

Abstract
Background
Lynch syndrome (LS) is caused by germline mismatch repair (MMR) gene mutations. De novo MMR gene mutations are rare, and somatic mosaicism in LS is thought to be infrequent. We describe the first case of somatic mosaicism by a de novo MLH1 mutation for a patient diagnosed with a rectosigmoid adenocarcinoma at age 31.
Methods
Twelve years after initial colorectal cancer diagnosis, tumor tissue of the patient was tested with sensitive next generation sequencing (NGS) analysis for the presence of somatic MMR mutations.
Results
In tumor tissue, an inactivating MLH1 mutation (c.518_519del; p.(Tyr173Trpfs*18)) was detected, which was also present at low level in the blood of the patient. In both parents, as well as the patient's sisters, the mutation was not present.
Conclusion
We show that low‐level mosaicism can be detected by using high‐coverage targeted NGS panels on constitutional and/or tumor DNA. This report illustrates that by using sensitive sequencing techniques, more cases of genetic diseases driven by mosaic mutations may be identified, with important clinical consequences for patients and family members.
Keywords: Lynch syndrome, MLH1, mosaicism
1. INTRODUCTION
Heterozygous germline mutations in MMR genes cause Lynch syndrome (LS), an autosomal dominant condition which predisposes to various types of cancer including colorectal cancer (CRC) and endometrial cancer (Hampel et al., 2008, 2006). In contrast to other hereditary CRC syndromes, for example Familial Adenomatous Polyposis, de novo germline mutations in MMR genes are described (Plasilova et al., 2006; Stulp et al., 2006) but appear to be rare. Only 2.3% of MMR gene mutation carriers are reported to have a de novo mutation (Win et al., 2011). Biallelic somatic MMR gene aberrations have been described more frequently, and account for 50%–70% of microsatellite instable (MSI) tumors without causal germline mutations or promotor methylation (Geurts‐Giele et al., 2014; Haraldsdottir et al., 2014; Mensenkamp et al., 2014). Somatic mosaicism is the presence of two or more populations of cells with different genotypes in one individual, and can cause the development of neoplasia if involving a gene related with oncogenesis (Hall, 1988). Somatic mosaicism in LS is thought to be very infrequent; to our knowledge, only two cases have been described thus far. Pastrello et al. (2009) describe somatic mosaicism in a LS patient caused by reversion of an inherited mutation in MLH1 (OMIM 120436), this mutation was present in >80% of both blood and tissues. Furthermore, Sourrouille et al. (2013) describe a patient with somatic mosaicism due to a de novo mutation in MSH2 (OMIM 609309), which was detected in normal colonic tissue but not in the blood. This mosaic MSH2 mutation apparently had germline transmission since this mutation was also present in the patient's son. Here, we report the first case of somatic mosaicism by a de novo MLH1 mutation.
Our male index patient was diagnosed with a rectosigmoid adenocarcinoma at the age of 31. The patient had no relevant medical history. He has two healthy sisters and no offspring. Both his father and mother did not develop any tumor during life and died at 85 and 80 years of age, respectively. A brother of his mother had bladder cancer at age 67, and grandfather from mother's side died from liver cancer diagnosed at age 66.
Because of his young age, he was referred for genetic counseling to the Familial Cancer Clinic. Immunohistochemistry (IHC) of the MMR proteins was performed on the tumor tissue, which showed normal nuclear expression of MSH2 and MSH6 but absence of nuclear expression of MLH1 and PMS2. Molecular analyses showed MSI without MLH1 promotor methylation in the tumor tissue. Mutation scanning of the four MMR genes by denaturing gradient gel electrophoresis (DGGE) of DNA isolated from blood showed no indication for the presence of a germline mutation. The bladder tumor of the index patient's uncle was also examined but did not show any signs of MSI (IHC was not possible). Based on these results, the patient was considered to be Lynch‐like. Therefore he and his sisters underwent colonoscopy every 2 years, no polyps were identified. Additionally, surveillance for extra‐colonic tumors was advised to him and his sisters. One sister decided to have her ovaries and uterus preventively removed at 46 years of age. In both organs, no signs of neoplastic growth were observed.
Recently, all Lynch‐like patients of the Familial Cancer Clinic at the Netherlands Cancer Institute are being evaluated for somatic MMR gene mutations, as the presence of a somatic MMR gene mutation can indicate a sporadic origin of the tumor. Therefore, last year the index patient visited the Familial Cancer Clinic again, 12 years after the initial CRC diagnosis. Subsequently, the tumor was tested with next generation sequencing (NGS) for the presence of somatic MMR mutations.
2. MATERIALS AND METHODS
2.1. Ethical compliance
The index patient was counseled by the Familial Cancer Clinic and provided informed consent.
2.2. Somatic mutation analysis of the MMR genes
Normal and tumor tissues of the index patient were manually microdissected from five to ten hematoxylin‐stained sections of formalin‐fixed paraffin‐embedded (FFPE) tissue, after which proteinase K and 5% Chelex 100 resin was added for DNA isolation, as previously described (van Lier et al., 2010). DNA concentrations were measured with the Qubit 2.0 Fluorometer and 10 ng DNA input was used for mutation analysis. Mutation analysis with the Ion S5 XL system (Ion Torrent) was performed with suppliers' materials and protocols (Thermo Fisher Scientific). A custom‐made primer panel was designed using the ampliseq designer. This panel targets the open reading frame including the intron–exon boundaries of MLH1 (NM_000249.3), MSH2 (NM_000251.2), MSH6 (OMIM 600678, NM_000179.2), and PMS2 (OMIM 600259, NM_000535.7), with a coding sequence coverage of 97%, 94%, 95%, and 81% respectively, and hotspots for BRAF (OMIM 164757, NM_004333.5; exon 11 and 15), POLD1 (OMIM 174761, NM_002691.3; exon 12), and POLE (OMIM 174762, NM_006231.3; exon 3 and 13). Furthermore, single nucleotide polymorphisms in and around the MLH1, MSH2, MSH6, and PMS2 genes are included to detect copy number variations, as previously described (Dubbink et al., 2016). Primer sequences are available on request.
Libraries were made using the Ion AmpliSeq Library Kit 2.0‐384 LV according to the Ion Ampliseq Library Preparation User Guide. Template was performed with the Ion 510/520/530 Chef kit and sequencing was performed on a 530 chip using the Ion S5 XL system. Data were analyzed using SeqPilot version 4.2.2 (JSI medical systems).
2.3. Sanger sequencing of MLH1
DNA isolation from blood of the index patient and his two sisters was performed using DNAzol (Thermo Fischer Scientific). DNA from normal FFPE tissue of the deceased parents was isolated using QIA Amp DNA purification kit (Qiagen). Sanger sequencing for MLH1 exon 6 was performed using standard procedures.
3. RESULTS
In the tumor, an inactivating MLH1 mutation (c.518_519del; p.(Tyr173Trpfs*18); see Figure 1) was identified and loss of the wild‐type MLH1 allele was seen, indicating biallelic MLH1 inactivation. This explains the MSI phenotype and the absence of nuclear expression of MLH1 and PMS2 in the tumor cells. Additionally, five missense variants of unknown significance were detected in tumor tissue only, one in MSH2 (c.2198C>A; p.(Ala733Asp)) and four in MSH6 (c.821G>A; p.(Ser274Asn), c.1730G>A; p.(Arg577His), c.2419G>A; p.(Glu807Lys) and c.3163G>A; p.(Ala1055Thr)).
Figure 1.
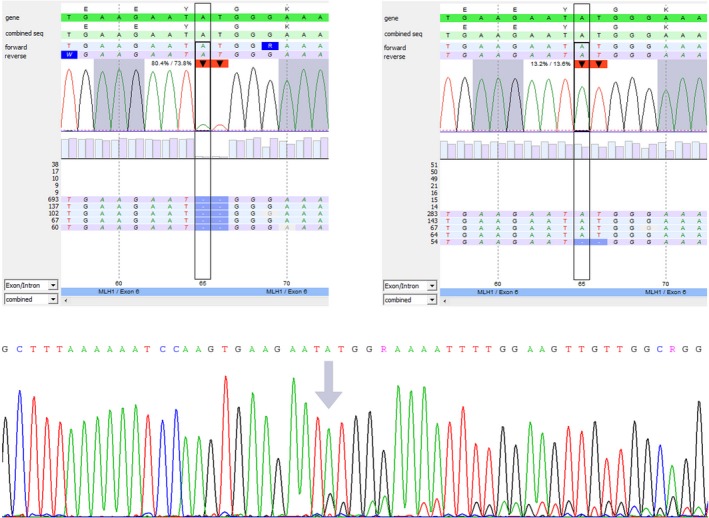
MLH1ª c.518_519del; p.(Tyr173Trpfs*18) analysis. (a) NGS analysis of the colon adenocarcinoma showing the mutation in 80% of the reads. (b) NGS analysis of DNA isolated from blood showing the mutation in 13% of the reads. (c) Sanger sequencing of DNA isolated from blood confirming the presence of the MLH1 mutation (arrow indicates start of the deletion). ªOMIM 120436, NM_000249.3. NGS, next generation sequencing
Surprisingly, the MLH1 mutation was also detected by NGS at low frequencies in DNA isolated from several normal tissues, including smooth muscle near the tumor, epithelium from the small intestine and lymph nodes without metastasis (see Table 1). NGS showed the presence of the inactivating MLH1 mutation with a variant allele frequency of 13% in the blood sample, a level that would not generally be detectable by DGGE. These results suggested mosaicism of the MLH1 mutation in at least two germ layers. To rule out reversion of an inherited MLH1 mutation as described before (Pastrello et al., 2009), normal tissue of the deceased parents was tested using Sanger sequencing for the presence of the MLH1 mutation. In both parents, the mutation was not detected, indicating the appearance of a post‐zygotic de novo mutation in the index patient. As expected, the mutation was also not identified in the DNA isolated from blood of the patient's two sisters. Therefore his sisters were dismissed from further LS surveillance.
Table 1.
Different tissues from the rectosigmoid resection of the index patient (including part of the bladder and ileum), as well as DNA isolated from blood, were included for somatic MMR gene sequencing analysis
| Tissue | Neoplastic cell content as estimated by a GE pathologist | Variant allele frequency (VAF) of MLH1 a c.518_519del; p.(Tyr173Trpfs*18) |
|---|---|---|
| Colon adenocarcinoma | 70% | 80%b |
| Colon muscle near adenocarcinoma | 0% | 14% |
| Small intestine muscle | 0% | 16% |
| Small intestine epithelium | 0% | 14% |
| Lymph node | 0% | 14% |
| Blood sample (in duplo) | Not applicable | 13% |
Abbreviation: MMR, mismatch repair.
OMIM 120436, NM_000249.3.
VAF indicative for loss of the wild‐type allele.
It is thought that somatic mosaicism gives a milder phenotype than full LS depending on the extend of the mosaicism in the cell types prone for malignant transformation (Hall, 1988). However, as the degree of mosaicism can vary strongly, even within an organ or cell type (Forsberg, Gisselsson, & Dumanski, 2017), it is impossible to estimate the patients' risk for various types of cancer based on the mutation rate. Therefore, the index patient is now considered to be a LS patient and is advised the regular LS surveillance. As the patient does not have offspring, we cannot check for gonadal mosaicism. After the above results were obtained, the patient continued the LS surveillance program.
4. DISCUSSION
As described previously for other mosaic cases, this report highlights that, as former gene scanning techniques in clinical genetics laboratories will be replaced by deep sequencing techniques, more cases of genetic diseases driven by mosaic mutations may be identified, with important clinical consequences for patients and family members. If no pathogenic mutation is detected in the blood, mutational analysis of tumor tissue might help to identify low‐level mosaicism. All neoplastic cells will most likely harbor the pathogenic mutation, and loss of the wild‐type allele, a common event for tumor suppressor genes, enriches for the pathogenic mutation even further. If a mutation is detected in tumor tissue, the surrounding normal tissue and constitutional DNA can be screened for the presence of this specific mutation with very sensitive techniques (like NGS), enabling detection of low‐frequency mutations.
When routine mutation analysis of constitutional DNA from patients with a clear clinical suspicion for a tumor syndrome is negative, one should consider to perform high‐coverage targeted NGS of the indicated genes to detect possible mosaicism. Furthermore, sequencing of tumor tissue can guide detection of a low‐level mosaic variant in the germline. This diagnostic strategy will result in improved health care both for the index patient and his or her family members.
CONFLICT OF INTEREST
The authors declare no conflict of interests.
Geurts‐Giele WR, Rosenberg EH, Rens AV, Leerdam MEV, Dinjens WN, Bleeker FE. Somatic mosaicism by a de novo MLH1 mutation as a cause of Lynch syndrome. Mol Genet Genomic Med. 2019;7:e699 10.1002/mgg3.699
REFERENCES
- Dubbink, H. J. , Atmodimedjo, P. N. , van Marion, R. , Krol, N. M. G. , Riegman, P. H. J. , Kros, J. M. , … Dinjens, W. N. M. (2016). Diagnostic detection of allelic losses and imbalances by next‐generation sequencing: 1p/19q co‐deletion analysis of gliomas. The Journal of Molecular Diagnostics, 18, 775–786. 10.1016/j.jmoldx.2016.06.002 [DOI] [PubMed] [Google Scholar]
- Forsberg, L. A. , Gisselsson, D. , & Dumanski, J. P. (2017). Mosaicism in health and disease—clones picking up speed. Nature Reviews Genetics, 18, 128–142. 10.1038/nrg.2016.145 [DOI] [PubMed] [Google Scholar]
- Geurts‐Giele, W. R. R. , Leenen, C. H. M. , Dubbink, H. J. , Meijssen, I. C. , Post, E. , Sleddens, H. F. B. M. , … Dinjens, W. N. M. (2014). Somatic aberrations of mismatch repair genes as a cause of microsatellite‐unstable cancers. The Journal of Pathology, 234, 548–559. 10.1002/path.4419 [DOI] [PubMed] [Google Scholar]
- Hall, J. G. (1988). Review and hypotheses: somatic mosaicism: observations related to clinical genetics. American Journal of Human Genetics, 43, 355–363. [PMC free article] [PubMed] [Google Scholar]
- Hampel, H. , Frankel, W. L. , Martin, E. , Arnold, M. , Khanduja, K. , Kuebler, P. , … de la Chapelle, A. (2008). Feasibility of screening for Lynch syndrome among patients with colorectal cancer. Journal of Clinical Oncology, 26, 5783–5788. 10.1200/JCO.2008.17.5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel, H. , Frankel, W. , Panescu, J. , Lockman, J. , Sotamaa, K. , Fix, D. , … de la Chapelle, A. (2006). Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Research, 66, 7810–7817. 10.1158/0008-5472.CAN-06-1114 [DOI] [PubMed] [Google Scholar]
- Haraldsdottir, S. , Hampel, H. , Tomsic, J. , Frankel, W. L. , Pearlman, R. , de la Chapelle, A. , & Pritchard, C. C. (2014). Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology, 147(1308–1316), e1301 10.1053/j.gastro.2014.08.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensenkamp, A. R. , Vogelaar, I. P. , van Zelst–Stams, W. A. G. , Goossens, M. , Ouchene, H. , Hendriks–Cornelissen, S. J. B. , … Ligtenberg, M. J. L. (2014). Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch‐repair deficiency in Lynch syndrome‐like tumors. Gastroenterology, 146, 643–646. 10.1053/j.gastro.2013.12.002 [DOI] [PubMed] [Google Scholar]
- Pastrello, C. , Fornasarig, M. , Pin, E. , Berto, E. , Pivetta, B. , & Viel, A. (2009). Somatic mosaicism in a patient with Lynch syndrome. American Journal of Medical Genetics A, 149A, 212–215. 10.1002/ajmg.a.32620 [DOI] [PubMed] [Google Scholar]
- Plasilova, M. , Zhang, J. , Okhowat, R. , Marra, G. , Mettler, M. , Mueller, H. , & Heinimann, K. (2006). A de novo MLH1 germ line mutation in a 31‐year‐old colorectal cancer patient. Genes, Chromosomes & Cancer, 45, 1106–1110. 10.1002/gcc.20374 [DOI] [PubMed] [Google Scholar]
- Sourrouille, I. , Coulet, F. , Lefevre, J. H. , Colas, C. , Eyries, M. , Svrcek, M. , … Soubrier, F. (2013). Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Familial Cancer, 12, 27–33. 10.1007/s10689-012-9568-9 [DOI] [PubMed] [Google Scholar]
- Stulp, R. P. , Vos, Y. J. , Mol, B. , Karrenbeld, A. , de Raad, M. , van der Mijle, H. J. , & Sijmons, R. H. (2006). First report of a de novo germline mutation in the MLH1 gene. World Journal of Gastroenterology, 12, 809–811. 10.3748/wjg.v12.i5.809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lier, M. G. F. , Wagner, A. , Van Leerdam, M. E. , Biermann, K. , Kuipers, E. J. , Steyerberg, E. W. , … Dinjens, W. N. M. (2010). A review on the molecular diagnostics of Lynch syndrome: a central role for the pathology laboratory. Journal of Cellular and Molecular Medicine, 14, 181–197. 10.1111/j.1582-4934.2009.00977.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win, A. K. , Jenkins, M. A. , Buchanan, D. D. , Clendenning, M. , Young, J. P. , Giles, G. G. , … Lindor, N. M. (2011). Determining the frequency of de novo germline mutations in DNA mismatch repair genes. Journal of Medical Genetics, 48, 530–534. 10.1136/jmedgenet-2011-100082 [DOI] [PMC free article] [PubMed] [Google Scholar]