

Abstract
Background
Site‐1 Protease (S1P) is a Golgi‐resident protein required for the activation of regulatory proteins that drive key cellular functions, including, the unfolded protein response (UPR) and lipid and cholesterol biosynthesis. While disruptions in S1P function have been widely characterized in animal models, to date, the implications of disrupted S1P function in human disease states are not completely known.
Methods
The patient and both parents underwent whole exome and mitochondrial DNA sequencing, and Sanger sequencing was used to confirm the mutation. Western blotting and immunofluorescence studies were performed on either proband‐derived fibroblasts or on an established cell line to assess protein expression and cellular localization of the mutated S1P protein. Quantitative real‐time PCR and luciferase reporter assays were used to examine activation of S1P target pathways in the context of the S1P mutation.
Results
We describe a female patient with a de novo heterozygous missense mutation in the transmembrane domain of S1P (p. Pro1003Ser). The patient presented to our neuromuscular clinic with episodic, activity‐induced, focal myoedema and myalgias with hyperCKemia. Her clinical phenotype was complex and included gastrointestinal hypomotility, ocular migraines, and polycystic ovary syndrome. Molecular analysis using proband‐derived fibroblasts and cell lines harboring the Pro1003Ser mutation demonstrated increased activation of UPR and lipid and cholesterol regulatory pathways and localization of S1P Pro1003Ser in the Golgi.
Conclusion
These findings suggest a critical function for S1P in several human organ systems and implicate an important role for S1P in various human disease states.
Keywords: ER stress, hyperCKemia, MBTPS1, myoedema, PCOS, Site‐1 Protease, SREBP
1. INTRODUCTION
Site‐1 Protease (S1P; also known as subtilisin/kexin‐isozyme 1 or PCSK8) is a membrane‐bound serine protease required for the activation of key regulators of cellular functions, most notably, the unfolded protein response (UPR), lysosomal biogenesis, and lipid and cholesterol biosynthesis (Marschner, Kollmann, Schweizer, Braulke, & Pohl, 2011; Raggo et al., 2002; Sakai et al., 1998; Stirling & O’hare, 2006; Ye et al., 2000). Disruptions in these cellular functions are implicated in several human diseases and restoring their normal function is a common therapeutic focus (Cohen, Horton, & Hobbs, 2011; Goldstein, Hazzard, Schrott, Bierman, & Motulsky, 1973; Hong, Kim, Kim, & Park, 2017; Hotamisligil, 2010). S1P is encoded by the MBTPS1 gene (OMIM accession number 603355) and exists as an inactive precursor in the endoplasmic reticulum (ER) (Elagoz, Benjannet, Mammarbassi, Wickham, & Seidah, 2002; Espenshade, Cheng, Goldstein, & Brown, 1999; Seidah et al., 1999). After a series of autocatalytic cleavage events, the mature S1P localizes to the Golgi where it can proteolytically cleave its substrates, the most well‐known being the sterol regulatory element‐binding protein family (SREBPs) of transcription factors, essential regulators of lipid and cholesterol homeostasis, and ATF6, a component of the UPR required to restore ER homeostasis (Elagoz et al., 2002; Espenshade et al., 1999; Sakai et al., 1998; Seidah et al., 1999; Ye et al., 2000).
Many animal studies suggest an important role for S1P in developmental, physiologic, and pathogenic functions (Brandl et al., 2009; Hawkins et al., 2008; Hay et al., 2007; Kondo et al., 2018; Rutschmann et al., 2012; Uchida et al., 2016; Yang et al., 2001). These observations along with S1P's regulation of signaling pathways involved in insulin resistance, nonalcoholic fatty liver disease, and persistent viral infection has made S1P the focus of pharmacological development efforts (Hawkins et al., 2008; Hay et al., 2007; Uchida et al., 2016). While these animal studies underscore an important role for S1P in health and disease, to date, the impact of S1P disruption on human health is not clearly understood.
Here, we describe a 24‐year old female patient with a novel heterozygous de novo mutation in MBTPS1 (NM_003791.3 c.3007C>T) that corresponds to a mutation in the highly conserved transmembrane domain of S1P (NP_003782.1 p.Pro1003Ser). The mutated MBTPS1 transcript is expressed at levels similar to the wild‐type transcript. The patient exhibited a complex phenotype suggestive of disrupted metabolism that includes muscle fatigue and hyperCKemia precipitated by moderate physical activity. Additional clinical complications include ovarian cysts, small fiber neuropathy, and chronic constipation. Our functional analysis of the mutant protein indicates Golgi localization of the protein and increased activation of its target pathways.
2. METHODS
2.1. Editorial policies and ethical considerations
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Written informed consent was obtained from the patient and her parents for exome and mitochondrial DNA sequencing and the patient provided written informed consent to participate in research studies to investigate the clinical significance of the genetic variant as approved by the Institutional Review Board (IRB) of Washington University in St. Louis.
2.2. Whole exome and mitochondrial DNA sequencing
Exome sequencing was performed by GeneDx (Gaithersburg, MD) using Agilent SureSelect XT2 All Exon V4 Kit and Illumina HiSeq 2000 100 bp paired‐end reads. Sequence was aligned to the UCSC build hg19 reference sequence. Mean depth of coverage of known protein‐coding RefSeq genes was 89× with a quality threshold of 97.9%. For the MBTPS1 gene (gDNA NG_033017.1, cDNA NM_003791.3), 94.1% of the coding region was covered at a minimum of 10X by exome sequencing. GeneDx's XomeAnalyzer was used to evaluate sequence changes between the proband, parental samples, and reference. Sanger sequencing was used for confirmation of the reported mutation.
2.3. Transcript characterization
Genomic DNA and RNA were extracted from patient and healthy donor fibroblasts using RNA‐Bee (Tel‐Test, Inc.) and cDNA was synthesized from RNA using SuperScript III (Invitrogen). To assess possible allele‐specific expression of the c.3007 mutant allele, sets of PCR primers were designed that would amplify a genomic DNA or cDNA region that includes the MBTPS1 c.3007C>T variant. The genomic DNA primers were located in adjacent introns, while the cDNA primers were located in adjacent exons, spanning two exon–exon junctions. Both the genomic and cDNA‐derived PCR products were sequenced on an Applied Biosystems 3130xl capillary sequencer and the Sanger trace files were compared with Applied Biosystems Sequence Analysis software. Illumina adaptors were ligated onto the ends of the patient and control cDNA‐derived PCR products and deep next‐generation sequencing was performed using an Illumina Miseq instrument (Illumina).
2.4. Cell culture
All cells were maintained at 37°C with 5% CO2. Primary skin fibroblasts were derived from patient and healthy donor skin biopsies under an approved IRB protocol at Washington University School of Medicine (protocol #: 201103019). Human fibroblasts were cultured in high‐glucose DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin‐streptomycin (PS). Patient and donor fibroblasts were used at matched passage numbers no greater than passage 10. The S1P‐deficient SRD‐12B cell line, a generous gift from Drs. Michael Brown and Joseph Goldstein of the University of Texas Southwestern Medical Center at Dallas and Dr. Peter J. Espenshade of the Johns Hopkins University School of Medicine, (Rawson, Cheng, Brown, & Goldstein, 1998) was maintained in Medium B (DMEM/F12, 5% FBS, 1% PS, 5 μg/ml cholesterol, 1 mmol/L sodium mevalonate, 20 μmol/L sodium oleate). Medium A (DMEM/F12, 1% PS) was supplemented with 5% lipoprotein‐deficient serum (LPDS) where indicated.
2.5. Cell growth assay
On Day 0, SRD‐12B cells were plated in 6 cm2 dishes at 2 × 105 in Medium B, transfected using Lipofectamine 2000 (Invitrogen) with wild‐type S1P, S1P Pro1003Ser, or empty vector on Day 1, and shifted to Medium A with 5% LPDS on Day 2. Medium was changed every two days and on Day 7, cells were rinsed in PBS, fixed in ice‐cold methanol at −20°C, and stained with 5% crystal violet.
2.6. Luciferase reporter assay
On Day 0, SRD‐12B cells were plated in 12‐well plates at 50% confluency in Medium B. On Day 1, wells were transfected in quadruplicate using 3 μl FuGENE HD (Promega), 50 μl Optimem Media (Gibco), 1.25 μg FAS‐Luc, 0.12 μg Renilla, and either 0.25 μg wild‐type S1P or S1P Pro1003Ser constructs per well. After 9–10 hr, cells were washed twice with 1× PBS and fed DMEM/F12 for 16 hr. Luciferase activities were then measured using the Promega Dual‐Glo Luciferase Assay kit and the ratio between firefly and Renilla luciferase activities was determined.
2.7. Immunofluorescence
Human skin fibroblasts were transfected using Lipofectamine 2000 with either wild‐type S1P or S1P Pro1003Ser constructs. After 24 hr, cells were fixed in paraformaldehyde and incubated with anti‐FLAG, anti‐KDEL, and anti‐GM130 antibodies followed by appropriate secondary antibodies. Cells were DAPI stained and visualized by fluorescence microscopy.
2.8. Statistical analysis
Data are expressed as mean ± SEM. Student's t tests were used to determine significant differences. p < 0.05 was considered statistically significant. Additional methods can be found in Supplemental Material.
3. RESULTS
The proband presented to our neuromuscular clinic for evaluation of recurrent episodes of hyperCKemia with muscle fatigue, swelling, and myoedema. During her first reported episode, she presented with elevated levels of creatine kinase (8,517 U/L), aspartate aminotransferase (212 U/L), and alanine aminotransferase (145 U/L), despite not exercising for over a week prior to the incident. Her interictal serum creatine kinase levels were normal. Subsequent episodes of hyperCKemia developed within hours to days of moderate exercise. After the first episode she developed persistent muscle fatigue that was consistently exacerbated by heavy activity and exercise, but no objective motor deficits were evident on exam. Prior to her first episode of hyperCKemia, the patient had been highly athletic in high school and college, having competed in track and volleyball. As an infant, the patient had normal motor development and was born at full term. She sustained a broken kneecap and hip from a volleyball injury at age 20 but had no major musculoskeletal injuries or impairments. Additional clinical complications included small fiber and autonomic/enteric neuropathy complicated by chronic constipation, gastroparesis, gastrointestinal hypomotility and chronic nausea and vomiting, unexplained fevers with leukocytosis, ocular migraines, pelvic inflammatory disease, and polycystic ovary syndrome based on the Rotterdam criteria (Rotterdam ESHRE/ASRM‐Sponsored PCOS Consensus Workshop Group, 2004).
Muscle histology revealed normal skeletal muscle structure, lipid content, fiber type distribution, and mitochondrial respiratory chain function (Figure S1a–d). Electron microscopy showed the patient's skeletal muscle had subtle disorganization of ultrastructure consisting of subsarcolemmal collections of mitochondria with scattered fat globules (Figure S1e) and mildly abnormal size and shape (Figure S1f); however, mitochondrial enzymatic activities were normal.
Whole exome and mitochondrial DNA sequencing of the proband and her parents revealed a heterozygous de novo missense mutation in exon 23 of the MBTPS1 (NM_003791.3 c.3007C>T) gene that corresponded to a single amino acid substitution in the transmembrane domain of S1P (NP_003782.1 p.Pro1003Ser) (Figure 1a). Proline 1003 is highly conserved across both vertebrate and invertebrate species (Figure 1b). The MBTPS1 c.3007C>T mutation was not identified in the ~6,500 individuals of European and African American ancestry in the NHLBI Exome Sequencing Project. This variant was not found among healthy controls based on ExAc and GnomAD; however, a mutation in the same codon (p.Pro1003His) was found in 1/108,864 European alleles by GnomAD. Substitution of a hydrophobic proline with a polar serine at residue 1003 within the transmembrane domain of S1P was predicted to be deleterious by PolyPhen‐2 (HDIV score 0.993; probably damaging), Align GVGD (score Class 65; most likely to interfere with function), and MutationTaster (p = 0.999; disease causing), but not by SIFT (score 0.19; tolerated) (Adzhubei et al., 2010; Kumar, Henikoff, & Ng, 2009; Mathe et al., 2006; Schwarz, Cooper, Schuelke, & Seelow, 2014; Tavtigian et al., 2006). MBTPS1 transcript levels in proband‐derived skin fibroblasts were similar to levels measured in control fibroblasts isolated from an individual without an S1P mutation (Figure 1c). Comparison of the Sanger sequencing trace files of the patient's genomic and cDNA‐derived PCR products at the MBTPS1 c.3007C>T locus showed approximately equal expression of the wild‐type and mutant transcripts (Figure 1d). Deep next generation sequencing of the patient cDNA‐derived PCR products confirmed this observation, with 7216x coverage and approximately equal allele balance (3,452 wild‐type alleles [47.8%] and 3,764 mutant alleles [52.2%]).
Figure 1.
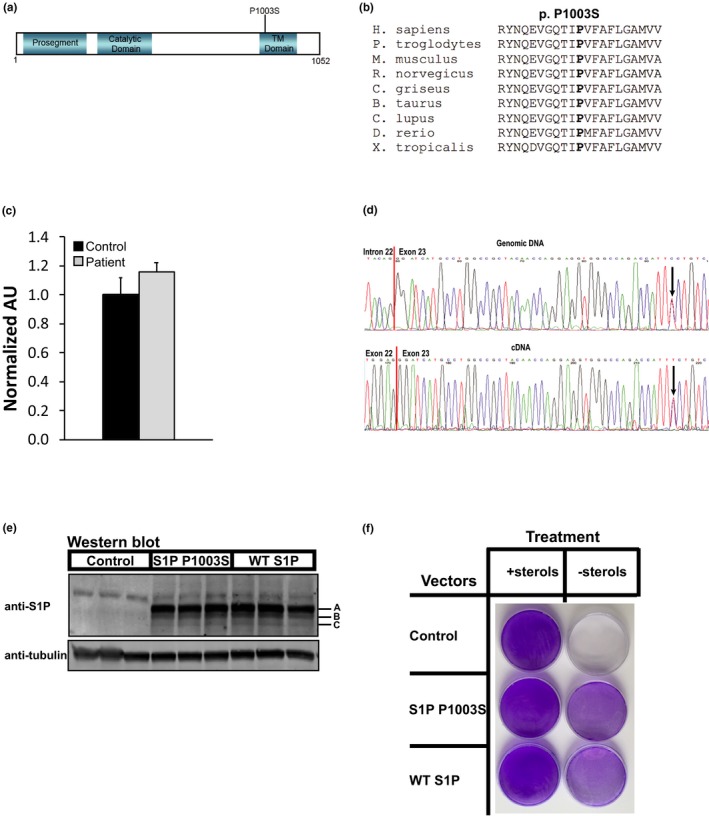
Genetic and functional analysis of S1P Pro1003Ser. (a) Schematic of S1P protein with the Pro1003Ser mutation and S1P protein domains indicated, TM, transmembrane domain. (b) Multispecies alignment of S1P amino acid sequences demonstrating the mutated proline 1003 residue is conserved (shown in bold). (c) mRNA expression levels of the MBTPS1 transcript in cultured control‐ and patient‐derived skin fibroblasts. n = 3. (d) Sanger sequencing trace files of patient genomic DNA (top) and cDNA (bottom)‐derived PCR products at the MBTPS1 c.3007C>T locus. Intron–exon boundary (genomic) and exon–exon junction (cDNA) are indicated. Arrows denote the variant location. (e) Western blot of whole‐cell lysates (60μg) from SRD‐12B cells transiently transfected with mock, WT S1P, or S1P Pro1003Ser plasmids after 24 hr. S1P‐A, B, and C forms are indicated. Blots were probed with S1P and α‐tubulin (loading control) antibodies. n = 3. (f) SRD‐12B cells were transfected as in (e) and grown either in medium supplemented with lipids and cholesterol (left column) or in lipid‐ and cholesterol‐free medium (right column) for 7 days followed by fixation in methanol and crystal violet staining, as reported previously (Rawson et al., 1998). Images are representative of three independent experiments
While disruption of S1P in mice decreases total plasma triglyceride and cholesterol levels (Yang et al., 2001), the patient's total plasma triglyceride and cholesterol levels were normal (Table S1) and her BMI was 20.7 kg/m2.
S1P must undergo multiple autocatalytic processing events in the ER before it can localize to the Golgi as an active mature protease (designated as C: 96 kDa estimated) (Espenshade et al., 1999; Ramos da Palma, Cendron, Seidah, Pasquato, & Kunz, 2015; Sakai et al., 1998). The ER‐resident S1P precursors exist as a proprotein (designated as A: 115 kDa estimated) and an intermediate cleaved protein (designated as B: 102 kDa estimated) (Espenshade et al., 1999; Ramos da Palma et al., 2015). To examine whether the variant MBTPS1 allele produced these three S1P forms, we transfected a Chinese hamster ovary cell line that lacks S1P, SRD‐12B, (Rawson et al., 1998) with plasmids encoding either human wild‐type (WT) S1P or S1P Pro1003Ser with a C‐terminal FLAG tag and visualized proteins via Western blot analysis. S1P Pro1003Ser‐transfected cells expressed all three forms of S1P as did WT S1P‐transfected cells (Figure 1e).
Because S1P is required for SREBP‐dependent lipid and cholesterol biosynthesis, SRD‐12B cells, which lack S1P, are lipid and cholesterol auxotrophs and must be supplemented with lipids and cholesterol to survive (Rawson et al., 1998). To test the functionality of S1P Pro1003Ser, we performed a cell complementation assay with SRD‐12B cells transfected with human S1P Pro1003Ser or WT S1P and grew cells in the absence of lipids and cholesterol. While mock transfected SRD‐12B cells failed to grow under these conditions, as reported previously (Rawson et al., 1998), SRD‐12B cells that expressed S1P Pro1003Ser grew in the absence of supplemented lipids and cholesterol similar to WT S1P‐expressing cells (Figure 1f).
To further examine the functionality of S1P Pro1003Ser, we tested the mutant's ability to activate its substrate pathways, SREBP and ATF6, using S1P Pro1003Ser patient‐derived fibroblasts and control fibroblasts from an individual without an S1P mutation. Treatment of sterol‐depleted cells with the HMG‐CoA reductase inhibitor mevastatin (compactin) promotes transcription of SREBP1a and 2 target genes (Brown, Faust, Goldstein, Kaneko, & Endo, 1978; Shimomura, Shimano, Horton, Goldstein, & Brown, 1997). We examined whether S1P Pro1003Ser patient‐derived fibroblasts respond to mevastatin treatment. S1P Pro1003Ser fibroblasts showed a dosage‐dependent increase in the expression of SREBP1 and 2 target genes that was higher than the expression levels of treated control fibroblasts (Figure 2a). Even in the absence of mevastatin, expression levels of SREBP1 and 2 target genes were elevated in S1P Pro1003Ser fibroblasts compared to control fibroblasts (Figure 2a). We next tested whether S1P Pro1003Ser could activate ATF6, a transcription factor required to restore ER homeostasis during ER stress, by treating fibroblasts with tunicamycin to induce ER stress. S1P Pro1003Ser patient fibroblasts treated with tunicamycin had higher expression levels of spliced XBP‐1 and the ATF6 target genes Grp78 and CHOP than control fibroblasts (Figure 2b). To determine whether S1P Pro1003Ser could directly activate SREBPs, we performed a luciferase reporter assay using an SREBP1‐response fatty acid synthase luciferase promoter construct. SRD‐12B cells expressing S1P Pro1003Ser exhibited higher levels of SREBP1‐dependent promoter activity than cells expressing WT S1P (Figure 2c).
Figure 2.
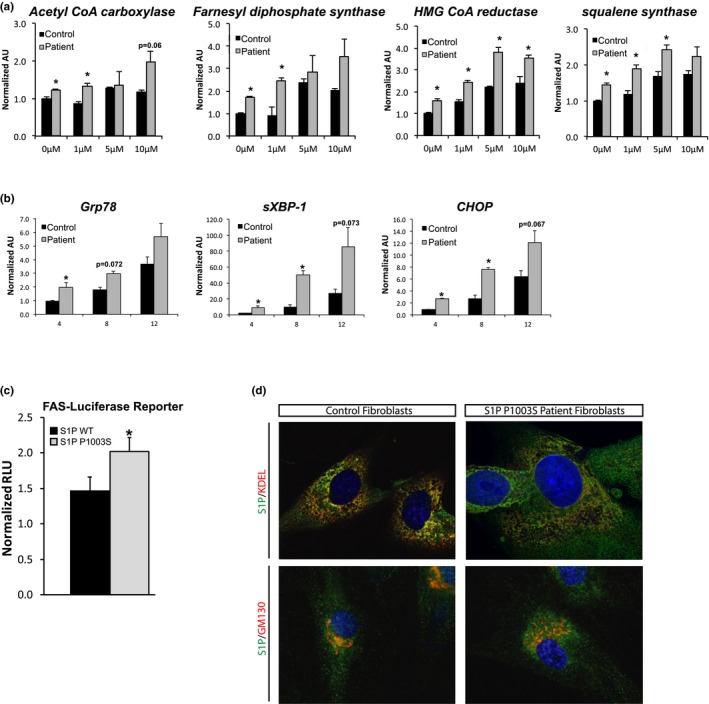
S1P Pro1003Ser has enhanced activity and accumulates in the Golgi. mRNA expression levels in cultured control‐ and patient‐derived skin fibroblasts of (a) SREBP1 and 2 target genes after 4 hr of treatment with the indicated concentrations of mevastatin and (b) UPR and ATF6 target genes following treatment with 1 µg/ml of tunicamycin for 4, 8, and 12 hr. (c) SRD‐12B cells transiently transfected with FAS‐Luc, Renilla, and WT S1P or S1P Pro1003Ser plasmids as indicated, after 10 hr, cells were treated with DMEM/F12 for 16 hr. Luciferase activities were measured and the ratio between firefly and Renilla luciferase activities was determined. (d) Immunofluorescence of S1P, ER marker KDEL, and Golgi marker GM130 in S1P Pro1003Ser patient and control fibroblasts. Cells were transfected with FLAG‐tagged WT S1P or S1P Pro1003Ser as indicated. Representative images are shown. All data are expressed as mean S.E. of three to five independent experiments, *p < 0.05
The intracellular localization of mature S1P and its substrates to the Golgi is an important regulatory mechanism that governs S1P function (DeBose‐Boyd et al., 1999; Marschner et al., 2011; Nohturfft, DeBose‐Boyd, Scheek, Goldstein, & Brown, 1999; Raggo et al., 2002; Sakai et al., 1998; Shen, Chen, Hendershot, & Prywes, 2002; Stirling & O’hare, 2006; Ye et al., 2000). To begin to understand the mechanism behind the enhanced activity of S1P Pro1003Ser, we investigated whether S1P Pro1003Ser localization was altered in the patient's fibroblasts. Control and patient fibroblasts were transfected with either FLAG‐tagged human WT S1P or S1P Pro1003Ser, respectively, and double‐labeled with anti‐FLAG and antibodies against KDEL‐bearing proteins (ER marker) and GM130 (Golgi marker) (Figure 2d). Similar to WT S1P, S1P Pro1003Ser localized to both the ER and the Golgi. This result was also observed when performed in SRD‐12B cells transfected with WT S1P and S1P Pro1003Ser (Figure S3).
4. DISCUSSION
In conclusion, we describe a patient with a de novo heterozygous variant in the gene MBTPS1 that produces a missense mutation in the transmembrane domain of S1P. Deep next generation sequencing showed that the mutant MBTPS1 transcript is expressed at levels similar to the wild‐type transcript in patient‐derived skin fibroblasts. The patient presented with a complex phenotype that included episodic hyperCKemia, focal myoedema, ocular migraines, and polycystic ovary syndrome. To date, associations between these clinical manifestations and S1P have not been described.
Our functional analysis showed the MBTPS1 variant produces a stable S1P Pro1003Ser protease that is able to complement lipid and cholesterol biosynthetic capacities in cells that lack S1P and is abundant in both the ER and the Golgi. S1P Pro1003Ser exhibited enhanced activation of SREBP and UPR pathways compared to WT S1P, suggesting this mutant protein has an altered function, possibly a gain‐of‐function.
Reports have shown S1P can exist as multiple shed ectodomain species (Elagoz et al., 2002; Kim et al., 2018). While S1P Pro1003Ser produced the unprocessed precursor, an intermediate cleaved product, and the mature protease, we were unable to detect shed ectodomain species. This may be due to differences in culture conditions—indeed it has been shown that production of some shed S1P species requires specific conditions (e.g., increased Caspase‐2 expression, mouse models of nonalcoholic steatohepatitis) (Kim et al., 2018) and/or due to differences in methodology (Elagoz et al. visualized shed S1P most abundantly in culture media via radiolabeling coupled with immunoprecipitation, while we performed standard Western blots of whole‐cell lysates).
S1P functions in concert with Site‐2 Protease (S2P) to activate substrates (Espenshade & Hughes, 2007). Loss‐of‐function S2P mutations are well documented as contributors to human disease including skeletal dysplasia (Aten et al., 2010; Bornholdt et al., 2013; Haghighi et al., 2013; Lindert et al., 2016; Naiki et al., 2012; Nakayama et al., 2011; Oeffner et al., 2009; Zhang et al., 2016). A recent study reported a case of human skeletal dysplasia in a pediatric patient with an S1P deficiency, resulting in disrupted ER stress, but retained lipid homeostasis (Kondo et al., 2018). Based on our thorough clinical analysis, such S2P‐ and S1P‐associated syndromes were not present in the proband, most likely due to the loss‐of‐function nature of those previously identified mutations.
The S1P Pro1003Ser patient phenotype is complex and includes conditions that effect several organs including skeletal muscle and the ovary. These observations suggest that S1P may play a critical role in the functions of these organs in ways that are not yet understood, either via well‐known S1P substrates or yet to be identified substrates. Our observations underscore the importance of S1P in human physiology and shed light on a potential role for S1P in a diverse array of human disorders and organ systems.
DISCLOSURE STATEMENT
Rita Brookheart and Brian Finck hold a provisional patent related to this work (provisional patent application #62/638,531).
Supporting information
ACKNOWLEDGMENTS
We thank the patient and her family for their participation in this study. We also thank Nat Wolins for providing the lysozyme‐GFP‐KDEL construct and the Washington University Center for Cellular Imaging for assistance with immunofluorescence experiments. This work was supported by the National Institutes of Health grant P30 DK056341 Nutrition Obesity Research Center (R.T.B).
Schweitzer GG, Gan C, Bucelli RC, et al. A mutation in Site‐1 Protease is associated with a complex phenotype that includes episodic hyperCKemia and focal myoedema. Mol Genet Genomic Med. 2019;7:e733 10.1002/mgg3.733
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aten, E. , Brasz, L. C. , Bornholdt, D. , Hooijkaas, I. B. , Porteous, M. E. , Sybert, V. P. , … den Dunnen, J. T. (2010). Keratosis Follicularis Spinulosa Decalvans is caused by mutations in MBTPS2. Human Mutation, 31(10), 1125–1133. 10.1002/humu.21335 [DOI] [PubMed] [Google Scholar]
- Bornholdt, D. , Atkinson, T. P. , Bouadjar, B. , Catteau, B. , Cox, H. , De Silva, D. , … Fuer Humangenetik, Z. (2013). Genotype‐phenotype correlations emerging from the identification of missense mutations in MBTPS2. Human Mutation, 34, 587–594. 10.1002/humu.22275 [DOI] [PubMed] [Google Scholar]
- Brandl, K. , Rutschmann, S. , Li, X. , Du, X. , Xiao, N. , Schnabl, B. , … Beutler, B. (2009). Enhanced sensitivity to DSS colitis caused by a hypomorphic Mbtps1 mutation disrupting the ATF6‐driven unfolded protein response. Proceedings of the National Academy of Sciences, 106(9), 3300–3305. 10.1073/pnas.0813036106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, M. S. , Faust, J. R. , Goldstein, J. L. , Kaneko, I. , & Endo, A. (1978). Induction of 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML‐236B), a competitive inhibitor of the reductase. The Journal of Biological Chemistry, 253(4), 1121–1128. [PubMed] [Google Scholar]
- Cohen, J. C. , Horton, J. D. , & Hobbs, H. H. (2011). Human fatty liver disease: Old questions and new insights. Science, 332(6037), 1519–1523. 10.1126/science.1204265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBose‐Boyd, R. A. , Brown, M. S. , Li, W. P. , Nohturfft, A. , Goldstein, J. L. , & Espenshade, P. J. (1999). Transport‐dependent proteolysis of SREBP: Relocation of site‐1 protease from Golgi to ER obviates the need for SREBP transport to Golgi. Cell, 99(7), 703–712. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/10619424. [DOI] [PubMed] [Google Scholar]
- Elagoz, A. , Benjannet, S. , Mammarbassi, A. , Wickham, L. , & Seidah, N. G. (2002). Biosynthesis and cellular trafficking of the convertase SKI‐1/S1P: Ectodomain shedding requires SKI‐1 activity. The Journal of Biological Chemistry, 277(13), 11265–11275. 10.1074/jbc.M109011200 [DOI] [PubMed] [Google Scholar]
- Espenshade, P. J. , Cheng, D. , Goldstein, J. L. , & Brown, M. S. (1999). Autocatalytic processing of site‐1 protease removes propeptide and permits cleavage of sterol regulatory element‐binding proteins. Journal of Biological Chemistry, 274(32), 22795–22804. 10.1074/jbc.274.32.22795 [DOI] [PubMed] [Google Scholar]
- Espenshade, P. J. , & Hughes, A. L. (2007). Regulation of sterol synthesis in eukaryotes. Annual Review of Genetics, 41, 401–427. 10.1146/annurev.genet.41.110306.130315 [DOI] [PubMed] [Google Scholar]
- Goldstein, J. L. , Hazzard, W. R. , Schrott, H. G. , Bierman, E. L. , & Motulsky, A. G. (1973). Hyperlipidemia in coronary heart disease. I. Lipid levels in 500 survivors of myocardial infarction. The Journal of Clinical Investigation, 52(7), 1533–1543. 10.1172/JCI107331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi, A. , Scott, C. A. , Poon, D. S. , Yaghoobi, R. , Saleh‐Gohari, N. , Plagnol, V. , & Kelsell, D. P. (2013). A missense mutation in the MBTPS2 gene underlies the X‐linked form of Olmsted syndrome. The Journal of Investigative Dermatology, 133(2), 571–573. 10.1038/jid.2012.289 [DOI] [PubMed] [Google Scholar]
- Hawkins, J. L. , Robbins, M. D. , Warren, L. C. , Xia, D. , Petras, S. F. , Valentine, J. J. , … Harwood, H. J. ( 2008). Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element‐binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. The Journal of Pharmacology and Experimental Therapeutics, 326(3), 801–808. 10.1124/jpet.108.139626 [DOI] [PubMed] [Google Scholar]
- Hay, B. A. , Abrams, B. , Zumbrunn, A. Y. , Valentine, J. J. , Warren, L. C. , Petras, S. F. , … Harwood, H. J. (2007). Aminopyrrolidineamide inhibitors of site‐1 protease. Bioorganic & Medicinal Chemistry Letters, 17(16), 4411–4414. 10.1016/j.bmcl.2007.06.031 [DOI] [PubMed] [Google Scholar]
- Hong, J. , Kim, K. , Kim, J.‐H. , & Park, Y. (2017). The role of endoplasmic reticulum stress in cardiovascular disease and exercise. International Journal of Vascular Medicine, 2017, 9 10.1155/2017/2049217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil, G. S. (2010). Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell, 140(6), 900–917. 10.1016/j.cell.2010.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. Y. , Garcia‐Carbonell, R. , Yamachika, S. , Zhao, P. , Dhar, D. , Loomba, R. , … Karin, M. (2018). ER stress drives lipogenesis and steatohepatitis via caspase‐2 activation of S1P. Cell, 175(1), 133–145; e15. 10.1016/j.cell.2018.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, Y. , Fu, J. , Wang, H. , Hoover, C. , McDaniel, J. M. , Steet, R. , … Xia, L. (2018). Site‐1 protease deficiency causes human skeletal dysplasia due to defective inter‐organelle protein trafficking. JCI Insight, 3(14). 10.1172/JCI.INSIGHT.121596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4(7), 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Lindert, U. , Cabral, W. A. , Ausavarat, S. , Tongkobpetch, S. , Ludin, K. , Barnes, A. M. , … Shotelersuk, V. (2016). MBTPS2 mutations cause defective regulated intramembrane proteolysis in X‐linked osteogenesis imperfecta. Nature Communications, 7, 11920 10.1038/ncomms11920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschner, K. , Kollmann, K. , Schweizer, M. , Braulke, T. , & Pohl, S. (2011). A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science (New York, N.Y.), 333(6038), 87–90. 10.1126/science.1205677 [DOI] [PubMed] [Google Scholar]
- Mathe, E. , Olivier, M. , Kato, S. , Ishioka, C. , Hainaut, P. , & Tavtigian, S. V. (2006). Computational approaches for predicting the biological effect of p53 missense mutations: A comparison of three sequence analysis based methods. Nucleic Acids Research, 34(5), 1317–1325. 10.1093/nar/gkj518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naiki, M. , Mizuno, S. , Yamada, K. , Yamada, Y. , Kimura, R. , Oshiro, M. , … Wakamatsu, N. (2012). MBTPS2 mutation causes BRESEK/BRESHECK syndrome. American Journal of Medical Genetics Part A, 158A(1), 97–102. 10.1002/ajmg.a.34373 [DOI] [PubMed] [Google Scholar]
- Nakayama, J. , Iwasaki, N. , Shin, K. , Sato, H. , Kamo, M. , Ohyama, M. , … Arinami, T. (2011). A Japanese case of ichthyosis follicularis with atrichia and photophobia syndrome with an MBTPS2 mutation. Journal of Human Genetics, 56(3), 250–252. 10.1038/jhg.2010.163 [DOI] [PubMed] [Google Scholar]
- Nohturfft, A. , DeBose‐Boyd, R. A. , Scheek, S. , Goldstein, J. L. , & Brown, M. S. (1999). Sterols regulate cycling of SREBP cleavage‐activating protein (SCAP) between endoplasmic reticulum and Golgi. Proceedings of the National Academy of Sciences of the United States of America, 96(20), 11235–11240. 10.1073/PNAS.96.20.11235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeffner, F. , Fischer, G. , Happle, R. , König, A. , Betz, R. C. , Bornholdt, D. , … Grzeschik, K.‐H. (2009). IFAP syndrome is caused by deficiency in MBTPS2, an intramembrane zinc metalloprotease essential for cholesterol homeostasis and ER stress response. The American Journal of Human Genetics, 84(4), 459–467. 10.1016/J.AJHG.2009.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raggo, C. , Rapin, N. , Stirling, J. , Gobeil, P. , Smith‐Windsor, E. , O’Hare, P. , & Misra, V. (2002). Luman, the cellular counterpart of herpes simplex virus VP16, is processed by regulated intramembrane proteolysis. Molecular and Cellular Biology, 22(16), 5639–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos da Palma, J. , Cendron, L. , Seidah, N. G. , Pasquato, A. , & Kunz, S. (2015). Mechanism of folding and activation of subtilisin kexin isozyme‐1(SKI‐1)/site‐1 protease (S1P). Journal of Biological Chemistry, 1(1), jbc.M115.677757 10.1074/jbc.M115.677757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawson, R. B. , Cheng, D. , Brown, M. S. , & Goldstein, J. L. (1998). Isolation of cholesterol‐requiring mutant Chinese hamster ovary cells with defects in cleavage of sterol regulatory element‐binding proteins at site 1. The Journal of Biological Chemistry, 273(43), 28261–28269. 10.1074/JBC.273.43.28261 [DOI] [PubMed] [Google Scholar]
- Rotterdam ESHRE/ASRM‐Sponsored PCOS Consensus Workshop Group (2004). Revised 2003 consensus on diagnostic criteria and long‐term health risks related to polycystic ovary syndrome. Fertility and Sterility, 81(1), 19–25. [DOI] [PubMed] [Google Scholar]
- Rutschmann, S. , Crozat, K. , Li, X. , Du, X. , Hanselman, J. C. , Shigeoka, A. A. , … Beutler, B. (2012). Hypopigmentation and maternal‐zygotic embryonic lethality caused by a Hypomorphic Mbtps1 mutation in mice. G3; Genes|genomes|genetics, 2(4), 499–504. 10.1534/g3.112.002196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai, J. , Rawson, R. B. , Espenshade, P. J. , Cheng, D. , Seegmiller, A. C. , Goldstein, J. L. , & Brown, M. S. (1998). Molecular identification of the sterol‐regulated luminal protease that cleaves SREBPs and controls lipid composition of animal cells. Molecular Cell, 2(4), 505–514. [DOI] [PubMed] [Google Scholar]
- Schwarz, J. M. , Cooper, D. N. , Schuelke, M. , & Seelow, D. (2014). MutationTaster2: mutation prediction for the deep‐sequencing age. Nature Methods, 11(4), 361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- Seidah, N. G. , Mowla, S. J. , Hamelin, J. , Mamarbachi, A. M. , Benjannet, S. , Touré, B. B. , … Marcinkiewicz, M. (1999). Mammalian subtilisin/kexin isozyme SKI‐ 1: A widely expressed proprotein convertase with a unique cleavage specificity and cellular localization. Proceedings of the National Academy of Sciences of the United States of America, 96(4), 1321–1326. 10.1073/PNAS.96.4.1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. , Chen, X. , Hendershot, L. , & Prywes, R. (2002). ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Developmental Cell, 3(1), 99–111. 10.1016/S1534-5807(02)00203-4 [DOI] [PubMed] [Google Scholar]
- Shimomura, I. , Shimano, H. , Horton, J. D. , Goldstein, J. L. , & Brown, M. S. (1997). Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein‐1 in human and mouse organs and cultured cells. The Journal of Clinical Investigation, 99(5), 838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling, J. , & O’hare, P., (2006). CREB4, a transmembrane bZip transcription factor and potential new substrate for regulation and cleavage by S1P. Molecular Biology of the Cell, 17(1), 413–426. 10.1091/mbc.E05-06-0500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian, S. V. , Deffenbaugh, A. M. , Yin, L. , Judkins, T. , Scholl, T. , Samollow, P. B. , … Thomas, A. (2006). Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. Journal of Medical Genetics, 43(4), 295–305. 10.1136/jmg.2005.033878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida, L. , Urata, S. , Ulanday, G. E. L. , Takamatsu, Y. , Yasuda, J. , Morita, K. , & Hayasaka, D. (2016). Suppressive effects of the Site 1 Protease (S1P) inhibitor, PF‐429242, on Dengue virus propagation. Viruses, 8(2), 46 10.3390/v8020046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Goldstein, J. L. , Hammer, R. E. , Moon, Y. A. , Brown, M. S. , & Horton, J. D. (2001). Decreased lipid synthesis in livers of mice with disrupted Site‐1 protease gene. Proceedings of the National Academy of Sciences of the United States of America, 98(24), 13607–13612. 10.1073/pnas.201524598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, J. , Rawson, R. B. , Komuro, R. , Chen, X. , Davé, U. P. , Prywes, R. , … Goldstein, J. L. (2000). ER stress induces cleavage of membrane‐bound ATF6 by the same proteases that process SREBPs. Molecular Cell, 6(6), 1355–1364. [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Wang, Y. , Cheng, R. , Ni, C. , Liang, J. , Li, M. , & Yao, Z. (2016). Novel MBTPS2 missense mutation causes a keratosis follicularis spinulosa decalvans phenotype: mutation update and review of the literature. Clinical and Experimental Dermatology, 41(7), 757–760. 10.1111/ced.12889 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials