

Abstract
Air pollution is a heterogeneous environmental toxicant that impacts humans throughout their life. We introduce Caenorhabditis elegans as a valuable air pollution model with its short lifespan, medium-throughput capabilities, and highly conserved biological pathways that impact healthspan. We exposed developmental and adult life stages of C. elegans to airborne nano-sized particulate matter (nPM) produced by traffic emissions and measured biological and molecular endpoints that changed in response. Acute nPM did not cause lethality in C. elegans, but short-term exposure during larval stage 1 caused delayed development. Gene expression responses to nPM exposure overlapped with responses of mouse and cell culture models of nPM exposure in previous studies. We showed further that the skn-1/Nrf2 antioxidant response has a role in the development and hormetic effects of nPM. This study introduces the worm as a new resource and complementary model for mouse and cultured cell systems to study air pollution toxicity across the lifespan.
Keywords: Caenorhabditis elegans, Air pollution, nPM, skn-1, Development
Epidemiological studies show that air pollution is associated with multiple chronic health hazards of older age including Alzheimer’s disease (AD), ischemic heart disease and stroke, lung cancer, and chronic obstructive pulmonary diseases—all of which decrease life expectancy (1–5). Understanding the underlying mechanisms between air pollution and these diseases requires modeling both air pollution and the resulting biological responses.
Traffic-related air pollution (TRAP) particles are a complex environmental toxicant consisting of a variety of inflammogens and toxicants derived from vast heterogeneous sources. It is estimated that ambient particulate matter (PM < 2.5 micron diameter) contributes to 3–4 million deaths (around 7.6% of total global death) and 103.1 million (4.2%) global disability-adjusted life year annually (1). The mortality number is apart from 3.5 million deaths attributed to indoor air pollution such as solid fuels for cooking and heating (6). It is essential that gerontological research understand the effects of environmental toxicants over organismal lifespan, the association with common aging phenotypes, and also identify the biomarkers that can predict these effects (reviewed in (7)).
The nano-sized subfraction of TRAP (nano-sized particulate matter [nPM]) has consistent toxic effects in rodent and cell models (8). Biochemical and cell assays of air pollution toxicity, while widely used, are not good predictors of in vivo responses for multicellular organisms (eg, Dithiothreitol [DTT], ascorbic acid [AA]-glutathione [GSH], and MTT (9). Exposure of mouse models to reaerosolized nPM results diverse systemic and organ-specific localized effects that involve different biological networks such as oxidative stress and antioxidant responses, innate immunity, and the nervous system (10–13). These responses were dependent on the dosage of PM samples and the developmental stage of the exposure. Mouse models are a valuable tool to study air pollution toxicity, but they are limited as a biological model; low reproduction yield, long lifespan, expense, and ethical considerations in these animals can reduce the feasibility of this model and also the statistical power of any experiments.
Caenorhabditis elegans is a valuable model for TRAP toxicology with a potential for much higher throughput than rodents (14). Humans and worms share several basic physiological and stress response processes with homologs in most human genes (60%–80%), including multiple signal transduction pathways (15). Easy maintenance, large scale production, small size, body transparency, full genomic characterization, complete cell lineage map, and mutant libraries make the worm an ideal model for gene network and environment interactions (16). While there are limitations such as lack of specific organ equivalents, a smaller immune response system, and large differences in overall lifespan, C. elegans allows medium-throughput whole organism-level assays with multiple endpoints (eg, development, reproduction, feeding, lifespan, locomotion) (15). Worms have been used to assess the toxicity of terrestrial environmental samples (eg, soils, sludge, river sediments (17,18), pesticides (eg, Glyphosate, Paraquat, Endosulfan and Dichlorvos for neurotoxicity, DNA damage, sterility and embryonic lethality) (19), metal toxicity (eg, Ag, Cd, Pb, Fe), lifespan, fertility, growth (20), nanoparticles (21), drugs (22), toxins (eg, nicotine) (23–25), as well as other bioreactive molecules including NaAsO2, NaF, caffeine, and DMSO (26). While airborne bacteria have been tested in C. elegans (17), its responses to TRAP have not been studied. Considering the usefulness of C. elegans in diverse toxicology models, this study introduces C. elegans as a multicellular model organism for air pollution toxicity.
Our prior mouse and cell culture studies show that nPM can induce oxidative stress, systemic inflammation, and selective neuroinflammation in different brain regions as well as exacerbate amyloidogenesis processes in AD mouse models (5,10–13). We also noticed that there is an interaction between age and air pollution-mediated toxicity (10). Here we examined different nPM dosages, as well as developmental and lifespan effects in C. elegans to evaluate the similarities of this model to higher organisms in several cell survival pathways and Alzheimer Aβ-related genes. In contemplating the response of cytoprotective transcription factor SKN-1/Nrf2 in C. elegans to nPM, we also investigated the association of SKN-1 in an nPM-mediated developmental delay phenotype as well as changes in lifespan.
Methods
C. elegans Strain Maintenance
Caenorhabditis elegans were maintained at 20°C unless otherwise noted. Strains used were Bristol N2 (wild type), LG335 skn-1(zu135)/nT1[qIs51(myo-2::GFP;pes-10::GFP;F22B7.9::GFP)], and CL2166 dvIs19 [(pAF15)gst-4p::GFP::NLS] III. Some strains were provided by the CGC, funded by NIH Office of Research Infrastructure Programs (P40 OD010440). LG335 was a gift from the Leonard Guarente laboratory. Escherichia coli strain OP50 was used for all non-RNAi experiments and for general C. elegans maintenance. For synchronization prior to plating, gravid adults were treated with a solution of bleach and hypochlorite to harvest eggs; then, eggs were washed and rocked overnight in M9 solution, allowing the hatching and L1 arrest/synchronization (27).
Airborne Nano-Sized Particle Collection
Ambient nano-sized particles (diameter < 0.18 µm) were collected on 8 × 10 inch commercially available Zeflour PTFE filters (Pall Life Sciences, Ann Arbor, MI) using a High-Volume Ultrafine Particle (HVUP) Sampler (28) operating at a sampling flow rate of 400 liters/min flow rate at the Particle Instrumentation Unit (PIU) of University of Southern California located within 150 m downwind of a major freeway (I-110). Gravimetric mass (nPM mass concentration) was determined from pre- and post-weighing the filters under controlled temperature (22–24°C) and relative humidity (40%–50%) conditions. The filter-deposited nPM was eluted by sonication into ultrapure deionized (milli-Q) water (11) providing the concentrated slurry suspension used for these exposures. A portion of the aqueous suspension was chemically characterized. After acid digestion, samples were analyzed by high resolution inductively coupled plasma sector field mass spectrometry (SF-ICPMS). Another portion was analyzed using a Sievers 900 Total Organic Carbon Analyzer to determine total organic carbon (TOC) content (Supplementary Figure S1). This characterized suspension was used in all the experiments of this study.
RNA Interference
The E. coli strain HT115 (DE3), harboring either the empty L4440 plasmid (“Control RNAi”) or the skn-1 RNAi plasmid (dsRNA production of skn-1 sequence - Ahringer Library), was grown 16–18 hours at 37°C overnight. Cultures were seeded onto RNAi plates (normal NGM plates with 5 mM isopropyl-β-D-thiogalactoside (IPTG) and 50 μg/mL carbenicillin) and left overnight to generate dsRNA for experiments (maintained at 20°C during and after dsRNA generation). To optimize RNAi of skn-1 in offspring, P0 worms were plated on bacteria expressing skn-1 RNAi for 12, 18, 24, or 48 hours (Supplementary Figure S2); this was done to further reduce maternally-deposited skn-1 mRNA transcripts as well as to deposit skn-1 RNAi in the F1 generation prior to hatching. To control developmental and RNAi timing, these adults were placed in 15°C for the duration of skn-1 RNAi exposure. Adult exposure for 24 or 48 hours caused 90%–100% of dead F1 eggs. 18 hours was chosen as an acceptable 20%–50% egg death while decreasing skn-1 transcripts in living animals to ~70% (Supplementary Figure S2).
Optimization of Air Pollution Exposure Model
Two routes of nPM exposure (liquid or chronic exposure on growth medium plates [chronic exposure data not shown]) and duration (1, 2, 4, 8, 24 hours) were tested in larval stage 1 or 4 (L1 or L4) C. elegans, as well as solvents (M9 buffer or K medium). 1-hour exposure in diluted nPM at different dosages (1–200 μg nPM/mL) with M9 was chosen for further experiments as it was the fastest exposure in inducing phenotypes without compromising developmental timing. For treatments, worms were washed into an Eppendorf tube, brought to a known volume, and nPM was added to each tube to achieve the listed concentration. Worms were incubated at 20°C for all experiments, unless otherwise indicated. Worms were gently rocked 1 hour for even distribution of nPM. After exposure, worms were washed once before plating at time 0 in post-exposure time. Worms treated as “L1s” were treated immediately at the synchronized developmental stage (prior to feeding).
Size Analysis
Following exposure of L1 stage for 1 hour to nPM and re-plating, worms were incubated at 20°C for 72 hours. Body size was analyzed by area using ImageJ (average of width × length).
Pharyngeal Pumping
Worms were treated with 200 μg/mL nPM for 1 hour at L1 stage, then allowed to recover for 24 hours on regular agar food plates described above (~100–150 worms/plate). These plates were then directly placed under a 10× objective, allowed to settle for 1 minute to prevent changes in pumping due to plate movement/vibrations, and individual worms were then followed and recorded using the Movie Recorder in the ZEN 2 software at 6–8 ms exposure (Zeiss Axio Imager) for 10–15 seconds (making sure to keep the pharynx in frame). Slowed-down (~4×) movies were later analyzed for pharyngeal pumping rate. Worms without pharyngeal pumping (dead, lethargus) during recording were excluded (estimated <5% of total worms observed).
Fluorescent Analysis of gst-4p::GFP Labeled C. elegans
gst-4p::GFP animals were treated with the listed nPM concentration (0–200 μg/mL) for 1 hour and then imaged 4 or 24 hours after exposure. Recovered worms were then plated into 10–12 μL droplets of M9 solution directly onto slides and 10mM sodium azide was added. Once animals were paralyzed, cover slips were slowly placed at a diagonal angle on top of the M9-immersed animals to prevent bursting/flattening. Slides were then immediately taken to image whole worms using an oil-based 40× objective (Numerical aperture 1.4) with DIC and GFP filters (Zeiss Axio Imager, an epifluorescence microscope). Animals were imaged to have as much of the pharynx, germline, and intestine in focus as possible. As prolonged exposure to sodium azide can eventually induce gst-4:GFP (29), each plate was imaged within 20–30 minutes, and a new plate was made after this time if more worms were needed - both done to avoid inflation of the GFP signal. Later, fluorescence was measured (the “measure” command) on the imaged worms via ImageJ using the polygon selection tool surrounding both the whole animal and a section containing no worm(s) next to the animal (within the same image) as background (Whole worm autofluorescence background was not considered in the analysis). Corrected total cell fluorescence (CTCF) was then determined using Microsoft Excel. CTCF = Integrated Density – (Area of selected cell X Mean fluorescence of background readings).
Lifespan
C. elegans were treated with 50 μg/mL nPM at either L1 stage or at L1 and Day 2 adult (~96 hours). Synchronized L1 animals (after nPM exposure) were plated on control RNAi or skn-1 RNAi. Worms were re-plated each day of reproduction (Day 1–5) to avoid overcrowding from progeny. Individuals were checked each day for survival by prodding with a platinum wire to verify touch responsivity. Individual worms were censored from survival analysis for gross morbidity (bursting, vulval protrusion, crawling off plate, etc.).
Statistics
Size and gst-4p::GFP analysis results were compared using analysis of variance followed by Tukey post hoc with calculating multiplicity adjusted p values by Graphpad Prism 7. In qPCR time points and the experiments with only two groups, pairwise comparison was done with T distribution tests in Graphpad Prism 7. Lifespan was analyzed by log-rank and Wilcoxon tests in JMP software. Heatmap and dendrogram analysis was done in R by the CompexHeatmap package of Bioconductor using Euclidean distance matrix and complete linkage for clustering of the genes. Factorial models were made from size, gst-4p::GFP, and skn-1 RNAi data to analyze the significance of interaction terms using R.
Quantitative Real-Time PCR
L1 or L4 worms were exposed to 50 μg/mL nPM for 1 hour, and then worms were collected and washed in M9 solution at 0 (immediately after 1hr exposure), 1, 2, 4, or 8 hours after exposure. “Populations” in figure legends refer to biological replicates of 500–1,000 worms. Worms were pelleted at collection, 500 µl of TriZol reagent (Zymo) was added. RNA was extracted using TriZol method (11) with an additional step of treatment with DNase, lysis by T&C lysis and protein precipitation by MPC reagent (Epicenter, United States) for purification. RNA concentration and purity were assessed by O.D. 230, 260, and 280 by spectrophotometry before qRT-PCR with gene-specific primers in Supplementary Table S1. The data was normalized to ama-1 as housekeeping gene. For Knockdown experiment, skn-1 RNAi was administered to P0 adults, and F1 progeny were analyzed (as “RNA interference” above).
Results
Acute nPM Exposure Drives a Reduction in C. elegans Body Size
Different exposure methods were tested to determine the acute lethal dosage 50 (LD50) of nPM samples. Larval stage 1 and 4 (L1 and L4, chosen to represent near the beginning and end of development) wild type N2-Bristol worms (~100 per replicate; at least 3 replicates/group) were exposed to a range of nPM concentrations (0.5–200 μg/mL) for 1, 4, or 24 hours in liquid culture or on growth medium plates. Caenorhabditis elegans did not show any acute mortality at the highest dose in any of tested conditions during 3 days after exposure.
However, nPM exposure did affect development in a dose-dependent manner (Figure 1A). Exposure of L1 stage animals to nPM in M9 buffer (10, 50 and 200 μg/mL) for only 1 hour showed dose-dependent size reduction (13%–26% of the worm’s area) in Day 1. This size difference was not due to lack of food intake because pharyngeal pumping rates were not impaired (Figure 1B). Since nPM at 50 μg/mL caused the same size reduction as 200 μg/mL, the lower dose was used for further experiments.
Figure 1.

Acute nPM exposure of L1 worms results in adult worm size reduction. (A) Dose-dependent size changes (mean ± SEM) of Day 1 adult animal body area following 1 hour exposure to nPM in L1 stage (n = 3*15 to 22 /group). The average area of the control group was 0.06 mm2 with average length of 0.97 mm. (B) Pharyngeal pumping rate of animals (n = 3*~7/group) analyzed at 24 hours post-exposure of the L1 stage wildtype animals to 200 μg/mL nPM. Statistical tests include analysis of variance followed by Tukey post hoc with correction for multiple statistical hypothesis testing for size and t test for pharyngeal pumping. Adjusted p-values: <.05 (*), <.01(**), <.001(***), <.0001(****).
skn-1 is Activated in Response to nPM Exposure
Given the size impairment under nPM exposure, we were interested in the underpinning biological response—including any defense mechanisms that were activated. TRAP nPM is a heterogeneous environmental toxicant consisting of multiple elements and water-soluble organic carbons with high oxidant activity (Supplementary Figure S1). We chose to examine SKN-1/Nrf2, a cytoprotective transcription factor, due to its established antioxidant defense response (30) and role in development (31); specifically, we used the used the reporter gst-4p::GFP (GFP linked to Glutathione S-transferase, a target of SKN-1) as an estimate of activation of SKN-1 in animals treated with nPM. In the gst-4p::GFP strain, both L1 and L4 had dose-dependent increases in response as early as 4 hours post-exposure to nPM. By 18 hours post-exposure of L1 stage and 24 hours post-exposure of L4 stage, we observed reduction of GFP signal in most of the nPM exposed animals (all groups except for 10 μg/mL nPM exposed L1s) (Figure 2A, Supplementary Figure S3) suggesting a rapid return of reversal of the gene expression changes (perhaps compensatorily) post-SKN-1 activation.
Figure 2.

nPM-mediated SKN-1 response underlies developmental and lifespan effects of TRAP particulate matter. (A) Dose-response gst-4 protein responses to nPM in L1 and L4 gst-4p::GFP strain (n = 3 × 3 to 7/group). Representative images of L1 animals are presented in front of the graph. (B) Examining the interaction of different life stages on nPM (50 μg/mL) mediated hormesis effects (n = 3 × 52–101/group) and the role of skn-1 response in this process until 90% survival in wild type non-exposed animals (n = 2–3 × 47–53/group) (Summary statistics in Supplementary Table S2). L: Larval stage, D: Day adult. Multivariate model: GST-4 ~ nPM × Life stage × Time post-exposure, F = 21.36, adjusted R2 = 0.42, p < .0001****. (nPM):(Time) interaction, p = .003**, β = −0.0002 (C) Analysis of adult skn-1(zu135) mutants and wild type worms exposed to nPM during development (n = 3×13 to 27/group). Multivariate model: size ~ skn-1×nPM, F = 37.72, adjusted R2 = 0.35, p < .0001****, skn-1:nPM interaction: p = .03*, β = 0.24. Statistical tests: Survival data was analyzed by Log-Rank and Wilcoxon tests. Size: Univariate ANOVA followed by Tukey post hoc with correction for multiple statistical hypothesis testing and also Multivariate ANOVA for interaction was used for size and gst-4p::GFP results. Adjusted p-values: <.05 (*), <.01(**), <.001(***), <.0001(****). Please refer to Supplementary Table S3 for summary statistics of multivariate models.
To determine the role of SKN-1 activation in response to nPM stress, we down-regulated SKN-1 by RNAi before exposure to nPM. First, we tested the effects of short-term nPM exposure on lifespan at different life stages to try to understand if certain exposure windows were more important for any lifespan effects. Contrary to the negative survival effects observed in cell lines (9), dual nPM exposure (50 μg/mL) during development (L1 and L3), or triple nPM exposure (50 μg/mL) during reproduction stage (D1, D3 and D5) had a modest increase of the lifespan. Increase of exposure intervals by including of both developmental and reproduction stages removed this lifespan difference. We then examined if dual nPM exposure (50 μg/mL) would have the same outcome in skn-1 knockdown worms. While worms fed with control RNAi had a modest increase in mean lifespan (1.1-day, p = .015 Log-rank test, analysis in 90% of total lifespan), reducing skn-1 expression by RNAi ablated this increase in lifespan (Figure 2B, Supplementary Table S2). In general, skn-1 knockdown lead to decrease of total lifespan regardless of nPM exposure (Supplementary Figure S4). While our confidence in the observed lifespan change is limited by the number of animals used (n = 200–300 per replicate), we are confident in our finding that the high nPM concentrations utilized do not negatively impact C. elegans lifespan.
We investigated skn-1 knockout (skn-1(zu135)) for its response to nPM. While L1 exposure of wild type worms to 50 μg/mL nPM reduced Day 1 adult size by 20%, skn-1(zu135) worms did not show this decrease in size (Figure 2C). In both conditions, skn-1(zu135) worms were smaller overall compared to wild type.
Gene Expression Changes Induced by nPM Exposure
To understand molecular responses to nPM, selected mRNAs were measured by qPCR to analyze a panel of genes involved in C. elegans stress responses, development, vitellogenesis, innate immunity, amyloid processing, and TGF-β signaling pathway in L1 or L4 (Figure 3, Supplementary Figures S5–S8). In general, animals exposed at L1 had larger nPM-mediated mRNA changes, particularly in the first hours of exposure compared to L4 animals; moreover, the genes with largest nPM responses in L1 stage did not change in L4 stage exposed animals (eg, gst-4, daf-2, apl-1, sel-12).
Figure 3.

mRNA response to nPM varies with developmental stage. (A) Heatmap showing the mRNA changes of the selected genes in L1 and L4 stage animals exposed to 50 μg/mL nPM for 1 hour (n = 4–5 replicates, ~500 animals/replicate). RNA levels were followed for 8 hours post-exposure in L1 stage animals and 4 hours post-exposure in L4 stage animals. Responses are clustered based on Euclidian distance using complete linkage method. The heatmap is annotated with the time point, treatment, life stage, average of mRNA changes and the function of target genes. Significant changes are shown in the heatmap. (B) mRNA changes of Alzheimer homolog genes in C. elegans to nPM. The t-test was used to compare nPM and controls at each time. p-values: <.05 (*), <0.01(**), <0.001(***), <0.0001(****).
In L1 stage animals, 1-hour exposure to nPM immediately (0 hour post-exposure) changed the expression of several genes (Figure 3, Supplementary Figures S6–S8). Some of the observed changes at this time included heat shock responses (eg, hsp-4 [expression/control = 0.5-fold]), hormone receptor and development (eg, daf-2 [0.58], daf-12 [0.61]), vitellogenin (eg, vit-6 [0.64]) and Alzheimer amyloid processing genes (eg, apl-1 [0.64], lrp-1 [0.68] and sel-12 [0.55]) and TGF-β signaling pathway (eg, daf-7 [0.53]). At 1 hour post-exposure, the expression of most genes returned to baseline (pre-exposure) levels (exceptions were hsf-1 [0.6], hsp-4 [0.47] and daf-2 [0.82]). This shift continued and lead to upregulation of several genes at 2 hours post-exposure of L1 second in response to nPM. These responses consisted of skn-1 antioxidant target genes (eg, gst-4 [3.5]), metal response genes (eg, cdr-1 [5.7]), innate immune response (eg, abf-2 [2.8]) and amyloid processing genes (eg, sel-12 [2.9]). At 8 hours post-exposure, these changes returned to base line or were decreased compared to controls. Metal-sensing genes (eg, aip-1 [0.54]) and heat shock responses (eg, hsf-1 [0.46]) were among the genes with lower mRNA levels compared to controls at 8 hours post-exposure (Figure 3).
In L4 stage animals, nPM exposure did not significantly alter the expression of most genes examined. However, the gene expression followed the trend observed in L1 exposed animals with increased expression at 1 hour post-exposure and return to baseline at 4 hours post-exposure (Figure 3). nPM caused decrease of tol-1 mRNA (−0.86) at this time. These data suggest that transcriptional responses to nPM exposure are dependent on developmental stage.
The Role of skn-1 Transcriptional Activity in Response to nPM Exposure
Given the responsiveness of gst-4 to nPM, we further studied the role of SKN-1 activity in nPM-mediated toxicity. The expression of skn-1 and its downstream genes was targeted by skn-1 RNAi at L1, which blocked gst-4 response in first 2 hours after nPM exposure (Figure 4); gst-4 was the only gene with significant nPM and the associated interaction with skn-1 or time based on multivariate regression analysis. At 0 hour post-exposure, all SKN-1 targets were lower (skn-1 [0.7], gst-4 [0.58], gcs-1 [0.47] and ugt-11 [0.25]) versus negative control RNAi. Control animals had significant gst-4 [1.56] mRNA increase at 0 hour post-exposure to nPM, which was not observed for skn-1 RNAi. Contrary to skn-1-related genes after RNAi, nPM still induced some innate immune responses, for example, abf-2 [0.7 nPM/control at 0 hour post-exposure]. Moreover skn-1 RNAi slightly increased abf-2 response (+30%) at 2 hours post-exposure regardless of nPM. For the amyloid processing genes, skn-1 RNAi decreased sel-12 (−40%) with no further response to nPM at 0 hour post-exposure. At 2 hours post-exposure, nPM still decreased sel-12 mRNA in worms fed Control RNAi (−20%), but not when fed skn-1 RNAi. Taken together with the gst-4::GFP expression data, it appears that SKN-1 may have a role in mediating the transcriptional responses to nPM exposure.
Figure 4.

SKN-1 mediates some transcriptional responses of nPM. RNA responses to 1 hour nPM (50 μg/mL nPM) after skn-1 RNAi (n = 3–5 replicates, ~500 animals/replicate). Pairwise t-test was used to compare Control RNAi vehicle controls with skn-1 RNAi or nPM versus controls at each time. Multivariate coefficients of the gst-4 expression regressed on nPM, skn-1 RNAi, time and the associated interactions: gst-4 was the only gene with significant nPM and nPM:skn-1 interaction. p-values: <.05 (*), <0.01(**), <0.001(***), <0.0001(****).
Discussion
This study introduces C. elegans as a valuable short-lived model with potential for medium-throughput for genetic and toxicological studies of air pollution toxicity in humans. Most notable are the transcriptomic responses to nPM, a toxic subfraction of the ultrafine air pollution PM. We identified several conserved genes that show developmental sensitivity to nPM. While nPM does not cause lethality in adult C. elegans up to 200 μg/mL, developmental exposure to nPM does reduce worm size and alter mRNA levels, dependent on SKN-1. Despite the observed reduction in worm size under nPM exposure, the time from which an animal hatches from an egg to laying its first egg is roughly the same (egg to egg time) (Supplementary Figure S3). As such, development is not markedly perturbed. Genes responsive to nPM include skn-1, the C. elegans homolog of the mammalian NRF2, which has fundamental roles in air pollution toxicity (9). In addition, 50 μg/mL of nPM given twice early in the C. elegans life cycle caused a mild increase in lifespan (1.2 days in mean lifespan), again dependent on SKN-1.
Although C. elegans lacks a respiratory system, exposing animals to cigarette smoke in smoking chambers for 3 hours impaired intestinal bacterial clearance and gene expression changes (32); we observe similar gene expression changes following nPM exposure (eg, tir-1: -3.24, and hso-16.2: 2.14-fold change). In the insect Drosophila, chronic exposure to air pollution decreased lifespan by 50% (33), and a similar exposure chamber was also used for exposing C. elegans to cigarette smoke (32). Future studies using this methodology will allow comparisons between liquid and aerosolized delivery of nPM. It is possible that suspended particles could limit the toxic effects of nPM, and exposure to aerosolized particles should be considered for future experiments. Using lifelong liquid cultures to maintain worms could also be used to increase exposure of nPM.
While most collected nPM has a cell cytotoxicity 50 (CC50) of 10–20 μg/mL (9), our data show that there is no acute lethality to nPM even at concentrations as high as 200 μg/mL, which have no obvious counterpart in human exposure. This lack of toxicity at high levels of nPM suggests that C. elegans may be highly resistant against nPM, through a currently unknown mechanism. It is possible that molecular defense pathways, such as those we observe, are important for this resistance, or perhaps its outer cuticle simply provides a strong barrier against the toxicity. While 200 μg/mL is an extremely high dose compared to normal human exposure, it may be prudent in the future to test “mega-doses” (eg, >500 μg/mL) to arrive at a true LD50 in C. elegans. However, our collection protocol hits a technical limitation around 200 μg/mL, and even with those concentrations, the volume becomes limiting in that it is difficult to do many follow-up experiments using the same nPM batch (as we have done here), requiring constant re-characterization of different nPM batches. We are interested in attempting higher concentrations to find a true LD50 if we find a way to make it technically feasible in the future.
Our study examined the role of skn-1/Nrf2 in the observed physiological and molecular changes of nPM. SKN-1/Nrf2 is a transcription factor well-known for affecting both development (31) and cytoprotection/detoxification (30), among other genes. Exposure of young adult mice (3 months), but not older adult mice (18 months), to nPM induced Nrf2-dependent phase II detoxifying enzymes such as GCLC and GCLM in lung, liver and brain samples (34). These age differences further show the different susceptibility of life stages to air pollution toxicity. Down-regulation of skn-1/Nrf2 in C. elegans did not cause lethality in response to nPM. However, skn-1 knockout animals no longer had a reduction in size under nPM exposure (albeit were smaller overall). It is possible this developmental effect may involve sel-12, as sel-12 mRNA was decreased by skn-1 RNAi - similar to size reduction in these animals. In view of SKN-1 targets, nPM specifically activated gst-4 with minimal change in gcs-1 expression. Genome wide screening of the genes associated with skn-1-mediated detoxifying responses showed involvement of alternative pathways (eg, apb-2 and csn-2) in activation of gcs-1, regardless of skn-1 activation (35). Thus gcs-1 may be regulated independently of other Phase 2 genes. It is worth noting that while animals lacking skn-1 mRNA transcripts experience embryonic lethality, it is unclear whether progeny that do survive the transgenerational RNAi exposure are “sicker” than wild type, or if their survival indicates that enough skn-1 mRNA was around to reach a “survival threshold” where they are healthy enough (observationally, they do not seem “sick”). In addition, while we focused on the effects of skn-1, it is likely that other defense-associated transcription factors could be implicated in these gene expression changes or otherwise. While each individual experiment on skn-1-nPM interactions yielded modest, albeit significant changes, in sum, our data strongly suggests a role for skn-1 under nPM exposure.
We must consider if C. elegans is an appropriate model for human air pollution toxicity, because humans have more complex circulatory and immune systems and 100-fold longer lifespans. Our previous studies of air pollution toxicity in mouse and cell culture models nonetheless show overlap in oxidative stress, inflammation and amyloidogenesis pathways with these C. elegans findings (5). This study investigates our previous findings of air pollution toxicity based on prior mouse and cell culture models. Specifically, we used TRAP ultrafine PM (<0.2 µm dia.) in this study. Ultrafine PM may be more toxic compared to PM2.5 (36,37), but is not currently regulated or monitored by the United States Environmental Protection Agency.
Air pollution and oxidative stress are strongly associated (38–40). Previously, we showed only 5 hours exposure to air pollution sufficed for oxidative damage of membrane lipids in olfactory epithelium of exposed mice, assayed as 4-HNE (13). Oxidative stress (eg, gst-4) was a prominent response to nPM. The other consistent air pollution response in cell cultures, mice, and humans is inflammation. Microarray analysis of the primary mixed glial culture responses to nPM showed MyD88 dependent activation of TLR4, suggesting this pathway as an important upstream sensors of air pollution leading to inflammation (41). Exposed mice had higher levels of brain TNFα depending on the duration of nPM exposure (8,11). Humans exposed to diesel exhaust also show rapid systemic inflammatory responses (42). Notably, TLR-associated gene homologs (tol-1: TLR4, abf-2: downstream of tol-1) were among the earliest responses of C. elegans to nPM.
Air pollution is recently recognized as an environmental risk factor for Alzheimer’s disease (AD) and accelerated cognitive decline (5,43). In mouse and cell models, exposure to nPM increased production of the Aβ peptide (5). Caenorhabditis elegans also shows similar effects of air pollution in the responsiveness of its homologous amyloid processing genes to nPM. Several amyloid-related genes are associated with C. elegans development; for example, inactivation of apl-1/APP results in penetrant lethality during the L1 to L2 transition due to molting defects (44). On the other hand, overexpression of apl-1/APP results in penetrant L1 lethality, shortened body length and morphological, locomotive, and reproductive effects (44,45). sel-12/PSEN is one of the genes regulating apl-1/APP cleavage and trafficking (44). The expression of both apl-1/APP (0 hour: 0.64) and sel-12/PSEN (0 hour: 0.55, 2 hours: 2.88) is significantly changed in L1 stage following nPM exposure, suggesting the importance of apl-1 and sel-12 expression for the developmental responses to nPM. In addition, air pollution is linked with developmental changes in humans; for example, childhood obesity is associated with maternal exposure to ambient air polycyclic aromatic hydrocarbons during pregnancy (46). Moreover, we showed that prenatal exposure of mice to nPM alters neuronal differentiation and depression-like responses (12). Lack of sel-12 lead to elevated endoplasmic reticulum (ER) mitochondrial Ca2+ signaling and oxidative stress due to mitochondrial superoxide production (47). Thus, sel-12/PSEN might be a part of antioxidant response to nPM.
We initially hypothesized exposure to nPM might be fatal for C. elegans, especially during development. Instead, we found that short-term exposure to air pollution could cause a hormetic increase of lifespan. This effect is apparently dose dependent, since exposing the worms for multiple times during development and reproduction ablated this effect of air pollution. Hormesis, wherein small doses of a toxin or stress can induce increases in health and lifespan, is well established in eukaryotes (48), including C. elegans (49). While modest, our data suggest the hormetic effect of air pollution might be dependent on a functional skn-1 response, indicating its importance on later lifespan in addition to its importance in physical development. If exposure to nPM is administered more chronically, perhaps by air exposure chambers, longer or more constant doses may eventually cause decreases in lifespan.
Future studies will further identify signaling pathways in responses to air pollution and hopefully help to identify drug and/or diet intervention strategies to counteract these ambient toxins. Systematic study of the life stages of C. elegans will identify critical periods of vulnerability to acute and chronic air pollution exposure. Caenorhabditis elegans could productively complement rodent models for studies of air pollution toxicity throughout the life cycle.
Funding
We thank the CGC, funded by National Institutes of Health (P40 OD010440) for some strains, and WormBase. This work was supported by National Institutes of Health grants T32AG052374 (A.H.); R01AG051521 (C.E.F.); R21AG05020 (C.E.F.); Cure Alzheimer’s Fund, (C.E.F.); R01GM109028 (S.P.C.), F31AG051382 (H.M.D.) and T32AG000037 (H.M.D).
Author Contributions
Conceptualization, S.P.C., C.E.F., and T.E.M.; Methodology, A.H., H.M.D., and S.P.C.; nPM collection and characterization: F.S. and C.S.; Investigation, A.H., H.M.D., N.S., and S.P.C.; Writing, A.H., H.M.D., and S.P.C.; Supervision, Project Administration, and Funding Acquisition, S.P.C. and C.E.F.
Conflict of Interest
None reported.
Supplementary Material
{kind=link}
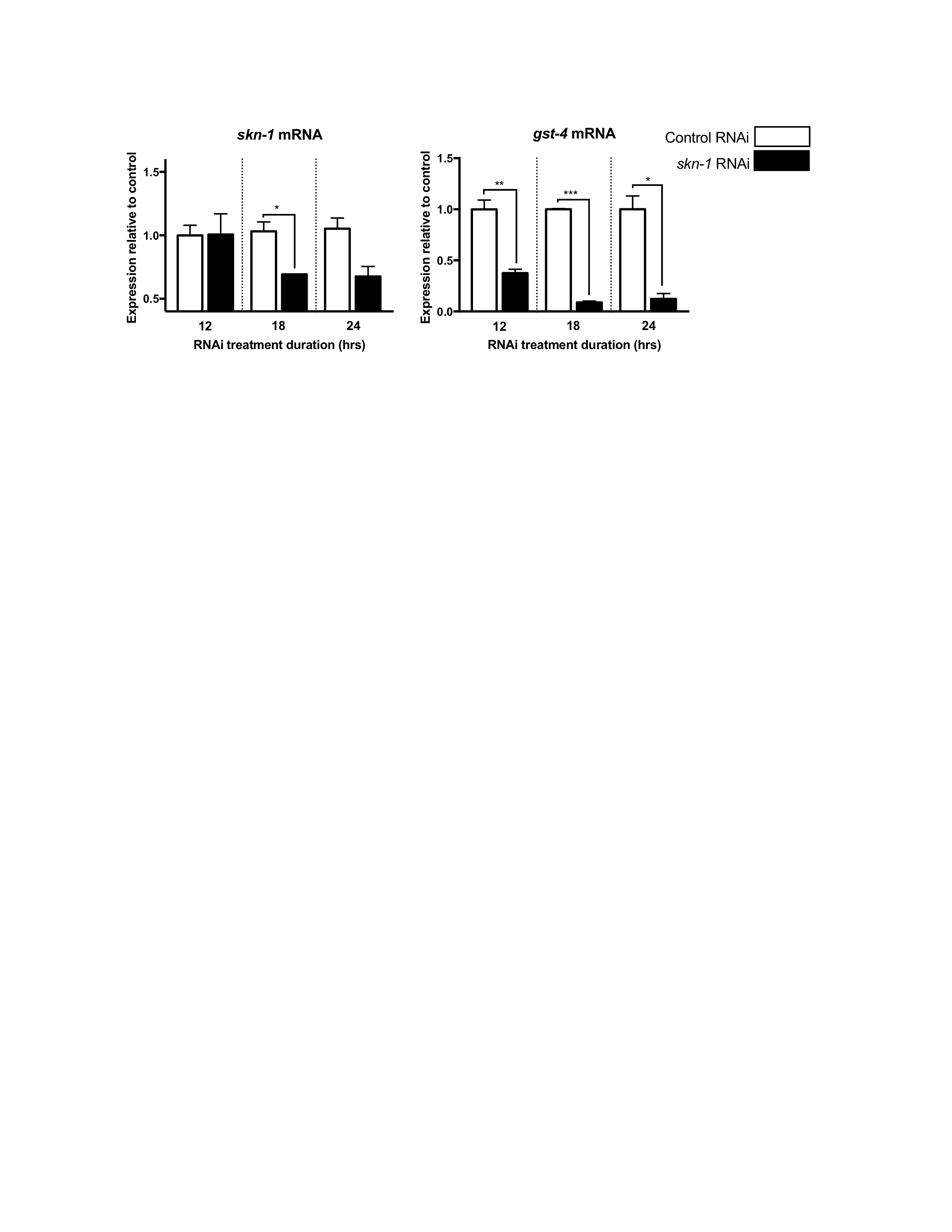{kind=link}
{kind=link}
{kind=link}
{kind=link}
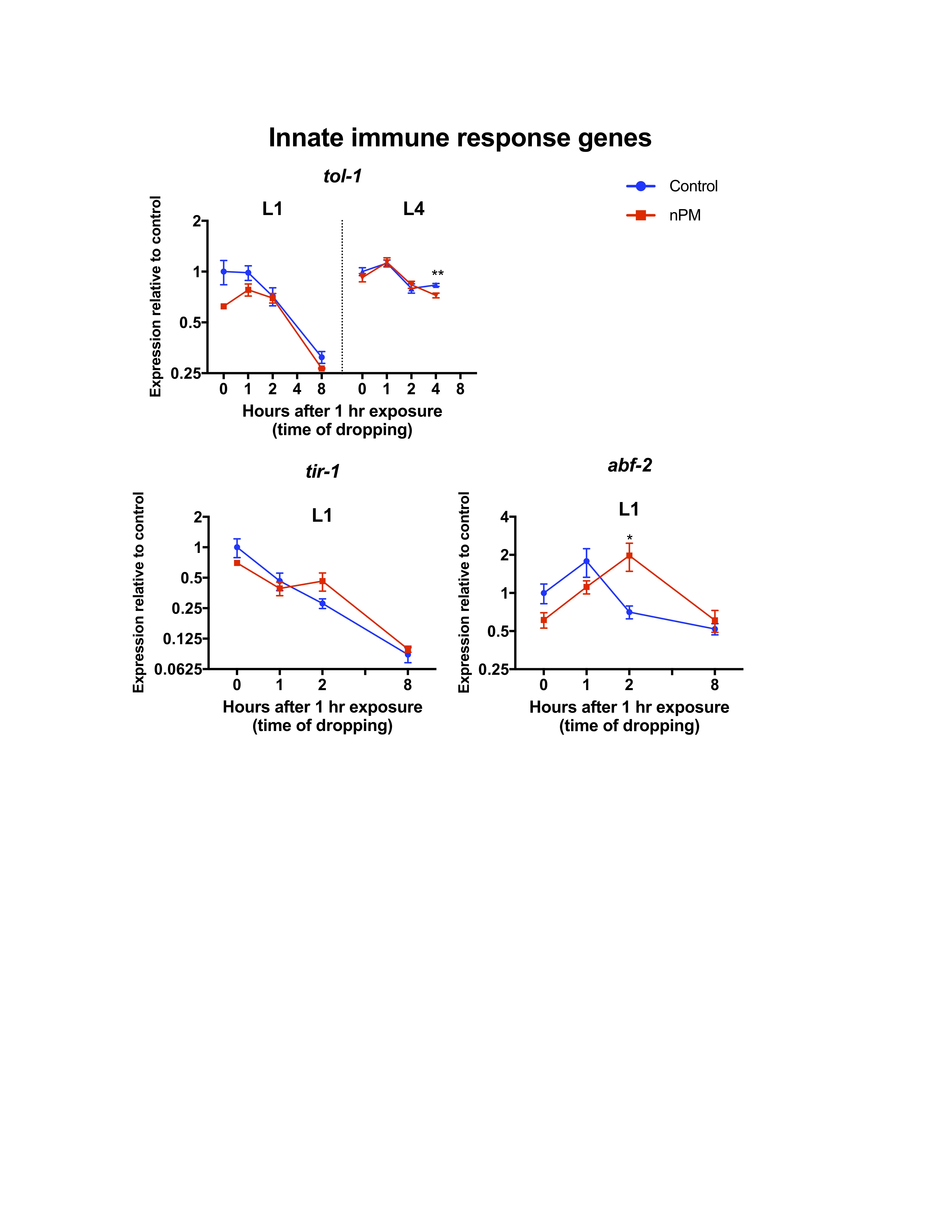{kind=link}
{kind=link}
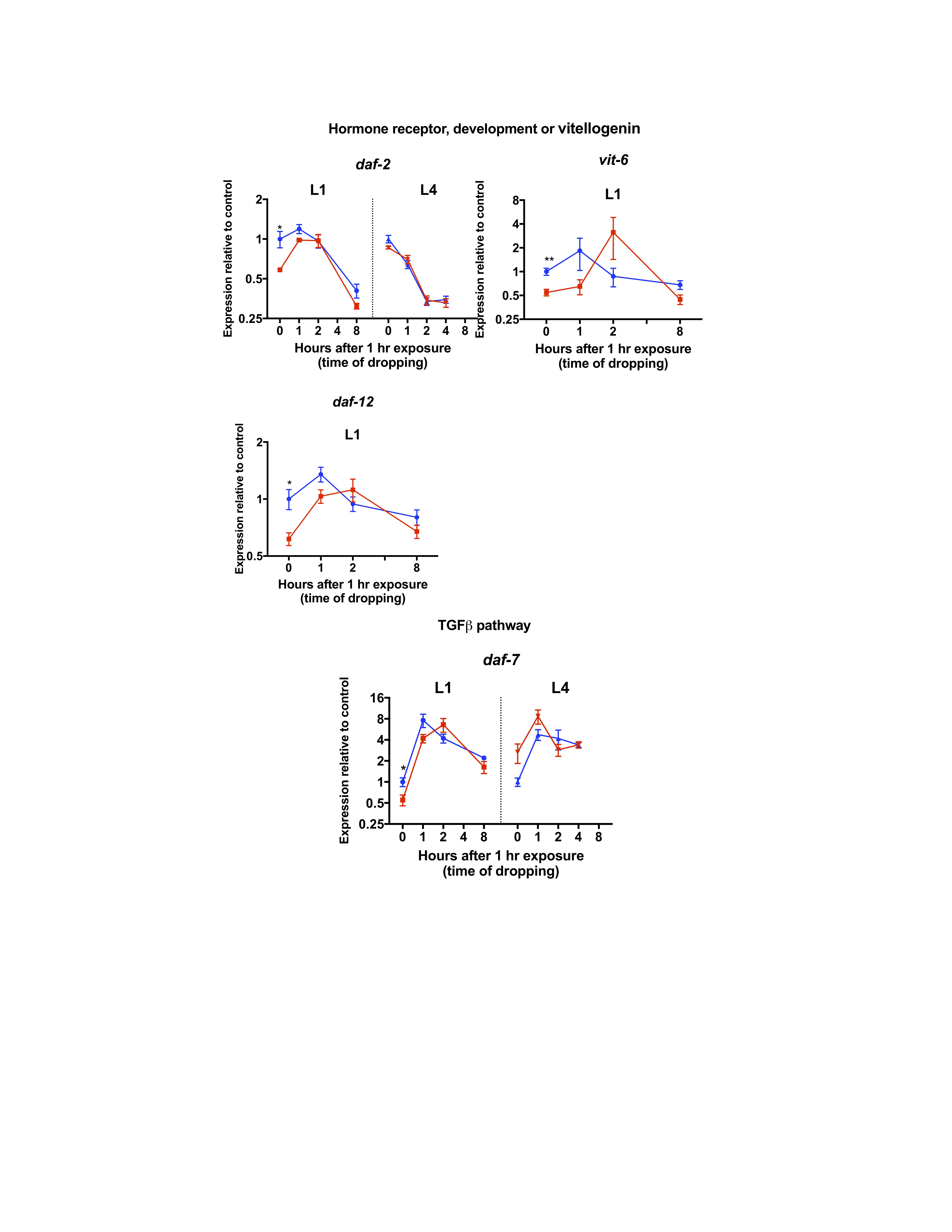{kind=link}
References
- 1. Burnett RT, Pope CA III, Ezzati M, et al. An integrated risk function for estimating the global burden of disease attributable to ambient fine particulate matter exposure. Environ Health Perspect. 2014;122:397–403. doi: 10.1289/ehp.1307049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Casanova R, Wang X, Reyes J, et al. A voxel-based morphometry study reveals local brain structural alterations associated with ambient fine particles in older women. Front Hum Neurosci. 2016;10:495. doi: 10.3389/fnhum.2016.00495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wolf K, Popp A, Schneider A, et al. ; KORA-Study Group. Association between long-term exposure to air pollution and biomarkers related to insulin resistance, subclinical inflammation, and adipokines. Diabetes. 2016;65:3314–3326. doi: 10.2337/db15-1567 [DOI] [PubMed] [Google Scholar]
- 4. Hamra GB, Guha N, Cohen A, et al. Outdoor particulate matter exposure and lung cancer: a systematic review and meta-analysis. Environ Health Perspect. 2014;122:906–911. doi: 10.1289/ehp/1408092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cacciottolo M, Wang X, Driscoll I, et al. Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl Psychiatry. 2017;7:e1022. doi: 10.1038/tp.2016.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lelieveld J, Evans JS, Fnais M, Giannadaki D, Pozzer A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature. 2015;525:367–371. doi: 10.1038/nature15371 [DOI] [PubMed] [Google Scholar]
- 7. Sorrentino JA, Sanoff HK, Sharpless NE. Defining the toxicology of aging. Trends Mol Med. 2014;20:375–384. doi: 10.1016/j.molmed.2014.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng H, Davis DA, Hasheminassab S, Sioutas C, Morgan TE, Finch CE. Urban traffic-derived nanoparticulate matter reduces neurite outgrowth via TNFα in vitro. J Neuroinflammation. 2016;13:19. doi: 10.1186/s12974-016-0480-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Forman HJ, Finch CE. A critical review of assays for hazardous components of air pollution. Free Radic Biol Med. 2018;117:202–217. doi: 10.1016/j.freeradbiomed.2018.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Woodward NC, Pakbin P, Saffari A, et al. Traffic-related air pollution impact on mouse brain accelerates myelin and neuritic aging changes with specificity for CA1 neurons. Neurobiol Aging. 2017;53:48–58. doi: 10.1016/j.neurobiolaging.2017.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morgan TE, Davis DA, Iwata N, et al. Glutamatergic neurons in rodent models respond to nanoscale particulate urban air pollutants in vivo and in vitro. Environ Health Perspect. 2011;119:1003–1009. doi: 10.1289/ehp.1002973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Davis DA, Bortolato M, Godar SC, et al. Prenatal exposure to urban air nanoparticles in mice causes altered neuronal differentiation and depression-like responses. PLoS One. 2013;8:e64128. doi: 10.1371/journal.pone.0064128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng H, Saffari A, Sioutas C, Forman HJ, Morgan TE, Finch CE. Nanoscale particulate matter from urban traffic rapidly induces oxidative stress and inflammation in olfactory epithelium with concomitant effects on brain. Environ Health Perspect. 2016;124:1537–1546. doi: 10.1289/EHP134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leung MC, Williams PL, Benedetto A, et al. Caenorhabditis elegans: an emerging model in biomedical and environmental toxicology. Toxicol Sci. 2008;106:5–28. doi: 10.1093/toxsci/kfn121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kaletta T, Hengartner MO. Finding function in novel targets: C. elegans as a model organism. Nat Rev Drug Discov. 2006;5:387–398. doi: 10.1038/nrd2031 [DOI] [PubMed] [Google Scholar]
- 16. Antoshechkin I, Sternberg PW. The versatile worm: genetic and genomic resources for Caenorhabditis elegans research. Nat Rev Genet. 2007;8:518–532. doi: 10.1038/nrg2105 [DOI] [PubMed] [Google Scholar]
- 17. Duclairoir Poc C, Groboillot A, Lesouhaitier O, Morin JP, Orange N, Feuilloley MJ. Caenorhabditis elegans: a model to monitor bacterial air quality. BMC Res Notes. 2011;4:503. doi: 10.1186/1756-0500-4-503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Anbalagan C, Lafayette I, Antoniou-Kourounioti M, et al. Transgenic nematodes as biosensors for metal stress in soil pore water samples. Ecotoxicology. 2012;21:439–455. doi: 10.1007/s10646-011-0804-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Anbalagan C, Lafayette I, Antoniou-Kourounioti M, et al. Use of transgenic GFP reporter strains of the nematode Caenorhabditis elegans to investigate the patterns of stress responses induced by pesticides and by organic extracts from agricultural soils. Ecotoxicology. 2013;22:72–85. doi: 10.1007/s10646-012-1004-2 [DOI] [PubMed] [Google Scholar]
- 20. Hunt PR, Olejnik N, Sprando RL. Toxicity ranking of heavy metals with screening method using adult Caenorhabditis elegans and propidium iodide replicates toxicity ranking in rat. Food Chem Toxicol. 2012;50:3280–3290. doi: 10.1016/j.fct.2012.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao Y, Wu Q, Tang M, Wang D. The in vivo underlying mechanism for recovery response formation in nano-titanium dioxide exposed Caenorhabditis elegans after transfer to the normal condition. Nanomedicine. 2014;10:89–98. doi: 10.1016/j.nano.2013.07.004 [DOI] [PubMed] [Google Scholar]
- 22. Liu S, Saul N, Pan B, Menzel R, Steinberg CE. The non-target organism Caenorhabditis elegans withstands the impact of sulfamethoxazole. Chemosphere. 2013;93:2373–2380. doi: 10.1016/j.chemosphere.2013.08.036 [DOI] [PubMed] [Google Scholar]
- 23. Saul N, Chakrabarti S, Stürzenbaum SR, Menzel R, Steinberg CE. Neurotoxic action of microcystin-LR is reflected in the transcriptional stress response of Caenorhabditis elegans. Chem Biol Interact. 2014;223:51–57. doi: 10.1016/j.cbi.2014.09.007 [DOI] [PubMed] [Google Scholar]
- 24. Smith MA Jr, Zhang Y, Polli JR, et al. Impacts of chronic low-level nicotine exposure on Caenorhabditis elegans reproduction: identification of novel gene targets. Reprod Toxicol. 2013;40:69–75. doi: 10.1016/j.reprotox.2013.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moore CE, Lein PJ, Puschner B. Microcystins alter chemotactic behavior in Caenorhabditis elegans by selectively targeting the AWA sensory neuron. Toxins (Basel). 2014;6:1813–1836. doi: 10.3390/toxins6061813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sprando RL, Olejnik N, Cinar HN, Ferguson M. A method to rank order water soluble compounds according to their toxicity using Caenorhabditis elegans, a complex object parametric analyzer and sorter, and axenic liquid media. Food Chem Toxicol. 2009;47:722–728. doi: 10.1016/j.fct.2009.01.007 [DOI] [PubMed] [Google Scholar]
- 27. Stiernagle T. Maintenance of C. elegans. WormBook. 2006;1–11. doi: 10.1895/wormbook.1.101.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Misra C, Kim S, Shen S, Sioutas C. Design and evaluation of a high-flow rate, very low pressure drop impactor for separation and collection of fine from ultrafine particles. J Aerosol Sci. 2002;33:736–752. [Google Scholar]
- 29. Shore DE, Carr CE, Ruvkun G. Induction of cytoprotective pathways is central to the extension of lifespan conferred by multiple longevity pathways. PLoS Genet. 2012;8:e1002792. doi: 10.1371/journal.pgen.1002792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. An JH, Blackwell TK. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 2003;17:1882–1893. doi: 10.1101/gad.1107803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bowerman B, Eaton BA, Priess JR. skn-1, a maternally expressed gene required to specify the fate of ventral blastomeres in the early C. elegans embryo. Cell. 1992;68:1061–1075. doi: 10.1016/0092-8674(92)90078-Q [DOI] [PubMed] [Google Scholar]
- 32. Green RM, Gally F, Keeney JG, et al. Impact of cigarette smoke exposure on innate immunity: a Caenorhabditis elegans model. PLoS One. 2009;4:e6860. doi: 10.1371/journal.pone.0006860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang X, Chen M, Zhong M, et al. Exposure to concentrated ambient PM2.5 shortens lifespan and induces inflammation-associated signaling and oxidative stress in Drosophila. Toxicol Sci. 2017;156:199–207. doi: 10.1093/toxsci/kfw240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang H, Liu H, Davies KJ, et al. Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments. Free Radic Biol Med. 2012;52:2038–2046. doi: 10.1016/j.freeradbiomed.2012.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Crook-McMahon HM, Oláhová M, Button EL, Winter JJ, Veal EA. Genome-wide screening identifies new genes required for stress-induced phase 2 detoxification gene expression in animals. BMC Biol. 2014;12:64. doi: 10.1186/s12915-014-0064-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang X, Staimer N, Gillen DL, et al. Associations of oxidative stress and inflammatory biomarkers with chemically-characterized air pollutant exposures in an elderly cohort. Environ Res. 2016;150:306–319. doi: 10.1016/j.envres.2016.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stone V, Miller MR, Clift MJD, et al. Nanomaterials versus ambient ultrafine particles: an opportunity to exchange toxicology knowledge. Environ Health Perspect. 2017;125:106002. doi: 10.1289/EHP424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brown DM, Donaldson K, Borm PJ, et al. Calcium and ROS-mediated activation of transcription factors and TNF-alpha cytokine gene expression in macrophages exposed to ultrafine particles. Am J Physiol Lung Cell Mol Physiol. 2004;286:L344–L353. doi: 10.1152/ajplung.00139.2003 [DOI] [PubMed] [Google Scholar]
- 39. Baulig A, Singh S, Marchand A, et al. Role of Paris PM(2.5) components in the pro-inflammatory response induced in airway epithelial cells. Toxicology. 2009;261:126–135. doi: 10.1016/j.tox.2009.05.007 [DOI] [PubMed] [Google Scholar]
- 40. Gilmour PS, Brown DM, Lindsay TG, Beswick PH, MacNee W, Donaldson K. Adverse health effects of PM10 particles: involvement of iron in generation of hydroxyl radical. Occup Environ Med. 1996;53:817–822. doi: 10.1136/oem.53.12.817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Woodward NC, Levine MC, Haghani A, et al. Toll-like receptor 4 in glial inflammatory responses to air pollution in vitro and in vivo. J Neuroinflammation. 2017;14:84. doi: 10.1186/s12974-017-0858-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Salvi S, Blomberg A, Rudell B, et al. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am J Respir Crit Care Med. 1999;159:702–709. doi: 10.1164/ajrccm.159.3.9709083 [DOI] [PubMed] [Google Scholar]
- 43. Zhang X, Chen X, Zhang X. The impact of exposure to air pollution on cognitive performance. Proc Natl Acad Sci USA. 2018;115:9193–9197. doi: 10.1073/pnas.1809474115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hornsten A, Lieberthal J, Fadia S, et al. APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability. Proc Natl Acad Sci USA. 2007;104:1971–1976. doi: 10.1073/pnas.0603997104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ewald CY, Raps DA, Li C. APL-1, the Alzheimer’s Amyloid precursor protein in Caenorhabditis elegans, modulates multiple metabolic pathways throughout development. Genetics. 2012;191:493–507. doi: 10.1534/genetics.112.138768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rundle A, Hoepner L, Hassoun A, et al. Association of childhood obesity with maternal exposure to ambient air polycyclic aromatic hydrocarbons during pregnancy. Am J Epidemiol. 2012;175:1163–1172. doi: 10.1093/aje/kwr455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sarasija S, Laboy JT, Ashkavand Z, Bonner J, Tang Y, Norman KR. Presenilin mutations deregulate mitochondrial Ca(2+) homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. Elife. 2018;7:1–30. doi: 10.7554/eLife.33052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gems D, Partridge L. Stress-response hormesis and aging: “that which does not kill us makes us stronger”. Cell Metab. 2008;7:200–203. doi: 10.1016/j.cmet.2008.01.001 [DOI] [PubMed] [Google Scholar]
- 49. Cypser JR, Johnson TE. Multiple stressors in Caenorhabditis elegans induce stress hormesis and extended longevity. J Gerontol A Biol Sci Med Sci. 2002;57:B109–B114. doi: 10.1093/gerona/57.3.B109 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.