

Abstract
Background
Recently, increasing innovations improved the accuracy of next generation sequencing (NGS) data. However, the validation of all NGS variants increased the cost and turn‐around time of clinical diagnosis, and therefore limited the further development of clinical applications. We aimed to comprehensively assess the necessity of validating NGS variants.
Methods
Validation data of 7,601 NGS variants involving 1,045 genes were collected from 5,190 clinical samples and sequenced by one of five targeted capture panels and two NGS chemistries, respectively. These genes and variants were widely distributed in 24 human chromosomes and mitochondrial genome. Variants validation was firstly processed by Sanger sequencing. If validation results were unavailable or inconsistent with NGS calls, another validation test would be performed by mass spectrometry genotyping.
Results
A total of 6,939 high quality NGS variants with ≥35 × depth coverage and ≥35% heterozygous ratio were 100% confirmed by a secondary methodology. 5,775 heterozygous variants were separated from 760 homozygous variants and 404 hemizygous variants by 80% heterozygous ratio. A total of 1.5% (98/6,939) of NGS variants were validated by mass spectrometry genotyping.
Conclusion
Considering of the above comprehensive assessment, a new variant with high quality from a well‐validated capture‐based NGS workflow can be reported directly without validation.
Keywords: NGS, quality threshold, Sanger sequencing, target enrichment, validation
1. INTRODUCTION
The past dozen years have witnessed a new era in DNA sequencing technologies. Next‐generation sequencing (NGS) performs sequencing of thousands to millions of DNA fragments simultaneously. This reduces the sequencing cost per base by several orders of magnitude. NGS technologies are nowadays being widely applied in diagnosis of genetic disorders (Goodwin, McPherson, & McCombie, 2016).
The high‐quality NGS data are essential for the clinical application. It is undisputed that sufficiently validated tests should be undertaken before using a new NGS workflow in a clinical diagnostic screening process (Matthijs et al., 2016). However, there is still a significant debate about whether variants identified by NGS should be validated using a secondary technology, such as traditional Sanger sequencing with increasing cost and turn‐around time. Several commercial and academic laboratories have reported with small amounts of validated samples that clinical NGS variants meeting threshold quality metrics were unnecessarily validated (Baudhuin et al., 2015; Judkins et al., 2015; Sikkema‐Raddatz et al., 2013; Strom et al., 2014). Recently, another two studies have been done with thousands of variants (5,800 and 7,845, respectively), by using a unique NGS sequencing platform with small gene sets (Beck, Mullikin, Program, & Biesecker, 2016; Mu, Lu, Chen, Li, & Elliott, 2016). The prior studies were not sufficient to conclude that variants validation will be insignificant or less significant for a large scale NGS application in clinical diagnosis.
In this study, we addressed a comprehensive and extensive assessment of NGS variants validation, which incorporated two NGS platforms, two validation methodologies, five targeted capture panels, 5,190 samples, 1,045 genes, and 7,601 variants. The variants included 5,956 SNVs (single‐nucleotide variants) and 1,645 indels (insertion/deletion). The GC content of variant flanking sequence was ranged from 20% to 84%. The variants were distributed in 22 autosomes, X and Y chromosomes, and mitochondrial genome. According to the rich data from capture‐based NGS, a significant quality metrics with 100% validation rate was addressed with at least 35 × depth coverage and more than 35% heterozygous ratio in our study.
2. MATERIALS AND METHODS
2.1. Ethical compliance
This study was approved by the institutional review board of BGI (NO. BGI‐IRB: 18030).
2.2. Sample and DNA Isolation
Blood samples were collected from individuals after obtaining the informed consent for this study. Genomic DNAs extraction was carried out using MagPure Buffy Coat DNA Midi KF Kit (Magen, China) according to the manufacturer's instruction. The quality and quantity of DNA samples were determined by NanoDrop spectrophotometer (Thermo Fisher Scientific, USA). DNA samples with absorbance ratio A260/A280 between 1.6 and 2.1 and total amount greater than 2 μg were acceptable and used in following steps.
2.3. NGS library preparation and sequencing
Briefly, 1μg genomic DNA was fragmented into 150–250 bp using a Covaris S220 series sonicator (Covaris, USA), followed by end repair, A‐tailing, and adapter ligation before pre‐capture amplification. Amplification products were purified by Agencourt AMPure XP beads. In‐solution targeted sequence capture was performed by either SeqCap EZ Choice Library (Roche, cat: 06266304001) or Seq Cap EZ Hybridization and Wash Kit (MGI, cat: 5634253001) with different customized capture probe panels according to patients’ clinical conditions (Table 1). After enrichment and quantification, DNA libraries were sequenced on a HiSeq2500 sequencer (Illumina, USA) as 90‐bp paired‐end reads or a BGISEQ‐500 sequencer (BGI, China) as 50‐bp paired‐end reads.
Table 1.
Information of the five capture panels used in this study
| Panel name | Targeted region (kb) | Number of genes | Number of exons | Panel provider | Average coverage | Average depth | ||
|---|---|---|---|---|---|---|---|---|
| HiSeq2500 | BGISEQ‐500 | HiSeq2500 | BGISEQ‐500 | |||||
| HCa | 839 | 115 | 1,734 | Roche | 99.20 | 99.18 | 426 | 261 |
| HD | 1,565 | 313 | 4,899 | Roche | 92.75 | 91.69 | 393 | 159 |
| 4.8M | 4,760 | 1,130 | 17,108 | Roche | 99.84 | 99.78 | 294 | 210 |
| C2181 | 5,199 | 2,182 | 31,632 | Roche | 99.84 | 99.84 | 234 | 176 |
| Exo | 58,882 | 18,979 | 197,748 | BGI | 99.45 | 99.74 | 123 | 122 |
HC, panel for hereditary cancer; HD, panel for hereditary deafness; 4.8M and C2181, two panels for Mendelian disease; Exo, human exome.
2.4. Bioinformatics analysis and variants selection
The pipeline of bioinformatics analysis included reads filtering, reads mapping against the hg19 human reference genome, variants detection, and functional annotation as described previously in our studies (Liu et al., 2015; Shang et al., 2017). According to the variants interpretation guidelines of American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015), clinically relevant variants (pathogenic variants, likely pathogenic variants, and variants of unknown significance) were interpreted and selected for validation using a secondary technology, in which the SNVs and small indels (<100bp) were validated by Sanger sequencing and/or mass spectrometry genotyping.
The GC content was counted using the 100bp flanking sequence of each variant.
2.5. Sanger sequencing
The specific primers (BGI, China) were designed using Primer‐BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) (Table S1). All regions containing variants were amplified by PCR and sequenced by a 3730xl DNA Analyzer (Thermo Fisher Scientific, USA) according to standard procedures.
2.6. Mass spectrometry genotyping
Mass spectrometry genotyping was performed using the MassARRAY system (Agena Bioscience, USA) when the variant was not validated by Sanger sequencing. Single‐plex assay was designed by Assay Design Suite v2.0 (Agena Bioscience, USA) and synthesized by BGI (Table S2). According to standard procedures, 1 μl genomic DNA was required in each 5 μl PCR reaction mix. The data were collected by MassARRAY Typer software v4.0 (Agena Bioscience, USA). For quality control, a negative control without genomic DNA and a positive control using normal genomic DNA as PCR template were undertaken simultaneously with clinical samples. The normal genomic DNA was taken from a healthy Han Chinese with known genome data at database (http://yh.genomics.org.cn/index.jsp).
2.7. Data access
The 7,601 variants information are available in Table S3.
3. RESULTS
3.1. The distribution of NGS variants
Five customized capture probe panels and two sequence platforms were selected to extensively assess the validation of NGS variants (Table 1). The panels included different numbers of genes and exons, from 115 to 18,979 and from 1,743 to 197,748 respectively. Similar average coverages were acquired with different average depths between HiSeq2500 and BGISEQ‐500. Assessment of NGS variants validation was performed on 7,601 germline variants, including 5,956 SNVs and 1,645 indels, from 5,190 samples and 1,045 genes (Table 2). These variants and genes were distributed in different chromosomes of human genome including mitochondrial genome (Figure 1). GC content is a very important factor which influences the accuracy of NGS data (Matthijs et al., 2016). The GC content was counted using the 100 bp flanking sequence of each variant. The statistical results showed that GC content of these variants, ranging from 20% to 84%, was mainly between 30% and 50% but with two peaks at 70% and 75% (Figure 1). In total, all the above information addressed that the variants in this study included a wide range of panel sizes, sequence chemistries, variant types, samples, gene attributes, chromosome distributions, and GC contents, which provided essential foundation for an extensive assessment of NGS variants validation.
Table 2.
Variants distribution used in this study
| Panel | Sequence chemistry | Sample | Gene | Variant | SNV | indel |
|---|---|---|---|---|---|---|
| HCa | HiSeq2500 | 110 | 8 | 114 | 67 | 47 |
| HC | BGISEQ‐500 | 216 | 10 | 223 | 119 | 104 |
| HD | HiSeq2500 | 662 | 73 | 1,125 | 842 | 283 |
| HD | BGISEQ‐500 | 291 | 8 | 507 | 416 | 91 |
| 4.8M | HiSeq2500 | 1,328 | 231 | 1,882 | 1,471 | 411 |
| 4.8M | BGISEQ‐500 | 134 | 5 | 184 | 152 | 32 |
| C2181 | HiSeq2500 | 1,584 | 295 | 2,042 | 1,553 | 489 |
| C2181 | BGISEQ‐500 | 523 | 32 | 669 | 508 | 161 |
| Exo | HiSeq2500 | 154 | 203 | 379 | 368 | 11 |
| Exo | BGISEQ‐500 | 188 | 180 | 476 | 460 | 16 |
| Total | 5,190 | 1,045 | 7,601 | 5,956 | 1,645 |
HC, panel for hereditary cancer; HD, panel for hereditary deafness; 4.8M and C2181, two panels for Mendelian disease; Exo, human exome.
Figure 1.
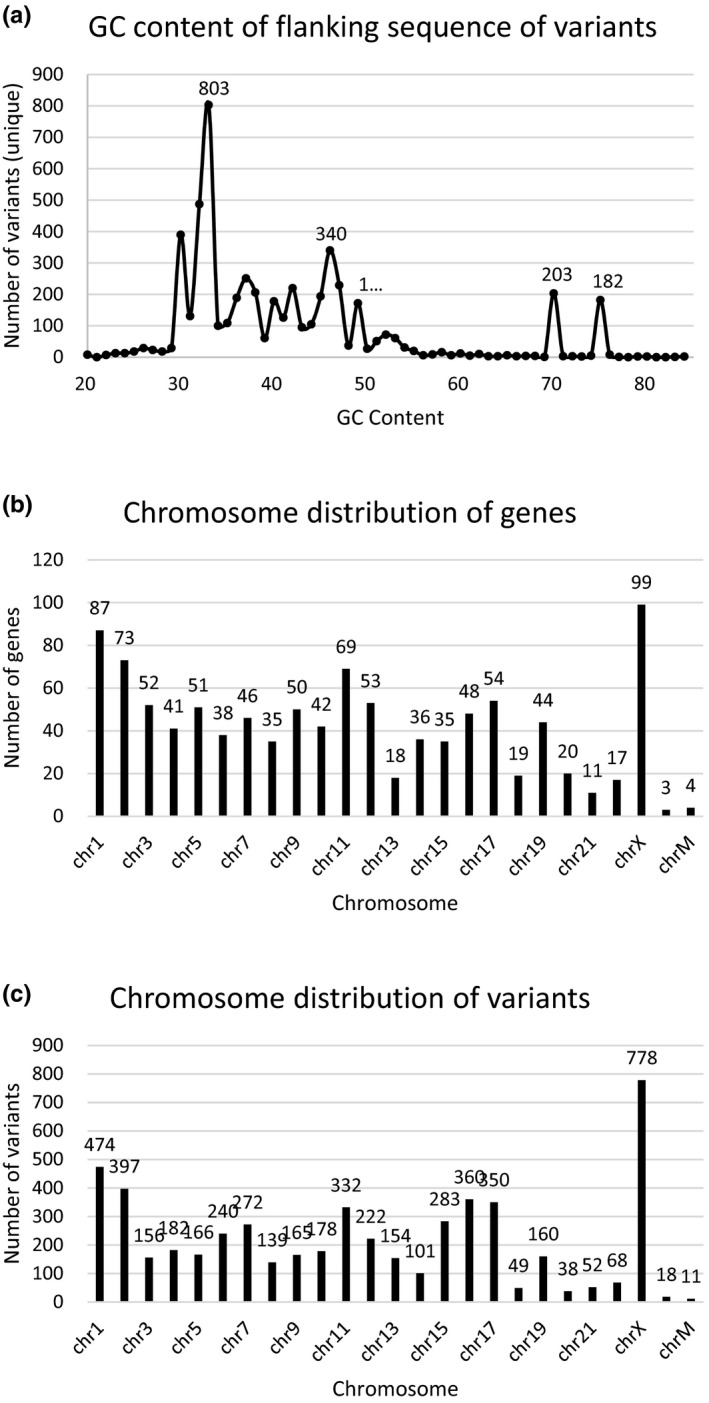
Characterization of variants and genes in this study. (a) The GC content distribution of the 5,345 unique variants. GC content was calculated using the 100 bp flanking sequence of each variant. (b) The chromosome distribution of the 5,345 unique variants. (c) The chromosome distribution of the 1,045 unique genes
3.2. Assessment of NGS variants validation
Among the 7,601 NGS variants analyzed, 6,939 variants with at least 35 × depth coverage and more than 35% heterozygous ratio as well as their zygosity were all confirmed by the secondary methodology (Figure 2). Specifically, 1,164 homozygous variants (including hemizygous variants) and 5,775 heterozygous variants were included and separated by a heterozygous ratio of 80% (Figure 2). The remaining 662 variants had a low‐quality score with either depth coverage less than 35 × or heterozygous ratio smaller than 35%, and only 64.4% (426/662) of them were confirmed by a secondary technology (Figure 2). Our further study suggested that variants with het ratio smaller than 35% and depth coverage more than 35 × contributed a higher proportion in true positive group than in false positive group (Table 3). A total of 37.9% (251/662) remaining variants had at least a homologous region, and had a higher false positive ratio when the depth coverage was more than 35 × and het ratio was smaller than 35%. Moreover, our study included 1,482 indels with high quality. There was no correlation between indels’ size and depth coverage (Figure S1). Collectively, the quality metrics of 35 × depth coverage and 35% heterozygous ratio were significant for the accuracy of the NGS variants, and the het ratio was more important than depth to decrease the false positive ratio.
Figure 2.
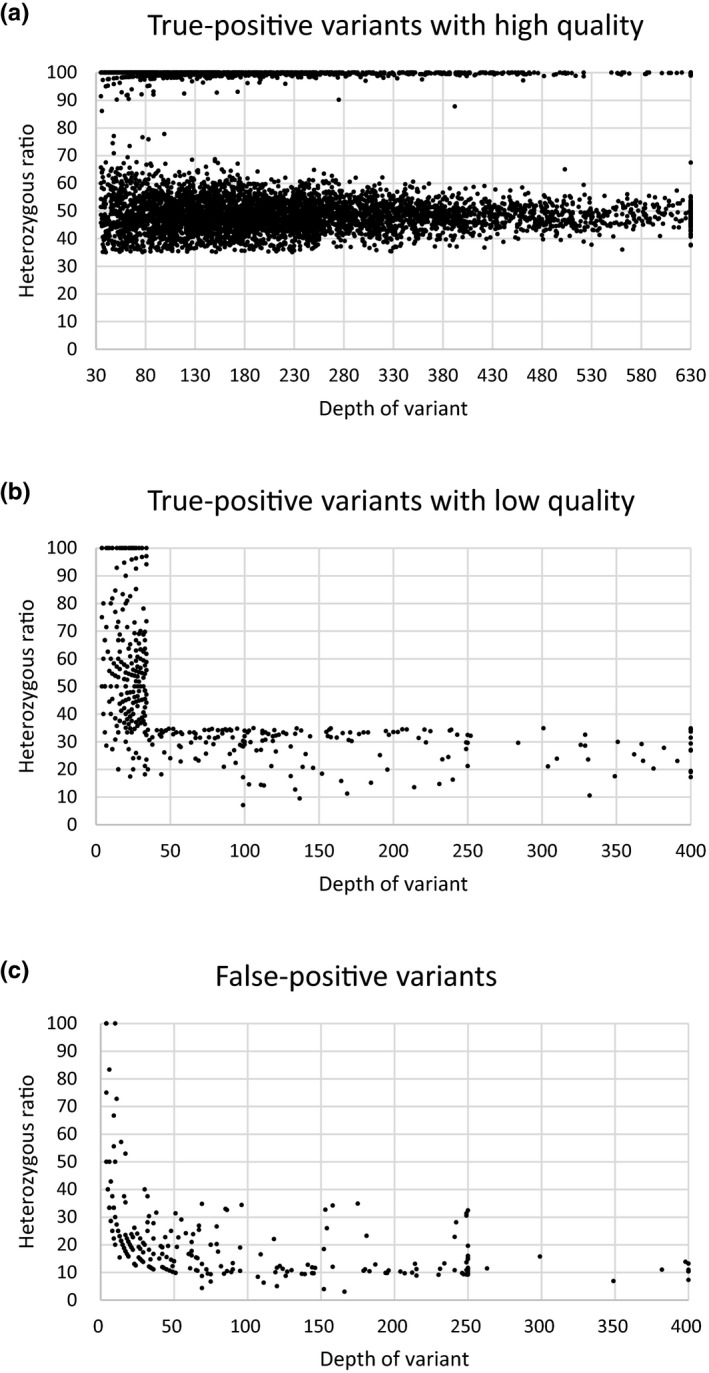
Validation results of NGS variants. The scattergrams show the NGS depth and heterozygous ratio of true‐positive variants with high quality (a), true‐positive variants with low quality (b), and false‐positive variants (c). The depth of variant was calculated as 630 (A) or 400 (B) (C), when it was up to 630 or 400, respectively
Table 3.
The NGS quality distribution of 662 variants with low‐quality score
| Total | With homology | |||||||
|---|---|---|---|---|---|---|---|---|
| True | True % | False | False % | True | True % | False | False % | |
| ≥35 × depth & <35% Het ratio | 171 | 40.1% | 142 | 60.2% | 64 | 40.8% | 73 | 77.7% |
| <35 × depth & <35% Het ratio | 28 | 6.6% | 71 | 30.1% | 14 | 8.9% | 14 | 14.9% |
| <35 × depth &≥35% Het ratio | 227 | 53.3% | 23 | 9.7% | 79 | 50.3% | 7 | 7.4% |
| Total | 426 | 100.0% | 236 | 100.0% | 157 | 100.0% | 94 | 100.0% |
Considering both the cost‐effectiveness and scientific rigor of validation, we validated the NGS variants by Sanger sequencing firstly. When the results were unavailable or inconsistent with NGS data, mass spectrometry genotyping was further used to confirm these variants. A total of 98 NGS variants with high quality were validated by mass spectrometry genotyping but not by Sanger sequencing, which indicated the advantage of NGS technology in testing these variants. Sanger sequencing is inaccurate to detect the homopolymeric region greater than seven bases. We found that there were 31 of 98 variants with a homopolymer at the100 bp flanking sequence (Table S4). Six of 98 variants were from FLG (filaggrin, OMIM: 135940) which has pseudogene. Further study revealed that the 200 bp flanking sequence of 11 variants was false mapped to human genome (Table S4). We did not find another characteristic to elucidate the failure of Sanger sequencing to confirm the other variants.
4. DISCUSSION
With the advantages of high‐throughput and low‐cost, NGS is becoming a new powerful tool of sequencing technology following Sanger DNA sequencing. Increased accessibility and flexibility have broadened NGS applications in both research and diagnostic fields (Goodwin et al., 2016). Recently, many revolutionary innovations contributed to the NGS technological advancement, especially the targeted capture sequencing, which decreases the cost of sequencing furthermore (Kozarewa, Armisen, Gardner, Slatko, & Hendrickson, 2015). However, much attention is paid to the accuracy of the NGS data because of the short‐read length. Ensuring high quality data are essential when using NGS (Nietsch et al., 2016).
Validation is a very effective and indispensable method to confirm the data quality of a new NGS workflow (Baudhuin et al., 2015; Judkins et al., 2015; Sikkema‐Raddatz et al., 2013; Strom et al., 2014). There was a recognized guideline for diagnostic NGS (Matthijs et al., 2016). For example, accuracy, sensitivity, specificity, and stability of a new NGS workflow could be obtained and accessible by testing some reference standards and comparing with other gene testing methods. Besides, there is another type of validation to confirm the data quality of a new variant from a validated and accessible NGS test. Several significant studies have focused on the necessity of the validation of NGS variant using Sanger sequencing, considering the increased cost and long turn‐around time (Baudhuin et al., 2015; Beck et al., 2016; Mu et al., 2016; Strom et al., 2014). In 2014, Strom and colleagues validated 110 SNVs from 144 clinical exome‐sequencing data by Sanger sequencing. One hundred and three of them were confirmed all with a quality score ≥Q500, while only one variant of the remaining seven variants with quality score <Q500 was not confirmed (Strom et al., 2014). Next year, Baudhuin and colleagues from Mayo Clinic evaluated the accuracy of targeted capture NGS results (797 SNVs and 122 indels from 84 samples) by Sanger sequencing. A total of 100% concordance between NGS and Sanger data was acquired with a much higher quality threshold of ≥100× coverage depth in 99.7% of targeted bases (Baudhuin et al., 2015). In 2016, Beck and colleagues from NIH, evaluated Sanger‐based validation of over 5,800 NGS‐derived SNVs from 19 genes in 684 samples, of which only two SNVs with low quality scores (Most Probable Genotype score <10) were truly discrepant. The study showed a much higher validation rate of 99.965% using a large number of variants but small coverage of human genome (Beck et al., 2016). Another large‐scale study was performed by Mu and colleagues from Ambry Genetics in the same year with 7,845 variants (6,912 SNVs and 933 indels) from a targeted capture NGS panel of 47 genes to assess the sensitivity and specificity of NGS test by Sanger sequencing (Mu et al., 2016). The data illustrated the variants with a conservative quality threshold of >100× coverage and >40% heterozygous ratio can be reported out without secondary confirmation (Mu et al., 2016). Although, different numbers of variants, genes, and samples with different quality metrics, the above four important studies showed the same conclusion that it is not necessary to validate a NGS variant with high quality score by a secondary methodology. However, there were at least three deficiencies of these four studies mentioned above as follows: first, limited human genome coverage (with 94, 117, 19, and 47 genes, respectively); second, less than two targeted capture panels in only one sequence chemistry (Illumina) in each study; and last, the quality metrics were either markedly high or complex to calculate.
In order to extensively assess the validation of NGS variants, we selected a data set from five targeted capture panels focused on hereditary cancer, hereditary hearing impairment, whole human exome, and Mendelian disease, respectively. The four small panels were provided by Roche, covering 115, 313, 1,130, and 2,181 genes, while the whole human exome panel (Exo) was made by BGI (Table 1). Furthermore, a new sequencer, BGISEQ‐500 using DNA nanoball and combinational probe anchor synthesis technologies, was applied in this study besides the popular Illumina sequencer HiSeq2500 (Huang et al., 2017). In our present study, 5,190 samples were chosen based on the criteria of at least one disease‐associated variant. The various panels, sequence chemistries, and samples made the conclusion of this study more confidence. Depth coverage and heterozygous ratio are the essential quality control parameters for NGS variants. Many important studies used these quality parameters because of the flexibility (Baudhuin et al., 2015; Mu et al., 2016). In our present study, 6,939 variants with at least 35 × depth coverage and more than 35% heterozygous ratio were confirmed by a secondary methodology, which included 5,775 heterozygous variants, 760 homozygous variants, and 404 hemizygous variants. In addition, considering of the whole genome distribution and large range of GC content of these variants, our quality threshold is more confident than that of the former studies. The lower depth coverage may be due to the higher stability and accuracy of our NGS workflow. A secondary variants validation test is often costly and time consuming. Additionally, without the validation of high quality NGS variants, the turn‐round time and fees will be reduced by 91.3% (6,939/7,601). Hence, the genetics diagnosis by NGS will be more accessible and affordable. Significantly, the NGS workflow should be validated scientifically and rigorously as the presentation from guidelines for diagnostic next‐generation sequencing (Matthijs et al., 2016), and the quality threshold for high‐confidence NGS should be defined before canceling the validation test of variants with clinical relevance. However, the variants with low quality or distributing in repetitive DNA sequence must be validated before reported for clinical applications.
As the gold standard for DNA sequencing, Sanger sequencing was selected to validate the NGS variants first. There were three different results, consistent, inconsistent, and unavailable, respectively. If the Sanger result was inconsistent with NGS or unavailable, we would confirm the result by mass spectrometry genotyping, which have a principle by short amplicon and single nucleotide extension. It is more scientific than change primers for a secondary Sanger sequencing test because of the technical limitation of Sanger sequencing (Beck et al., 2016). For example, the double peaks will influence the Sanger sequencing results when there is a homopolymeric region greater than seven bases between variant site and sequencing primer. However, the homopolymeric region cannot influence the accuracy of mass spectrometry genotyping. In our present study, 98.5% (6,841/6,939) of variants with high quality of capture‐based NGS call were confirmed by Sanger sequencing, and the remaining 98 variants validation results were from mass spectrometry genotyping. Accordingly, primers for Sanger sequencing should be specific and without homopolymer greater than 7 bp between the sequence primer and variant.
Although Heterozygous and Homozygous variants were separated by 80% heterozygous ratio in this study (Figure 2), the boundary of heterozygous ratio between Heterozygous and Homozygous variants was blurry even for some variants with a high depth coverage. Our study was incapable to explain the cause and find corresponding solutions. We supposed that index switching might contribute to this phenomenon (Owens, Todesco, Drummond, Yeaman, & Rieseberg, 2018). This will be improved in our future studies.
5. CONCLUSION
With the comprehensive and extensive assessment of 7,601 NGS variants validation from different NGS platforms, targeted capture panels, validation methodologies, samples, genes, chromosomes, and GC contents, we found variants with a quality threshold of ≥35× depth coverage and ≥35% heterozygous ratio were 100% true‐positive in this study. In consideration of both the labor and cost burden of validation test and increasing requirements for clinical genetic diagnosis and improved accuracy of NGS data, we suggest that a new variant with high quality from a well‐validated capture‐based NGS workflow does not require a secondary validation and can be reported directly.
CONFLICT OF INTEREST
Upon manuscript submission, all authors declare no conflict of interest.
Supporting information
Zheng J, Zhang H, Banerjee S, et al. A comprehensive assessment of Next‐Generation Sequencing variants validation using a secondary technology. Mol Genet Genomic Med. 2019;7:e748 10.1002/mgg3.748
Jianchao Zheng and Hongyun Zhang contributed equally to this work.
Funding information
This research was supported by Shenzhen Engineering Laboratory for Birth Defects Screening (DRC‐SZ [2011]861), Special Foundation for High‐level Talents of Guangdong (2016TX03R171), and Shenzhen Peacock Plan (No. KQTD20150330171505310).
Contributor Information
Zhiyu Peng, Email: pengzhiyu@genomics.cn.
Wei Wang, Email: wangw@genomics.cn.
Ye Yin, Email: yinye@genomics.cn.
REFERENCES
- Baudhuin, L. M. , Lagerstedt, S. A. , Klee, E. W. , Fadra, N. , Oglesbee, D. , & Ferber, M. J. (2015). Confirming variants in next‐generation sequencing panel testing by sanger sequencing. The Journal of Molecular Diagnostics, 17(4), 456–461. 10.1016/j.jmoldx.2015.03.004 [DOI] [PubMed] [Google Scholar]
- Beck, T. F. , Mullikin, J. C. , Program, N. C. S. , & Biesecker, L. G. (2016). Systematic evaluation of sanger validation of next‐generation sequencing variants. Clinical Chemistry, 62(4), 647–654. 10.1373/clinchem.2015.249623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin, S. , McPherson, J. D. , & McCombie, W. R. (2016). Coming of age: Ten years of next‐generation sequencing technologies. Nature Reviews Genetics, 17(6), 333–351. 10.1038/nrg.2016.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, J. , Liang, X. , Xuan, Y. , Geng, C. , Li, Y. , Lu, H. , … Gao, S. (2017). A reference human genome dataset of the BGISEQ‐500 sequencer. Gigascience, 6(5), 1–9. 10.1093/gigascience/gix024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judkins, T. , Leclair, B. , Bowles, K. , Gutin, N. , Trost, J. , McCulloch, J. , … Timms, K. (2015). Development and analytical validation of a 25‐gene next generation sequencing panel that includes the BRCA1 and BRCA2 genes to assess hereditary cancer risk. BMC Cancer, 15, 215 10.1186/s12885-015-1224-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozarewa, I. , Armisen, J. , Gardner, A. F. , Slatko, B. E. , & Hendrickson, C. L. (2015). Overview of target enrichment strategies. Current Protocols in Molecular Biology, 112, 7 21. 21–723. 10.1002/0471142727.mb0721s112 [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Wei, X. , Kong, X. , Guo, X. , Sun, Y. , Man, J. , … Yang, Y. (2015). Targeted next‐generation sequencing for clinical diagnosis of 561 mendelian diseases. PLoS ONE, 10(8), e0133636 10.1371/journal.pone.0133636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthijs, G. , Souche, E. , Alders, M. , Corveleyn, A. , Eck, S. , Feenstra, I. , … Bauer, P. (2016). Guidelines for diagnostic next‐generation sequencing. European Journal of Human Genetics, 24(10), 1515 10.1038/ejhg.2016.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthijs, G. , Souche, E. , Alders, M. , Corveleyn, A. , Eck, S. , Feenstra, I. … European Society of Human Genetics . (2016). Guidelines for diagnostic next‐generation sequencing. European Journal of Human Genetics, 24(1), 2–5. 10.1038/ejhg.2015.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu, W. , Lu, H. M. , Chen, J. , Li, S. , & Elliott, A. M. (2016). Sanger confirmation is required to achieve optimal sensitivity and specificity in next‐generation sequencing panel testing. The Journal of Molecular Diagnostics, 18(6), 923–932. 10.1016/j.jmoldx.2016.07.006 [DOI] [PubMed] [Google Scholar]
- Nietsch, R. , Haas, J. , Lai, A. , Oehler, D. , Mester, S. , Frese, K. S. , … Meder, B. (2016). The role of quality control in targeted next‐generation sequencing library preparation. Genomics Proteomics Bioinformatics, 14(4), 200–206. 10.1016/j.gpb.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens, G. L. , Todesco, M. , Drummond, E. B. M. , Yeaman, S. , & Rieseberg, L. H. (2018). A novel post hoc method for detecting index switching finds no evidence for increased switching on the Illumina HiSeq X. Molecular Ecology Resources, 18(1), 169–175. 10.1111/1755-0998.12713 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang, X. , Peng, Z. , Ye, Y. , Asan,, Zhang, X. , Chen, Y. , … Xu, X. (2017). Rapid targeted next‐generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine, 23, 150–159. 10.1016/j.ebiom.2017.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikkema‐Raddatz, B. , Johansson, L. F. , de Boer, E. N. , Almomani, R. , Boven, L. G. , van den Berg, M. P. , … Sinke, R. J. (2013). Targeted next‐generation sequencing can replace Sanger sequencing in clinical diagnostics. Human Mutation, 34(7), 1035–1042. 10.1002/humu.22332 [DOI] [PubMed] [Google Scholar]
- Strom, S. P. , Lee, H. , Das, K. , Vilain, E. , Nelson, S. F. , Grody, W. W. , & Deignan, J. L. (2014). Assessing the necessity of confirmatory testing for exome‐sequencing results in a clinical molecular diagnostic laboratory. Genetics in Medicine, 16(7), 510–515. 10.1038/gim.2013.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials