

Abstract
Background
Glycophosphatidylinositol‐anchored proteins (GPI‐APs) mediate several physiological processes such as embryogenesis and neurogenesis. Germline variants in genes involved in their synthesis can disrupt normal development and result in a variety of clinical phenotypes. With the advent of new sequencing technologies, more cases are identified, leading to a rapidly growing number of reported genetic variants. With this number expected to rise with increased accessibility to molecular tests, an accurate and up‐to‐date database is needed to keep track of the information and help interpret results.
Methods
We therefore developed an online resource (www.gpibiosynthesis.org) which compiles all published pathogenic variants in GPI biosynthesis genes which are deposited in the LOVD database. It contains 276 individuals and 192 unique public variants; 92% of which are predicted as damaging by bioinformatics tools.
Results
A significant proportion of recorded variants was substitution variants (81%) and resulted mainly in missense and frameshift alterations. Interestingly, five patients (2%) had deleterious mutations in untranslated regions. CADD score analysis placed 97% of variants in the top 1% of deleterious variants in the human genome. In genome aggregation database, the gene with the highest frequency of reported pathogenic variants is PIGL, with a carrier rate of 1/937.
Conclusion
We thus present the GPI biosynthesis database and review the molecular genetics of published variants in GPI‐anchor biosynthesis genes.
Keywords: genetic disorders, GPI biosynthesis, GPI‐anchored proteins, LOVD, PIG
1. INTRODUCTION
Glycophosphatidylinositol‐Anchor Proteins (GPI‐APs) are estimated to make up about 1% of the human proteome. They mediate physiologic processes including signaling, cell adhesion, and immune modulation, and play key roles notably in fertilization, embryogenesis, neurogenesis, and prion disease pathogenesis (Fujita & Kinoshita, 2012; Kinoshita, 2014). In vivo studies in mice have in fact shown that GPI‐APs are required for proper brain and embryonic development (McKean & Niswander, 2012; Nozaki et al., 1999).
The synthesis of the GPI‐anchor is an elaborate process that requires the sequential action of multiple mammalian proteins which have been named as the phosphatidylinositol glycan (PIG) enzymes. It begins in the cytoplasmic region of the endoplasmic reticulum (ER) and is initiated by PIG‐A, ‐Q, ‐Y, ‐C, ‐P, ‐H, which mediate the addition of phosphatidylinositol (PI) to N‐acetylglucosamine (GlcNAc). The resulting product, GlcNAC‐PI, is deacetylated by PIGL and flipped to the lumen of the ER where its inositol moiety is linked to an acyl chain by PIGW. Three mannose residues from dolichol‐phosphate‐mannose are sequentially added to the resultant GlcN‐(acyl)PI by PIGM/PIGX, PIGV and PIGB. The mannose residues are further modified by the addition of its ethanolamine‐phosphate side chain in reactions involving PIGN, PIGO/PIGF and PIGG/PIGF, to generate a mature GPI‐anchor. The synthesized GPI‐anchor is then coupled to a mature protein by a transamidase complex involving GPAA1, PIGK, PIGS, PIGU, and PIGT. To facilitate efficient transport to the Golgi, mature GPI‐anchor proteins are structurally remodeled by members of the post‐GPI‐attachment to proteins (PGAP) family of proteins. This process involves the removal of the acyl chain from inositol by PGAP1 as well as the elimination of ethanolamine‐phosphate from the second mannose unit by PGAP5. Within the Golgi, PGAP2 and PGAP3 modify the fatty acid structure within the GPI‐anchor to facilitate its association with lipid rafts and subsequent transport to the plasma membrane (Fujita & Kinoshita, 2012; Kinoshita, 2014). The final structure of a GPI‐AP can be seen in Figure 1.
Figure 1.
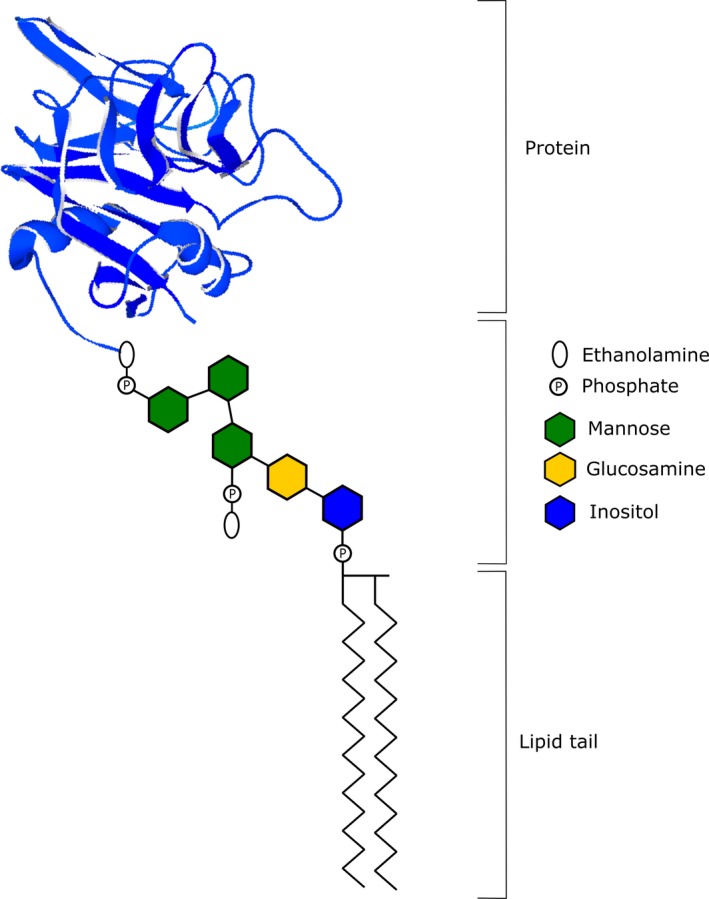
Final structure of a glycophosphatidylinositol‐anchored protein
Given the intricate and elaborate process involved in the production and coupling of GPI‐anchors to proteins, variants in genes implicated in the GPI‐anchor biosynthesis network are expected to result in missorting, defective transport, altered GPI‐AP expression, or secretion of mature protein without an anchor (Fujita & Kinoshita, 2012). This is supported by in vitro studies in which rescuing the variant with lentivirus expressing the wild‐type protein can restore GPI‐APs to a normal level (Nguyen et al., 2017). With the progress of sequencing technologies, an increasing number of germline variants encoding several GPI biosynthesis enzymes have been found and reported in the literature (PIGA, PIGQ, PIGY, PIGC, PIGP, PIGH, PIGL, PIGW, PIGM, PIGV, PIGN, PIGO, PIGG, PIGS, PIGT, GPAA1, PGAP1, PGAP3, and PGAP2 [OMIM accession numbers in Table 1]). Due to the great variability in the clinical consequences of these variants, determining the right diagnosis, which is critical to providing the correct treatment plan or counseling, still proves to be a challenge for both clinicians and scientists. We therefore developed a web resource that catalogs all currently published variants (www.gpibiosynthesis.org) in the goal of making these data widely accessible and easy to find. This online platform also contains a section for families to initiate online discussion on inherited GPI disorders.
Table 1.
GPI biosynthesis genes and their associated disease caused by germline variants
| Gene | Gene OMIM # | Disease | Disease OMIM # | Affected individuals |
|---|---|---|---|---|
| GPAA1 | 603048 | Glycosylphosphatidylinositol biosynthesis defect 15 (GPIBD15) | 617810 | 10 |
| PGAP1 | 611655 | Mental retardation, autosomal recessive 42 (MRT42) | 615802 | 11 |
| PGAP2 | 615187 | Hyperphosphatasia with mental retardation syndrome 4 (HPMRS4) | 614207 | 17 |
| PGAP3 | 611801 | Hyperphosphatasia with mental retardation syndrome 3 (HPMRS3) | 615716 | 48 |
| PIGA | 311770 | Multiple congenital anomalies‐hypotonia‐seizures syndrome 2 (MCAHS2) | 300868 | 43 |
| PIGC | 601730 | Glycosylphosphatidylinositol biosynthesis defect 16 (GPIBD16) | 617816 | 3 |
| PIGG | 616918 | Mental retardation, autosomal recessive 53 | 616917 | 8 |
| PIGH | 600154 | Glycosylphosphatidylinositol biosynthesis defect 17 | 618010 | 4 |
| PIGL | 605947 | CHIME syndrome (Zunich neuroectodermal syndrome) | 280000 | 15 |
| PIGM | 610273 | Glycosylphosphatidylinositol biosynthesis defect 1 (GPIBD1) | 610293 | 4 |
| PIGN | 606097 | Multiple congenital anomalies, hypotonia, seizures syndrome 1 (MCAHS1), Fryns syndrome | 614080, 229850 | 33 |
| PIGO | 614730 | Hyperphosphatasia with mental retardation syndrome 2 (HPMRS2) | 614749 | 17 |
| PIGP | 605938 | Epileptic encephalopathy, early infantile, 55 (EIEE55) | 617599 | 2 |
| PIGQ | 605754 | Epileptic encephalopathy, early infantile, EIEE | None | 2 |
| PIGS | 610271 | Glycosylphosphatidylinositol biosynthesis defect 18 | 618143 | 6 |
| PIGT | 610272 | Multiple congenital anomalies‐hypotonia‐seizures syndrome 3 (MCAHS3) | 615399, 615398 | 18 |
| PIGV | 610274 | Hyperphosphatasia with mental retardation syndrome 1 (HPMRS1) | 239300 | 27 |
| PIGW | 610275 | Hyperphosphatasia with mental retardation syndrome 5 (HPMRS5) | 616025 | 4 |
| PIGY | 610662 | Hyperphosphatasia with mental retardation syndrome 6 (HPMRS6) | 616809 | 4 |
2. MATERIALS AND METHODS
2.1. Database structure, content, and functionality
The GPI biosynthesis disorder database (http://www.gpibiosynthesis.org/) is an online tool that contains information for families, clinicians, and scientists. The “Families” tab gives the opportunity for family members to join patient discussion groups concerning GPI biosynthesis defects as well as to participate in ongoing research studies. The “Clinician and Scientist” menu contains a list of genes involved in GPI biosynthesis, each with links to published variants and patients. The links provide direct access to the Locus‐Specific Databases (LSDBs) of the corresponding gene created in the LOVD database (www.LOVD.nl) (Figure 2a).
Figure 2.
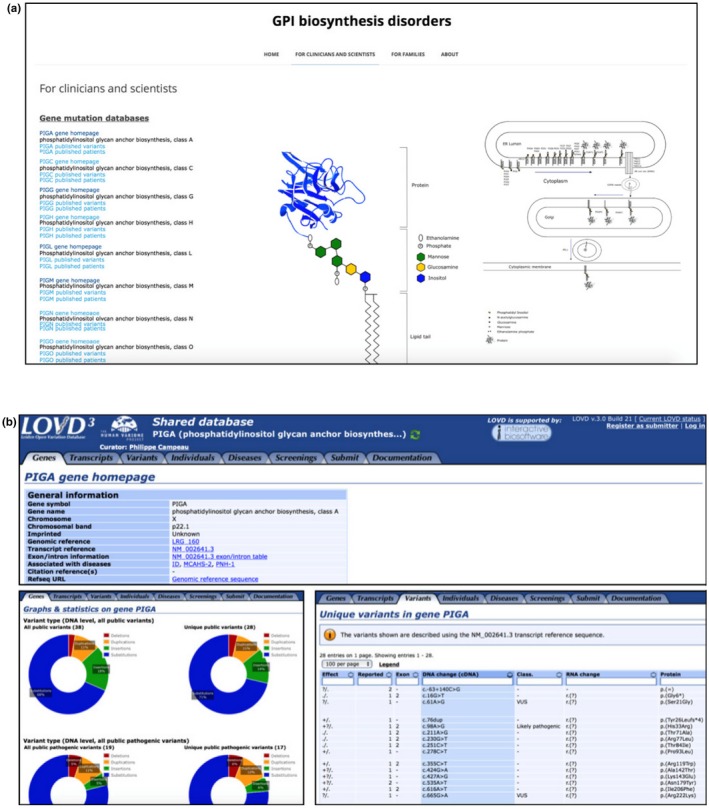
(a) Screenshot of the www.gpibiosynthesis.org webpage displaying the content of the “For clinicians and scientists” tab. Links for each glycophosphatidylinositol biosynthesis gene (seen on the left side) lead directly to the LOVD shared database of the chosen gene. (b) Multiple screenshots of the LOVD database displaying the different sections and tabs (Gene homepage, Gene graphs, and Unique variants shown here). Clicking on the different links shows more information about the gene as well as detailed description on the reported variants such as the exon location, changes in the DNA and protein level, as well as references to articles where they were published
The LOVD database contains publicly available variant data on many genes, including those involved in GPI‐anchor biosynthesis. All currently published variants have been added to this database by curators who are experts in the field, making LSDBs a reliable source of information (Vihinen, den Dunnen, Dalgleish, & Cotton, 2012). The variants follow the Human Genome Variation Society (HGVS) nomenclature, and new variants or additional LSDBs are created and linked to the homepage as soon as variant data become publicly available.
Each LSDB contains many features that allow easy visualization of variant data in the gene. The user can view or retrieve specific data by assessing the different tabs (genes, transcripts, variants, individuals, diseases, or screening) (Figure 2b). The “Genes” tab, for example, provides access to general information about the gene of interest including gene symbol, gene name, chromosome, chromosomal location, genomic reference, transcript reference, associated diseases, reported public DNA variants and number of individuals with public variants. The genes tab also provides a quick access to the graphical display utility that allows users to view graphs and statistics for each GPI‐anchor biosynthesis gene including variant location, variant type (deletion, duplication, insertion, substitution), and its effect on the protein (frameshift, missense, stop and silent variant). Users can also have access and view variants in other platforms such as the UCSC and ensemble genome browsers, and the NCBI sequence viewer. Moreover, additional information can be obtained through links to other resources such as HGNC, Entrez, Pubmed articles, OMIM gene and diseases, HGMD, GeneCards and GeneTest. Data can be exported with one click and be sent to ClinVar.
2.2. Data sources and curation
Relevant articles reporting patients with variants in GPI biosynthesis genes were searched in the Pubmed database (http://www.ncbi.nlm.nih.gov/pubmed/) and selected for further analysis. In this work, we reviewed in February 2019 a total of 107 published papers that describe 276 patients with germline variants in a homozygous or a compound heterozygous state in GPI genes. Variants that were not present in the LSDBs were added, resulting in a total of 192 unique public variants identified in the PIGA, PIGQ, PIGY, PIGC, PIGP, PIGH, PIGL, PIGW, PIGM, PIGV, PIGN, PIGO, PIGG, PIGS, PIGT, GPAA1, PGAP1, PGAP3, and PGAP2 genes.
2.3. Variants classification and analysis
The classification of the variants was done according to their HGVS nomenclature. The classification data from LOVD include nonpublic variants into their graphs which are not included in the present analysis.
For variant analysis, all the missense, nonsense, and splice variants were selected (156 variants out of 192) and annotated by wANNOVAR (http://wannovar.wglab.org), a web = based version of the annotation tool ANNOVAR. This analysis allowed the computation of different pathogenicity scores such as SIFT, PolyPhen‐2, and MutationTaster as well as the extraction of the allele frequencies from major population genomics projects such as the 1,000 genomes project, ExAC, ESP6500si, and Genome Aggregation Database (gnomAD).
Our population analysis of the GPI variants was done with data from the gnomAD database. First, the information was downloaded for each gene, then the data for the 74 variants present in the gnomAD database were extracted from the data files and compiled. For each variant, we looked at their exomic frequency, their genomic frequency as well as their overall frequency.
3. RESULTS AND DISCUSSION
3.1. Gene variants and diseases
The GPI biosynthesis disorder database currently contains a total of 276 individuals with germline variants in GPI genes. Based on OMIM classification and data from the GPI database, variants in these genes can give rise to various disorders including, but not limited to, hyperphosphatasia with mental retardation syndrome (HPMRS), multiple congenital anomalies‐hypotonia‐seizures syndrome (MCAHS), CHIME syndrome, early infantile epileptic encephalopathy (EIEE), Glycosylphosphatidylinositol biosynthesis defect (GPIBD), and multiple congenital and CNS abnormalities (Ng & Freeze, 2015). Clinical data retrieved from the literature revealed that 48 individuals, which represent the most patients reported for a gene, suffer from HPMRS type 3 (OMIM #615716) which is caused by a variant in the PGAP3 gene and 43 patients have defects in the PIGA gene, causing MCAHS type 2 (OMIM #300868). In contrast, only two were reported to have a variant in the PIGQ gene which causes EIEE, as well as in the PIGP gene which leads to EIEE type 55 (OMIM #617599) (Table 1).
A compilation of the clinical characteristics of these affected patients revealed that 99% of the patients with defects in GPI‐AP biosynthesis genes had intellectual deficiency (ID) and developmental delay (DD), and 77% suffered from seizures. Symptoms that appeared to be less common include, but are not limited to, cranial shape anomalies, deafness, ophthalmological anomalies, hand and feet anomalies, and abnormal levels of alkaline phosphatase (Figure 3). Phenotypic analysis also revealed that some clinical characteristics were more prominent in patients with variants in a certain GPI gene more than others. The majority of patients with variants in PIGL, for example, was reported to have colobomas (Knight Johnson, Schaefer, Lee, Hu, & Del Gaudio, 2017), an ophthalmological anomaly that was not present in patients with variants in other GPI genes, and patients with PIGV variants appeared to have nail anomalies more than others (Bellai‐Dussault, Nguyen, Baratang, Jimenez‐Cruz, & Campeau, 2019). Efforts are being pursued by other research groups to develop computer‐assisted facial photo analysis for phenotypic comparisons between affected individuals (Knaus et al., 2018).
Figure 3.
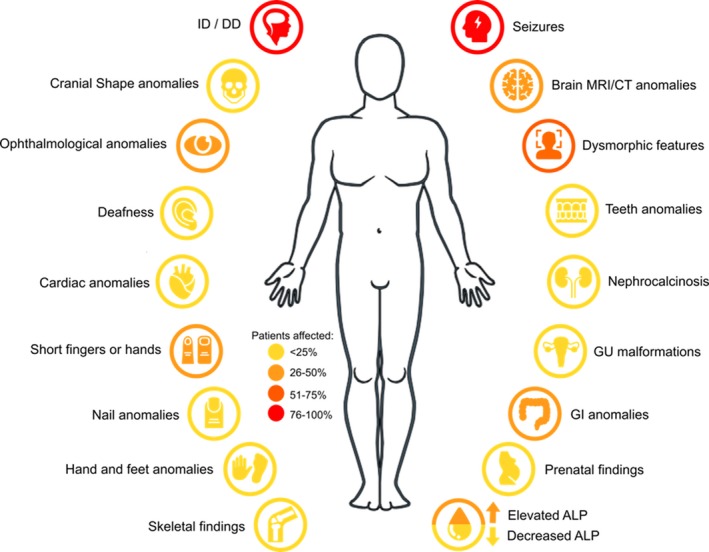
Main clinical phenotypes reported in patients with variants in glycophosphatidylinositol biosynthesis genes. ALP: alkaline phosphatase; CT: computed tomography; DD: developmental delay; GI: gastrointestinal; GU: genitourinary; ID: intellectual deficiency; MRI: magnetic resonance imaging
3.2. Variant location
An analysis of the 192 unique variants contained in the GPI biosynthesis disorder database and its associated LSDBs revealed that 89% of the variants are located in the coding region of their respective gene and the majority of the genes in this study (GPAA1, PGAP1, PGAP3, PIGA, PIGG, PIGL, PIGN, PIGO, PIGQ, PIGS, and PIGT) has variants in a splice site. Variants in the 5’UTR were only found in genes PIGY and PIGM, and one variant in the 3’UTR position was only found in the PGAP3 gene (Figure 4a).
Figure 4.
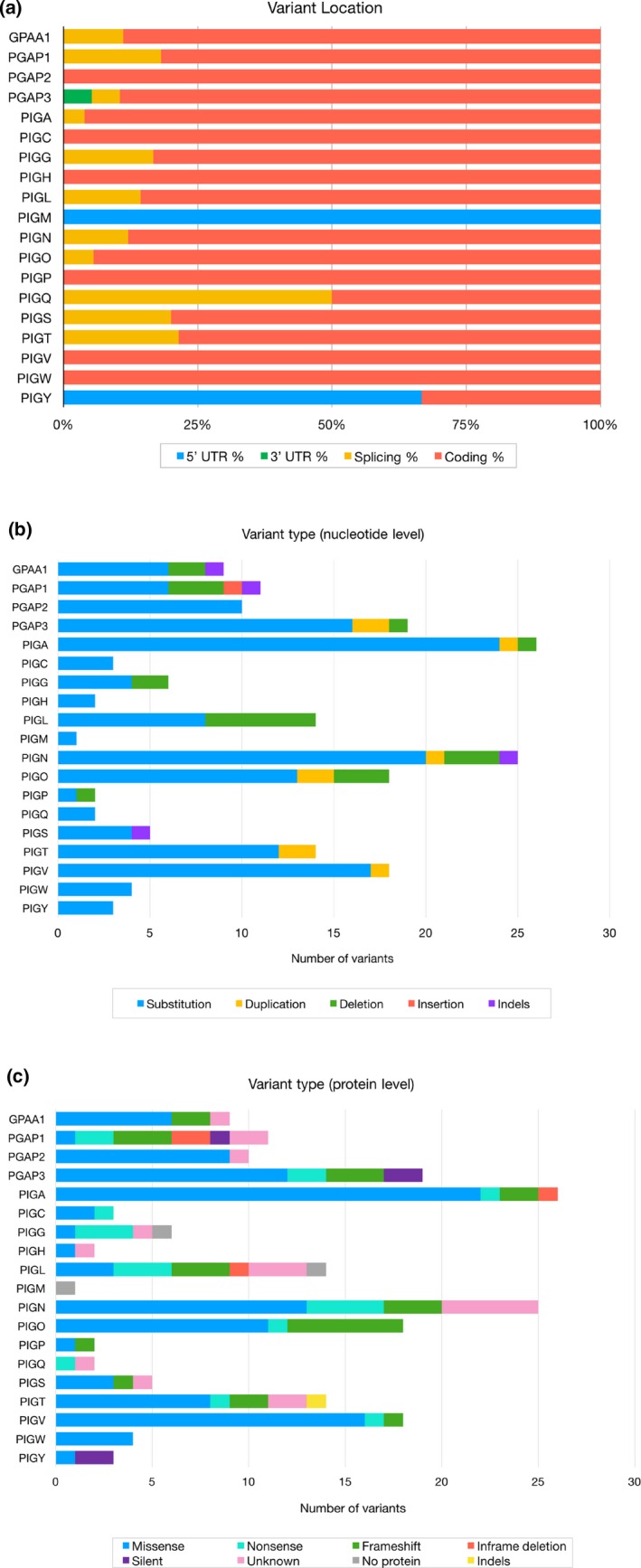
(a) Genomic location of 192 unique variants in glycophosphatidylinositol biosynthesis genes reported in the literature. (b) Types of nucleotide changes of the analyzed variants. (c) Types of changes seen at the protein level as reported in the literature
Variants can therefore occur in coding and noncoding regions of the genes. One of the clinically observed effects of variant location is its impact on the patient's phenotype. An example of this are the four patients reported by Ilkovski et al. with variants in PIGY. Two of the patients had variants in the coding region which manifested into a multisystemic disease including seizures, cataracts, and severe developmental delay. These individuals eventually passed away at an early age. The other two patients, on the other hand, had variants in the promoter region of the gene and presented less severe phenotypes such as moderate developmental delay and microcephaly (Ilkovski et al., 2015).
Furthermore, variants in splice sites can have various consequences at the nucleotide and amino acid level and can lead to various effects such as frameshifts and exon skipping. In our cohort, 75% of the splice variants were seen in the latter part of GPI biosynthesis (from PIGN to PGAP3) where side chains are added to the mannose residues and where the GPI structures are further remodeled to generate a mature and functional GPI‐anchor. An example of such a variant is one found in the PIGO gene where a splice site variant (NM_032634.3:c.3069 + 5G>A, p.Val952Aspfs) resulted in the skipping of exon 9 causing a frameshift followed with a premature stop codon. This variant led to an abnormal production of the GPI‐anchor and consequently a reduced level of GPI‐APs at the cell surface (Krawitz et al., 2012).
3.3. Types of variants
We examined the types of variants both at the DNA and at the protein level and our analysis has shown that the most frequent variant type at the nucleotide level was substitution variants (81.2%), followed by deletions (11.5%), duplications (4.7%), indels (2.1%) and insertions (0.5%). A distribution of these variant types for each gene is shown in Figure 6b. Certain variants can exist in a homozygous or a compound heterozygous state as is the case with the PIGV missense variant NM_017837.3:c.1022C > A (Horn et al., 2014). Interestingly, this variant is considered a mutational hotspot as it was found in >60% of the patients with PIGV variants. Another frequent variant found in the GPI biosynthesis database is a heterozygous missense variant in PIGL (NM_004278.3:c.500T > C) which was present in 80% of the patients with PIGL variants. Recognizing the areas that are more frequently mutated than others in inherited GPI disorders (IGDs) may provide hints into the molecular mechanism of these diseases.
Figure 6.
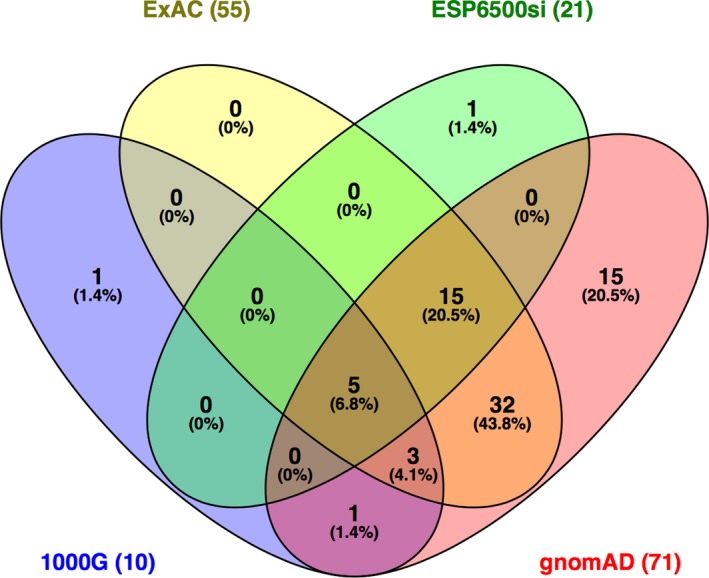
Venn diagram showing the number of variants reported by four different databases (ExAC, ESP6500si, 1000G, gnomAD) through the wANNOVAR tool. The gnomAD database included the most variants compared to the three others. The diagram was produced with Venny 2.1 (http://bioinfogp.cnb.csic.es/tools/venny/index.html)
We then examined the types of variants at the amino acid level as one variant type at the DNA level can lead to several kinds of changes in the protein product (Figure 4c). We found that the most frequent type of variant at the amino acid level was missense variants, representing about 59% of the variants (Figure 7). Other protein changes include frameshifts (14%), nonsense (10%) and silent variants (3%). Inframe deletions (2%) ranged from a single amino acid to large deletions of a few exons. For instance, in the PIGL gene, the variant (NM_ 004278.3:c.426 + 6654_660+3131del) leads to the skipping of three exons (Knight Johnson et al., 2017), and in PIGN, (NM_176787.4:c.324_549 + 196del) and (NM_176787.4:c.329_549 + 1908del) are predicted to result in a null allele as they lead to a deletion spanning 2–3 exons (Alessandri et al., 2018).
Figure 7.
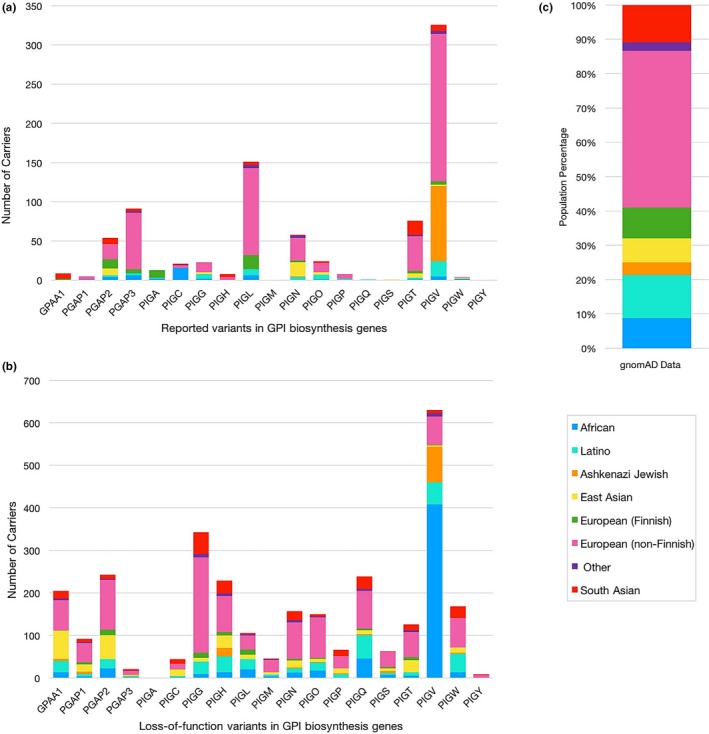
Populational distribution of individuals carrying reported variants in glycophosphatidylinositol (GPI) biosynthesis genes according to the gnomAD database. (a) Number of variant carriers for each GPI biosynthesis gene according to its population. Reported PIGM and PIGY variants are not found in gnomAD. (b) Number of carriers for loss‐of‐function variants in GPI biosynthesis genes. (c) Population composition of the gnomAD data
Variants can also lead to no protein being produced (2%). In the literature, entire gene deletions of either the PIGL or the PIGG gene were reported in two patients (Chi et al., 2012; Makrythanasis et al., 2016). The deletion of the whole PIGL gene had an important effect as the patient presented many abnormalities including colobomas, mental retardation, and craniofacial dysmorphism (Tinschert, Anton‐Lamprecht, Albrecht‐Nebe, & Audring, 1996). In the case of PIGG, the affected patient had severe developmental delay, but the authors discovered, through functional analysis, that the deletion of the whole gene does not cause decreased expression of GPI‐APs at the cell surface nor an impaired GPI‐AP structure. This is explained by the role of the PIGG protein during biosynthesis: in normal cells, PIGG transfers an EtNP to the second mannose of the GPI‐anchor, but this side chain is eventually removed by the PGAP5 protein later in the process (Makrythanasis et al., 2016).
Thus, the degree of severity of each variant and its clinical manifestation depend on various factors such as the location of the variant, the type of variant at the nucleotide level, the nature of the amino acid changes as well as the role of the gene in the biosynthesis pathway. Note that the protein consequences of 9% of the variants are undetermined as the predicted effects were not assessed in the articles.
3.4. Variant pathogenicity
The pathogenicity of the variants was evaluated by looking at the computed scores from three different prediction tools: PolyPhen‐2, SIFT, and MutationTaster (Table S1). PolyPhen‐2 outputs a probabilistic score between 0 and 1 for which the higher values indicate a higher probability of a variant to be damaging (Adzhubei et al., 2010). Scores over a value of 0.957 are considered probably damaging by ANNOVAR. Fifty‐nine percent of the analyzed variants are considered as probably damaging.
SIFT is similar to PolyPhen‐2 although in this case, the lower the score, the higher the probability of the variant being damaging. Scores below 0.05 are considered damaging variants (Kumar, Henikoff, & Ng, 2009). In our cohort, 65% of the variants are predicted as damaging.
MutationTaster works slightly differently from the other two tools as it makes a prediction between the four following cases: A for an automatic disease‐causing variant, D for a disease‐causing variant, N for a polymorphism and P for an automatic polymorphism. The “automatic” nomenclature corresponds to a variant already known to be disease‐causing or benign from public databases. The score, ranging between 0 and 1, is in fact a probability value of the prediction being accurate. An article comparing different online pathogenicity prediction tools stated MutationTaster to have perfect sensitivity but low specificity as it can correctly predict 100% of the credibly pathogenic variants the authors studied, but it does not accurately predict benign variants (Walters‐Sen et al., 2015). In our set, MutationTaster predicted 91% of the variants to be disease‐causing. When looking at all three pathogenicity scores, 92% of the variants have been predicted to be damaging by at least one of the tools. The pathogenic status of several variants remains undetermined (Table S1).
Since the percentages of damaging variants predicted by each tool are variable, we also looked at the CADD score. This score is computed by looking at the results from other prediction tools and harmonizing them. The CADD score is a classification system where the score indicates to what fraction of the top deleterious variants in the human genome the analyzed variant belongs to. Ninety‐seven percent of our variants for which the score was obtained had a CADD score over 20, placing them in the top 1% of deleterious variants in the human genome.
In order to better assess the pathogenicity in this set of variants, we compared the scores of the GPI biosynthesis variants obtained by Polyphen‐2, SIFT, and MutationTaster, as well as the CADD score to those computed for Clinvar's benign variants and likely benign missense variants in those genes, and for gnomAD's missense found in this same family of genes. For the gnomAD variants, we chose variants only seen once in the population to avoid CADD training circularity. To stay consistent across all programs, damaging was considered similarly to probably damaging, automatic disease‐causing, and disease‐causing. Tolerated was considered similarly to benign, polymorphism, and automatic polymorphism. When no score is reported, the variant is listed under No Prediction.
Overall, Clinvar benign and likely benign variants showed the lowest rate of pathogenicity prediction for variants in GPI biosynthesis genes. When comparing scores, T‐tests show that the CADD scores of the reported set of variants are significantly different from the Clinvar and gnomAD sets (Figure 5a). A similar observation can be made when comparing the scores calculated by the three other programs (Figure 5b). The gnomAD set of variants shows a pathogenicity level similar to Clinvar's set of variants when looking at the CADD scores, but its level tends to be closer to the reported set of variants when looking at the predictions from other tools. This analysis shows the variability of pathogenicity scores between different online prediction tools. While it may appear more convenient to use online programs as it reduces the need to perform functional studies, careful analysis must be taken as they may not accurately predict the actual pathogenic status of the variant.
Figure 5.
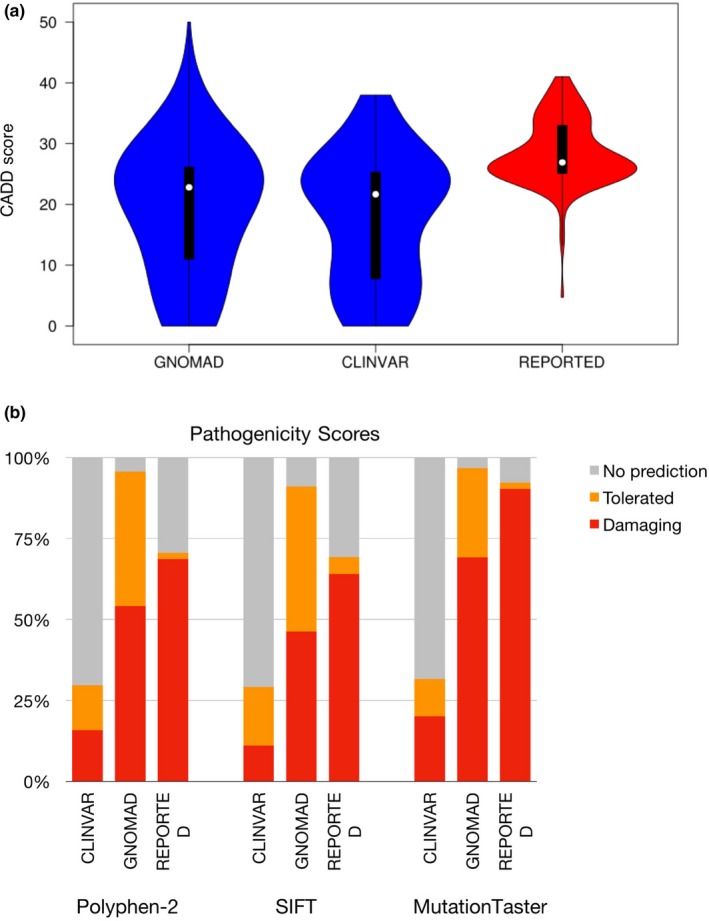
(a) Violin plot comparing the CADD scores of variants in the glycophosphatidylinositol (GPI) biosynthesis genes for Clinvar's benign variants, gnomAD's missense singletons and 145 reported variants. T‐tests suggest statistical difference between the reported variants and the Clinvar variants (ρ < 2.2e‐16) as well as with gnomAD's singletons (ρ = 1.99e‐06). (b) Comparison of the pathogenicity predictions by Polyphen‐2, SIFT, and MutationTaster for Clinvar's benign variants, gnomAD's missense singletons and the GPI biosynthesis genes‐reported variants. Any prediction considered as damaging or potentially damaging was included in the damaging category. The tolerated category includes anything predicted as tolerated, benign, probably benign. The NO prediction category consists of the variants for which the tool could not compute a prediction
3.5. Variant frequency
The frequency for all variants, excluding those located in intronic and untranslated regions, were analyzed with four databases: the 1000 Genomes Project (1000G), the Exome Aggregation Consortium (ExAC), the NHLBI Exome Sequencing Project (ESP6500si), and the gnomAD through the wANNOVAR tool. From an input of 156 variants, only 73 variants showed a frequency in either one of these four databases (Table S2). Of these, gnomAD gave the highest output, showing frequencies for 71/156 (46%) of the variants, followed by ExAC which gave frequencies for 55/156 (35%) variants (Figure 6). 1000G and ESP65000si only gave frequencies for 10/156 (6%) and 21/156 (13%) of the variants, respectively. This is not surprising as gnomAD contains both exome and genome sequencing data which include part of the data from ExAC, whereas the 1000G and ESP6500si data sets only consist of sequencing information from smaller cohorts of individuals (Lek et al., 2016).
Assuming each individual is a carrier for only one GPI variant, we obtain an estimated carrier frequency of 0.61% (1/162) from a total count of 873 mutated alleles in 1,41,456 individuals represented in the gnomAD database, for the 74 variants analyzed. This frequency represents a rough estimate of how often deleterious variants in any GPI biosynthesis genes occur in the whole population. It is important to note that this calculation does not include disease‐causing alleles that may have been missed due to poor coverage of exomes, as well as any unpublished data.
Due to the fact that prior discoveries from sequencing studies have revealed genetic variants that are more commonly found in certain populations, we decided to look at the frequency of the variants in the gnomAD database to see which population between Africans, Ashkenazi Jews, East Asians, Finnish Europeans, non‐Finnish Europeans, Latinos, South Asians, and other populations showed the highest frequency for our set of variants.
Of the 192 variants reported in GPI biosynthesis genes, only 74 (39%) were found in the gnomAD database. None of the reported PIGM and PIGY variants were in the database. For each of the 74 variants, we determined which population had the highest allele frequency. These numbers do not take into account gnomAD LoF (loss‐of‐function) variants never reported in patients, but many of which would be deleterious. Other gnomAD LoF variants might not actually affect splicing significantly or truncating variants at the end of a protein might not actually lead to a loss‐of‐function, so we feel that gnomAD predicted LoF variants should be treated separately (see later). For 24 of the 74 published variants found in gnomAD (33%), the variants were found most frequently in the non‐Finnish European population, with frequencies in that cohort ranging between 1/100,000 and 1/100. This was followed with the South Asian population which had highest frequencies for 11/74 variants, the Latino population (10/74), the East Asian population (10/74), the African and other populations (7/74 and 6/74 respectively), the Finnish European population (5/74), and finally, the Ashkenazi Jewish population which was however the population with the highest frequency of a single published PIGV variant (NM_017837.3:c.1369C > T, p.(Leu457Phe)) where one individual out of 54 is a carrier (Table S3). This variant, however, was later shown to be benign through experiments done on PIGV deficient Chinese hamster ovary cells (Howard et al., 2014).
We then looked at the total number of carriers for each gene instead of focusing on specific variants in order to examine the distribution of variants in any single GPI biosynthesis gene across the population. Figure 7a shows that for 11 out of 17 GPI biosynthesis genes, a majority of the carriers are from non‐Finnish European descent. This goes accordingly with the composition of the gnomAD data. The remaining genes have the most carriers in the African population (PIGC and PIGW), the Finnish European population (PIGA), the Latino population (PIGQ), the East Asian population (PIGS), and in the South Asian population (GPAA1). Carriers of PIGH variants were found in the South Asian and non‐Finnish European population at the same amount. Taking into account that Figure 7a includes the aforementioned benign PIGV variant which is heavily represented in the Ashkenazi Jewish and non‐Finnish European populations, we can say that PIGL is the GPI biosynthesis gene showing the highest number of variant carriers with PGAP3 coming in second. If we look at all LoF variants in the 19 GPI biosynthesis genes presented in this work excluding those that were reported in patients with IGDs, 690 variants are found in gnomAD with a total of 2,938 mutated alleles. No LoF variant is present for PIGA. Most carriers are of non‐Finnish European descent representing 0.8% of the total number of individuals in gnomAD, followed by the African population due to a high frequency of the PIGV variant NM_017837:c.101C > T, p.Pro34Leu present in this group (Figure 7b). The next group with the highest number of carriers is the Latino population, followed by the East Asian, South Asian, Ashkenazi Jewish and Finnish European populations. Individuals considered as “other” have the least number of carriers.
A limitation of this work is the small number of reported individuals with variants. Population trends may be more apparent or different in larger population genetic studies. Also, only a minority of variants had detailed information in public databases. Nonetheless, comparison of the frequencies on published GPI variants from exome and genome sequencing data shows aspects that may be interesting to population genetics and to scientists. It can also aid clinicians in disease prediction if a particular variant is more commonly reported in the population.
4. CONCLUSION
GPI‐APs play a role in several developmental processes. Variants in GPI biosynthesis genes can therefore lead to different types of diseases with many clinical abnormalities ranging in severity. These diseases are rare which makes it difficult to pinpoint their cause. The advent and robustness of NGS technologies have facilitated the identification of variants in several genes involved in GPI biosynthesis, and the amount of genetic information is expected to increase with the rapid progress of sequencing technologies. Robust and trustworthy databases are therefore needed to organize all this information. With this in mind and with the goal of centralizing all reported variants and making it widely and easily accessible to clinicians, scientists as well as to families, we developed an online platform (http://www.gpibiosynthesis.org/) to integrate the LOVD Locus‐specific databases for GPI biosynthesis genes. We aim to keep it up‐to‐date through ongoing curation and maintenance, and regularly updating the data with new publications.
This web resource allows the user to search for any publicly reported variant on genes involved in this biosynthesis pathway as it compiles all published variants, and through the different tabs, the user can instantly view or retrieve specific data on a particular variant. These features of the GPI biosynthesis webpage, combined with those of the LOVD platform, make this online database a helpful tool for health professionals and scientists to rapidly assess if a certain variation seen in the patient has already been reported, if it is pathogenic, if the effects of the variant are known both at the molecular and at the phenotypic level, and if it is common in a certain population. Further, this information can guide the clinician into choosing the right therapeutic intervention and/or counseling for the patient by linking genetic information to clinical characteristics. In the same view, if a variant requires further analysis due to the effect being unknown or the variant being novel, these new discoveries can expand the database to provide a more complete view of all variants in GPI biosynthesis genes. This database can therefore prove to be a useful tool for scientists, clinicians, as well as families looking to learn more about these genes involved in the biosynthesis of GPI‐APs.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Supporting information
ACKNOWLEDGEMENTS
We thank the Canadian Institutes of Health Research for funding (grant RN324373).
Baratang NV, Jimenez Cruz DA, Ajeawung NF, Nguyen TTM, Pacheco‐Cuéllar G, Campeau PM. Inherited glycophosphatidylinositol deficiency variant database and analysis of pathogenic variants. Mol Genet Genomic Med. 2019;7:e743 10.1002/mgg3.743
REFERENCES
- Adzhubei, I. A. , Schmidt, S. , Peshkin, L. , Ramensky, V. E. , Gerasimova, A. , Bork, P. , … Sunyaev, S. R. (2010). A method and server for predicting damaging missense mutations. Nature Methods, 7(4), 248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessandri, J. L. , Gordon, C. T. , Jacquemont, M. L. , Gruchy, N. , Ajeawung, N. F. , Benoist, G. , … Thevenon, J. (2018). Recessive loss of function PIGN alleles, including an intragenic deletion with founder effect in La Reunion Island, in patients with Fryns syndrome. European Journal of Human Genetics, 26(3), 340–349. 10.1038/s41431-017-0087-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellai‐Dussault, K. , Nguyen, T. T. M. , Baratang, N. V. , Jimenez‐Cruz, D. A. , & Campeau, P. M. (2019). Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clinical Genetics, 95(1), 112–121. 10.1111/cge.13425 [DOI] [PubMed] [Google Scholar]
- Chi, Z. , Nie, L. , Peng, Z. , Yang, Q. , Yang, K. , Tao, J. , … Zhao, Y. (2012). RecQL4 cytoplasmic localization: Implications in mitochondrial DNA oxidative damage repair. International Journal of Biochemistry & Cell Biology, 44(11), 1942–1951. 10.1016/j.biocel.2012.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita, M. , & Kinoshita, T. (2012). GPI‐anchor remodeling: Potential functions of GPI‐anchors in intracellular trafficking and membrane dynamics. Biochimica Et Biophysica Acta, 1821(8), 1050–1058. 10.1016/j.bbalip.2012.01.004 [DOI] [PubMed] [Google Scholar]
- Horn, D. , Wieczorek, D. , Metcalfe, K. , Barić, I. , Paležac, L. , Ćuk, M. , … Krawitz, P. (2014). Delineation of PIGV mutation spectrum and associated phenotypes in hyperphosphatasia with mental retardation syndrome. European Journal of Human Genetics, 22(6), 762–767. 10.1038/ejhg.2013.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard, M. F. , Murakami, Y. , Pagnamenta, A. T. , Daumer‐Haas, C. , Fischer, B. , Hecht, J. , … Krawitz, P. M. (2014). Mutations in PGAP3 impair GPI‐anchor maturation, causing a subtype of hyperphosphatasia with mental retardation. American Journal of Human Genetics, 94(2), 278–287. 10.1016/j.ajhg.2013.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilkovski, B. , Pagnamenta, A. T. , O'Grady, G. L. , Kinoshita, T. , Howard, M. F. , Lek, M. , … Clarke, N. F. (2015). Mutations in PIGY: Expanding the phenotype of inherited glycosylphosphatidylinositol deficiencies. Human Molecular Genetics, 24(21), 6146–6159. 10.1093/hmg/ddv331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, T. (2014). Biosynthesis and deficiencies of glycosylphosphatidylinositol. Proceedings of the Japan Academy, Series B, 90(4), 130–143. 10.2183/pjab.90.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus, A. , Pantel, J. T. , Pendziwiat, M. , Hajjir, N. , Zhao, M. , Hsieh, T.‐C. , … Krawitz, P. M. (2018). Characterization of glycosylphosphatidylinositol biosynthesis defects by clinical features, flow cytometry, and automated image analysis. Genome Medicine, 10(1), 3 10.1186/s13073-017-0510-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Johnson, A. , Schaefer, G. B. , Lee, J. , Hu, Y. , & Del Gaudio, D. (2017). Alu‐mediated deletion of PIGL in a Patient with CHIME syndrome. American Journal of Medical Genetics. Part A, 173(5), 1378–1382. 10.1002/ajmg.a.38181 [DOI] [PubMed] [Google Scholar]
- Krawitz, P. M. , Murakami, Y. , Hecht, J. , Krüger, U. , Holder, S. E. , Mortier, G. R. , … Horn, D. (2012). Mutations in PIGO, a member of the GPI‐anchor‐synthesis pathway, cause hyperphosphatasia with mental retardation. American Journal of Human Genetics, 91(1), 146–151. 10.1016/j.ajhg.2012.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. , & Ng, P. C. (2009). Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4(7), 1073–1081. 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makrythanasis, P. , Kato, M. , Zaki, M. S. , Saitsu, H. , Nakamura, K. , Santoni, F. A. , … Murakami, Y. (2016). Pathogenic Variants in PIGG cause intellectual disability with seizures and hypotonia. American Journal of Human Genetics, 98(4), 615–626. 10.1016/j.ajhg.2016.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKean, D. M. , & Niswander, L. (2012). Defects in GPI biosynthesis perturb Cripto signaling during forebrain development in two new mouse models of holoprosencephaly. Biology Open, 1(9), 874–883. 10.1242/bio.20121982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng, B. G. , & Freeze, H. H. (2015). Human genetic disorders involving glycosylphosphatidylinositol (GPI) anchors and glycosphingolipids (GSL). Journal of Inherited Metabolic Disease, 38(1), 171–178. 10.1007/s10545-014-9752-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, T. T. M. , Murakami, Y. , Sheridan, E. , Ehresmann, S. , Rousseau, J. , St‐Denis, A. , … Campeau, P. M. (2017). Mutations in GPAA1, Encoding a GPI transamidase complex protein, cause developmental delay, epilepsy, cerebellar atrophy, and osteopenia. American Journal of Human Genetics, 101(5), 856–865. 10.1016/j.ajhg.2017.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki, M. , Ohishi, K. , Yamada, N. , Kinoshita, T. , Nagy, A. , & Takeda, J. (1999). Developmental abnormalities of glycosylphosphatidylinositol‐anchor‐deficient embryos revealed by Cre/loxP system. Laboratory Investigation, 79(3), 293–299. [PubMed] [Google Scholar]
- Tinschert, S. , Anton‐Lamprecht, I. , Albrecht‐Nebe, H. , & Audring, H. (1996). Zunich neuroectodermal syndrome: Migratory ichthyosiform dermatosis, colobomas, and other abnormalities. Pediatric Dermatology, 13(5), 363–371. 10.1111/j.1525-1470.1996.tb00702.x [DOI] [PubMed] [Google Scholar]
- Vihinen, M. , den Dunnen, J. T. , Dalgleish, R. , & Cotton, R. G. (2012). Guidelines for establishing locus specific databases. Human Mutation, 33(2), 298–305. 10.1002/humu.21646 [DOI] [PubMed] [Google Scholar]
- Walters‐Sen, L. C. , Hashimoto, S. , Thrush, D. L. , Reshmi, S. , Gastier‐Foster, J. M. , Astbury, C. , & Pyatt, R. E. (2015). Variability in pathogenicity prediction programs: Impact on clinical diagnostics. Mol Genet Genomic Med, 3(2), 99–110. 10.1002/mgg3.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials