholar]
- 12b. Huang H.-C., Li Z.-J., Wang B., Chen X., Li Y.-S., J. Polym. Sci. Part A 2018, 56, 203–212; [Google Scholar]
- 12c. Huang H.-C., Wang B., Zhang Y.-P., Li Y.-S., Polym. Chem. 2016, 7, 5819–5827; [Google Scholar]
- 12d. Chuang W.-J., Huang Y.-T., Chen Y.-H., Lin Y.-S., Lu W.-Y., Lai Y.-C., Chiang M. Y., Hsu S. C. N., Chen H.-Y., RSC Adv. 2016, 6, 33014–33021; [Google Scholar]
- 12e. Chen Y.-H., Chen Y.-J., Tseng H.-C., Lian C.-J., Tsai H.-Y., Lai Y.-C., Hsu S. C. N., Chiang M. Y., Chen H.-Y., RSC Adv. 2015, 5, 100272–100280; [Google Scholar]
- 12f. Altaf C. T., Wang H., Keram M., Yang Y., Ma H., Polyhedron 2014, 81, 11–20; [Google Scholar]
- 12g. Liu Y., Dong W.-S., Liu J.-Y., Lia Y.-S., Dalton Trans. 2014, 43, 2244–2251; [DOI] [PubMed] [Google Scholar]
- 12h. Chen H.-L., Chuang H.-J., Huang B.-H., Lin C.-C., Inorg. Chem. Commun. 2013, 35, 247–251; [Google Scholar]
- 12i. Yu X.-F., Zhang C., Wang Z.-X., Organometallics 2013, 32, 3262–3268; [Google Scholar]
- 12j. Hsiao M.-W., Lin C.-C., Dalton Trans. 2013, 42, 2041–2051; [DOI] [PubMed] [Google Scholar]
- 12k. Chuang H.-J., Su Y.-C., Ko B.-T., Lin C.-C., Inorg. Chem. Commun. 2012, 18, 38–42. [Google Scholar]
- 13.
- 13a. Piesik D. F. J., Range S., Harder S., Organometallics 2008, 27, 6178–6187; [Google Scholar]
- 13b. Neculai D., Roesky H. W., Neculai A. M., Magull J., Schmidt H. G., Noltemeyer M., J. Organomet. Chem. 2002, 643–644, 47–52; [Google Scholar]
- 13c. H. F. Holtzclaw Jr. , Collman J. P., Alire R. M., J. Am. Chem. Soc. 1958, 80, 1100–1103. [Google Scholar]
- 14. Scheiper C., Dittrich D., Wölper C., Bläser D., Roll J., Schulz S., Eur. J. Inorg. Chem., 2014, 2230–2240. [Google Scholar]
- 15. Durand E., Clemancey M., Quoineaud A.-A., Verstraete J., Espinat D., Lancelin J.-M., Energy Fuels 2008, 22, 2604–2610. [Google Scholar]
- 16. 1: [C34H52N4O4Zn2], M=711.53, 0.230 x 0.180 x 0.050, monoclinic, space group P21/c; a=9.8530(6) Å, b=17.8299(10) Å, c=19.6519(12) Å; α=90°, β=92.240(2)°, γ=90°, V=3449.8(4) Å3; Z=4; μ=1.432 mm−1; ρ calc=1.370 g e ⋅ cm−3; 82400 reflections (θ max=30.645°), 10544 unique (R int=0.0393); 438 parameters; largest max./min in the final difference Fourier synthesis 0.927 e ⋅ Å−3/−0.444 e ⋅ Å−3; max./min. transmission 0.75/0.62; R 1=0.0379 (I>2σ(I)), wR 2=0.0850 (all data). 2: [C34H52N4O4Zn2], M=711.53, 0.458×0.194×0.100 mm, triclinic, space group P ; a=8.6700(9) Å, b=10.4507(10) Å, c=11.2159(11) Å; α=67.038(4)°, β=72.090(5)°, γ=81.040(5)°, V=889.64(16) Å3; Z=1; μ=1.388 mm−1; ρ calc=1.328 g × cm−3; 54063 reflections (θ max=33.39 °), 6580 unique (R int=0.0280); 258 restraints, 280 parameters; largest max./min in the final difference Fourier synthesis 0.863 e ⋅ Å−3/ −0.463 e⋅Å−3; max./min. transmission 0.75/0.62; R 1=0.0239 (I >2σ(I)), wR 2=0.0634 (all data). 3: [C38H60N4O4Zn2], M=767.64, 0.189×0.128×0.078 mm, monoclinic, space group P21/c; a=12.0348(9) Å, b=10.2358(8) Å, c=15.5503(11) Å; α=90°, β=97.039(4)°, γ=90°, V=1901.1(2) Å3; Z=2; μ=1.341 mm−1; ρ calc=1.341 g e ⋅ cm−3; 61470 reflections (θ max=33.167 °), 7035 unique (R int=0.0338); 222 parameters; largest max./min in the final difference Fourier synthesis 0.561 e⋅Å−3/ −0.413 e⋅Å−3; max./min. transmission 0.75/0.68; R 1=0.0520 (I>2σ(I)), wR 2=0.0736 (all data). 4: [C30H44N4O4Zn2], M=655.43, 0.250×0.180×0.130 mm, monoclinic, space group P21 /c; a=9.4005(4) Å, b=17.0978(8) Å, c=10.5776(5) Å; α=90°, β=116.049(2)°, γ=90°, V=1527.41(12) Å3; Z=2; μ=1.611 mm−1; ρ calc=1.425 g⋅cm−3; 28881 reflections (θ max=30.525 °), 4582 unique (R int=0.0201); 185 parameters; largest max./min in the final difference Fourier synthesis 0.552 e ⋅ Å−3/ −0.276 e ⋅ Å−3; max./min. transmission 0.75/0.63; R 1=0.0216 (I>2σ(I)), wR 2=0.0604 (all data). 5: [C18H28N2O2Zn], M=369.79, dark red crystal, 0.440×0.360×0.280 mm, monoclinic, space group P21; a=8.323(2) Å, b=12.343(3) Å, c=8.800(2) Å; α=90°, β=91.073(6)°, γ=90°, V=903.9(4) Å3; Z=2; μ=1.369 mm−1; ρ calc=1.359 g⋅cm−3; 15057 reflections (θ max=30.586 °), 5318 unique (R int=0.0242); 1 restraints, 215 parameters; Flack parameter 0.022(3),24 largest max./min in the final difference Fourier synthesis 0.413 e ⋅ Å−3/ −0.244 e ⋅ Å−3; max./min. transmission 0.75/0.52; R 1=0.0182 (I>2σ(I)), wR 2=0.0482 (all data). 6: [C36H56N4O4Zn2], M=739.58, 0.371×0.200×0.160 mm, Tetragonal Space group I41 /a; a=25.6258(16) Å, b=25.6258(16) Å, c=11.1739(7) Å; α=90°, β=90°, γ=90°, V=7337.7 Å3; Z=8; μ=1.349 mm−1; ρ calc=1.339 g⋅cm−3; 87544 reflections (θ max=33.223 °), 7011 unique (R int=0.0258); 214 parameters; largest max./min in the final difference Fourier synthesis 0.461 e⋅Å−3/ −0.375 e⋅Å−3; max./min. transmission 0.75/0.61; R 1=0.0238 (I>2σ(I)), wR 2=0.0650 (all data). 7: [C40H64N4O4Zn2], M=795.69, 0.265×0.230×0.094 mm, monoclinic, space group P21/c; a=9.9064(12) Å, b=14.1071(16) Å, c=14.2769(15) Å; α=90°, β=94.676(5)°, γ=90°, V=1988.6(4) Å3; Z=2; μ=1.250 mm−1; ρ calc=1.329 g⋅cm−3; 109018 reflections (θ max=46.702 °), 13517 unique (R int=0.0378); 231 parameters; largest max./min in the final difference Fourier synthesis 0.742 e ⋅ Å−3/ −0.393 e ⋅ Å−3; max./min. transmission 0.75/0.62; R 1=0.0278 (I>2σ(I)), wR 2=0.0796 (all data). 8: [C32H48N4O4Zn2⋅C7H8], M=775.61, 0.430 x 0.320 x 0.180 mm, monoclinic, space group P21; a=10.6474(5) Å, b=15.1340(6) Å, c=12.4806(6) Å; α=90°, β=103.622(2)°, γ=90°, V=1954.52(15) Å3; Z=2; μ=1.270 mm−1; ρ calc=1.318 g⋅cm−3; 48987 reflections (θ max=30.689°), 11048 unique (R int=0.0229); 1 restraints, 451 parameters; Flack parameter 0.023(3),[26] largest max./min in the final difference Fourier synthesis 0.287 e ⋅ Å−3/−0.247 e ⋅ Å−3; max./min. transmission 0.75/0.60; R 1=0.0202 (I >2σ(I)), wR 2=0.0522 (all data). CCDC-1061215 (1), −1900055 (2), −1900056 (3), −1900057 (4), −1061216 (5), −1900058 (6), −1900059 (7) and −1900060 (8) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 17. Addison A. W., Rao T. N., Reedijk J., van Rijn J., Verschoor G. C., Dalton Trans. 1984, 1349–1356. [Google Scholar]
- 18.Gaussian 16, Revision A.03, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- 19.
- 19a. Obot I. B., Obi-Egbedi N. O., Corros. Sci. 2010, 52, 657–660; [Google Scholar]
- 19b. Zhan C. G., Nicholes J. A., Dixon D. A., J. Phys. Chem. A 2003, 107, 4184–4195. [Google Scholar]
- 20. Zell M. T., Padden B. E., Paterick A. J., Thakur K. A. M., Kean R. T., Hillmyer M. A., Munson E. J., Macromolecules 2002, 35, 7700–7707. [Google Scholar]
- 21.
- 21a. Chisholm M. H., Gallucci J. C., Phomphrai K., Inorg. Chem. 2005, 44, 8004–8010; [DOI] [PubMed] [Google Scholar]
- 21b. Cai C.-X., Amgoune A., Lehmann C. W., Carpentier J.-F., Chem. Commun. 2004, 330–331; [DOI] [PubMed] [Google Scholar]
- 21c. Ovitt T. M., Coates G. W., J. Am. Chem. Soc. 2002, 124, 1316–1326; [DOI] [PubMed] [Google Scholar]
- 21d. Chisholm M. H., Huffman J. C., Phomphrai K., Dalton Trans. 2001, 222–224; [Google Scholar]
- 21e. Thakur K. A. M., Kean R. T., Hall E. S., Kolstad J. J., Lindgren T. A., Doscotch M. A., Siepmann J. I., Munson E. J., Macromolecules 1997, 30, 2422–2428. [Google Scholar]
- 22.
- 22a. Dove A. P., Gibson V. C., Marshall E. L., Rzepa H. S., White A. J. P., Williams D. J., J. Am. Chem. Soc. 2006, 128, 9834–9843; [DOI] [PubMed] [Google Scholar]
- 22b. Chisholm M. H., Eilerts N. W., Huffman J. C., Iyer S. S., Pacold M., Phomphrai K., J. Am. Chem. Soc. 2000, 122, 11845–11854. [Google Scholar]
- 23. Sheldrick G. M., Acta Crystallogr., Sect. A 1990, 46, 467–473. [Google Scholar]
- 24. Sheldrick G. M., SHELX’ ‘A Short History of, Acta Crystallogr., Sect. A 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]
- 25. ShelXle, A Qt GUI for SHELXL, C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Cryst 2011, 44, 1281–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flack H. D., Acta Cryst. A 1983, 39, 876–881. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary