

 A catalytic C(sp2)–F bond carboxylation with CO2 is reported; the reaction is enabled by dual photoredox/palladium catalysis and involves a crucial fluoroalkenyl radical intermediate.
A catalytic C(sp2)–F bond carboxylation with CO2 is reported; the reaction is enabled by dual photoredox/palladium catalysis and involves a crucial fluoroalkenyl radical intermediate.
Abstract
The catalytic C–F bond carboxylation of organofluorines with CO2 gas remains a challenging problem in synthetic chemistry. Here, we describe a selective defluorinative carboxylation of gem-difluoroalkenes through photoredox/palladium dual catalysis. The C–F bond activation is enabled by single electron reduction through photoredox catalysis to generate a fluorovinyl radical, which subsequently participates in an unprecedented palladium-catalyzed carboxylation. This novel C–F functionalization proved applicable to a wide range of substituted gem-difluoroalkenes, providing a rapid access to valuable α-fluoroacrylic acids.
Introduction
C–(pseudo)halide bond carboxylation that allows the reductive coupling of organic (pseudo)halides and CO2 is of pivotal importance from the viewpoint of preparation of structurally diversified carboxylic acids as well as CO2 incorporation.1 After the pioneering studies of Yamamoto, various catalytic carboxylation procedures for the C–X bond, such as I, Br, Cl and OTf, have been well established, thus enabling smooth incorporation of CO2 into important chemical architectures (Scheme 1A).2 Despite the impressive chemical yields and selectivity achieved in these systems, catalytic C–F bond carboxylation has been largely underdeveloped, primarily because of the lack of an efficient protocol to effect the essential C–F bond scission.3 In view of the growing demand for fluorinated carboxylic acids and their derivatives in medicinal chemistry and materials science,4 the development of selective C–F bond carboxylation is especially appealing owing to the strategic advantage of utilizing the easily available and less expensive polyfluoro organic building blocks.5
Scheme 1. Rational design of catalytic C–F bond carboxylation. (A) Classical catalytic C–X bond carboxylation. (B) Recent development in photoredox enabled C(sp2)–F bond activation. (C) Established synthetic methods for a-fluoroacrylic acid/ester. (D) Our photoredox/Pd catalyzed C–F carboxylation of gem-difluoroalkenes.

Existing methods for C–(pseudo)halide bond carboxylation mostly rely on oxidative addition to low-valence transition-metals to generate an organometallic intermediate which is capable of undergoing carboxylation under a CO2 atmosphere. For example, the Tsuji and Fujihara group6 and the Martin group7 have developed a series of protocols based on Ni-catalysis with Mn as the reducing agent. Very recently, the Iwasawa group also reported an exciting breakthrough in this field, wherein the employed tertiary amine acted as the reductant under photoredox conditions to reduce the high-valence palladium intermediate, thus guaranteeing the whole catalytic cycle.8 This was subsequently followed by König's photoredox/Ni dual catalysis enabled carboxylation of arylbromide/triflate, wherein Hantzsch ester is used as the terminal reductant.9 Although these methods are highly effective for C–(pseudo)halide carboxylation, extension to C–F analogs has proven extremely challenging because C–F bonds are far more stable than the heavier congeners and thus recalcitrant to undergo the imperative oxidative addition process.3a Notably, most of the currently available carboxylation reactions for C–F bond activation depend on the use of stoichiometric amounts of alkaline(earth) metals.10 During the last several years, the photoredox catalysis has been recognized as an effective strategy for C–F bond activation through a single electron transfer process.11 For instance, the Weaver laboratory has reported a number of photoredox catalyzed reactions that provide single C–F bond alkylation/alkenylation/arylation products from polyfluoroarenes.11a–f Furthermore, the Hashmi group developed selective defluorinative α-aminoalkylation of gem-difluoroalkenes (Scheme 1B).11g While this activation mode has been employed in the context of radical coupling reactions, reports on the interception of these fluorine-containing carbon radicals by transition-metals to form versatile organometallic species are still scarce. Given that photoredox/transition-metal dual catalysis has emerged as a powerful strategy in synthetic chemistry,12 we envisioned the possibility of merging C-centered radical formation by photoinduced C–F bond activation with transition-metal catalyzed carboxylation. Also of note is that amines could serve as potential reductants for the regeneration of active transition-metal catalysts under photoredox catalysis, enabling mild and user-friendly reaction conditions. With our continuing interest in synthetic elaboration of gem-difluoroalkenes,13 we would like to report herein that these molecules can be smoothly coupled with the notoriously inert electrophile CO2 by photoredox/Pd dual catalysis, thereby establishing a conceptually new and direct C–F carboxylation for the straightforward access to a broad array of structurally privileged α-fluoroacrylic acids distinct from the existing methods (Scheme 1C and D).14
Results and discussion
We initially explored the C–F bond carboxylation of biphenyl substituted gem-difluoroalkene 1a with an Ir-based photocatalyst and different co-catalysts under the irradiation of blue LEDs (λ = 459 nm), with DIPEA and Cs2CO3 as the terminal reductant and base, respectively (Table 1). Although nickel proved to be a very effective catalyst for the C–C coupling in combination with photocatalysts,15 it failed to provide the desired product in this specific case. Replacing nickel with cobalt was also unproductive (entry 2).16 To our delight, with Pd(PPh3)4 as the co-catalyst the defluorinative carboxylation of 1a proceeded to give the desired product 2a in 72% yield and the ratio of Z and E-isomers was determined to be 90 : 10 by NMR spectroscopy (entry 3).17,18 Further screening of photocatalysts proved PC 1 as the optimal choice (entries 3 to 7). Encouragingly, with the combination of PdCl2 and PPh3, the desired carboxylation occurred smoothly, giving the product in an improved yield (87%, entry 8). Subsequently, phosphine ligands were examined, however, only diminished yields were obtained (entries 9 to 11). Interestingly, in the absence of any ligand, PdCl2 and even Pd/C could afford the desired product, albeit in lower yields (Table S4,† entries 13 and 14).19 Conducting the reaction for a prolonged reaction time resulted in full conversion, delivering 2a in higher yield (entry 12). To our surprise, the defluorinative aminoalkylation of 1a was not observed when Et3N was employed instead of iPr2NEt, although it is a competent substrate in Hashmi's work (entry 13).11e In contrast, N,N-dimethylaniline led to 40% (Z/E = 81 : 19) of defluoroaminoalkylation product, while the carboxylation product was only formed in 11% yield (entry 14). This result indicated the intermediacy of transient fluorovinyl radical species, presumably via SET reduction of gem-difluoroalkenes under the present photocatalysis reaction conditions. Considering that the palladium/phosphine complex has been frequently demonstrated as an effective single electron donor under photo-irradiation,20 the exact role of the Ir-photocatalyst in this reaction was examined. However, omitting the iridium complex shuts down the reaction completely. Moreover, visible-light-driven metal-free carboxylation of alkenes through either radical–radical coupling21 or radical-polar crossover scenario was reported.22 Control experiments in the absence of either the Pd-catalyst or light irradiation unequivocally supported the vital importance of both parameters for the success of this transformation (Table S10†).
Table 1. Optimization of the reaction conditions a .
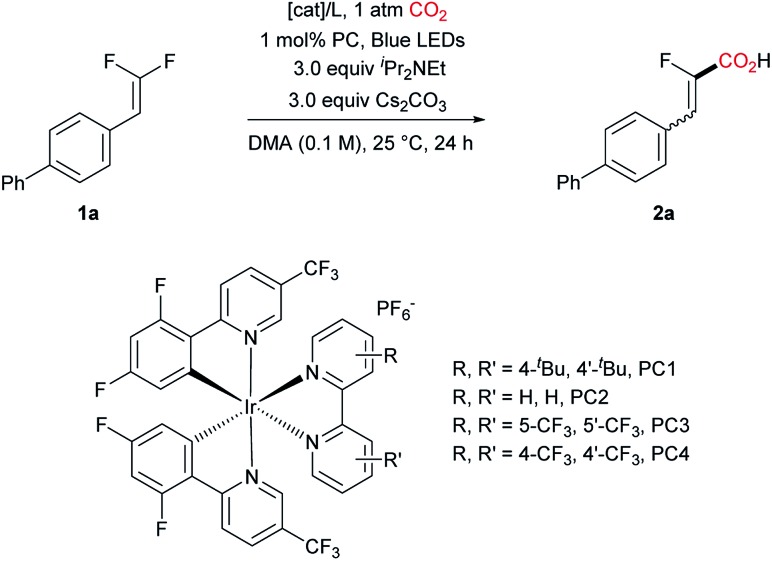
| ||||
| Entry | [Cat]/L | PC | Yield/% | Z/E |
| 1 | NiBr2/bipyridyl | PC 1 | Trace | — |
| 2 | CoCl2/neocuproine | PC 1 | Trace | — |
| 3 | Pd(PPh3)4/none | PC 1 | 71 | 90/10 |
| 4 | Pd(PPh3)4/none | PC 2 | 48 | 88 : 12 |
| 5 | Pd(PPh3)4/none | PC 3 | 11 | 82 : 18 |
| 6 | Pd(PPh3)4/none | PC 4 | 65 | 86 : 14 |
| 7 | Pd(PPh3)4/none | PC 5 fac-Ir(ppy)3 | 0 | — |
| 8 | PdCl2/PPh3 | PC 1 | 87 | 92 : 8 |
| 9 | PdCl2/PCy3 | PC 1 | 58 | 93 : 7 |
| 10 | PdCl2/Xphos | PC 1 | 63 | 92 : 8 |
| 11 | PdCl2/Xantphos | PC 1 | 52 | 86 : 14 |
| 12 b | PdCl2/PPh3 | PC 1 | 100(97) c | 92 : 8 |
| 13 d | PdCl2/PPh3 | PC 1 | 74 | 92 : 8 |
| 14 e | PdCl2/PPh3 | PC 1 | 11 | 73 : 27 |
aUnless otherwise noted, the reactions were carried out with 0.2 mmol scale.
bThe reaction was run for 48 h.
cThe isolated yield of corresponding methyl ester is indicated in the parentheses.
dEt3N was used instead of iPr2NEt.
eMe2NPh was used instead of iPr2NEt.
With the optimal conditions in hand, we subsequently investigated the generality of the present C–F carboxylation reaction (Scheme 2). Initially, various biaryl motifs were studied (2b–e). For substrates with electron-donating groups on the biphenyl backbone, the reaction tends to afford the desired product with decreased yield (2b). We surmise that this consequence may be ascribed to the relatively low reduction potential of the corresponding fluorovinyl radical intermediate, which in turn resulted in an attenuated interaction with either the Pd-catalyst or photocatalyst for the subsequent carboxylation process. By contrast, the electron-withdrawing group (OCF3) worked well in this reaction and furnished the product 2c in high yield, suggesting that an electron-deficient substrate may be preferred in this reaction. Extending the conjugation of the biaryl structure led to good yield and excellent stereoselectivity (2d). Replacing the biphenyl group with a relatively electron-rich heteroaryl-phenyl scaffold resulted in a significant decline in the reaction efficiency (2e) which is consistent with the aforementioned substitution effect. Furthermore, substrates possessing fused aromatics including naphthyl (2f and 2g), dibenzofuran (2h), benzothiophene (2i) and indole (2j) were all competent in this reaction, producing carboxylic acids in moderate to good yields. Remarkably, the nice compatibility of heterocyclic substrates underpins the synthetic potential of this methodology in the modification of complex molecules. Next, substrates bearing a single aromatic ring were examined. Specifically, a range of electron-withdrawing groups, such as fluoride, trifluoromethyl, amide, sulfone, ester and cyanide were all accommodated, leading to the formation of carboxylic acids (2k–r) in moderate to good yields. Importantly, in the case of using a substrate with a fluoroaryl motif, the reaction occurred uneventfully with Ar–F remaining intact (2k). It's worth mentioning that alkyldifluoroalkenes failed to couple with carbon dioxide, indicating the essentiality of aromatic conjugation for the carboxylation reaction (see ESI†). Pleasingly, gem-difluoroalkenes containing two substituents were amenable in the current transformation, albeit requiring a prolonged reaction time. A variety of functional groups are tolerated, such as long carbon chain (2t), alkene (2u), ether (2v), and carbamate (2x).
Scheme 2. Substrate scope. See ESI† for experimental details. Isolated yields are indicated.

Having demonstrated the scope of gem-difluoroalkenes for carboxylation, the relevant carbonyl electrophiles were also examined. Aromatic ketones were found to be amenable in the reaction, however, a defluorinative alkenylation product derived from further dehydration was obtained (Scheme 3, see Fig. S2† for the proposed mechanism). Interestingly, arylbromide was tolerated under the current conditions, implying a reaction pathway distinct from Iwasawa's Ar–Br/Cl carboxylation.8
Scheme 3. Addition to aromatic ketones.

To gain more insight with respect to the reaction mechanism, a set of control experiments were implemented, including the attempt with 1-bromo-1-fluoro-difluoroalkene (Scheme 4a) and reactions with a radical scavenger (Scheme 4b). As expected, the C–Br carboxylation did not occur with 1.0 equiv. of Pd(PPh3)4, suggesting that the traditional two-electron oxidative addition may not operate in the present catalytic system. Moreover, either adding TEMPO or BHT suppressed the reaction, indicating that a radical intermediate is involved in the catalytic cycle. The engagement of a fluoroalkenyl radical intermediate was further substantiated by the formation of hydrodefluorination product 5a with Hantzsch ester as the hydrogen donor (Scheme 4c). However, a markedly discrepant stereoselectivity was obtained, which implicates the configurational lability of the fluoroalkenyl radical intermediate and the different kinetic reactivity profile of such radical species compared with that of the fluoroalkenyl palladium intermediate.23 In order to exclude the carbanion-associated pathway, we performed the reaction in the presence of D2O under a nitrogen atmosphere (Scheme 4d). As expected, no deuterated product D1-5a was detected, thus ruling out the possibility of carboxylation occurring with the fluorovinyl carbanion intermediate. Additionally, the reduction potential of 1a was measured (Ered1/2 = –0.67 V vs. SCE), showing that the reduced form of PC 1 (E1/2[IrIII/IrII] = –1.37 V vs. SCE)24 could serve as the operative reductant for the single electron reduction of 1a (see ESI† for details). This result is also in accordance with Hashmi's report,11g hence tending to support the photoinduced C–F activation hypothesis.
Scheme 4. Control experiments.

Based on the experimental results and previous reports, a working model that accounts for the selective C–F carboxylation of gem-difluoroalkenes is tentatively proposed (Scheme 5). Single electron reduction of gem-difluoroalkenes with PC 1, which is generated via reductive quenching of excited PC* by iPr2NEt, occurs to generate fluoroalkenyl radical A. Further capture of the fluoroalkenyl radical by Pd(0) complex B produces a putative Pd(i) intermediate C, which subsequently undergoes CO2 coordination and migratory insertion to afford the carboxyl-Pd(i) complex E. The formation of carboxylate 2 arises from SET between PC* and E, whereby Pd(0) is regenerated as well.25 With regard to the Z/E selectivity, we speculate that under these metallaphotoredox conditions the combination of the fluorovinyl radical with the Pd-catalyst is a reversible process, thus enabling a facile interconversion of Z- and E-fluoroalkenyl-Pd, and it is the kinetically more favored carboxylation with E-fluoroalkenyl-Pd that ultimately leads to the observed stereoselectivity.
Scheme 5. Mechanistic proposal.

Conclusions
Through the photoredox/Pd dual catalysis, an efficient and selective C–F bond carboxylation of gem-difluoroalkenes was achieved. The methodology represents a rare example of catalytic C–F bond carboxylation as well as unprecedented reactivity of gem-difluoroalkenes to provide diverse classes of α-fluoroacrylic acids and 2-fluoro-1,3-diene derivatives, which are difficult to access using existing protocols. This method expands the boundaries for carboxylation of organic (pseudo)halides, allowing the carboxylation reaction to proceed at the inherently inert C–F site. We envision that by using a similar dual catalysis strategy the carboxylation of otherwise challenging substrates could be achieved successfully.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support of the “Thousand Talents Plan” Youth Program, the “Jiangsu Specially-Appointed Professor Plan”, the National Natural Science Foundation of China (21801131 and 21871138), and the Natural Science Foundation of Jiangsu Province (BK20170992 and BK20170984).
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc01336a
References
- (a) Börjesson M., Moragas T., Gallego D., Martin R. ACS Catal. 2016;6:6739–6749. doi: 10.1021/acscatal.6b02124. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tortajada A., Juliá-Hernández F., Bçrjesson M., Moragas T., Martin R. Angew. Chem., Int. Ed. 2018;57:15948–15982. doi: 10.1002/anie.201803186. [DOI] [PubMed] [Google Scholar]; (c) Artz J., Müller T. E., Thenert K. Chem. Rev. 2018;118:434–504. doi: 10.1021/acs.chemrev.7b00435. [DOI] [PubMed] [Google Scholar]
- For selected examples, see: ; (a) Osakada K., Sato R., Yamamoto T. Organometallics. 1994;13:4645–4647. [Google Scholar]; (b) Correa A., Martin R. J. Am. Chem. Soc. 2009;131:15974–15975. doi: 10.1021/ja905264a. [DOI] [PubMed] [Google Scholar]; (c) Tran-Vu H., Daugulis O. ACS Catal. 2013;3:2417–2420. doi: 10.1021/cs400443p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lee K. N., Lei Z., Nagai M.-Y. J. Am. Chem. Soc. 2017;139:5003–5006. doi: 10.1021/jacs.7b01373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Rebih F., Andreini M., Moncomble A., Harrison-Marchand A., Maddaluno J., Durandetti M. Chem.–Eur. J. 2016;22:3758–3763. doi: 10.1002/chem.201503926. [DOI] [PubMed] [Google Scholar]; (f) Correa A., León T., Martin R. J. Am. Chem. Soc. 2014;136:1062–1069. doi: 10.1021/ja410883p. [DOI] [PubMed] [Google Scholar]
- (a) Kiplinger J. L., Richmond T. G., Osterberg C. E. Chem. Rev. 1994;94:373–431. [Google Scholar]; (b) Amii H., Uneyama K. Chem. Rev. 2009;109:2119–2183. doi: 10.1021/cr800388c. [DOI] [PubMed] [Google Scholar]; (c) Whittlesey M. K., Peris E. ACS Catal. 2014;4:3152–3159. [Google Scholar]
- (a) Gillis E. P., Eastman K. J., Hill M. D., Donnelly D. J., Meanwell N. A. J. Med. Chem. 2015;58:8315–8359. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]; (b) Jaeger E. P., Jurs P. C., Stouch T. R. E. Eur. J. Med. Chem. 1993;28:275–290. [Google Scholar]; (c) Cracowski J.-M., Montembault V., Bosc D., Ameduri B., Odobel F., Fontaine L. J. Polym. Sci., Part A: Polym. Chem. 2009;47:1403. [Google Scholar]; (d) Handbook of Fluoropolymer Science and Technology, ed. D. W. Smith, S. T. Iacono and S. S. Iyer, John Wiley & Sons, Inc., Hoboken, NJ, 2014. [Google Scholar]; (e) Han Y. T., Yang S.-M., Wang X.-Y., Li F.-N. Arch. Pharmacal Res. 2014;37:440–451. doi: 10.1007/s12272-013-0228-x. [DOI] [PubMed] [Google Scholar]; (f) Vivier D., Soussia I. B., Rodrigues N., Lolignier S., Devilliers M., Chatelain F. C., Prival L., Chapuy E., Bourdier G., Bennis K., Lesage F., Eschalier A., Busserolles J., Ducki S. J. Med. Chem. 2017;60:1076–1088. doi: 10.1021/acs.jmedchem.6b01285. [DOI] [PubMed] [Google Scholar]
- (a) Ahrens T., Kohlmann J., Ahrens M., Braun T. Chem. Rev. 2015;115:931–972. doi: 10.1021/cr500257c. [DOI] [PubMed] [Google Scholar]; (b) Liu C., Zhang B. Chem. Rec. 2016;16:667–687. doi: 10.1002/tcr.201500248. [DOI] [PubMed] [Google Scholar]
- (a) Fujihara T., Nogi K., Xu T., Terao J., Tsuji Y. J. Am. Chem. Soc. 2012;134:9106–9109. doi: 10.1021/ja303514b. [DOI] [PubMed] [Google Scholar]; (b) Nogi K., Fujihara T., Terao J., Tsuji Y. J. Org. Chem. 2015;80:11618–11623. doi: 10.1021/acs.joc.5b02307. [DOI] [PubMed] [Google Scholar]
- (a) Börjesson M., Moragas T., Martin R. J. Am. Chem. Soc. 2016;138:7504–7507. doi: 10.1021/jacs.6b04088. [DOI] [PubMed] [Google Scholar]; (b) Moragas T., Gaydou M., Martin R. Angew. Chem., Int. Ed. 2016;55:5053–5057. doi: 10.1002/anie.201600697. [DOI] [PubMed] [Google Scholar]; (c) Wang X., Liu Y., Martin R. J. Am. Chem. Soc. 2015;137:6476–6479. doi: 10.1021/jacs.5b03340. [DOI] [PubMed] [Google Scholar]; (d) Moragas T., Cornella J., Martin R. J. Am. Chem. Soc. 2014;136:17702–17705. doi: 10.1021/ja509077a. [DOI] [PubMed] [Google Scholar]; (e) Liu Y., Cornella J., Martin R. J. Am. Chem. Soc. 2014;136:11212–11215. doi: 10.1021/ja5064586. [DOI] [PubMed] [Google Scholar]
- Shimomaki K., Murata K., Martin R., Iwasawa N. J. Am. Chem. Soc. 2017;139:9467–9470. doi: 10.1021/jacs.7b04838. [DOI] [PubMed] [Google Scholar]
- Meng Q.-Y., Wang S., König B. Angew. Chem., Int. Ed. 2017;56:13426–13430. doi: 10.1002/anie.201706724. [DOI] [PubMed] [Google Scholar]
- (a) Beck C. M., Park Y.-J., Crabtree R. H. Chem. Commun. 1998:693–694. [Google Scholar]; (b) Gilman H., Soddy T. S. J. Org. Chem. 1957;22:1121–1122. [Google Scholar]; (c) Martinet P., Sauvêtre R., Nonnant J.-F. J. Organomet. Chem. 1989;367:1–10. [Google Scholar]; (d) Yamauchi Y., Fukuhara T., Hara S., Senboku H. Synlett. 2008;3:438–442. [Google Scholar]; (e) Yus M., Herrera R. P., Guijarro A. Tetrahedron Lett. 2003;44:5025–5027. [Google Scholar]
- (a) Senaweera S. M., Singh A., Weaver J. D. J. Am. Chem. Soc. 2014;136:3002–3005. doi: 10.1021/ja500031m. [DOI] [PubMed] [Google Scholar]; (b) Senaweera S., Weaver J. D. J. Am. Chem. Soc. 2016;138:2520–2523. doi: 10.1021/jacs.5b13450. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Khaled M. B., Mokadem R. K. E., Weaver J. D. J. Am. Chem. Soc. 2017;139:13092–13101. doi: 10.1021/jacs.7b06847. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; (d) Singh A., Kubik J. J., Weaver J. D. Chem. Sci. 2015;6:7206–7212. doi: 10.1039/c5sc03013g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Singh A., Fennell C. J., Weaver J. D. Chem. Sci. 2016;7:6796–6802. doi: 10.1039/c6sc02422j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Priya S., Weaver J. D. J. Am. Chem. Soc. 2018;140:16020–16025. doi: 10.1021/jacs.8b09156. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Xie J., Yu J., Rudolph M., Rominger F., Hashmi A. S. K. Angew. Chem., Int. Ed. 2016;55:9416–9421. doi: 10.1002/anie.201602347. [DOI] [PubMed] [Google Scholar]; (h) Xie J., Rudolph M., Rominger F., Hashmi A. S. K. Angew. Chem., Int. Ed. 2017;56:7266–7270. doi: 10.1002/anie.201700135. [DOI] [PubMed] [Google Scholar]
- For selected reviews on photoredox and transition-metal dual catalysis, see: ; (a) Levin M. D., Kim S., Toste F. D. ACS Cent. Sci. 2016;2:293–301. doi: 10.1021/acscentsci.6b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Goddard J.-P., Ollivier C., Fensterbank L. Acc. Chem. Res. 2016;49:1924–1936. doi: 10.1021/acs.accounts.6b00288. [DOI] [PubMed] [Google Scholar]; (c) Skubi K. L., Blum T. R., Yoon T. P. Chem. Rev. 2016;116:10035–10074. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hopkinson M. N., Tlahuext-Aca A., Glorius F. Acc. Chem. Res. 2016;49:2261–2272. doi: 10.1021/acs.accounts.6b00351. [DOI] [PubMed] [Google Scholar]; (e) Twilton J., Le C., Zhang P., Shaw M. H., Evans R. W., MacMillan D. W. C. Nat. Rev. Chem. 2017;1:0052. [Google Scholar]
- (a) Tian P., Feng C., Loh T.-P. Nat. Commun. 2015;6:7472–7478. doi: 10.1038/ncomms8472. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tian P., Wang C.-Q., Cai S.-H., Song S., Ye L., Feng C., Loh T.-P. J. Am. Chem. Soc. 2016;138:15869–15872. doi: 10.1021/jacs.6b11205. [DOI] [PubMed] [Google Scholar]; (c) Cai S.-H., Ye L., Wang D.-X., Wang Y.-Q., Lai L.-J., Zhu C., Feng C., Loh T.-P. Chem. Commun. 2017;53:8731–8734. doi: 10.1039/c7cc04131d. [DOI] [PubMed] [Google Scholar]; (d) Zhu C., Song S., Zhou L., Wang D.-X., Feng C., Loh T.-P. Chem. Commun. 2017;53:9482–9485. doi: 10.1039/c7cc04842d. [DOI] [PubMed] [Google Scholar]; (e) Tang H.-J., Lin L.-Z., Feng C., Loh T.-P. Angew. Chem., Int. Ed. 2017;56:9872–9876. doi: 10.1002/anie.201705321. [DOI] [PubMed] [Google Scholar]; (f) Zhou L., Zhu C., Bi P., Feng C. Chem. Sci. 2019;10:1144–1149. doi: 10.1039/c8sc04162h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Liu H., Ge L., Wang D.-X., Chen N., Feng C. Angew. Chem., Int. Ed. 2019;58:3918–3922. doi: 10.1002/anie.201814308. [DOI] [PubMed] [Google Scholar]
- For the synthesis of α-fluoroacrylic acids and derivatives, see: ; (a) Rousée K., Bouillon J.-P., Couve-Bonnaire S., Pannecoucke X. Org. Lett. 2016;18:540–543. doi: 10.1021/acs.orglett.5b03571. [DOI] [PubMed] [Google Scholar]; (b) Bouazzaoui O., Rousée K., Mulengi J. K., Pannecoucke X., Bouillon J.-P., Couve-Bonnaire S., Eur. J. Org. Chem., 2018. , 3705 –3715 , , and references cited therein . [Google Scholar]
- (a) Tellis J. C., Kelly C. B., Primer D. N., Jouffroy M., Patel N. R., Molander G. A. Acc. Chem. Res. 2016;49:1429–1439. doi: 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Matsui J. K., Gutiérrez-Bonet Á., Rotella M., Alam R., Gutierrez O., Molander G. A. Angew. Chem., Int. Ed. 2018;57:15847–15851. doi: 10.1002/anie.201809919. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Li J., Luo Y., Cheo H. W., Lan Y., Wu J. Chem. 2019;5:192–203. [Google Scholar]; (d) Meng Q.-Y., Wang S., Huff G. S., König B. J. Am. Chem. Soc. 2018;140:3198–3201. doi: 10.1021/jacs.7b13448. [DOI] [PubMed] [Google Scholar]
- (a) Cartwright K. C., Tunge J. A. ACS Catal. 2018;8:11801–11806. [Google Scholar]; (b) Hou J., Ee A., Feng W., Xu J.-H., Zhao Y., Wu J. J. Am. Chem. Soc. 2018;140:5257–5263. doi: 10.1021/jacs.8b01561. [DOI] [PubMed] [Google Scholar]; (c) Thullen S. M., Rovis T. J. Am. Chem. Soc. 2017;139:15504–15508. doi: 10.1021/jacs.7b09252. [DOI] [PubMed] [Google Scholar]
- (a) Kalyani D., McMurtrey K. B., Neufeldt S. R., Sanford M. S. J. Am. Chem. Soc. 2011;133:18566–18569. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Osawa M., Nagai H., Akit M. Dalton Trans. 2007:827–829. doi: 10.1039/b618007h. [DOI] [PubMed] [Google Scholar]; (c) Maestri G., Malacria M., Derat E. Chem. Commun. 2013;49:10424–10426. doi: 10.1039/c3cc46355a. [DOI] [PubMed] [Google Scholar]; (d) Zhou C., Li P., Zhu X., Wang L. Org. Lett. 2015;17:6198–6201. doi: 10.1021/acs.orglett.5b03192. [DOI] [PubMed] [Google Scholar]; (e) Cheng W.-M., Shang R., Yu H.-Z., Fu Y. Chem.–Eur. J. 2015;21:13191–13195. doi: 10.1002/chem.201502286. [DOI] [PubMed] [Google Scholar]
- The configuration of 2a is determined according to a similar structure to that reported in ref. 13a
- We appreciate one of the reviewers’ suggestion on the possible heterogeneous catalysis for this reaction as implied by the productive reaction with Pd/C as the catalyst. However, the lower yields under ligandless conditions also stress the important role of supporting ligands and demonstrate the homogeneous catalysisWe appreciate one of the reviewers’ suggestion on the possible heterogeneous catalysis for this reaction as implied by the productive reaction with Pd/C as the catalyst. However, the lower yields under ligandless conditions also stress the important role of supporting ligands and demonstrate the homogeneous catalysis as a more favorable modeas a more favorable mode
- (a) Parasram M., Gevorgyan V. Chem. Soc. Rev. 2017;46:6227–6240. doi: 10.1039/c7cs00226b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ratushnyy M., Parasram M., Wang Y., Gevorgyan V. Angew. Chem., Int. Ed. 2018;57:2712–2715. doi: 10.1002/anie.201712775. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chuentragool P., Yadagiri D., Morita T., Sarkar S., Parasram M., Wang Y., Gevorgyan V. Angew. Chem., Int. Ed. 2019;58:1794–1798. doi: 10.1002/anie.201812398. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhou W.-J., Cao G.-M., Shen G., Zhu X.-Y., Gui Y.-Y., Ye J.-H., Sun L., Liao L.-L., Li J., Yu D.-G. Angew. Chem., Int. Ed. 2017;56:15683–15687. doi: 10.1002/anie.201704513. [DOI] [PubMed] [Google Scholar]
- (a) Yeung C. S. Angew. Chem., Int. Ed. 2019;58:5492–5502. doi: 10.1002/anie.201806285. [DOI] [PubMed] [Google Scholar]; (b) Tan F., Yin G. Chin. J. Chem. 2018;36:545–554. [Google Scholar]; (c) Seo H., Katcher M. H., Jamison T. F. Nat. Chem. 2017;9:453–456. doi: 10.1038/nchem.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Seo H., Liu A., Jamison T. F. J. Am. Chem. Soc. 2017;139:13969–13972. doi: 10.1021/jacs.7b05942. [DOI] [PubMed] [Google Scholar]
- (a) Hou J., Ee A., Cao H., Ong H.-W., Xu J.-H., Wu J. Angew. Chem., Int. Ed. 2018;57:17220–17224. doi: 10.1002/anie.201811266. [DOI] [PubMed] [Google Scholar]; (b) Fan X., Gong X., Ma M., Wang R., Walsh P. J. Nat. Commun. 2018;9:4936–4943. doi: 10.1038/s41467-018-07351-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yatham V. R., Shen Y., Martin R. Angew. Chem., Int. Ed. 2017;56:10915–10919. doi: 10.1002/anie.201706263. [DOI] [PubMed] [Google Scholar]; (d) Ju T., Fu Q., Ye J.-H., Zhang Z., Liao L.-L., Yan S.-S., Tian X.-Y., Luo S.-P., Li J., Yu D.-G. Angew. Chem., Int. Ed. 2018;57:13897–13901. doi: 10.1002/anie.201806874. [DOI] [PubMed] [Google Scholar]
- (a) Curran D. P., Porter N. A. and Giese B., Stereochemistry of Radical Reactions: Concepts, Guidelines and Synthetic Applications, Wiley-VCH Verlag, Weinheim, Germany, 1996. [Google Scholar]; (b) Wille U. Chem. Rev. 2013;113:813–853. doi: 10.1021/cr100359d. [DOI] [PubMed] [Google Scholar]
- Lowry M. S., Goldsmith J. I., Slinker J. D., Rohl R., Pascal R. A., Malliaras G. G., Bernhard S. Chem. Mater. 2005;17:5712–5719. [Google Scholar]
- We acknowledge one of the reviewers for the suggestion on the possible Pd(i)/Pd(ii) catalytic cycle. For detailed discussion, see ESI.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.