

Abstract
Background
Alport Syndrome (AS) is a progressive hereditary glomerular disease. It is often accompanied by sensorineural hearing loss and ocular abnormalities and can sometimes develop into end stage renal disease (ESRD), which is caused by mutations in the genes encoding the collagen type IV family of proteins.
Methods
This study analyzed the association between the clinical data of seven AS families and genes and the disease progression of different mutation types, including COL4A3 (OMIM 120070),COL4A4 (OMIM 120131), and COL4A5 (OMIM303630).
Results
A total of six new pathogenic mutation sites, one complex heterozygous mutation at COL4A3, and a combined mutation of COL4A5 and INF2 (OMIM 610982) were identified in this study. It was revealed that the clinical manifestations of X‐linked AS caused by mutations in the COL4A5 gene were more severe in males than in females. In addition, the difference in patient phenotype can be attributed to the location of gene mutations affecting the protein domain or functional domain. Our data suggested that the gene deletion and nonsense mutations had a high risk for progression to ESRD.
Conclusion
Our results revealed the spectrum of type IV collagen genes, which contribute to the enrichment of database resources and has important implications in the diagnosis, prognosis, and guiding treatment of AS.
Keywords: Alport Syndrome, Genotype‐phenotype correlation, prognostic impact
1. INTRODUCTION
Alport Syndrome (AS) is a progressive hereditary glomerular disease with a high degree of family genetic predisposition. It is caused by mutations in type IV collagen genes, encoding the chains of collagen type IV. Extrarenal signs might be present, including sensorineural hearing loss and ocular anomalies.(Grunfeld, 1985)Genetic mutations in AS cause damage to the glomerular basement membrane(GBM), including preventing the synthesis of type IV collagen and/or the formation of type IV collagen network structures, thereby leading to various clinical manifestations in AS.(Hudson, 2004).
There are three common inheritance patterns in AS patients: X‐linked recessive inheritance and dominant inheritance (approximately 65%), autosomal recessive inheritance (approximately 15%), and autosomal dominant inheritance (approximately 20%)(Fallerini et al., 2014; Moriniere et al., 2014). X‐linked inheritance accounts for the majority of AS cases, with most cases caused by mutations in the COL4A5 gene on the X chromosome and a few cases caused by mutations in the COL4A6 (OMIM 303631) gene, which is adjacent to the 5'‐end of the COL4A5 gene (Kashtan, 1995; Uliana et al., 2011) For X‐linked AS, almost all carriers have different degrees of hematuria, and the clinical phenotypes are different; due to the deactivation of the X chromosome and the complementation of two X chromosomes in females, the symptoms in female patients are milder than those in males (Jais et al., 2003; Yamamura et al., 2017) Because the α6 chain (type IV collagen) is not expressed in the basement membrane(Ninomiya et al., 1995), mutations at the junction of the 5'‐end of COL4A5 and COL4A6 cause AS accompanied by leiomyosarcoma(Dahan et al., 1995; Zhou et al., 1993). In addition, AS caused by COL4A3 or COL4A4 gene defects accounts for approximately 15% of AS patients and is inherited in an autosomal recessive manner. The severity of symptoms between female and male patients is rather consistent (Mochizuki, 1994). Furthermore, approximately 20% of patients with AS are of autosomal dominant inheritance, which is caused by mutations in the COL4A3 or COL4A4 genes. This disease type has similar clinical and pathological characteristics to the X‐linked type but has a slower progression of impaired renal function and is less commonly accompanied by visual and hearing impairment (Pochet, Bobrie, Landais, Goldfarb, & Grunfeld, 1989). Some studies have shown that mutations in the COL4A3 or COL4A4 genes cause thin basement membrane nephropathy (TBMN, benign familial hematuria), which is clinically characterized by persistent or intermittent asymptomatic microscopic hematuria while rarely showing progressive proteinuria and progressing to ESRD (Longo et al., 2002; Voskarides et al., 2007; Voskarides et al., 2007). In contrast, Voskarides reported higher percentages of patients who had TBMN and progressed to FSGS and ESRD (Voskarides et al., 2007). Taken together, AS has genetic heterogeneity and phenotypic diversity due to different mutation sites, the intrinsic link between genotype and phenotype will help predict the clinical progression and prognosis of affected individuals. This study reported seven AS families, analyzed the potential association between clinical phenotypes and gene mutation sites, and studied the intrinsic correlation between gene mutations at relevant sites and AS phenotype, progression, and clinical prognosis.
2. METHODS
2.1. Ethical compliance
Ethical approval for the study was granted through the Chinese PLA General Hospital Medical Ethics Committee (2012–001). All participants in this study signed a consent form indicating that they had been fully informed about the nature of the interview, as well as the likely uses of their data.
2.2. Clinical characteristics
According to the diagnostic criteria of AS(Kashtan, 2004; Pirson, 1999; Savige et al., 2018), seven families with confirmed or suspected AS in our department were selected as study subjects, and clinical biological samples of the probands and other family members of the seven families were collected; clinical data of family members within three generations in the family were compiled. The clinical information the of probands and other family members, included physical examination of eye and ear lesions and routine examinations such as urine and renal function examinations; renal histopathological data collection; light microscopy and electron microscopy examination; and collagen type IV expression in the basement membranes of renal tissues. Data screening and bioinformatics analysis were conducted for data on targeted gene capture and high‐throughput sequencing from DNA samples for core family members. This study passed review by the ethics committee of the PLA General Hospital, and signed informed consent was obtained from all patients.
2.3. Gene analysis
2.3.1. Genomic DNA extraction
Two milliliters of peripheral venous blood was collected from each core family members of the seven families; samples were collected with the anticoagulation agent ethylenediaminetetraacetic acid (EDTA). Genomic DNA was extracted using a whole‐blood DNA extraction kit (Qiagen, Germany).
2.3.2. Whole‐genome DNA library preparation
Three micrograms of DNA were taken from each of the study objects, and upon dilution, ultrasonic fragmentation was performed using a Covris‐S220 ultrasound system (Covaris, USA) to generate fragments approximately 150 bp in size. A whole‐genome library was prepared using the standard Illumina library construction kit (MyGenostics, China; NEB, USA). The DNA library was captured using the GenCap liquid‐phase capture kit (developed by MyGenostics), and a fluorescence real‐time quantitative PCR instrument (ABI7500) was used for quantitation and quality control.
2.3.3. Targeted gene capture and high‐throughput sequencing
The GenCap technology for liquid‐phase targeted gene capture (MyGenostics, Beijing, China) was used to capture the exons of pathogenic genes in the pathogenic kidney disease panel, including ACTN4 (OMIM 604638), B2M (OMIM 109700), WT1 (OMIM 607102), FGA (OMIM 134820), LMX1B (OMIM 602575), NPHS1 (OMIM 602716), PTPRO (OMIM 600579), COL4A3, CD2AP (OMIM 604241), GLA (OMIM 300644), LYZ (OMIM 153450), NPHS2 (OMIM 604766), SMARCAL1 (OMIM 606622), APOL1 (OMIM 603743), APOA1 (OMIM 107680), COL4A4, COQ6 (OMIM 614647), INF2, MYH9 (OMIM 160775), PAX6 (OMIM 607108), TRPC6 (OMIM 603652), ARHGDIA (OMIM 601925), COL4A5, DGKE(OMIM 601440), LAMB2 (OMIM 150,325), MYO1E (OMIM 601479), PLCE1 (OMIM 608414) and COL4A6. Double‐stranded sequencing was performed with an Illumina HiSeq 2000 next‐generation sequencer with a read length of 100 bp.
2.3.4. Data screening and bioinformatics analysis
After the target region was sequenced, the original data were subjected to label splitting to obtain the original sequence of each sample, and the adapter and low‐quality data (quality value less than or equal to 40 bp) in the sequencing data were removed. The GenBank reference sequences used for COL4A3, COL4A4, COL4A5, and INF2 were NM_000091.4, NM_000092.4, NM_000495.4,and NM_022489.3, respectively. The resultant sequence data were aligned against the reference genome(hg19) using BWA software; the polymorphic sites of the alignment data were detected with GATK software for each sample, and the data of single nucleotide polymorphisms (SNPs) and insertion and deletion mutations (InDels) were counted and statistically analyzed. Meanwhile, statistical analysis of sequencing depth, homogeneity, and probe specificity was performed. SNP and InDel were identified by the 1,000 Genomes Project( 1000G, http://www.1000genomes.org), ExAC_ALL (Exome Aggregation Consortium, http://exac.broadinstitute.org/) and in‐house exome database of 1,000 health controls. The mutations were detected through filtering steps using these databases at a frequency level of < 1%. And effects of filtered mutations were predicted with a multitude of software, including SIFT and Ployphen2.
2.3.5. Sanger sequencing validation
Primers for DNA fragments that required sequencing to identify gene mutations were synthesized, and the DNA fragments were amplified by polymerase chain reaction (PCR). Fragments were then sequenced by the Sanger sequencing method using an ABI3730xl sequencer (Applied Biosystems, USA). The sequencing results were compared with the results of targeted capture sequencing.
3. RESULTS
The clinical characteristics of the seven AS families with an established molecular diagnosis are listed in Table 1. The mutations in suspected AS patients are summarized in Table 2, as well as the frequency of novel and known mutations. The mutational sites were not present or extremely low frequency in 1,000 Genome Project, ExAC_ALL and in‐house database.
Table 1.
Clinical characteristics of the 7 Alport's families with an established molecular diagnosis
| Family code |
Individual No. (gender) |
Gene |
Disease‐ causing variant |
State | Inheritence | Phenotype | Kidney Repalcement therapy | Other signs | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age at oneset (years) | Age of ESRD (date) | Hearing Loss | OcularLesions | GBM Splitting | Hematuria |
Hypertension (mmHg) |
Proteinuria | ||||||||
| 1 | III 1 (F) | COL4A5 | c.443C > T | Het | XD | 36 | ‐ | ‐ | ‐ | NA | ++ | 145/105 | ± | ‐ | NA |
| III 2 (M) | Hemi | 34 | ‐ | ‐ | ‐ | NA | ++ | 140/100 | ± | ‐ | NA | ||||
| 2 | III 2 (M) | COL4A5 | c.1289C > A | Hemi | XD | 27 | 30 | ‐ | ‐ | NA | NA | NA | ++++ | HD | IgA, Poor clinical results after hormone therapy |
| INF2 | c.550G > A | Het | AD | ||||||||||||
| 3 | III 1 (F) | COL4A5 | c.2366_2383del | Het | XD | 17 | 20a | + | ‐ | NA | NA | 150/90 | NA | HD | NA |
| 4 | III 1 (M) | COL4A5 | c.3188G > A | Het | Isolated case | 24 | ‐ | ‐ | ‐ | + | ++ | 120/80 | +++ | ‐ | NA |
| 5 | III 2 (M) | COL4A5 | c.4473T > A | Hemi | Isolated case | 18 | ‐b | + | ‐ | NA | + | 120/80 | ++ | ‐ | NA |
| 6 | III 2 (M) | COL4A3 | c.971G > A | Het | Isolated case | 30 | ‐ | + | NA | + | ‐ | ‐ | ++++ | ‐ | Poor clinical results after hormone therapy |
| c.4411_4412del | Het | Isolated case | |||||||||||||
| 7 | III2(M) | COL4A4 | c.3782G > A | Het | AD | 34 | ‐ | ‐ | ‐ | NA | +++ | ‐ | 4.3g/day | ‐ | ACEI |
Abbreviations: AD, Autosomal dominant; ESRD, end‐stage renal disease; GBM, glomerula basement membrane; NA, not available; XD, X‐linked dominant; XR, X‐linked recessive.
ESRD age of brother
Scr > 200mmol/l
Table 2.
The mutations in suspected AS patients
|
Family Code |
Chr | Genomic coordinate (GRCh37) | Gene | Accession. Version numbers | Exon | Mutation Type | Nucleotide Change | Protein Change | Characterisic | State | De no | 1000G | ExAC_ALL | inhouse | Pathogenic | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 1 | X | 107,815,045 | COL4A5 | ‐ | E8 | missense | c.443C > T | p.Pro148Leu | Hematuria | Het/Hemi | √ | 0 | 0.00005854 | 0 | + | ||||||||
| Inoue (1999) | COL4A5 | Frameshift | c.440del | p.Gly147fs | Heavy proteinuria | 0 | 0 | 0 | + | ||||||||||||||
| Martin (1998) | COL4A5 | c.446del | p.Gly149fs | ESRD(20) | 0 | 0 | 0 | + | |||||||||||||||
| Nagel (2005) | COL4A5 | c.446dup | p.Gly150fs | ESRD | 0 | 0 | 0 | + | |||||||||||||||
| Family 2 | X | 107,834,411 | COL4A5 | NM_000495 | E20 | missense | c.1289C > A | p.Ala430Asp | ESRD(30) | Hemi | ‐ | 0.00238411 | 0.0047 | 0 | ± | ||||||||
| Knebelmann (1996) | COL4A5 | Nonconserved X residue | NA | Benign | |||||||||||||||||||
| Family 3 | X | 107,850,093–107850110 | COL4A5 | ‐ | E29 | Deletion | c.2366_2383del | p.789_795del | ESRD(20) | Het | √ | 0 | 0 | 0 | + | ||||||||
| King (2006) | COL4A5 | Missense | c.2360G > T | p.Gly787Val | NA | 0 | 0 | 0 | + | ||||||||||||||
| Plant (1999) | COL4A5 | c.2386G > A | p.Gly796Arg | ESRD(<43), HL | 0 | 0 | 0 | + | |||||||||||||||
| Family 4 | X | 107,869,521 | COL4A5 | NM_000495 | E36 | Missense | c.3188G > A | p.Gly1063Asp | Proteinuria, Hematuria | Het | ‐ | 0 | 0 | 0 | + | ||||||||
| Nagel (2005) | COL4A5 | C.3188G > T | p.Gly1063Thr | NA | 0 | 0 | 0 | + | |||||||||||||||
| Family 5 | X | 107,930,887 | COL4A5 | NM_000495 | E47 | Nossense | c.4473T > A | p.Tyr1491X | Hearing loss Proteinuria, Hematuria, Scr 200mmol/l | Hemi | √ | 0 | 0 | 0 | ± | ||||||||
| Family 6 | 2 | 228,121,096 | COL4A3 | NM_000091 | E17 | Missense | c.971G > A | p.G324D |
Heavy proteinuria (10g/day) |
Het | √ | 0.000199681 | 0.00002489 | 0 | + | ||||||||
| 2 | 228,172,583 228,172,585 | COL4A3 | NM_000091 | E48 | Frameshift | c.4411_4412del | p.S1472Ffs37 | Het | √ | 0 | 0 | 0 | + | ||||||||||
| Vega(2003) | 2 | NA | COL4A3 | E1 | Deletion | 40_63del24 |
Proteinuria (4.3g/day), Ocular loss |
NA | 0 | 0 | 0 | + | |||||||||||
| 2 | NA | COL4A3 | E48 | Frameshift | 4316delC | NA | 0 | 0 | 0 | + | |||||||||||||
| Family 7 | 2 | 227,896,696 | COL4A4 | NM_000092 | E40 | Missense | c.3782G > A | p.G1261E |
Proteinuria (1.28g/day), Hematuria |
Het | √ | 0 | 0 | 0 | + | ||||||||
*, Their frequency; √, The novel mutations
3.1. Clinical characteristics and genetic analysis of the COL4A5 mutant family
Pedigrees and COLA5 re‐sequencing chromatograms is in the Figure 1. Among the five families with the COL4A5 mutations, for Family 1, gene analysis suggested a mutation NM_000495.4(COL4A5): c.443C > T in exon 8, and family members carrying the mutations had a mild clinical manifestation, with persistent microscopic hematuria only as the main characteristic. It has been reported that missense mutations in exon 8 of COL4A5 occur mostly in highly conserved Gly‐X‐Y sequences(Inoue et al., 1999; Martin et al., 1998; Nagel, Nagorka, & Gross, 2005) and the clinical symptoms of the families with mutations were severe, showing progressive renal impairment (two cases progressed to ESRD)(Table2). In Family 2, the disease progression rate of the diseased individuals was fast, and the proband and his/her mother and elder sister developed ESRD within a short period of time after identification; renal biopsy pathology and electron microscopy showed that AS was combined with IgA nephropathy, and gene analysis indicated mutations in COL4A5 and INF2 were NM_000495.4(COL4A5): c.1289C > A and NM_022489.3(INF2): c.550G > A(Brown et al., 2010), respectively. Knebelmann et al. (1996) reported that the nonconserved X residue of this mutant type IV collagen α5 chain was benign, with low risk of disease incidence. However, Family 2 had serious clinical symptoms and rapid progression, with all having early onset ESRD, as well as a high degree of family genetic predisposition. Previous reports described (Boyer et al., 2011; Brown et al., 2010) INF gene‐related mutations related to focal segmental glomerulosclerosis, and the impact of different genetic mutations on disease progression is worthy of further discussion. For Family 3, the mutation found in this family was compared with the previously reported case in which exon 29 was mutated (King, Flinter, & Green, 2006). In this family, the NM_000495.4(COL4A5): c.2366_2388del mutation caused a change in the corresponding amino acid sequence, causing irregular proliferation, and basement membrane rupture; the rate of progression was fast and was manifested as early onset ESRD. In contrast, compared with progression to ESRD, missense mutations in the previous report had a decreased progression rate and can manifest as adult‐type ESRD or life‐long renal failure symptoms. Proband 4 had hematuria and proteinuria as the main clinical manifestations. Nagel et al. (2005) reported this mutation site NM_000495.4(COL4A5): c.3188G > T, which is located in the numerous repetitive triplet sequence (Gly‐X‐Y) in the collagen region. The patient had no clear family history and had a spontaneous mutation; combined with the pathological results of the renal biopsy in the proband, it was considered that the mutation in the COL4A5 gene caused a basement membrane abnormality, leading to AS. In Family 5, the proband had proteinuria and persistent microscopic hematuria at onset and progressive renal dysfunction as the main manifestations; in addition, gross hematuria could appear upon infection of the upper respiratory tract. The homozygous mutation is a nonsense mutation, and the patient's mother was a heterozygous mutant with no relevant clinical manifestations. The mutation site NM_000495.4(COL4A5): c.4473T > A is located in the NC1 domain, and because of the lack of previous reports on this mutation site, it is a newly discovered site.
Figure 1.

(a) Pedigrees and COLA5 re‐sequencing chromatograms(family 1 and family 2. Arrows point to the mutations. Family 1 has one novel mutations, c.443C > T in the figure 1a;#, INF2 re‐sequencing chromatograms of Family 2. (b) Pedigrees and COLA5 re‐sequencing chromatograms. Arrows point to the mutations (family 3, family 4, and family 5). Family 3 has one novel mutations, c.2366_2383del in the figure 1b; Family 5 has one novel mutations, c.4473T > A in the figure 1b
3.2. Clinical characteristics and genetic analysis of the COL4A3 mutant family
Pedigrees and COLA3 re‐sequencing chromatograms is in the Figure 2. In Family 6, the proband with a COL4A3 gene mutation was diagnosed with the onset of a large amount of proteinuria (5 g–10 g/24 hr) at 3 months pregnant, and the urinary protein was significantly reduced upon hormone treatment (approximately 1 g). There was no clear family history, and the postpartum renal biopsy suggested the following: one visible glomerulus, segmental mild hyperplasia in glomerular mesangial cells and the mesangial matrix, thin basement membrane, open capillary lumen, multifocal renal tubule atrophy (30%), and the presence of red blood cells and a small amount of protein casts in the renal tubule cavity. Multifocal interstitial fibrosis and infiltration of focal foam cells, lymphocytes and mononuclear cells were also present. Renal tubule interstitial lesions occurred. Electron microscopy showed AS with no obvious family history. Patient genetic analysis indicated a missense mutation NM_000091.4(COL4A3): c.971G > A in exon 17 combined with a frameshift mutation NM_000091.4(COL4A3): c.4411_4412del in exon 47, thus showing a compound heterozygous mutation. The parents each carried mutations at the two sites and were diagnosed with AS; the two mutation sites in this patient have not been reported. The mutations in the proband were from both parents. At the age of 60, the physical examination of the proband's father showed that urinary occult blood was positive, whereas other parameters examined were normal. No obvious abnormalities were observed in creatinine, urea, and kidney ultrasound‐related renal function indexes in the proband's mother.
Figure 2.
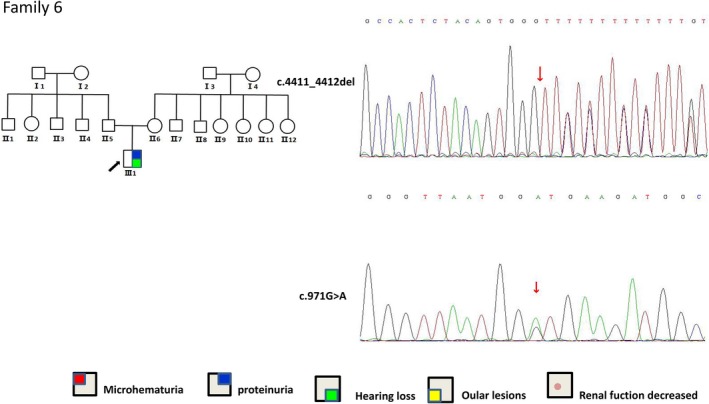
Pedigrees and COLA3 re‐sequencing chromatograms (family 6). Arrows point to the mutations. Family 6 has two novel mutations, c.971G > A and c.4411_4412del
3.3. Clinical characteristics and genetic analysis of the COL4A4 mutant family
Pedigrees and COLA4 re‐sequencing chromatograms is in the Figure 3. In Family 7, the proband with a COL4A4 gene mutation had hematuria and proteinuria at onset, accompanied by progressive renal functional insufficiency, and had a high degree of family genetic predisposition; the renal biopsy showed five full incidents of glomerulosclerosis (25%), two glomerular capsule adhesions (10%), and no segmental sclerosis or crescent bodies. The patient was diagnosed with focal glomerulosclerosis, and proteinuria was not relieved after treatment with angiotensin‐converting enzyme inhibitor (ACEI) drugs. The proband did not have mutations in other genes (TRPC6, ACTN4, WT1, CD2AP, INF2, NPHS1, NPHS2, and PLCE1) related to familial focal segmental glomerulosclerosis (FSGS), and gene analysis suggested a heterozygous mutation NM_000092.4(COL4A4): c.3782G > A in exon 40, which is a putative new mutation. A diagnosis of autosomal recessive Alport syndrome (ARAS) was made by combining the COL4A4 heterozygous mutation with the clinical characteristics of the family. A number of studies have shown that disease progression caused by COL4A3 or COL4A4 heterozygous mutations is slower than that of X‐linked AS (Pochet et al., 1989), but this family had a high degree of family aggregation, and many people in the family had renal insufficiency.
Figure 3.

Pedigrees and COLA4 re‐sequencing chromatograms (family 7). Arrows point to the mutations. Family 7 has one novel mutation, c. 3,782 G > A
4. DISCUSSION
X‐linked AS caused by mutations in the COL4A5 gene is a hereditary progressive glomerular disease often accompanied by sensorineural deafness and visual impairment (Grunfeld, 1985). As a hereditary disease, AS is divided into two clinical phenotypes: early onset and late onset. The pathological characteristics mainly include irregular thickening and rupture of the basement membrane and the deposition of immune complexes accompanied by the loss of type IV collagen. Type IV collagen molecules consist of three αchains, and six different type IV collagen chains are encoded by six different genes distributed among three chromosomes (Hudson, 2004). The COL4A5 gene is approximately 250 kb in length and contains 51 exons; the COL4A5 encoded type IV collagen α5 chain contains 1685 amino acids, including the signal peptides, composed of 26 residues (encoded by exon 1), the collagen domain, composed of 1,430 amino acid residues (encoded by exons 27–14), and the noncollagen domain (NC1), composed of 229 C‐terminal residues. Since the collagen domain contains a myriad of repetitive triplet glycine‐X‐Y (Gly‐X‐Y), the proportion of glycine residue substitution in this region is high (Bekheirnia et al., 2010).
X‐linked AS was first reported in 1990, and nearly 120 mutation sites had been reported as of 1996. Thus far, the number of COL4A5 related mutation sites has increased to more than 800, and most of the mutations are mainly deletions of small DNA fragments and allelic mutations (Crockett et al., 2010). Two large‐scale studies conducted in Europe and the USA elucidated the natural course of AS disease and the genotype‐phenotype relationship (Bekheirnia et al., 2010; Jais et al., 2003). With 195 European families and 175 American families included, these two studies showed that AS patients with different genetic mutation types have different risks of developing ESRD prior to the age of 30. The risk of large‐fragment deletions, nonsense mutations or frameshift mutations is approximately 90%, the risk of splicing site mutations ranks second (approximately 70%), and the risk of missense mutations is minimal (approximately 50%)(Jais et al., 2003); the mean ages of ESRD in patients with missense mutations, splicing site mutations, and truncation mutations are 37 years, 28 years, and 25 years, respectively (Bekheirnia et al., 2010). To predict kidney prognosis, it is important to determine whether these mutations are truncating or nontruncating transcript (Horinouchi et al., 2018). We searched the Alport syndrome COL4A5 variant database (Crockett et al., 2010) and HGMD database, and retrieved the relevant published articles. At the same time, our study compared the published symptoms with the symptoms of our patients (as shown in Table2). In this study, Family 3(NM_000495.4(COL4A5): c.2366_2388del, frameshift mutation) and Family 5 (NM_000495.4(COL4A5): c.4473T > A, nonsense mutation) were characterized by rapid progression of renal function impairment and hearing impairment, consistent with the above findings. AS is defined as progressive hereditary glomerulus nephritis with a high degree (accounting for approximately 1/3‐2/3 of cases) of familial genetic predisposition. Nagel et al. considered the percentage of X‐linked AS gene mutations in families with no clear family history to be 60%. Family 4 had no clear family history, and the mutation was located in the Gly‐X‐Y repetitive sequences of the COL4A5 collagen domain. Whether the Gly‐X‐Y mutation can indicate poor disease progression remains controversial, so the combination of a spontaneous mutation and the mutation site at Gly‐X‐Y is not predictive of disease prognosis, for which long‐term follow‐up is required.
In most patients with COL4A5 mutations, all α‐3/4/5 chains are deleted, so it is speculated that the TBMN gene defect may be similar to that observed in AS. Previous studies have confirmed that TBNM is associated with COL4A3 and COL4A4 related mutations.(Buzza, Wilson, & Savige, 2001; Torra, Tazon‐Vega, Ars, & Ballarin, 2004). However, some studies have concluded that some male patients with COL4A5 mild mutations may also exhibit TBMN in adulthood (Demosthenous et al., 2012; Pierides, Voskarides, Kkolou, Hadjigavriel, & Deltas, 2013). Thus, TBMN patients are considered to be "carriers" of ARAS (Mochizuki, 1994). In our study, the clinical symptoms caused by the NM_000495.4(COL4A5): c.443C > Tmutation in COL4A5 located on the X chromosome also consisted of persistent microscopic hematuria, and it is speculated that the collagen defect produced by the abnormal gene product of this mutation may affect the normal structure of the glomerular basement membrane (GBM). However, the NM_000495.4(COL4A5): c.443C > T mutation on COL4A5 was different from the previously reported relevant mutation at exon 8 in that the C443T mutation on COL4A5 only leads to persistent microscopic hematuria, indicating that the COL4A5 mutation at this site only hindered, to a certain degree, expression of the α‐3, α‐4, and α‐5 chains. This study may also indicate that mutation of the non‐Gly‐X‐Y sequence in the α5‐chain collagen domain is related to the NM_000495.4(COL4A5): c.443C > T mutation in COL4A5, and the phenotype is milder, with only a small possibility of progression to ESRD.
ARAS accounts for 20%‐30% of AS patients and is caused by heterozygous mutations in the COL4A3 and COL4A4 genes. ARAS presents no sex differences (Mochizuki, 1994) and has a slower progression than X‐linked AS; previous studies have also suggested that it is rare for patients diagnosed with ARAS to have a normal or partial α‐5 chain. Different types of gene mutations contribute to the variability in GBM structure (Hudson, 2004): nonsense mutations or the insertion of stop codons make formation of the type IV collagen network difficult, whereas most missense mutations can only cause abnormalities in the type IV collagen 3, 4, and 5 chains. In many previous studies and case reports of ARAS, most patients maintained normal renal function at the age of 40. Zhang et al. (2012) reported that 17%‐34% of ARAS patients had COL4A3/COL4A4 complex mutations. Kamijo et al. (2017) reported a 41‐year‐old male patient with ARAS, and gene analysis for this patient suggested that an insertion mutation (NM_000091.4(COL4A3): c.933 + 1G>A) occurred in exon 16 and that a deletion mutation (NM_000091.4(COL4A3): c 3650_3657del8) occurred in exon 42. The basement membrane α5 chain and α2 chain were positive in this patient, which was unlike the commonly reported cases where only the α2 chain was positive while the α5 chain had a disordered formation. The 41‐year‐old male patient only showed urine occult blood as positive and had no other symptoms of progressive renal impairment. Compared with the case reported by Kamijo et al. (2017), our patient was different in that there were two COL4A3 gene mutations (NM_000091.4(COL4A3): c.971G > A and NM_000091.4(COL4A3): c.4411_4412del), with each mutation from one of the parents. Both of the parents had no relevant clinical manifestations, and the family history was also negative. Thus, the formation of α3/4/5 chains and the abnormality of the basement membrane in our patient were the outcomes of the simultaneous action of these two mutations but could not be attributed solely to the missense mutation or the deletion; it was a complex heterozygous mutation, and the associated symptom was more severe than that with the simple missense mutation or with the single‐base deletion mutation.
Previous studies have focused on the relationship between FSGS and AS and on that between IgA nephropathy and AS (Gast et al., 2016; Hines et al., 2018; Kamiyoshi et al., 2016; Lin et al., 2014; Malone et al., 2014; Xie et al., 2015), suggesting that AS has variable clinical phenotypes, AS patients who have proteinuria or AS patients who have proteinuria and whose renal biopsy results are consistent with changes in FSGS or in IgA nephropathy are sometimes misdiagnosed with a primary nephropathy such as FSGS. In recent years, several researches and cases reported that AS patients with COL4A5 mutations who have coinherited with COL4A3/COL4A4, MYO1E and LAMA5 mutations could exacerbated phenotypes (Lennon et al., 2015; Voskarides et al., 2018). These clinical symptoms occur mostly in the late disease stages and are thought to be secondary changes caused by primary GBM defects resulting from abnormalities in the collagen reticular structure (Braunisch et al., 2018). For these misdiagnosed patients, if treatment is conducted according to the classic glucocorticoid‐based regimen for FSGS, the treatment effect is not significant, and the patient may be considered to have hormone‐resistant nephrotic syndrome. Combined with the analyses of Family 2 and Family 7 in this study, the pathological results of a renal biopsy of the proband in Family 2 showed AS complicated with IgA nephropathy in which proteinuria and other clinical manifestations were more significant compared with previous cases with mutations at this site. Previous studies (Knebelmann et al., 1996) suggest that the mutation in Family 2 has a high frequency of disease incidence in normal people, and the mutation site is located in a nonconserved sequence. This mutation leads to nonserious AS clinical symptoms, which does not agree with the clinical phenotype of the family in this study. It is, however, worth noting that Family 2 in this study also had an INF2 mutation, which is associated with focal segmental glomerulosclerosis(FSGS)(Brown et al., 2010), the clinical manifestation of which is progressive renal dysfunction with a fast progression rate. By reviewing the literature, INF2 and COL4A5 mutations are not associated in terms of proteomics and genomics and could cause glomerular disease clinical manifestations, but these mutations have an additive effect on the clinical manifestations of nephropathy, compared with those of single gene‐causing nephropathy (Brown et al., 2010). In this study, AS in Family 7 was caused by a dominant genetic heterozygous mutation in the COL4A4 gene. The renal biopsy results suggested focal sclerosing nephritis, in which the onset includes hematuria and proteinuria. Multiple cohort studies (Gast et al., 2016; Kamiyoshi et al., 2016; Voskarides et al., 2007; Voskarides, Patsias, Pierides, & Deltas, 2008; Voskarides, Pierides, & Deltas, 2008; Xie et al., 2015) have revealed the correlation between FSGS and AS or TBMN, and the association of AS with collagen‐associated gene mutations (COL4A3/COL4A4) is easily misdiagnosed as FSGS due to their clinical symptom similarities. These studies suggest that some of the unexplained familial hereditary nephritis may be AS, and with the development of gene sequencing technology, the diagnostic ratio of AS will increase. Based on the above analysis, it was shown that AS patients have different phenotypes. On the one hand, this is because of secondary changes that result from the collagen reticular abnormalities caused by collagen‐related gene mutations (Malone et al., 2014). On the other hand, it is speculated that there may be other secondary factors affecting the disease phenotype (Kamiyoshi et al., 2016), and some AS patients are thus easily misdiagnosed as primary nephropathy (such as FSGS) or hormone‐resistant nephrotic syndrome. Taken together, the primary nature of both FSGS and AS is controversial (Voskarides et al., 2007).
5. CONCLUSIONS
Although AS has no hotspot mutations and cannot be diagnosed by design kits, in recent years, due to the rapid development of next‐generation sequencing exon capture technology, the clarification of gene mutation types, enrichment of AS gene mutation databases and provision of accurate genetic counseling have become hot issues of concern. In this study, by collecting AS familial data, improving kidney disease clinical data and obtaining gene detection results, eight AS mutation sites (two frameshift mutations, five missense mutations, and one nonsense mutation) were found, of which six were new mutations. In Family 2, we found the AS patient with INF2 mutation could have an additive effect on the clinical manifestations of nephropathy. Family 6 had a complex heterozygous mutation at COL4A3 (including one missense mutation and one frameshift mutation); these mutations enriched the AS gene pathogenic sites and database resources. The intrinsic relationship between the genetic mutations of AS families and their clinical phenotypic characteristics were analyzed, which may further reveal that the clinical manifestations of X‐linked AS in males were more commonly severe than those in females and that the risk of gene segmental deletion and nonsense mutation progressing to ESRD was high. However, these findings need to be confirmed in further studies with larger sample sizes. Due to its genetic heterogeneity and phenotypic diversity, AS is easily misdiagnosed as a primary nephropathy such as FSGS, and gene sequencing analysis provides the necessary methods for diagnosis and treatment. Summarizing the characteristics of AS gene mutations and the phenotypic and genotypic heterogeneity allows for the provision of database resources for the diagnosis, treatment and prognosis of AS.
CONFLICT OF INTEREST
The authors declare they have no conflict of interest.
AUTHORS’ CONTRIBUTIONS
Contributed to the conception and design: SLS, PF, QGL, and XMC. Analyzed and interpreted the data: SLS, FP, TW, PL, QGL, and XMC. Provided study patients: SLS FP, XYW, QGL, and XMC. Wrote and revised the manuscript: SLS, FP, QGL, and XMC. All authors reviewed the manuscript and approved the final version of the manuscript.
ACKNOWLEDGMENTS
This work was supported by Beijing Nova Program(Z161100004916129) and National Natural Science Foundation of China(81570597,81770664 and 81830019 ). The authors admitted that the National Clinical Medicine Research Center for Chronic Nephropathy and the Department of Nephrology at the Chinese PLA General Hospital, supported the data provided for this study.
Shang S, Peng F, Wang T, et al. Genotype‐phenotype correlation and prognostic impact in Chinese patients with Alport Syndrome. Mol Genet Genomic Med. 2019;7:e741 10.1002/mgg3.741
Shunlai Shang, Fei Peng and Tao Wang are equal contributors
Contributor Information
Qinggang Li, Email: lqgbj301@126.com.
Xiang M. Chen, Email: xmchen301@126.com
REFERENCES
- Bekheirnia, M. R. , Reed, B. , Gregory, M. C. , McFann, K. , Shamshirsaz, A. A. , Masoumi, A. , & Schrier, R. W. (2010). Genotype‐phenotype correlation in X‐linked Alport syndrome. Journal of the American Society of Nephrology, 21(5), 876–883. 10.1681/asn.2009070784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer, O. , Benoit, G. , Gribouval, O. , Nevo, F. , Tete, M. J. , Dantal, J. , … Antignac, C. (2011). Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. Journal of the American Society of Nephrology, 22(2), 239–245. 10.1681/asn.2010050518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunisch, M. C. , Büttner‐Herold, M. , Günthner, R. , Satanovskij, R. , Riedhammer, K. M. , Herr, P.‐M. , … Hoefele, J. (2018). Heterozygous COL4A3 variants in histologically diagnosed focal segmental glomerulosclerosis. Frontiers in Pediatrics, 6, 171 10.3389/fped.2018.00171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, E. J. , Schlöndorff, J. S. , Becker, D. J. , Tsukaguchi, H. , Tonna, S. J. , Uscinski, A. L. , … Pollak, M. R. (2010). Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nature Genetics, 42(1), 72–76. 10.1038/ng.505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzza, M. , Wilson, D. , & Savige, J. (2001). Segregation of hematuria in thin basement membrane disease with haplotypes at the loci for Alport syndrome. Kidney International, 59(5), 1670–1676. 10.1046/j.1523-1755.2001.0590051670.x [DOI] [PubMed] [Google Scholar]
- Crockett, D. K. , Pont‐Kingdon, G. , Gedge, F. , Sumner, K. , Seamons, R. , & Lyon, E. (2010). The Alport syndrome COL4A5 variant database. Human Mutation, 31(8), E1652–1657. 10.1002/humu.21312 [DOI] [PubMed] [Google Scholar]
- Dahan, K. , Heidet, L. , Zhou, J. , Mettler, G. , Leppig, K. A. , Proesmans, W. , … David, A. (1995). Smooth muscle tumors associated with X‐linked Alport syndrome: Carrier detection in females. Kidney International, 48(6), 1900–1906. 10.1038/ki.1995.489 [DOI] [PubMed] [Google Scholar]
- Demosthenous, P. , Voskarides, K. , Stylianou, K. , Hadjigavriel, M. , Arsali, M. , Patsias, C. , … Deltas, C. (2012). X‐linked Alport syndrome in Hellenic families: Phenotypic heterogeneity and mutations near interruptions of the collagen domain in COL4A5. Clinical Genetics, 81(3), 240–248. 10.1111/j.1399-0004.2011.01647.x [DOI] [PubMed] [Google Scholar]
- Fallerini, C. , Dosa, L. , Tita, R. , Del Prete, D. , Feriozzi, S. , Gai, G. , … Ariani, F. (2014). Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clinical Genetics, 86(3), 252–257. 10.1111/cge.12258 [DOI] [PubMed] [Google Scholar]
- Gast, C. , Pengelly, R. J. , Lyon, M. , Bunyan, D. J. , Seaby, E. G. , Graham, N. , … Ennis, S. (2016). Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrology, Dialysis, Transplantation, 31(6), 961–970. 10.1093/ndt/gfv325 [DOI] [PubMed] [Google Scholar]
- Grunfeld, J. P. (1985). The clinical spectrum of hereditary nephritis. Kidney International, 27(1), 83–92. 10.1038/ki.1985.14 [DOI] [PubMed] [Google Scholar]
- Hines, S. L. , Agarwal, A. , Ghandour, M. , Nabeel, A. , Mohammad, A. N. , & Atwal, P. S. (2018). Novel variants in COL4A4 and COL4A5 are rare causes of FSGS in two unrelated families. Human Genome Variation, 5, 15 10.1038/s41439-018-0016-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horinouchi, T. , Nozu, K. , Yamamura, T. , Minamikawa, S. , Omori, T. , Nakanishi, K. , … Iijima, K. (2018). Detection of Splicing Abnormalities and Genotype‐Phenotype Correlation in X‐linked Alport Syndrome. Journal of the American Society of Nephrology, 29(8), 2244–2254. 10.1681/asn.2018030228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, B. G. (2004). The molecular basis of Goodpasture and Alport syndromes: Beacons for the discovery of the collagen IV family. Journal of the American Society of Nephrology, 15(10), 2514–2527. 10.1097/01.asn.0000141462.00630.76 [DOI] [PubMed] [Google Scholar]
- Inoue, Y. , Nishio, H. , Shirakawa, T. , Nakanishi, K. , Nakamura, H. , Sumino, K. , … Yoshikawa, N. (1999). Detection of mutations in the COL4A5 gene in over 90% of male patients with X‐linked Alport's syndrome by RT‐PCR and direct sequencing. American Journal of Kidney Diseases, 34(5), 854–862. 10.1016/s0272-6386(99)70042-9 [DOI] [PubMed] [Google Scholar]
- Jais, J. P. , Knebelmann, B. , Giatras, I. , De Marchi, M. , Rizzoni, G. , Renieri, A. , … Gubler, M. C. (2003). X‐linked Alport syndrome: Natural history and genotype‐phenotype correlations in girls and women belonging to 195 families: A "European Community Alport Syndrome Concerted Action" study. Journal of the American Society of Nephrology, 14(10), 2603–2610. 10.1097/01.ASN.0000090034.71205.74 [DOI] [PubMed] [Google Scholar]
- Kamijo, M. , Kitamura, M. , Muta, K. , Uramatsu, T. , Obata, Y. , Nozu, K. , … Nishino, T. (2017). A case of mild phenotype Alport syndrome caused by COL4A3 mutations. CEN Case Rep, 6(2), 189–193. 10.1007/s13730-017-0273-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyoshi, N. , Nozu, K. , Fu, X. J. , Morisada, N. , Nozu, Y. , Ye, M. J. , … Iijima, K. (2016). Genetic, clinical, and pathologic backgrounds of patients with autosomal dominant alport syndrome. Clinical Journal of the American Society of Nephrology, 11(8), 1441–1449. 10.2215/cjn.01000116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashtan, C. E. (1995). Clinical and molecular diagnosis of Alport syndrome. Proceedings of the Association of American Physicians, 107(3), 306–313. [PubMed] [Google Scholar]
- Kashtan, C. E. (2004). Diagnosis of Alport syndrome. Kidney International, 66(3), 1290–1291; author reply 1291. doi:10.1111/j.1523‐1755.2004.884_6.x. [DOI] [PubMed] [Google Scholar]
- King, K. , Flinter, F. A. , & Green, P. M. (2006). A two‐tier approach to mutation detection in the COL4A5 gene for Alport syndrome. Human Mutation, 27(10), 1061 10.1002/humu.9453 [DOI] [PubMed] [Google Scholar]
- Knebelmann, B. , Breillat, C. , Forestier, L. , Arrondel, C. , Jacassier, D. , Giatras, I. , … Antignac, C. (1996). Spectrum of mutations in the COL4A5 collagen gene in X‐linked Alport syndrome. American Journal of Human Genetics, 59(6), 1221–1232. [PMC free article] [PubMed] [Google Scholar]
- Lennon, R. , Stuart, H. M. , Bierzynska, A. , Randles, M. J. , Kerr, B. , Hillman, K. A. , … Woolf, A. S. (2015). Coinheritance of COL4A5 and MYO1E mutations accentuate the severity of kidney disease. Pediatric Nephrology(Berlin, Germany), 30(9), 1459–1465. 10.1007/s00467-015-3067-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, F. , Bian, F. , Zou, J. , Wu, X. , Shan, J. , Lu, W. , … Gale, D. P. (2014). Whole exome sequencing reveals novel COL4A3 and COL4A4 mutations and resolves diagnosis in Chinese families with kidney disease. BMC Nephrology, 15, 175 10.1186/1471-2369-15-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo, I. , Porcedda, P. , Mari, F. , Giachino, D. , Meloni, I. , Deplano, C. , … De Marchi, M. (2002). COL4A3/COL4A4 mutations: From familial hematuria to autosomal‐dominant or recessive Alport syndrome. Kidney International, 61(6), 1947–1956. 10.1046/j.1523-1755.2002.00379.x [DOI] [PubMed] [Google Scholar]
- Malone, A. F. , Phelan, P. J. , Hall, G. , Cetincelik, U. , Homstad, A. , Alonso, A. S. , … Gbadegesin, R. A. (2014). Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney International, 86(6), 1253–1259. 10.1038/ki.2014.305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, P. , Heiskari, N. , Zhou, J. , Leinonen, A. , Tumelius, T. , Hertz, J. M. , … Tryggvason, K. (1998). High mutation detection rate in the COL4A5 collagen gene in suspected Alport syndrome using PCR and direct DNA sequencing. Journal of the American Society of Nephrology, 9(12), 2291–2301. [DOI] [PubMed] [Google Scholar]
- Mochizuki, T. , Lemmink, H. H. , Mariyama, M. , Antignac, C. , Gubler, M.‐C. , Pirson, Y. , … Reeders, S. T. (1994). Identification of mutations in the α3(IV) and α4(IV) collagen genes in autosomal recessive Alport syndrome. Nature Genetics, 8(1), 5 10.1038/ng0994-77 [DOI] [PubMed] [Google Scholar]
- Morinière, V. , Dahan, K. , Hilbert, P. , Lison, M. , Lebbah, S. , Topa, A. , … Heidet, L. (2014). Improving mutation screening in familial hematuric nephropathies through next generation sequencing. Journal of the American Society of Nephrology, 25(12), 2740–2751. 10.1681/asn.2013080912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel, M. , Nagorka, S. , & Gross, O. (2005). Novel COL4A5, COL4A4, and COL4A3 mutations in Alport syndrome. Human Mutation, 26(1), 60 10.1002/humu.9349 [DOI] [PubMed] [Google Scholar]
- Ninomiya, Y. , Kagawa, M. , Iyama, K. , Naito, I. , Kishiro, Y. , Seyer, J. M. , … Sado, Y. (1995). Differential expression of two basement membrane collagen genes, COL4A6 and COL4A5, demonstrated by immunofluorescence staining using peptide‐specific monoclonal antibodies. Journal of Cell Biology, 130(5), 1219–1229. 10.1083/jcb.130.5.1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierides, A. , Voskarides, K. , Kkolou, M. , Hadjigavriel, M. , & Deltas, C. (2013). X‐linked, COL4A5 hypomorphic Alport mutations such as G624D and P628L may only exhibit thin basement membrane nephropathy with microhematuria and late onset kidney failure. Hippokratia, 17(3), 207–213. [PMC free article] [PubMed] [Google Scholar]
- Pirson, Y. (1999). Making the diagnosis of Alport's syndrome. Kidney International, 56(2), 760–775. 10.1046/j.1523-1755.1999.00601.x [DOI] [PubMed] [Google Scholar]
- Pochet, J. M. , Bobrie, G. , Landais, P. , Goldfarb, B. , & Grunfeld, J. P. (1989). Renal prognosis in Alport's and related syndromes: Influence of the mode of inheritance. Nephrology, Dialysis, Transplantation, 4(12), 1016–1021. [PubMed] [Google Scholar]
- Savige, J. , Ariani, F. , Mari, F. , Bruttini, M. , Renieri, A. , Gross, O. , … Storey, H. (2018). Expert consensus guidelines for the genetic diagnosis of Alport syndrome. Pediatric Nephrology(Berlin, Germany), 10.1007/s00467-018-3985-4 [DOI] [PubMed] [Google Scholar]
- Torra, R. , Tazon‐Vega, B. , Ars, E. , & Ballarin, J. (2004). Collagen type IV (alpha3‐alpha4) nephropathy: From isolated haematuria to renal failure. Nephrology, Dialysis, Transplantation, 19(10), 2429–2432. 10.1093/ndt/gfh435 [DOI] [PubMed] [Google Scholar]
- Uliana, V. , Marcocci, E. , Mucciolo, M. , Meloni, I. , Izzi, C. , Manno, C. , … Salviati, L. (2011). Alport syndrome and leiomyomatosis: The first deletion extending beyond COL4A6 intron 2. Pediatric Nephrology(Berlin, Germany), 26(5), 717–724. 10.1007/s00467-010-1693-9 [DOI] [PubMed] [Google Scholar]
- Voskarides, K. , Damianou, L. , Neocleous, V. , Zouvani, I. , Christodoulidou, S. , Hadjiconstantinou, V. , … Deltas, C. (2007). COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. Journal of the American Society of Nephrology, 18(11), 3004–3016. 10.1681/asn.2007040444 [DOI] [PubMed] [Google Scholar]
- Voskarides, K. , Papagregoriou, G. , Hadjipanagi, D. , Petrou, I. , Savva, I. , Elia, A. , … Deltas, C. (2018). COL4A5 and LAMA5 variants co‐inherited in familial hematuria: Digenic inheritance or genetic modifier effect? BMC Nephrology, 19(1), 114 10.1186/s12882-018-0906-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskarides, K. , Patsias, C. , Pierides, A. , & Deltas, C. (2008). COL4A3 founder mutations in Greek‐Cypriot families with thin basement membrane nephropathy and focal segmental glomerulosclerosis dating from around 18th century. Genet Test, 12(2), 273–278. 10.1089/gte.2007.0110 [DOI] [PubMed] [Google Scholar]
- Voskarides, K. , Pierides, A. , & Deltas, C. (2008). COL4A3/COL4A4 mutations link familial hematuria and focal segmental glomerulosclerosis. glomerular epithelium destruction via basement membrane thinning? Connective Tissue Research, 49(3), 283–288. 10.1080/03008200802148280 [DOI] [PubMed] [Google Scholar]
- Xie, J. , Wu, X. , Ren, H. , Wang, W. , Wang, Z. , Pan, X. , … Chen, N. (2015). COL4A3 mutations cause focal segmental glomerulosclerosis. J Mol Cell Biol, 7(2), 184 10.1093/jmcb/mjv023 [DOI] [PubMed] [Google Scholar]
- Yamamura, T. , Nozu, K. , Fu, X. J. , Nozu, Y. , Ye, M. J. , Shono, A. , … Iijima, K. (2017). Natural history and genotype‐phenotype correlation in female X‐linked alport syndrome. Kidney International Reports, 2(5), 850–855. 10.1016/j.ekir.2017.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Wang, F. , Ding, J. , Zhang, H. , Zhao, D. , Yu, L. , … Wang, S. (2012). Genotype‐phenotype correlations in 17 Chinese patients with autosomal recessive Alport syndrome. American Journal of Medical Genetics. Part A, 158a(9), 2188–2193. 10.1002/ajmg.a.35528 [DOI] [PubMed] [Google Scholar]
- Zhou, J. , Mochizuki, T. , Smeets, H. , Antignac, C. , Laurila, P. , de Paepe, A. , … Reeders, S. T. (1993). Deletion of the paired alpha 5(IV) and alpha 6(IV) collagen genes in inherited smooth muscle tumors. Science, 261(5125), 1167–1169. [DOI] [PubMed] [Google Scholar]