

Abstract
S-Adenosyl-l-methionine (SAM) is universal in biology, serving as the second most common cofactor in a variety of enzymatic reactions. One of the main roles of SAM is the methylation of nucleic acids, proteins and metabolites. Methylation often imparts regulatory control to DNA and proteins, and leads to an increase in the activity of specialized metabolites such as those developed as pharmaceuticals. There has been increased interest in using SAM analogs in methyltransferase-catalyzed modification of biomolecules. However, SAM and its analogs are expensive and unstable, degrading rapidly under physiological conditions. Thus, the availability of methods to prepare SAM in situ are desirable. In addition, synthetic methods to generate SAM analogs suffer from low yields and poor diastereoselectivity. The chlorinase SalL from the marine bacterium Salinispora tropica catalyzes the reversible, nucleophilic attack of chloride at the C5′ ribosyl carbon of SAM leading to the formation of 5′-chloro-5′-deoxyadenosine (ClDA) with concomitant displacement of l-methionine. It has been demonstrated that the in vitro equilibrium of the SalL-catalyzed reaction favors the synthesis of SAM. In this chapter, we describe methods for the preparation of SalL, and the chemoenzymatic synthesis of SAM and SAM analogs from ClDA and l-methionine congeners using SalL. In addition, we describe procedures for the in situ chemoenzymatic synthesis of SAM coupled to DNA, peptide and metabolite methylation, and to the incorporation of isotopes into alkylated products.
Keywords: S-adenosylmethionine (SAM), chlorodeoxyadenosine synthase, chlorinase, methylation, isotope incorporation, chemoenzymatic
1. INTRODUCTION
S-Adenosyl-l-methionine (SAM or AdoMet) is ubiquitous in nature and the second most utilized enzyme substrate after ATP. SAM is not only a source of methyl groups to methylate a variety of substrates, including nucleic acid, proteins and small molecules (Cantoni, 1975; Thomsen, Vogensen, Buchardt, Burkart & Clausen, 2013), but also a source of methylene, amino, ribosyl and aminopropyl groups, in addition to 5′-deoxyadenosyl radicals (Fontecave, Atta & Mulliez, 2004). Remarkably, SAM has also recently been shown to serve as cofactor in coordinating pericyclic reactions via electrostatic catalysis (Ohashi, Liu, Hai, Chen, Tang, Yang, et al., 2017). Another role for SAM is to serve as a substrate in reactions catalyzed by SAM-dependent halogenases of the 5′-halo-5′-deoxyadenosine synthase class (Dong, Huang, Deng, Schaffrath, Spencer, O’Hagan & Naismith, 2004; Eustáquio, Pojer, Noel & Moore, 2008; Agarwal, Miles, Winter, Eustáquio, El Gamal & Moore, 2017).
Halogenated compounds are pervasive across all domains of life. Over 5,000 organohalogens are known. Organochlorines and organobromines are the most abundant with >2,000 known examples of each, whereas organoiodines are more scarce (<200) and organofluorines are very rare with only five rigorously identified compounds (Gribble, 2012; Chan & O’Hagan, 2012, and Wang, Zhou, Fredimoses, Liao & Liu, 2014). Halogenating enzymes employ various mechanistic strategies depending on the properties of the halogen atom and the reactivity of the organic substrate. Most halogenases activate chloride, bromide and iodide via oxidation by utilizing cofactors such as heme-iron, vanadium, flavin and Fe(II)/α-ketoglutarate (Agarwal et al., 2017). For fluoride, on the other hand, which evades oxidation due to its extreme electronegativity, an orthogonal strategy has been described that uses SAM as co-substrate in an SN2-type, nucleophilic substitution reaction (Figure 1). The only fluorinase described thus far (FlA) catalyzes the first committed step in the biosynthesis of fluoroacetate and fluorothreonine in the soil bacterium Streptomyces cattleya in which fluoride attacks the C5′ electrophilic carbon of SAM, displacing L-methionine and generating 5′-fluoro-5′-deoxyadenosine (Dong et al., 2004).
Figure 1. Reaction catalyzed by SAM-dependent halogenases.
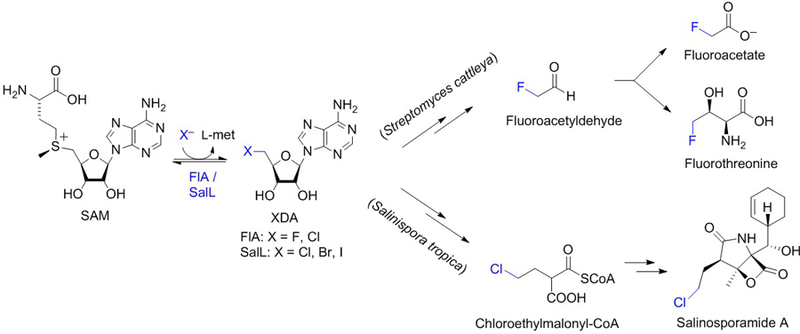
Fluorinase FlA from soil bacterium Streptomyces cattleya (Dong et al., 2004) and chlorinase SalL from marine bacterium Salinispora tropica (Eustáquio et al., 2008) catalyze reversible SN2-type, nucleophilic substitution reactions in which the halide attacks the C5′ ribosyl carbon of SAM, generating 5′-halo-5′-deoxyadenosine (XDA) and releasing l-methionine. FlA and SalL catalyze the first committed step in the biosynthesis of fluorometabolites and salinosporamide A, respectively.
Four years after the structural characterization of the fluorinase, the chlorinase SalL was reported from the marine bacterium Salinispora tropica (Eustáquio et al., 2008). SalL is analogous to fluorinase and the only other characterized SAM-dependent halogenase of the 5′-halo-5′-deoxyadenosine (XDA) synthase class (Figure 1). SalL catalyzes the first committed step in the biosynthesis of proteasome inhibitor salinosporamide A (Eustáquio et al., 2008 and 2009; Feling, Buchanan, Mincer, Kauffman, Jensen & Fenical, 2003). Both FlA and SalL can also catalyze the reverse reaction, i.e. synthesis of SAM from XDA. For FlA, the in vitro equilibrium lies in favor of FDA by a factor of three (Dong et al., 2004). That the forward reaction is favored by FlA is unsurprising given that the C-F bond is the strongest bond in organic chemistry. For chlorinase SalL, on the other hand, synthesis of SAM from ClDA and l-met is favored, with the relative enzyme efficiency (kcat/Km) for the reverse reaction (SAM synthesis) being several orders of magnitude higher than the forward halogenation reaction (Eustáquio et al., 2008). The fluorinase was also reported to favor the reverse reaction in vitro when using chloride as the halide (Deng, Cobb, McEwan, McGlinchey, Naismith, O’Hagan et al., 2006).
As a versatile methyl donor to biomolecules, there has been considerable interest in exploring the synthesis of SAM analogs of chemical and biological utility. For instance, non-natural SAM analogs could be used to modify cellular targets such as DNA, RNA, proteins and metabolites. Moreover, SAM itself is an expensive and unstable cofactor, degrading rapidly under physiological or alkaline conditions. Thus, the availability of methods to prepare SAM in situ are desirable.
Traditionally, SAM analogs have been chemically synthesized from S-adenosyl-l-homocysteine. However, this synthesis suffers from low yields and lack of diastereoselectivity (Dalhoff, Lukinavicius, Klimasauskas & Weinhold, 2006). Furthermore, SAM synthesis is challenging due to its inherent lability and tendency to racemize (Hoffman, 1986). Chemoenzymatic synthesis using chlorinase SalL and ClDA offers an alternative to chemical synthesis. In addition to being stable and commercially available, ClDA can also be synthesized economically from adenosine and SOCl2 (Sinhababu, Bartel, Pochopin & Borchardt 1985). Chemoenzymatic synthesis of SAM and analogs in situ could also be coupled with downstream applications such as methylation or alkylation by methyltransferases. This strategy can also be used for the facile incorporation of isotopes into alkylated products.
We have demonstrated the use of SalL for the diastereoselective synthesis of SAM and SAM congeners from ClDA and l-met analogs (See section 3). We have also coupled the SalL-catalyzed production of SAM with DNA and with metabolite methylation (Lipson, Thomsen, Moore, Clausen, La Clair & Burkart 2013). Importantly, we have demonstrated that in situ generated SAM analogs were recognized by rat protein arginine methyltransferase 1 (rPRMT1) which alkylated its peptide substrate (Thomsen, Vogensen, Buchardt, Burkart, & Clausen 2013).
In this chapter, we describe procedures for the preparation and assay of SalL, the SalL-mediated chemoenzymatic synthesis of SAM analogs from ClDA and l-met congeners, and the in situ chemoenzymatic synthesis of SAM coupled to DNA, peptide and metabolite methylation, and to the incorporation of isotopes into alkylated products.
2. Preparation and Assay of Chlorinase SalL
2.1. Purification of Recombinant SalL from Escherichia coli
Recombinant SalL containing a N-terminal His8 tag was obtained by gene expression in and protein purification from E. coli BL21(DE3) (Eustáquio et al., 2008).
2.1.1. Cloning of the salL Gene into an Expression Vector
The salL gene can be amplified by PCR using total genomic DNA isolated from S. tropica CNB-440 as a template and appropriate primers. The following are suggested primers that were used to clone salL into pHIS8 (Jez, Ferrer, Bowman, Dixon & Noel, 2000) by restriction/ligation, yielding plasmid pAEM7 (Eustáquio et al., 2008):
for: 5’-CGTGGTTCCCATGGCATGCAGCACAATCTCATTGC-3’ (NcoI site underlined)
rev: 5’-GCTCGAATTCAAGCTTGTCAGCTACCCGAGCACCG-3’ (HindIII site underlined)
The primers were designed to ensure that the ATG (Met start codon, bold) was in-frame with the His8 tag. The priming sites are in italics. Bases upstream of the restriction sites are random sequences to facilitate cutting of the PCR product with the respective restriction enzyme. salL was amplified by PCR using the Expand High Fidelity PCR system (Roche) following the manufacturer’s instructions and using 5% DMSO (v/v) and annealing temperature of 58 °C. Alternative, proofreading DNA polymerases include Q5® High Fidelity polymerase and Phusion® High Fidelity polymerase (New England Biolabs). The expected size of the PCR product when using primers above is 888 bp. Alternatively, synthetic DNA can be ordered from GenScript. The accession code for the salL gene (Strop_1026) of S. tropica CNB-440 is NC_009380.1 (position 1,162,700 to 1,163,551, complement). If using the primers above, digest the PCR product with NcoI and HindIII and ligate it into the same sites of pHIS8 to yield the pHIS8-salL expression vector (pAEM7). While the PCR product can be digested and cloned directly into pHIS8, we subcloned the PCR product into the pGEM-T Easy vector (Promega) by following the manufacturer’s instructions. The insert of the obtained plasmid (pAEM5) was sequenced and confirmed to contain no mistakes. Following digestion of pAEM5 with HindIII, NcoI and DraI (DraI cuts only the vector backbone), and isopropanol precipitation, the HindIII-NcoI salL fragment (868 bp) was cloned into the same sites of pHIS8. pHIS8 contains an N-terminal, 8xHis tag that will facilitate protein purification. Note that the pAEM7 salL expression plasmid used here contains native DNA from S. tropica rather than DNA which is codon-optimized for expression in E. coli.
2.1.2. Expression of salL in E. coli BL21(DE3) and Purification of Recombinant SalL Enzyme
2.1.2.1. Materials
Competent E. coli BL-21 (DE3) cells (e.g. Novagen)
salL expression vector (pAEM7)
Luria-Bertani (LB) agar (Becton Dickinson)
LB broth (Becton Dickinson)
Terrific Broth (TB) medium (Becton Dickinson)
Ni2+-NTA resin (Qiagen)
Lysis buffer: 50 mM sodium phosphate buffer (pH 8.0), 500 mM NaCl, 20 mM imidazole (pH 8.0), 20 mM β-mercaptoethanol, 10% (v/v) glycerol, and 1% (v/v) Tween-20, 1 mg/mL lysozyme
Wash buffer: 50 mM sodium phosphate buffer (pH 8.0), 500 mM NaCl, 20 mM imidazole (pH 8.0), 20 mM β-mercaptoethanol, and 10% (v/v) glycerol
Elution buffer: 50 mM sodium phosphate buffer (pH 8.0), 500 mM NaCl, 250 mM imidazole (pH 8.0), 20 mM β-mercaptoethanol, and 10% (v/v) glycerol
Storage buffer: 50 mM sodium phosphate buffer (pH 7.9), 20% glycerol
Disposable polypropylene column (e.g. from Qiagen)
Disposable PD-10 desalting columns (Sephadex G-25, GE Healthcare)
SDS-PAGE (e.g. 10% Bis-Tris NuPAGE gel with MOPS running buffer, ThermoFisher)
2.1.2.2. Procedure
Competent E. coli BL-21 (DE3) cells are commercially available (e.g. Novagen, cat. No. 69450–3). Follow the manufacturer’s instructions to introduce the salL-containing expression vector into E.coli BL-21 (DE3) by chemical transformation. Plate the transformation reaction on LB agar containing 50 mg/L kanamycin (pHIS8 contains a kanamycin-resistance gene) and incubate the plates overnight at 37 °C.
Inoculate a single colony onto 50 mL LB containing 50 mg/L kanamycin (use a 250-mL flask) and incubate the flask overnight at 37 °C, 200 rpm.
Prepare cryo stocks for long-term storage by mixing the E. coli BL21(DE3) culture containing the salL plasmid 1:1 with sterile glycerol 40%, giving a final glycerol concentration of 20%. Store cryo stocks at −80 °C.
Use the overnight LB culture to inoculate 1 L of TB media containing 50 mg/L kanamycin so that the starting A600 = 0.1 (10 to 20 mL of the overnight E. coli seed culture are expected to be necessary). Incubate the flask at 25 °C, 225 rpm until A600 = 0.8. Once an A600 of 0.8 is reached, induce cells with 0.25 mM IPTG (final concentration) and incubate overnight at 20 °C, 225 rpm. A 2.8-L Fernbach flask is recommended for cultivation of 1-L cultures.
Harvest cells by centrifugation (e.g. 15 min at 5,000 × g) and resuspend the cell pellet in ~20 mL of cold lysis buffer. Incubate on ice for 30 min.
Note: take care to keep the preparation cold by cooling centrifuges, buffers, and keeping the sample on ice.
Lyse cells by sonication on ice (e.g. 2 sec on, 2 sec off for 3 min total). Alternatively, a French press can be used for cell lysis.
Centrifuge the lysate at high speed (> 10,000 × g) for 30 min at 4 °C.
The clear lysate is then purified using Ni2+-NTA resin (Qiagen) and a modified batch purification method as briefly described below (refer to the manufacturer’s instructions “The QIAexpressionist” for more details).
Transfer the clear lysate to a clean, autoclaved beaker on ice, and add 5 mL Ni2+-NTA to the clear lysate. Mix gently using a magnetic stirrer for 1 hour. Alternatively, use a rotary shaker for mixing the sample contained in a conical tube.
If using a beaker for mixing, transfer the lysate / Ni-NTA suspension to a conical tube and centrifuge at 3,000 × g for 5 min at 4 °C to collect the Ni-NTA resin.
Discard the supernatant and wash the resin twice with 25 mL wash buffer each as following. Add 25 mL wash buffer to the Ni-NTA pellet, resuspend gently, and centrifuge at 3,000 × g for 5 min at 4 °C. Discard supernatant and repeat.
After the last wash, resuspend the Ni-NTA pellet in ~5 mL wash buffer and transfer the resin to an empty polypropylene column with the bottom outlet capped. Remove bottom cap and discard flow-through. Wash once with 25 mL cold wash buffer. Discard flow through.
Elute the His8-tagged SalL with 2× 5 mL elution buffer.
Assess the quality of the protein preparation using SDS-PAGE (e.g. 10% Bis-Tris NuPAGE gel with MOPS running buffer, ThermoFisher). The expected size of the His8-tagged protein is 32.4 kDa (Figure 2).
The expected yield is > 60 mg recombinant, soluble SalL per liter of culture. Protein yield can be calculated using the Bradford assay (Bradford 1976).
Desalting can be performed using PD-10 columns (Sephadex G-25): cut the bottom tip off the column and let the storage liquid flow (discard); equilibrate the column with 25 mL storage buffer; load sample in a total volume of 2.5 mL per PD-10 column; discard the flow through; elute with 3.5 mL storage buffer. Alternatively, dialysis can be used for desalting.
Figure 2. SDS-PAGE analysis of purified, recombinant SalL.
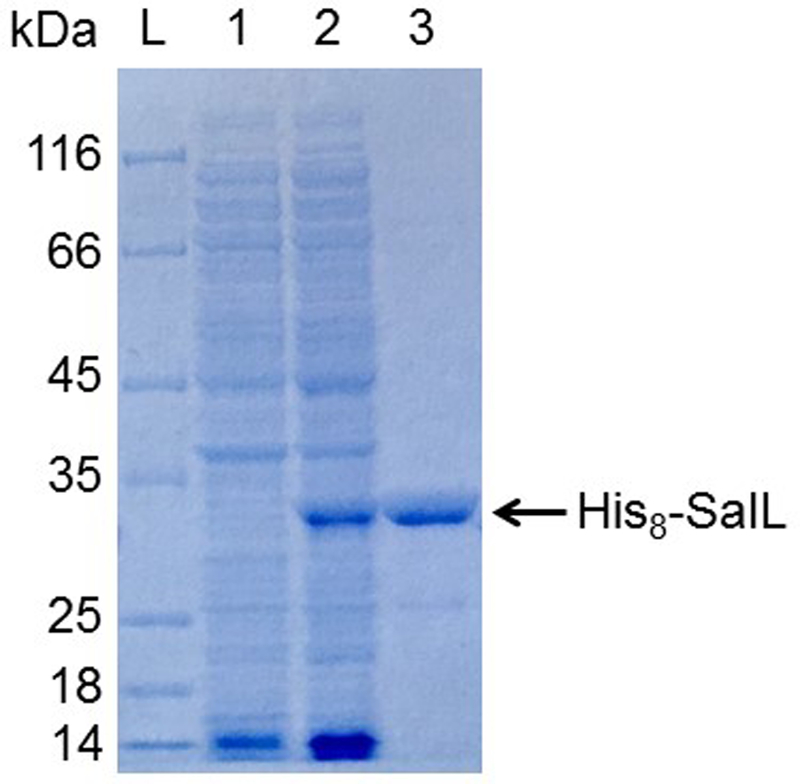
10% Bis-Tris NuPAGE gel with MOPS running buffer (ThermoFisher). L, Fisherbrand EZ-Run protein marker; 1, soluble protein before induction; 2, soluble protein after induction; 3, his8-SalL purified by Ni-NTA affinity chromatography.
2.2. SalL Activity Assay
The reaction catalyzed by chlorinase SalL is reversible. Chloride, bromide and iodide are accepted as the halide co-substrates in the reaction with SAM (forward reaction) to yield 5′-halo-5′-deoxyadenosine (XDA). Kinetic studies demonstrated that the reverse reaction (formation of SAM from XDA and l-met) is favored in vitro, with the relative enzyme efficiency (kcat/Km) of the reverse reaction being orders of magnitude higher than the forward reaction (Eustáquio et al., 2008). The activity of recombinant SalL can be investigated by a HPLC-based method as described below.
2.2.1. Reagents
S-adenosyl-l-methionine (Sigma-Aldrich)
KCl (analytical grade)
l-methionine (Sigma-Aldrich)
5′-Chloro-5′-deoxyadenosine (Sigma-Aldrich)
50 mM sodium phosphate buffer pH 7.9
Recombinant SalL (section 2.1)
2.2.2. Instrumentation and Solvents
Agilent HPLC 1200
C18 column (250 × 4.60 mm, 5 µm particle size)
Mobile phase A: 95% 50 mM KH2PO4; 5% acetonitrile
Mobile phase B: 80% 50 mM KH2PO4; 20% acetonitrile
Note: 50 mM KH2PO4 gives pH ~ 4.5; there is no need to adjust the pH.
2.2.3. Procedure
-
In order to test if your SalL preparation is active, set up two different reactions by incubating SalL (400 nM) with
- SAM (0.5 mM) and KCl (400 mM)
- l-methionine (10 mM) and ClDA (0.5 mM)
in 50 mM sodium phosphate buffer (pH 7.9) at 37°C for 1 hour. In addition, set up following controls: reactions with inactivated enzyme (the enzyme is first boiled for 5 min and then, the reagents are added) and boiled enzyme only (the enzyme is boiled for 5 min in buffer).
Boil the reaction mixture for 2 min at 95 °C and centrifuge for 30 min, 10,000 × g to remove precipitated protein. 10 µl of clear supernatant is then analyzed by HPLC using a C18 column (250 × 4.60 mm, 5 µm particle size) and 1 mL/min flow rate. After equilibrating the system for at least 15 min using 100 % mobile phase A, run following gradient: 100% A for 1 min, 0 to 100% B for 29 min, 100% to 0% B over 5 min, 100% A for 5 min. Inject also SAM and ClDA standards for comparison. The retention times obtained under the conditions described were ClDA (16.4 min) and SAM (18.1 min).
- Notes:
- SAM is unstable and SAM solutions should be freshly prepared
- Recombinant SalL purified from E. coli co-purifies with adenosine and ClDA. Thus, the boiled enzyme negative control may have a small peak for ClDA.
3. CHEMOENZYMATIC SYNTHESIS OF SAM ANALOGS
3.1. Overview of Chemoenzymatic Methods to Prepare SAM and SAM Analogs
SAM is biosynthesized by methionine adenosyltransferases (MAT) from methionine and adenosine triphosphate (ATP) (Cantoni, 1975; Figure 3). SAM is an essential cofactor for SAM-dependent methyltransferases to methylate various substrates including nucleic acids, proteins, lipids, and small molecules (Martin & McMillan 2002; Lin, 2011; Struck, Thompson, Wong & Micklefield, 2012; Bauerle, Schwalm, & Booker, 2015). While transmethylation produces small and modest chemical changes, these drastically affect molecular recognition of substrates by cellular targets to control processes such as epigenetic regulation of protein expression, neurotransmission, and natural products biosynthesis. The prevalence of SAM in cellular processes has inspired the development of analogs as chemical reporters for nucleic acid (Dalhoff Lukinavicius, Klimasauskas & Weinhold, 2006a; Dalhoff et al., 2006b; Lukinavicius, Lapiene, Stasevskij, Dalhoff, Weinhold & Klimasauskas 2007; Motorin, Burhenne, Teimer, Koynov, Willnow, Weinhold, et al., 2011; Vranken, Fin, Tufar, Hofkens, Burkart & Tor, 2016), protein (Peters, Willnow, Duisken, Kleine, Macherey, Duncan et al., 2010; Wang, Zheng, Yu, Deng & Luo, 2011; Islam, Zheng, Yu, Deng & Luo, 2011; Islam, Bothwell, Chen, Sengelaub, Wang, Deng & Luo, 2011; Wang, Islam, Liu, Zheng, Tang, Lailler et al., 2013; Wang & Luo, 2013; Islam, Chen, Wu, Bothwell, Blum, Zeng et al., 2013; Guo, Wang, Zheng, Chen, Blum, Deng et al., 2014), small molecule (Zhang, Weller, Thorson & Rajski, 2006; Stecher, Tengg, Ueberbacher, Remler, Schwab, Griengl et al., 2009; Lee, Sun, Zang, Kim, Alfaro & Zhou, 2010; Thomsen et al., 2013; Singh, Zhang, Huber, Sunkara, Hurley, Goff et al., 2014), and natural products methyltransferases (Zhang et al., 2006; Winter, Chiou, Bothwell, Xu, Garg, Luo et al., 2013).
Figure 3. Biosynthesis, utilization, and chemoenzymatic synthesis of S-adenosyl-l-methionine (SAM) analogs.
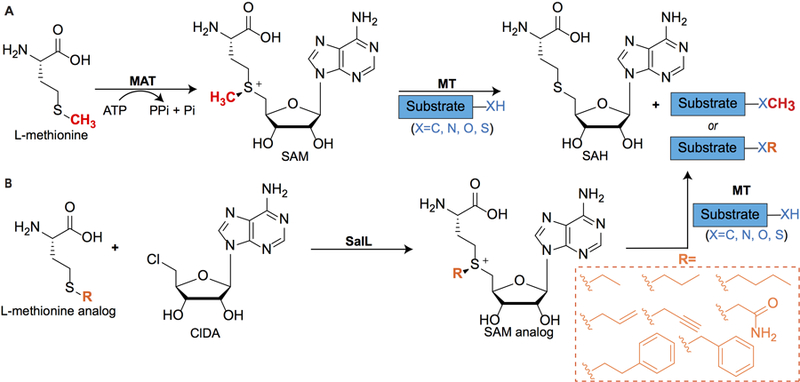
(A) Biosynthesis of SAM in vivo is catalyzed by l-methionine-adenosyltransferases (MAT). Subsequently, nucleic acid, protein, or small molecule substrates are methylated by SAM-dependent methyltransferases (MT). SAH, S-adenosyl-l-homocysteine. (B) Chemoenzymatic synthesis of SAM analogs in vitro using chlorinase SalL from Salinispora tropica. ClDA, 5′-chloro-5′-deoxyadenosine.
The preparation of SAM and its analogs is especially challenging due to inherent lability, tendency to racemize under experimental conditions, and rapid degradation at physiological pH (Hoffman, 1986). Chemical synthesis of SAM and SAM derivatives involves nucleophilic substitution between S-adenosyl-l-homocysteine (SAH or AdoCy) and an alkyl halide to produce a mixture of two sulfonium diastereomers, usually in variable yields (de la Haba, Jamieson, Mudd & Richards, 1959; Dalhoff et al., 2006; Stecher et al., 2009). The S,S diastereomer serves as the methyl donor, while the S,R diastereomer inhibits the methyltransferase (Borchardt & Wu, 1976; Khani-Oskouee, Jones, & Woodard, 1984). Additionally, analogs of SAM where sulfur is replaced with selenium are more reactive methyl donors due to the longer, weaker Se-C bonds and do not racemize (Iwig & Booker, 2004). Notably, select Se-adenosyl-L-selenomethionines (SeAM), i.e. propargyl SeAM, are more stable than their SAM analogs (Bothwell, Islam, Chen, Zheng, Blum, Deng et al., 2012; Bothwell & Luo, 2014). Overall, these early synthetic efforts ignited investigations in which non-natural SAM analogs could be used by endogenous methyltransferases to modify and profile various cellular targets.
To complement chemical synthesis of SAM analogs, we pioneered chemoenzymatic methods to expand the range of SAM analogs available for labeling methyltransferase substrates (Lipson et al., 2013; Thomsen et al., 2013; Vranken et al., 2016). We employed two homologous halogenases, SalL and FlA, to produce SAM and SAM analogs. As discussed earlier, SalL catalyzes the reaction of SAM with chloride to produce 5′-chloro-5′-deoxyadenosine (ClDA) and l-methionine via nucleophilic attack in Salinispora tropica (Figure 1). Similarly, Streptomyces cattleya uses fluorinase FlA to react fluoride with SAM to afford 5′-fluoro-5′-deoxyadenosine (FDA) and l-methionine. Based on Le Chatelier’s Principle, we hypothesized that under low salt concentrations and excess l-methionine, these halogenases could catalyze the biologically reversed reaction to produce SAM in situ (Figure 3) from easily prepared ClDA. Indeed, we observed SAM production by both SalL and FlA in various buffers, including Tris, HEPES, and phosphate buffers at pH 7–8, although SalL produced SAM in substantially higher yields than FlA. Furthermore, we generated a W190A point mutant of SalL that could accommodate larger alkyl substrates (Thomsen et al., 2013). Both wild type (WT) and mutant SalL generated alkyl, allylic, and benzylic SAM analogs, although the WT was most effective at generating small SAM analogs (methyl, ethyl), whereas both the WT and W190A mutant were equally effective at producing larger SAM analogs (propyl, butyl, allyl, and benzyl). Importantly, we demonstrated that our in situ generated SAM congeners were recognized by nucleic acid, peptide, and small molecule methyltransferases, and alkylated their respective substrates (Thomsen et al., 2013).
3.2. SalL-Mediated Chemoenzymatic Synthesis of SAM and SAM Analogs
3.2.1. Reagents
DMSO (Sigma-Aldrich)
HPLC grade acetonitrile (Fisher Scientific)
5′-chloro-5′-deoxyadenosine (ClDA) (Sigma-Aldrich)
l-methionine (Sigma-Aldrich)
S-ethyl-l-homocysteine (Sigma-Aldrich)
S-propyl-l-homocysteine (prepared as described by Thomsen et al., 2013)
S-butyl-l-homocysteine (Toronto Research Chemicals, Inc.)
S-allyl-l-homocysteine (prepared as described by Thomsen et al., 2013)
S-propargyl-l-homocysteine (prepared as described by Thomsen et al., 2013)
S-benzyl-l-homocysteine (prepared as described by Thomsen et al., 2013)
S-phenylethyl-l-homocysteine (prepared as described by Thomsen et al., 2013)
S-(2-amino-2-carboxylethyl)-l-homocysteine (Sigma-Aldrich)
S-(2-amino-2-oxoethyl)-l-homocysteine (prepared as described by Thomsen et al., 2013)
S-adenosyl-l-methionine (New England Biolabs)
3.2.2. Enzyme
SalL (expressed and purified as described above in section 2.1.2)
3.2.3. Stock Solutions and Buffers
300 mM ClDA in DMSO
150 mM l-methionine in 250 mM NaOH
15–150 mM l-methionine analog in 250 mM NaOH
32 mM SAM in 5 mM H2SO4 and 10% ethanol
50 mM sodium phosphate buffer, pH 6.8
400 mM sodium formate, pH 3.0
50 mM HEPES, pH 7.3
3.2.4. Analytical Instrumentation
Agilent 6230 ESI-TOF LC-MS
Agilent HPLC 1200
Agilent Poroshell 300SB (5 µm, 2.1×75 mm) column
3.2.5. Procedure
Equilibrate 200 µM ClDA and 3 µM SalL in 50 mM phosphate buffer, pH 6.8 at 37 ⁰C.
Add 1.5–15 mM l-methionine or analog from stock. Due to the poor solubility of the benzyl and phenylethyl analogs of l-methionine, these were added at a final concentration of 1.5 mM from a 15 mM stock solution in 250 mM NaOH. l-Methionine and all other analogs were added at a final concentration of 15 mM from a 150 mM stock solution in 250 mM NaOH. In all assays, the addition of l-methionine or analog was 10% of the total reaction volume, resulting in a final pH of 7.4.
Quench reactions at various time points (0–5 hours) by the addition of equal volumes of 400 mM sodium formate. Store quenched aliquots at 4 ⁰C until HPLC analysis.
-
Analyze aliquots using analytical reversed phase HPLC using a Synergi™ Hydro RP column (4 µm, 4.6×150 mm, Phenomenex) using the following gradient:
Buffer A: 10 mM NH4HCO2, pH 3.2
Buffer B: 90% CH3CN, 10 mM NH4HCO2, pH 3.2
0 min: 100% buffer A, 1 mL/min
5 min: 98% buffer A, 1.5 mL/min
10 min: 70% buffer A, 2 mL/min
12 min: 0% buffer A, 2 mL/min
Note: Complete conversion of ClDA (retention time = 11 min) into SAM (retention time = 4 min) was observed within 30 min.
Verify the identity of ClDA and SAM by collecting 0.1 mL fractions from the HPLC and analyze by LC-MS. Table 1 provides a summary of LC-MS data for select SAM analogs. See Thomsen, et al. 2013 for representative LC-MS traces and chromatograms.
Table 1.
LC-MS Retention Times and Chromatographic Analyses of Select SAM Analogs
| Analog | Retention Time | Observed Monoisotopic Mass |
|---|---|---|
| SAM | 4 min. | 399. 1555 |
| Ethyl-SAM | 5 min. | 413. 1671 |
| Propyl-SAM | 8.5, 8.9 min.a | 427.1784 |
| Butyl-SAM | 9.5 min. | 441.2366 |
| Allyl-SAM | 7.2, 7.9 min.a | 425.1862 |
| Benzyl-SAM | 10.4 min. | 475.2242 |
Mixture of diastereomers.
3.3. In situ Chemoenzymatic Synthesis of SAM Coupled to DNA Methylation by HhaI
HhaI is a DNA methyltransferase that methylates cytosine at C5 (Sankpal & Rao, 2002). In this assay, HhaI is coupled with in situ SAM production with methylation of lambda DNA.
3.3.1. Reagents
N6-methyladenine free lambda DNA (New England Biolabs)
3.3.2. Enzymes
SalL (expressed and purified as described above in section 2.1.2)
HhaI methyltransferase (New England Biolabs)
0.2 U/µL HinP1I restriction enzyme (New England Biolabs)
3.3.3. Stock Solutions and Buffers
300 mM ClDA in DMSO
150 mM l-methionine in 250 mM NaOH
HhaI buffer (5 mM 2-mercaptoethanol, 10 mM EDTA, 50 mM Tris•HCl, pH 7.5)
NEB4 buffer (10 mM KOAc, 4 mM Tris•HOAc, 2 mM MgOAc2, 0.2 mM DTT, pH 7.9)
3.3.4. Procedure
-
Equilibrate 50 ng/µL N6-methyladenine free lambda DNA, 4 U/µL HhaI, 200 µM ClDA, and 15 mM l-methionine in HhaI buffer at 37 ⁰C.
Note: The buffers used for DNA methyltransferase HhaI and SalL activity assays were incompatible. HhaI was inactive in 50 mM sodium phosphate buffer, pH 7.9, however, the activity of SalL was reduced by 30% in HhaI buffer. Thus, coupled assays were conducted in HhaI buffer.
Add SalL to a final concentration of 900 nM.
After 1 hour, quench reaction by heating at 80 ⁰C for 5 min.
Dilute reaction in an equal volume of HinP1I in 2x NEB4 buffer to afford a final concentration of 0.1 U/µL HinP1I.
Incubate 1 hour at 37 ⁰C.
Analyze lambda DNA cleavage by gel electrophoresis on 2% agarose gel.
3.4. In situ Chemoenzymatic Synthesis of SAM Coupled to Teicoplanin Methylation by MtfA
MtfA is a small molecule methyltransferase from the chloroeremomycin biosynthetic pathway in Amycolatopsis orientalis that catalyzes N-methylation of vancomycin-like glycopeptides (Shi et al., 2009). In this assay, MtfA is used to couple in situ SAM production with methylation of teicoplanin.
3.4.1. Reagents
Teicoplanin (TargetMol)
3.4.2. Enzymes
SalL (expressed and purified as described above in section 2.1.2)
MtfA methyltransferase (expressed and purified as described below)
3.4.3. Stock Solutions and Buffers
300 mM ClDA in DMSO
150 mM l-methionine (or [methyl-13C]-l-methionine) in 250 mM NaOH
50 mM HEPES, pH 7.3
3.4.4. Expression and Purification of MtfA Methyltransferase
One liter of 2x YT medium (Sigma-Aldrich) supplemented with 50 µg/mL kanamycin was inoculated with E. coli BL21 (DE3) cells transformed with mtfA/pET28a plasmid (obtained from Prof. Gerard D. Wright, McMaster University). The cells were cultured at 37 ⁰C to an OD600 of 0.6. The cells were cooled to 0 ⁰C on ice, induced with 100 µM isopropyl β-D-1-thiogalactopyranoside (IPTG), and grown for 20 h at 16 ⁰C. Cells were harvested via centrifugation at 2,000 × g, 30 min, 4 ⁰C and the pellet was resuspended in lysis buffer (50 mM HEPES, 500 mM NaCl, 20 mM imidazole, and 1 mM dithiothreitol (DTT), pH 7.5) and lysed by passage through a French pressure cell. The lysate was clarified by centrifugation at 4150 × g, 1.5 h, 4 ⁰C. His6-MtfA was purified using 2 mL Ni-NTA resin (GE Healthcare) that was equilibrated with lysis buffer. The column was washed with 40 mL of buffer A (50 mM HEPES, 1 M NaCl, 20 mM imidazole, 1 mM DTT, pH 7.5), followed by 40 mL of buffer B (50 mM HEPES, 400 mM NaCl, 30 mM imidazole, 1 mM DTT, pH 7.5). The protein was eluted with buffer C (50 mM HEPES, 400 mM NaCl, 200 mM imidazole, 1 mM DTT, pH 7.5). We obtained around 50 mg of MtfA per L of cell culture.Eluted proteins were visualized with 12% SDS-PAGE with Coomassie Brilliant Blue staining. Imidazole was removed using spin concentration into 20 mM HEPES, 20 mM NaCl, and 5 mM DTT, pH 7.5. Pure protein was stored at −20 ⁰C in 10 mM HEPES, 10 mM NaCl, 2.5 mM DTT, pH 7.5 containing 50% glycerol. The protein is stable as a glycerol stock at −20 ⁰C.
3.4.5. Procedure
Equilibrate a 50 µL mixture of 20 mM ClDA, 50 mM l-methionine, and 1.4 µM SalL in 32 mM phosphate buffer, pH 7.9 at 37 ⁰C.
After 1–3 h, add 250 µL of a solution containing: 1 mM teicoplanin, 1 mg/mL BSA, and 54 µM MtfA in 50 mM HEPES, pH 7.3.
Monitor the methylation of teicoplanin by removing aliquots at various times and analyzing by LC-MS using a C18 column (3 µm, 2.1×150 mm, Atlantis) and an isocratic mixture of 3:1 H2O/CH3CN (v/v) with 0.1% formic acid. Under these conditions, the retention times of teicoplanin and N-methyl-teicoplanin were 9 min and 9.5 min, respectively.
3.5. In situ Chemoenzymatic Synthesis of 13C-labeled SAM Coupled with Isotope Incorporation into Teicoplanin via Methylation by MtfA
We envisioned SalL-mediated in situ chemoenzymatic synthesis of SAM as a compelling and facile way to incorporate isotopes into biomolecules. Here we describe the use of this technology to incorporate 13C into teicoplanin for sensitive NMR identification (Lipson et al., 2013).
3.5.1. Reagents
d6-DMSO (Cambridge Isotope Laboratories, Item No. DLM-10–10)
[Methyl-13C]-l-methionine (Cambridge Isotope Laboratories, Item No. CLM-206-PK)
Teicoplanin (TargetMol)
3.5.2. Enzymes
SalL (expressed and purified as described above in section 2.1.2)
MtfA methyltransferase (expressed and purified as described in section 3.4.4)
3.5.3. Stock Solutions and Buffers
300 mM ClDA in d6-DMSO
300 mM [methyl-13C]-l-methionine in d6-DMSO
10 mM phosphate buffer, pH 7.5
3.5.4. Analytical Instrumentation
Varian VX500 NMR spectrometer equipped with an X-Sens cold probe
3.5.5. Procedure
Prepare 800 µL of a mixture of 350 nM SalL and 350 nM MtfA in 10 mM phosphate buffer, pH 7.5.
Prepare 200 µL of a mixture of 7 mM teicoplanin, 142 mM [methyl-13C]-l-methionine, and 14.2 mM ClDA in 10 mM phosphate buffer, pH 7.5 containing 50% d6-DMSO.
Transfer the mixture of teicoplanin, [methyl-13C]-l-methionine, and ClDA mixture to the mixture of SalL and MtfA.
Transfer the entire solution to a clean 5 mm NMR tube.
Collect 13C-NMR spectra on a Varian VX500 NMR spectrometer at various time points. For each time point, we acquired data for ≥ 64 scans with a delay time (d1) of 5 seconds. In our hands, we observed the appearance of [methyl-13C]-N-teicoplanin within 3–9 hours, as evidenced by a 13C-NMR signal at δ = 33.7 ppm.
3.6. In situ Chemoenzymatic Synthesis of SAM Coupled to RGG Peptide Methylation by rPRMT1
Protein arginine methyltransferase 1 (PRMT1) is responsible for the majority of arginine methylation in mammalian cells. In the assay described below, rPRMT1, a rat homolog that dimethylates arginine residues, is used to couple in situ SAM production with methylation of arginine residues in the RGG peptide. Transfer of ethyl, allyl, and benzyl congeners of L-methionine has also been observed (Thomsen et al., 2013). Overall, sterics likely influences transfer since we observed RGG allylation, but did not observe RGG propylation, although the corresponding SAM analogs have similar sizes and are formed in situ at similar rates. RGG allylation most likely occurs via an SN2′ mechanism.
3.6.1. Reagents
RGG peptide (expressed and purified as described by Thomsen et al., 2013)
3.6.2. Enzymes
SalL (expressed and purified as described above in section 2.1.2)
Rat protein arginine methyltransferase 1 (rPRMT1, expressed and purified as described by Thomsen et al., 2013)
3.6.3. Stock Solutions and Buffers
300 mM ClDA in DMSO
150 mM l-methionine in 250 mM NaOH
400 mM sodium formate, pH 3.0
50 mM phosphate buffer, pH 7.4
3.6.4. Procedure Peptide Methylation using rPRMT1
Equilibrate a mixture containing 200 µM ClDA, 15 mM l-methionine, 1 µM SalL, 10 µM RGG peptide, and 2 µM rPRMT1 in 50 mM phosphate buffer, pH 7.4 at 37 ⁰C.
After 12 hours, quench reaction by the addition of an equal volume of 400 mM sodium formate.
Assess the formation of methylated RGG peptide using LC-MS.
4. SUMMARY AND CONCLUSIONS
We described procedures for the chemoenzymatic synthesis of SAM and analogs that employ chlorinase SalL discovered from a marine actinomycete bacterium. Recombinant SalL can be purified from E. coli in relatively high yields, i.e. over 60 mg soluble enzyme per liter culture. The SalL-catalyzed synthesis of SAM analogs from l-methionine congeners and the stable and commercially available substrate 5′-chloro-5′-deoxyadenosine is diastereoselective. Moreover, we described methods for in situ synthesis of SAM coupled with DNA, protein and metabolite methylation, and with the incorporation of isotopes into biomolecules.
In addition to SAM-dependent halogenases, numerous groups have used methionine adenosyltransferase wild type and mutants that accommodate structural changes in l-methionine and ATP to generate stable SAM analogs that alkylate a variety of acceptor substrates (Wang et al., 2013; Singh et al., 2014; Huber, Wang, Singh, Johnson, Zhang, Sunkara et al., 2016; Huber, Johnson, Zhang, & Thorson, 2016). Overall, the development of chemoenzymatic methods and protein engineering efforts has expanded the structural scope of SAM analogs that can be generated for biological studies.
ACKNOWLEDGEMENTS
Financial support for this work was provided by the National Center for Advancing Translational Sciences, National Institutes of Health (NIH), under grant KL2TR002002 (to A.S.E.), by NIH GM095970 (to M.D.B) and by startup funds from the Department of Medicinal Chemistry and Pharmacognosy and the Center for Biomolecular Sciences, University of Illinois at Chicago (to A.S.E.). T.D.D. was supported by the NIH/NIGMS IRACDA K12 GM068524 award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Plasmid pAEM7 for the expression of salL was constructed in Prof. Bradley S. Moore’s laboratory (Scripps Institution of Oceanography). The authors also graciously thank Prof. Joseph P. Noel (Salk Institute for Biological Studies) for vector pHIS8, Prof. Gerard D. Wright (McMaster University) for the mtfA gene construct, Dr. Yongxuan Su (UC San Diego) for mass spectral analyses, and Drs. Anthony Mrse and Xuemei Huang (UC San Diego) for assistance with acquiring NMR spectral data.
REFERENCES
- Agarwal V, Miles ZD, Winter JM, Eustáquio AS, El Gamal AA, & Moore BS (2017). Enzymatic halogenation and dehalogenation reactions: pervasive and mechanistically diverse. Chemical Reviews, 117, 5619–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauerle MR, Schwalm EL, & Booker SJ (2015). Mechanistic diversity of radical S-adenosylmethionine (SAM)-dependent methylation. Journal of Biological Chemistry, 290, 3995–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchardt RT & Wu YS (1976). Potential inhibitors of S-adenosylmethionine-dependent methyltransferases V: role of the asymmetric sulfonium pole in the enzymatic binding of S-adenosyl-l-methionine. Journal of Medicinal Chemistry, 19, 1099–1103. [DOI] [PubMed] [Google Scholar]
- Bothwell IR, Islam K, Chen Y, Zheng W, Blum G, Deng H, & Luo M (2012). Se-adeonsyl-l-selenomethionine cofactor analogue as a reporter of protein methylation. Journal of the American Chemical Society, 134, 14905–14912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothwell IR & Luo M (2014). Large-scale, protection free synthesis of Se-adenosyl-l-selenomethionine analogues and their application as cofactor surrogates of methyltransferases. Organic Letters, 16, 3056–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Cantoni GL (1975). Biological methylation: selected aspects. Annual Review of Biochemistry, 44, 435–451. [DOI] [PubMed] [Google Scholar]
- Chan KK, & O’Hagan D (2012). The rare fluorinated natural products and biotechnological prospects for fluorine enzymology. Methods in Enzymology, 516, 219–235. [DOI] [PubMed] [Google Scholar]
- Dalhoff C, Lukinavicius G, Klimasauskas S, & Weinhold E (2006). Direct transfer of extended groups from synthetic cofactors by DNA methyltransferases. Nature Chemical Biology, 2, 31–32. [DOI] [PubMed] [Google Scholar]
- Dalhoff C, Lukinavicius G, Klimasauskas S, & Weinhold E (2006). Synthesis of S-adenosyl-l-methionine analogs and their use for sequence-specific transalkylation of DNA by methyltransferases. Nature Protocols, 1, 1879–1886. [DOI] [PubMed] [Google Scholar]
- Deng H, Cobb SL, McEwan AR, McGlinchey RP, Naismith JH, O’Hagan D, Robinson DA, & Spencer JB (2006). The fluorinase from Streptomyces cattleya is also a chlorinase. Angewandte Chemie, 45, 759–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O’Hagan D, & Naismith JH (2004). Crystal structure and mechanism of a bacterial fluorinating enzyme. Nature, 427, 561–565. [DOI] [PubMed] [Google Scholar]
- Eustáquio AS, Pojer F, Noel JP & Moore BS (2008). Discovery and characterization of a marine bacterial SAM-dependent chlorinase. Nature Chemical Biology, 4, 66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustáquio AS, McGlinchey RP, Liu Y, Hazzard C, Beer LL, Florova G, Alhamadsheh MM, Lechner A, Kale AJ, Kobayashi Y, Reynolds KA & Moore BS (2009). Biosynthesis of the salinosporamide A polyketide synthase substrate chloroethylmalonyl-coenzyme A from S-adenosyl-l-methionine. Proceedings of the National Academy of Sciences of the United States of America, 106, 12295–12300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feling RH, Buchanan GO, Mincer TJ, Kauffman CA, Jensen PR & Fenical W (2003). Salinosporamide A: a highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angewandte Chemie, 42, 355–357. [DOI] [PubMed] [Google Scholar]
- Fontecave M, Atta M, & Mulliez E (2004). S-adenosylmethionine: nothing goes to waste. Trends in Biochemical Sciences, 29, 243–249. [DOI] [PubMed] [Google Scholar]
- Gribble GW (2012). Recently discovered naturally occurring heterocyclic organohalogen compounds. Heterocycles, 84, 157–207. [Google Scholar]
- Guo H, Wang R, Zheng W, Chen Y, Blum G, Deng H, & Luo M (2014). Profiling substrates of protein arginine N-methyltransferase 3 with S-adenosyl-l-methionine analogues. ACS Chemical Biology, 9, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Haba G, Jamieson GA, Mudd SH, & Richards HH (1959). S-adenosylmethionine: the relation of configuration at the sulfonium center to enzymatic reactivity. Journal of the American Chemical Society, 81, 3975–3980. [Google Scholar]
- Hoffman JL (1986). Chromatographic analysis of the chiral and covalent instability of S-adenosyl-l-methionine. Biochemistry, 25, 4444–4449. [DOI] [PubMed] [Google Scholar]
- Huber TD, Johnson BR, Zhang J, & Thorson JS (2016). AdoMet analog synthesis and utilization: current state of the art. Current Opinion in Biotechnology, 42, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber TD, Wang F, Singh S, Johnson BR, Zhang J, Sunkara M, Van Lanen SG, Morris AJ, Philips GN Jr., & Thorson JS (2016). Functional AdoMet isosteres resistant to classical AdoMet degradation pathways. ACS Chemical Biology, 11, 2484–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam K, Zheng W, Yu H, Deng H, & Luo M (2011). Expanding cofactor repertoire of protein lysine methyltransferase for substrate labeling. ACS Chemical Biololgy, 6, 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam K, Bothwell I, Chen Y, Sengelaub C, Wang R, Deng H, & Luo M (2012). Bioorthogonal profiling of protein methylation using azido derivative of S-adenosyl-l-methionine. Journal of the American Chemical Society, 134, 5909–5915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam K, Chen Y, Wu H, Bothwell IR, Blum GJ, Zeng H, Dong A, Zheng W, Min J, Deng H, & Luo M (2013). Defining efficient enzyme-cofactor pairs for bioorthogonal profiling of protein methylation. Proceedings of the National Academy of Sciences of the United States of America, 110, 16778–16783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwig DF, & Booker SJ (2004). Insight into the polar ractivity of the onium chalcogen analogues of S-adenosyl-l-methionine. Biochemistry, 43, 13496–13509. [DOI] [PubMed] [Google Scholar]
- Jez JM, Ferrer JL, Bowman ME, Dixon RA & Noel JP (2000). Dissection of malonyl-coenzyme A decarboxylation from polyketide formation in the reaction mechanism of a plate polyketide synthase. Biochemistry, 39, 890–902. [DOI] [PubMed] [Google Scholar]
- Khani-Oskouee S, Jones JP, & Woodard RW (1984). Stereochemical course of the biosynthesis of l-aminocyclopropane-1-carboxylic acid. I. Role of the asymmetric sulfonium pole and the α-amino acid center. Biochemical and Biophysical Research Communications, 121, 181–187. [DOI] [PubMed] [Google Scholar]
- Lee BW, Sun HG, Zang T, Kim BJ, Alfaro JF, & Zhou ZS (2010). Enzyme-catalyzed transfer of a ketone group from an S-adenosylmethionine analogue: a tool for the functional analysis of methyltransferases. Journal of the American Chemical Society, 132, 3642–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H (2011). S-adenosylmethionine-dependent alkylation reactions: when are radical reactions used? Bioorganic Chemistry, 39, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson JM, Thomsen M, Moore BS, Clausen RP, La Clair JJ, & Burkart MD (2013). A tandem chemoenzymatic methylation of S-adenosyl-l-methionine, ChemBioChem, 14, 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavicius G, Lapiene V, Stasevskij Z, Dalhoff C, Weinhold E, & Klimasauskas S (2007). Targeted labeling of DNA by methyltransferase-directed transfer of activated groups (mTAG). Journal of the American Chemical Society, 129, 2758–2759. [DOI] [PubMed] [Google Scholar]
- Martin JL, & McMillan FM (2002). SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Current Opinion in Structural Biology, 12, 783–793. [DOI] [PubMed] [Google Scholar]
- Motorin Y, Burhenne J, Teimer R, Koynov K, Willnow S, Weinhold E, & Helm M (2011). Expanding the chemical scope of RNA: methyltransferases to site-specific alkynylation of RNA for click labeling. Nucleic Acids Research, 39, 1943–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi M, Liu F, Hai Y, Chen M, Tang MC, Yang Z, Sato M, Watanabe K, Houk KN, Tang Y (2017). SAM-dependent enzyme-catalysed pericyclic reactions in natural product biosynthesis. Nature, 549, 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters W, Willnow S, Duisken M, Kleine H, Macherey T, Duncan KE, Litchfield DW, Luscher B, & Weinhold E (2010). Enzymatic site-specific functionalization of protein methyltransferase substrates with alkynes for click labeling. Angewandte Chemie International Edition England, 49, 5170–5173. [DOI] [PubMed] [Google Scholar]
- Sankpal UT & Rao DN (2002). Structure, function, and mechanism of HhaI DNA methyltransferases. Critical Reviews in Biochemistry and Molecular Biology, 37, 167–197. [DOI] [PubMed] [Google Scholar]
- Shi R, Lamb SS, Zakeri B, Proteau A, Cui Q, Sulean T, Matte A, Wright GD, & Cygler M (2009). Structure and function of the glycopeptide N-methyltransferase MtfA, a tool for the biosynthesis of modified glycopeptide antibiotics. Chemistry & Biology, 16, 401–410. [DOI] [PubMed] [Google Scholar]
- Singh S, Zhang J, Huber TD, Sunkara M, Hurley K, Goff RD, Wang G, Zhang W, Liu C, Rohr J, Van Lanen SG, Morris AJ, & Thorson JS (2014). Facile chemoenzymatic strategies for the synthesis and utilization of S-adenosyl-l-methionine analogues. Angewandte Chemie International Edition, 53, 3965–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinhababu AK, Bartel RL, Pochopin N, & Borchardt RT (1985). Mechanism of action of S-adenosyl-l-homocysteine hydrolase. Measurement of kinetic isotope effects using adenosine-3′-d and S-adenosyl-l-homocysteine-3′-d as substrates. Journal of American Chemical Society, 107, 7628–7632. [Google Scholar]
- Stecher H, Tengg M, Ueberbacher BJ, Remler P, Schwab H, Griengl H, & Gruber-Khadjawi M (2009). Biocatalytic Friedel-Crafts alkylation using non-natural cofactors. Angewandte Chemie International Edition England, 48, 9546–9548. [DOI] [PubMed] [Google Scholar]
- Struck AW, Thompson ML, Wong LS, & Micklefield J (2012). S-adenosyl-methionine-dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. ChemBioChem, 13, 2642–2655. [DOI] [PubMed] [Google Scholar]
- Thomsen M, Vogensen SB, Buchardt J, Burkart MD, & Clausen RP (2013). Chemoenzymatic synthesis and in situ application of S-adenosyl-l-methionine analogs. Organic and Biomolecular Chemistry, 11, 7606–7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken C, Fin A, Tufar P, Hofkens J, Burkart MD, & Tor Y (2016). Chemoenzymatic synthesis and utilization of a SAM analog with an isomorphic nucleobase. Organic and Biomolecular Chemistry, 14, 6189–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Zheng W, Yu H, Deng H, & Luo M (2011). Labeling substrates of protein arginine methyltransferase with engineered enzymes and matched S-adenosyl-l-methionine analogues. Journal of the American Chemical Society, 133, 7648–7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Islam K, Liu Y, Zheng W, Tang H, Lailler N, Blum G, Deng H, & Luo M (2013). Profiling genome-wide chromatin methylation with engineered posttranslation apparatus within living cells. Journal of the American Chemical Society, 135, 1048–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, & Luo M (2013). A journey toward bioorthogonal profiling of protein methylation inside living cells. Current Opinion in Chemical Biology, 17, 729–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhou X, Fredimoses M, Liao S & Liu Y (2014). Naturally occurring organoiodines. RSC Advances, 4, 57350–57376. [Google Scholar]
- Winter JM, Chiou G, Bothwell IR, Xu W, Garg NK, Luo M, & Tang Y (2013). Expanding the structural diversity of polyketides by exploring the cofactor tolerance of an inline methyltransferase domain. Organic Letters, 15, 3774–3777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Weller RL, Thorson JS, & Rajski SR (2006). Natural product diversification using a non-natural cofactor analogue of S-adenosyl-l-methionine. Journal of the American Chemical Society, 128, 2760–2761. [DOI] [PubMed] [Google Scholar]