

Abstract
Over 100 years ago, Lewy bodies and Lewy neurites were defined as a pathological hallmark of Parkinson’s disease. Eighty years later, α-synuclein was found to be the primary component of these inclusions. Emerging evidence suggests that α-synuclein pathology propagates across interconnected networks throughout the nervous system in a prion-like manner. Pathologic α-synuclein seeds aggregation of native α-synuclein, resulting in the formation of insoluble inclusions. These seeds can propagate within the neuron and to interconnected neurons, resulting in the spread of pathology throughout the brain. Here, we discuss how the findings that α-synuclein pathology spreads throughout the nervous system has revolutionized our understanding about Parkinson’s disease pathogenesis and resulted in the development of novel therapeutic strategies to halt disease progression.
Introduction
More than 10 million people worldwide suffer Parkinson’s disease (PD). This neurodegenerative disorder is classically defined by its motor symptoms including tremor at rest, rigidity, slowness of movement and postural instability. Research and observations presented during past decade have led to the realization, however, that PD also leads to several non-motor symptoms. They include, e.g., depression, cognitive decline, anxiety, sleep behavior disorder, constipation, hyposmia, etc, which collectively represent a significant source of morbidity. More recently, it has become apparent that the clinical diagnosis is preceded, typically for 5–15 years, by a “prodromal” phase when non-motor symptoms are evident, but the classical motor deficits have yet to appear. Pharmacologic therapies that counteract the loss of the nigrostriatal dopamine are initially effective at alleviating the motor symptoms. However, these treatments do little to reduce the non-motor symptoms and do not prevent the progression of the disease. Eventually, patients experience less effective alleviation of motor symptoms on dopaminergic therapies, and the therapeutic benefits are limited by the emergence of side effects that include drug-induced dyskinesias. In advanced disease, falls and the development of dementia are significant sources of caregiver-burden and they are frequently the reasons for institutionalization. Thus, a major unmet medical need is the prevention of disease progression.
Pathologically, PD is characterized by loss of dopamine neurons in the substantia nigra pars compacta and proteinaceous inclusions called Lewy bodies and Lewy neurites that are present in numerous brain regions, which, as is described in more detail later, engage an increasing number of brain regions as the disease progresses. These inclusions were first described by Friedrich Heinrich Lewy in 1912, but it was unclear what they were made up of, and therefore their importance in PD was not fully realized until 85 years later (Spillantini et al., 1997; Goedert et al., 2017). Thus, twenty years ago, the discoveries that Lewy bodies are composed mostly of the protein, α-synuclein, and that missense mutations in α-synuclein cause PD opened up the PD field to understanding underlying pathogenic mechanisms (Polymeropoulos et al., 1997; Spillantini et al., 1997; Spillantini et al., 1998). The use of α-synuclein antibodies has facilitated the identification of Lewy pathology not only in the substantia nigra pars compacta, where they are associated with the loss of dopamine neurons, but also in several other tissues. Indeed, Lewy bodies and Lewy neurites are also found in several other brain regions and in the peripheral nervous system, and this Lewy pathology likely contributes to the non-motor symptoms of PD. Based on the systematic analyses of numerous autopsies, it has also been suggested that Lewy pathology progressively involves more regions of the nervous system as the disease advances (Braak et al., 2003c). Some of these post-mortem findings indicate that the olfactory bulb and the enteric nervous system are among the first areas affected by Lewy pathology and that gradually regions that are anatomically interconnected become engaged. Around a decade ago, it was suggested that the gradual increase in Lewy pathology, during the progression of PD, is due to prion-like propagation of α-synuclein aggregates. In this review, we will discuss 1) evidence from human patients and animal models that pathogenic α-synuclein spreads in a prion-like manner, 2) how pathologic α-synuclein templates conversion of normal α-synuclein to form amyloid inclusions, 3) the potential triggers that cause α-synuclein to misfold, 4) the cellular mechanisms of propagation and 5) how the prion-like hypothesis informs novel therapeutic strategies.
Evidence for PD as a prion-like disease
Studies that examine Lewy pathology from postmortem brains from PD patients from early stages of the disease to those with advanced stages of the disease, as well as people not diagnosed with a clinical syndrome but who display so-called “incidental” Lewy Body Disease (ILBD), show that Lewy pathology is first found in the olfactory bulb and enteric nervous system (Braak et al., 2003c). In the mildest, “Stage 1” cases, Lewy bodies and Lewy neurites are apparent only in the dorsal IX/X motor nucleus and the anterior olfactory nucleus. At stage 2, Lewy bodies and Lewy neurites appear in the raphe nucleus and locus coeruleus. It is not until stage 3 that Lewy bodies and Lewy neurites are found in the substantia nigra pars compacta, particularly the posterolateral subnucleus. By stage 4, Lewy bodies and Lewy neurites are found in the CA2 region of the hippocampus and the temporal mesocortex. By stages 5 and 6, Lewy bodies and Lewy neurites localize to higher order primary sensory and motor cortices. Appearance of inclusions coincides with onset of symptoms; the olfactory bulb is one of the earliest sites of inclusion formation and impaired olfaction is one of the earliest symptoms of PD, appearance of inclusions in the substantia nigra pars compacta coincides with the onset of motor symptoms, and appearance of inclusions in the temporal cortex coincides with cognitive decline. The most relevant and important findings of this study are that Lewy bodies and Lewy neurites do not seem to appear in the nervous at random; their appearance follows a predictable topographical pattern across connected neuronal networks. There are two major possible explanations that currently are being considered. Either there is selective vulnerability to the underlying disease process in the different brain regions, explaining why they are affected at progressive stages of the disease. Alternatively, there is a transmissible agent that moves between the affected brain regions. In their original publications, Braak and colleagues favored the idea that there was a transmissible agent that was transported between anatomically connected regions and caused the aggregation of α-synuclein. They speculated if this might be a “neurotropic virus”, although in one paper they took the idea one step further and considered the possibility that the transmissible agent might even be “prion”(Braak et al., 2003a).
Another staging system was developed that, in addition to PD patients and ILBD cases, also included cases with Dementia with Lewy Bodies (DLB) and Alzheimer’s disease with sparse predominantly limbic Lewy bodies (ADLB) (Beach et al., 2009). This staging system was developed because when an attempt was made to classify all of these cases according to the Braak staging system or the DLB consortium staging system, up to 50% of cases could not be classified. According to this classification, Stage I is olfactory bulb predominant Lewy pathology. From here the staging diverges to Stage IIa, which is brain stem predominant pathology, or Stage IIb in which most pathology is found in the limbic system. In Stage III, the abundance of Lewy pathology in the brainstem and limbic region is equivalent, and in Stage IV, cortical areas are involved. It has been hypothesized if a Lewy pathology travels from Stage I to Stage IIa, PD develops, and if Lewy pathology progresses from Stage I to Stage IIb, DLB develops (Beach et al., 2009).
Overall, both of the aforementioned staging systems demonstrate that Lewy pathology appears in a predictable topographical manner across connected neuronal networks. It is important to consider that it is possible that not all synucleinopathies initiate in either the olfactory bulb or vagal nerve. There may be neuron intrinsic (so called “cell-autonomous”) mechanisms within brain regions that cause α-synuclein to convert from a normal to abnormal, pathologic conformation. The observation that the affected brain regions are connected by long neural pathways (in particular unmyelinated fibers), as opposed to the nearest cells, makes it a compelling notion that the disease process spreads along axons. That said, not until the most recent decade has robust evidence emerged from several laboratory studies showing that misfolded variants of α-synuclein may indeed be the spreading agent.
Evidence for PD as a prion-like disease
An important advance in the understanding of PD pathogenesis and mechanisms that might contribute to disease progression is the emergence of evidence that α-synuclein aggregates can spread from one cell to another in prion-like manner. According to the prion hypothesis, small amounts of fibrillar α-synuclein act as seeds which can trigger the conversion of normal, soluble α-synuclein into insoluble α-synuclein Lewy bodies and Lewy neurites. These seeds can be released from neurons and taken up by neighboring neurons, thus inducing the spread of pathologic α-synuclein throughout the nervous system. The first clinical evidence to support this was that α-synuclein was found in human cerebrospinal fluid, suggesting that it can be released into the extracellular space (El-Agnaf et al., 2003). In addition, studies on fetal nigral grafts placed in the striatum of PD patients, who died over one decade after the surgery, showed that a subset of the pigmented neurons inside the grafts contained Lewy bodies (Kordower et al., 2008a; Kordower et al., 2008b; Li et al., 2008; Li et al., 2010; Kurowska et al., 2011). When these findings were reported in 2008, it was proposed that the grafted neurons developed Lewy bodies as a consequence of prion-like transfer of misfolded α-synuclein originating in the host brains (Brundin et al., 2008). Notably, at the time of death, over two decades had passed since the patients had been clinically diagnosed with PD, and therefore the synucleinopathy was already widespread in the brains of the graft recipients.
In vitro cell culture models support that α-synuclein pathology can spread in a prion-like manner. It was found that α-synuclein is secreted form cultured neurons into the surrounding medium (El-Agnaf et al., 2003; Lee et al., 2005). Further studies characterized some of the regulation of α-synculein release from cultured neurons, and went on to suggest that such extracellular α-synuclein might be taken up by surrounding neurons and trigger the activation of glia (Lee et al., 2010). However, these studies did not examine the conformation of α-synuclein released from the neurons which is critical, because it is likely the beta-sheet conformation that acts as a seed to initiate conversion of normal α-synuclein to form abnormal inclusions. The first evidence that amyloid fibrils of α-synuclein can seed corruption of normal α-synuclein to form phosphorylated, insoluble inclusions came from studies in which fibrils generated from recombinant protein were added to HEK293 or SH-SY5Y cells stably overexpressing α-synuclein (Luk et al., 2009; Nonaka et al., 2010). These findings provided a critical proof-of-concept that pathogenic templating of α-synuclein could occur. However, seeding in the cultured cells required the use of a transfection reagent (e.g. cationic lipid, Bioporter) to introduce the fibrils to the cells and induce efficient seeding. Therefore, it was surprising when a subsequent study demonstrated that fibrils can simply be added to primary neurons in a dish without any reagent and that these fibrils induced the endogenous α-synuclein to form inclusions and cause neuronal dysfunction (Volpicelli-Daley et al., 2011) (Fig. 1). This study in cultured primary neurons was the first demonstration that fibrillar seeds can corrupt endogenous proteins. In addition, the findings suggest that concentrated α-synuclein in neuronal presynaptic terminals is required for an efficient seeding to occur. This study also demonstrated when fibrils were to neurons from α-synuclein knockout mice, no pathology or phenotypes were triggered indicating, consistent with the prion-like hypothesis, that endogenous α-synuclein needs to be present for the propagation and seeding events to take place.
Fig. 17.1.
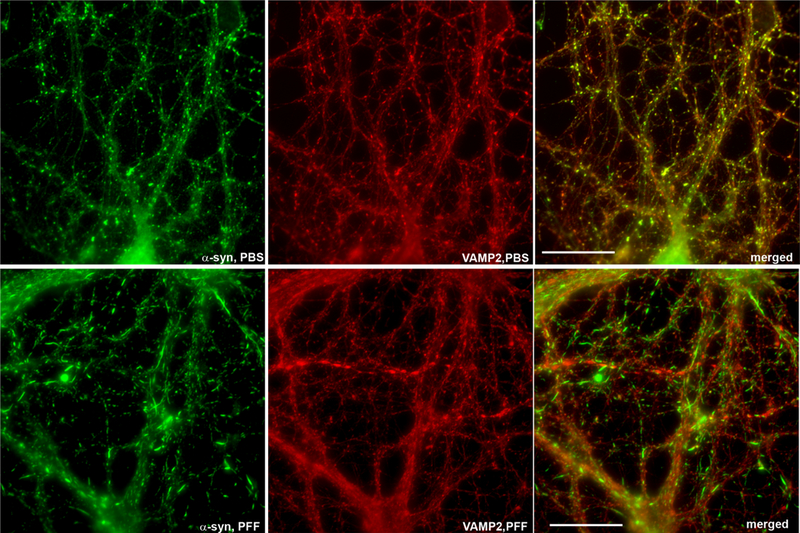
Redistribution of α-synuclein from the presynaptic terminal to Lewy neurite-like inclusions. Primary hippocampal neurons were exposed to preformed fibrils (PFF) of α-synuclein and fixed 14 days later. Immunofluorescence was performed using an antibody to total a-synuclein (green, top panels), phospho-Ser129-α-synuclein (green, bottom panels), or VAMP2 to show pre-synaptic terminals. In control, phosphate-buffered saline (PBS)-exposed neurons, total a-synuclein localizes to small puncta that colocalize with VAMP2 at the presynaptic terminal. After exposure to PFF, α-synuclein localizes to serpentine-like inclusions that resemble Lewy neurites found in Parkinson disease brains and shows minimal localization to the presynaptic terminal.
The first animal models of cell-to-cell transfer of α-synuclein focused on neural transplantation paradigms. Using species-specific antibodies, it was shown that rodent neural cells grafted into adult rodent brains, which overexpressed human α-synuclein, contained human α-synuclein (Desplats et al., 2009; Hansen et al., 2011; Kordower et al., 2011). These findings demonstrated that host-brain derived α-synuclein can be taken up by grafted neurons, akin to what was suggested to have occurred in the grafted PD patients that had been analyzed a few years earlier. Subsequently, it was reported that fibrils of recombinant α-synuclein, when injected in the rodent or primate brain, induce formation of inclusions from endogenous α-synuclein (Luk et al., 2012b; Luk et al., 2012a; Masuda-Suzukake et al., 2014; Paumier et al., 2015; Shimozawa et al., 2017). Importantly, injection of fibrils into the striatum, for example, primarily induce inclusion formation in neurons that directly project to the site of injection site, such as the substantia nigra pars compacta, amygdala and cortex. Sparse aggregates also appear in neuron populations that do not project directly to the injection site, such as the olfactory bulb and hippocampus (Luk et al., 2012b; Masuda-Suzukake et al., 2013; Masuda-Suzukake et al., 2014). Formation of α-synuclein inclusions in the substantia nigra leads to death of dopamine neurons, demonstrating two cardinal features of PD neuropathology, namely Lewy-like aggregates and the death of nigral neurons. As a consequence of the nigral pathology, the rodent develops motor deficits (Luk et al., 2012b; Paumier et al., 2015). Interestingly, hindlimb intramuscular injection of α-syn fibrils in young mice expressing human wild type or A53T synuclein, produces inclusions in the central nervous system, demonstrating that pathology can spread from inoculation of peripheral sites with abnormal α-syn (Sacino et al., 2014).
Because olfactory bulb pathology is a feature of several synucleinopathies, recent studies have highlighted how α-synuclein fibrils injected into the olfactory bulb of wild-type mice can trigger a slowly progressing (described up to one year) α-synucleinopathy throughout the olfactory system and interconnected brain regions (Rey et al., 2013; Rey et al., 2016b) (Fig. 2). The α-synuclein pathology eventually involves over 40 brain structures, some of which that are located two or more synapses away from the injection site, and include brain stem regions such as the substantia nigra, locus coeruleus and raphe nuclei. Mice with this progressive pathology also exhibit gradual development of olfactory deficits (Rey et al. 2016). The olfactory model is particularly interesting in the context or prodromal PD, because it replicates the olfactory deficits, α-synuclein aggregates in olfactory brain structures, in animals where very early and sparse signs of Lewy pathology, but no neurodegeneration, are observed in the substantia nigra.
Fig. 17.2.
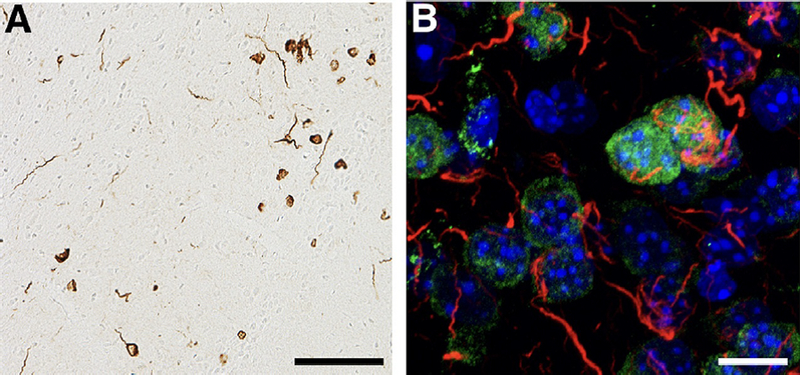
α-Synuclein pathology in mouse model of prodromal Parkinson disease. (A) Mice were injected with mouse preformed fibrils (PFF) of α-synuclein unilaterally into the olfactory bulb, and perfused 12 months later for immunohistochemistry. Phospho-Ser129-α-synuclein inclusions can be seen in the piriform cortex. There are abundant inclusions with the appearance of Lewy neurites and inclusions in the soma. Scale bar = 100 mm. (B) Mice were injected with mouse PFF of α-synuclein unilaterally into the olfactory bulb, and were perfused 1 month later. Abundant phospho-Ser129-α-synuclein inclusions (red) can be seen in the anterior olfactory nucleus. Lewy neurite-like inclusions are apparent, as well as inclusions juxtaposed to the nucleus (NeuN, green and DAPI, blue).
Injected brain extracts from old and symptomatic α-synuclein transgenic mice can accelerate the development of Lewy-like pathology and symptoms in younger mice from the same transgenic lines (Mougenot et al 2012). Along similar lines, but of greater clinical relevance, purified extracts of α-synuclein aggregates derived from PD or DLB brains induce inclusion formation in wild type mice, mice heterozygous for mutant human α-synuclein, and macaque primates (Masuda-Suzukake et al., 2013; Recasens et al., 2014). Interestingly, brain extracts from patients with multiple systems atrophy (MSA) are suggested to be more efficient, than those from PD brains, at inducing pathologic aggregates when injected into experimental animals (Watts et al., 2013; Prusiner et al., 2015). MSA is a synucleinopathy similar to PD, but unlike PD in which inclusions are intraneuronal, α-synuclein aggregates are found primarily in oligodendrocytes. MSA is typically more rapidly progressing than PD, with a life expectancy of about 9 years compared to 20 years for PD. Also of interest, oligodendrocytes express lower, albeit not negligible, levels of α-synuclein (Djelloul et al., 2015), and it is possible that the inclusions in the olgodendrocytes are a consequence of small α-synuclein seeds propagating from neurons. It is interesting to speculate that inclusions derived from brain extracts from patients with distinct synucleinopathies (PD, DLB or MSA) might be more or less potent due to possible structural differences in the aggregates. However, before such a conclusion can be made, it will be necessary to quantify the concentration of different types of α-synuclein assemblies in these extracts, and determine if preliminary reports of differences in seeding capacity are simply due to higher concentrations of α-synuclein aggregates in some disorders compared to others. It has not yet been possible to define precisely which molecular species of α-synuclein that most efficiently seeds aggregation of native α-synuclein. It remains possible that several different molecular forms of α-synuclein are approximately equipotent as nucleating agents, but that they are more or less likely to be taken up by the different types of cells that are present in the brain.
α-Synuclein misfolding and permissive templating
α-Synuclein can exist as an amphipathic alpha-helix, likely as a tetramer or multimer, an unfolded/disorded conformation, or beta-sheet, fibrillar conformation. Unfolded α-synuclein can become misfolded and interact with other α-synuclein molecules to form multimers with beta sheet structure. These multimers build to form amyloid fibrils which are the hallmark of Lewy bodies and Lewy neurites. Either fragments of these fibrils or smaller beta-sheet oligomers then act as a seed or “nucleus” to recruit monomeric α-synuclein to misfold and associate into pathogenic fibrils. It has been suggested that α-synuclein that has adopted an alpha-helical conformation and is associated with membranes does not convert to amyloid fibrils. The cytosolic, disordered conformation of α-synuclein, however, is susceptible to forming beta-sheets that stack to form amyloid fibrils (Burre et al., 2015). The amyloid fibril form is the lowest energy conformation for α-synuclein. Missense mutations in α-synuclein that cause PD (A30P, A53T, E46K, H50Q and G51D) reduce its association as a native multimeric alpha-helix complex, and increase its propensity to adopt a monomeric form (Dettmer et al., 2015). Reduced association of α-synuclein with membranes increases aggregate formation. Other studies show that α-synuclein may aggregate while associated with membranes, but the composition of the membrane and the extent to which the central hydrophobic within the NAC domain of α-synuclein is exposed appears to determine its propensity to aggregate (Galvagnion et al., 2015; Ysselstein et al., 2015; Galvagnion, In Press). For example, familial mutations of α-synuclein cause dissociation of the central hyrdrophobic region and increase aggregation at the membrane. Thus, within a neuron, α-synuclein likely exists in equilibrium between an alpha-helical/multimeric form and an unfolded from, and some factors induce misfolding, shifting α-synuclein to a beta-sheet, aggregate prone conformation. These aggregates then amplify within the neuron, and it is suggested that they are released under certain conditions, allowing the cell-to-cell propagation process to ensue.
While it is clear that misfolded seeds can template α-synuclein pathology, it is unclear what factors initiate the abnormal conversion of α-synuclein to pathologic aggregates (Fig. 3). As mentioned above, an extremely small proportion of PD patients carry one of the six known missense mutations in the α-synuclein gene, SNCA. These mutations can reduce its normal α-helical/tetrameric conformation, and also influence membrane association (Burre et al., 2015; Dettmer et al., 2015; Ysselstein et al., 2015).
Fig. 17.3.
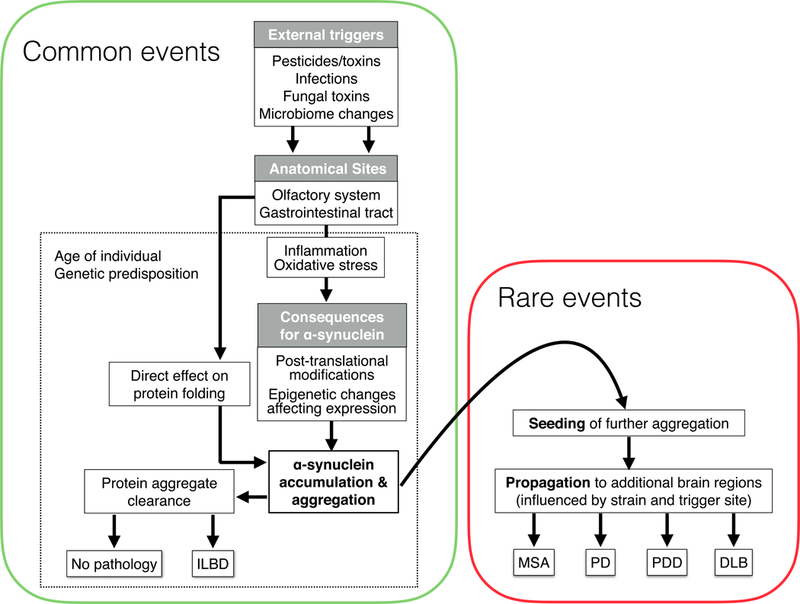
A schematic of events leading to formation of pathologic α-synuclein aggregates and their propagation throughout the nervous system. Events that we postulate can occur frequently and in large numbers of normal individuals are depicted in the green box, whereas events with rare occurrences, and associated with the development of α-synucleinpathies, are in the red box. DLB, dementia with Lewy bodies; ILBD, incidental Lewy body disease; MSA, multiple system atrophy; PD, Parkinson disease; PDD, Parkinson disease dementia.
Duplication, triplication and polymorphisms of the α-synuclein gene also cause a neurological syndrome that includes PD-like features. A modest, 1.5–2 fold, increase in α-synuclein expression as demonstrated with SNCA duplication, may increase levels of unfolded α-synuclein that is more easily converted into the abnormal beta-sheet, aggregated conformation (Farrer et al., 2004; Miller et al., 2004; Devine et al., 2011). Certain single nucleotide polymorphisms which are associated with a slight increase in PD are located close to SNCA, and studies in cultured neurons suggest that a polymorphism located in a distal enhancer of SNCA can lead to a small (1.06 times) increase in neuronal α-synuclein levels (Soldner et al., 2016). Levels of α-synuclein also increase in normal aging (Chu and Kordower, 2007), and this has been suggested to be due to decline of the lysosomal-autophagy system which normally degrades α-synuclein (Chu et al., 2009). These observations are consistent with the fact that high age is the greatest risk factor for the development of PD. Recent work suggests that increased expression of α-synuclein in neurons might be a protective response to viral or bacterial exposure (Beatman et al., 2015; Tomlinson et al., 2017). It is conceivable that increased α-synuclein expression due to exposure to a foreign microbe increases the likelihood that α-synuclein aggregates in neurons (Fig. 3).
Defects in the proteostasis machinery, beyond that seen in normal aging and mentioned above, could also influence α-synuclein levels and prevent degradation of aggregates. For example, α-synuclein probably toggles between normal, unfolded and misfolded conformations, but the misfolded conformations are efficiently cleared by the proteosome, or the autophagosome/lysosome machinery. In addition to changes occurring during normal aging, many genes shown to cause relatively rare inherited forms of PD play a role in protein degradation pathways such as leucine rich-repeat kinase 2, glucocerebrosidase, lysosomal type 5 P-type ATPase and Rab7L1 (Alegre-Abarrategui et al., 2009; Mazzulli et al., 2011; Dehay et al., 2012; Usenovic et al., 2012; MacLeod et al., 2013; Orenstein et al., 2013). Thus dysfunction in these proteins could lead to an abnormal accumulation of α-synuclein and the formation of α-synuclein aggregates. Indeed, it was recently shown that neuronal expression of the disease mutant G2019S-LRRK2, slightly increases total levels of α-synuclein (Volpicelli-Daley et al., 2016) which could be due to increases in translation of α-synuclein, or impaired chaperone-mediated autophagy.
Damage to mitochondria can induce formation of pathologic α-synuclein aggregates. The pesticide rotenone, inhibits complex I of mitochondria and produces reactive oxidative species (Greenamyre et al., 1992). Oxidatively modified α-synuclein is more prone to aggregation (Souza et al., 2000) (Fig. 3). Epidemiological evidence indicates that pesticide exposure increases the risk of developing PD (Paul et al., 2016). Because α-synuclein pathology appears to initiate in the gut and olfactory bulb, it has been hypothesized that exposure of olfactory epithelia or enteric mucosa to pesticides and toxins may increase production of reactive oxygen species, possibly triggering inflammation and initiating abnormal conversion of α-synuclein (Rey et al., 2016a).
A recent study showed that the gut microbiome can influence α-synuclein aggregation, providing further support that extrinsic factors in peripheral tissues might contribute to the abnormal conversion of α-synuclein and represent a starting point from which misfolded α-synuclein can propagate throughout the nervous system (Sampson et al., 2016). One of the earliest signs of PD is constipation and dysbiosis (Verbaan et al., 2007; Sharon et al., 2016; Houser and Tansey, 2017). Indeed, PD patients frequently exhibit intestinal inflammation (Devos et al., 2013) and demonstrate unique microbial signatures compared to control people (Scheperjans et al., 2015; Bedarf et al., 2017). Aggregates of α-synuclein appear in the enteric nervous system and vagal nerve early in the disease, in some cases 20 years before the diagnosis of PD (Braak et al., 2003b; Shannon et al., 2012; Stokholm et al., 2016). Vagotomized individuals have been suggested to have reduced risk for PD (Svensson et al., 2015; Liu et al., 2017). It is possible that inflammation in the gut, viral diseases such as hepatitis (Pakpoor et al., 2017), or exposure to toxins could cause the release of factors, e.g. short chain fatty acids, that could somehow influence aggregation of the nervous system (Fig. 3). It is also interesting to speculate that many individuals harbor α-synuclein aggregates in their enteric nervous system, but that the cellular degradation machinery prevents accumulation of these aggregates. Genetic susceptibility factors, gastrointestinal disease states could reduce lysosomal function causing an accumulation of α-synuclein aggregates that travel the long distance along the vagal nerve, resulting in Lewy pathology in the dorsal IX/X motor nucleus (considered one of the first sites affected by α-synuclein aggregates, see above). Once present in the dorsal IX/X motor nucleus, the Lewy pathology can propagate to the locus coerleus, substantia nigra pars compacta, and beyond, to cause PD. It is important to emphasize that the hypothesis that the gut is a starting point for PD pathology does not exclude that the olfactory system is also an initiation site for α-synuclein aggregation. Indeed, it is conceivable that different individuals who develop PD will in some cases exhibit pathology first in the olfactory bulb, while others show changes in the gut, and some might have simultaneous development of α-synuclein aggregates both in the olfactory and enteric pathways (e.g. due to an environmental insult affecting both locations) (Fig. 3).
α-Synuclein pathogenic strains
In transmissible spongiform encephalopathies or prion diseases, normal cellular prion protein PrpC is converted into a pathogenic form, PrPsc which forms beta-sheet, detergent-insoluble aggregates that are protease-resistant. Different conformations of PrPSc produce different lesion profiles in distinct brain regions with different clinical phenotypes. They include Creutzfeldt-Jakob disease, Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia and kuru. Furthermore, PrPSc from these different diseases show distinct biochemical and morphological conformations which could account for the different disease manifestations (Collinge and Clarke, 2007).
It has been hypothesized that α-synuclein can form aggregates with distinct pathological conformations, giving rise to the different synucleinopathies. Patients with DLB present with dementia and hallucinations, but also develop motor symptoms similar to PD in a later phase. Some PD patients progress to develop dementia relatively soon after diagnosis of the motor syndrome, and are then classified as Parkinson’s disease dementia (PDD), a condition which is virtually indistinguishable from DLB. The presence of neurocognitive symptoms is more likely when Lewy bodies are abundant in limbic and cortical brain regions (Dickson et al., 2009). The fact that PD, PDD and DLB all involve α-synuclein aggregates and yet have of distinct neuropathological profiles and functional deficits, add fuel to the idea that different strains of α-synuclein aggregates may exist and underpin the differences. The notion that differences in α-synuclein aggregates are important does not, however, stop with PD, PDD and DLB. For example, as mentioned above, MSA is characterized by α-synuclein aggregates primarily in oligodendrocytes called glial cytoplasmic inclusions (GCIs) (Papp et al., 1989), although neuronal inclusions can also be found in MSA. Unlike PD and DLB in which inclusions are found only in the cytoplasm, inclusions are found in the nucleus and cytoplasm in MSA. Little is known about the structural characteristics of α-synuclein aggregates in MSA, and how they differ from PD, PDD and DLB. As previously mentioned, brain homogenates prepared from PD and MSA brains differ in their capacity to trigger synucleinopathy in experimental settings (Watts et al., 2013; Prusiner et al., 2015), suggesting that α-synuclein aggregates from MSA brains may represent a distinct strain from those found in PD.
While the evidence for strains in α-synucleinopathies is not nearly as advanced as studies of prion diseases, it appears that different “strains” of relevance to PD and MSA can be created artificially. Natively unfolded α-synuclein can adopt different folding conformations that act as templates to recruit additional monomers leading to assembly and elongation of fibrillar assemblies with distinct conformations. For example, by varying the assembly conditions by altering the buffer, salt or pH, α-synuclein can generate fibrils with a classic cylindrical shape, or a flat, ribbon shape as demonstrated by electron microscopy (Bousset et al., 2013). The features of these distinct assemblies faithfully propagate in vitro and in cultured cells. The classic fibrillar α-synuclein generated with appears to be more toxic to dopamine neurons when injected into the rat substantia nigra pars compacta, whereas the ribbons are unique in that they seed α-synuclein aggregate formation in both neurons and oligodendrocytes, which appears to be relevant to MSA (Peelaerts et al., 2015). Despite the aggregates taking on unique and complex three-dimensional structures, complementary primary amino acid structure is still critical for cross-seeding of distinct assemblies. As an excellent example, even though human and mouse α-synuclein share 95% amino acid identity, human fibrils are not as efficient as seeding mouse monomers, or vice versa (Luk et al., 2016).
Patients with different α-synuclein point mutations also exhibit distinct clinical PD profiles, and it can be speculated that this is related to the preferred pathogenic conformation that the mutant protein adopts. For example, patients with the G51D α-synuclein mutation can have a very early onset of the disease (as early as 19 years old), severe dementia, hallucinations, psychiatric disturbances, and can exhibit α-synuclein inclusions in both neurons and oligodendrocytes (Kiely et al., 2013). Thus, single amino acid substitutions could change the structural properties of the α-synuclein assemblies that could cause distinct disease phenotypes.
Interestingly, α-synuclein fibrils appear to be able to convert from a strain that cannot induce tau aggregation into a strain that can seed tau aggregate formation (Guo et al., 2013). In test tube experiments, α-synuclein fibrils are generated in phosphate buffered saline from recombinant α-synuclein, 5% of these fibrils are added to a new fibrillization reaction with monomer and this process is repeated several times. The distinct conformers of α-synuclein show different electrophoretic mobility after proteinase K digestion. These findings suggests that, in the brain, repeated α-synuclein fibrillization as it passes from neuron to neuron may cause a conversion to a form that is able to induce tau aggregation. Aberrant α-synuclein aggregation could lead to a more generalized disruption of protein folding homeostasis, triggering aggregation of tau as has been shown for polyglutamine repeat expansion diseases (Gidalevitz et al., 2006).
Often in neurodegenerative disease, different protein aggregates can occur at the same time. For example, 50% of Alzheimer’s disease patients, a diseased characterized by Aβ plaques and neurofibrillary tangles composed of Tau, display α-synuclein inclusions, primarily in the amygdala (Hamilton, 2000). Parkinson’s disease dementia and DLB patients often display neurofibrillary tangles (Irwin et al., 2017).Tau is a microtubule associated protein that is the primary component of neurofibrillary tangles. MAPT (the gene locus encoding tau) consistently produces a strong signal in genome wide association studies of PD patients, although the contribution of tau to PD is unknown (Satake et al., 2009; Simon-Sanchez et al., 2009; Edwards et al., 2010). Tau colocalizes with α-synuclein in Lewy bodies in PD and DLB brains. While phospho-immunoreactive tau inclusions are present in sporadic PD, they are more abundant in familial PD patients with missense α-synuclein mutations (Duda et al., 2002; Ishizawa et al., 2003; Fujishiro et al., 2013). Phopsho-tau-immunoreactive inclusions can be found in multiple brain regions including the amygdala, nucleus basalis of Meynert, striatum, hippocampus, cortex and brainstem (Fujishiro et al., 2013). The presence of both α-synuclein and tau aggregates, particularly in the cortex, correlates with development of PD dementia (Irwin et al., 2012). The findings that different conformations of misfolded α-synuclein trigger tau aggregation and other do not, suggest these differences could contribute to why some patients develop dementia whereas others do not.
Further experiments, which compare the biochemical and morphological differences among aggregates from different synucleinopathies and their seeding capacities, are required to clarify if distinct α-synuclein strains exist. It is unknown whether different conformations of α-synuclein contribute to the distinct clinical and histological phenotypes of different synucleinopathies or whether post-translational modifications, such as phosphorylation, ubiquitination, nitration, or sumoylation, are crucial contributors to the differences in pathology induced by MSA and PD brain tissues (Giasson et al., 2000; Fujiwara et al., 2002; Tofaris et al., 2003; Kim et al., 2011; Krumova et al., 2011; Vicente Miranda et al., 2017). It is also conceivable that even within one disorder (e.g. PD) different types of α-synuclein conformations preferentially populate different brain regions, e.g. basal ganglia versus neocortex, and that this is due to how different α-synuclein assemblies interact with varying forms of lipids (which can show differential levels between brain regions). In the brainstem, Lewy bodies in neuronal soma exhibit a dense core surrounded by a halo. By contrast, in neocortex the Lewy bodies are spheroid, have more diffuse contours and do not display a compact halo. These neocortical α-synuclein aggregates have been suggested to represent a less mature form of Lewy bodies (Kosaka, 1978). In neurites, Lewy pathology can appear as long serpentine inclusions. Small proteinase K aggregates are also abundant in the cortex of DLB patients (Kramer and Schulz-Schaeffer, 2007). It is not clear yet, which form of aggregates is more toxic - it may be that the less dense inclusions and aggregates represent the toxic species and the dense, thioflavin T-positive, Lewy bodies are inert. The existence of different strains of α-synuclein aggregates also opens up the possibility that they govern which type (e.g. PD, PDD, DLB or MSA) of synucleinopathy develops. In line with this idea, the very initial misfolding event (e.g. occurring in the olfactory bulb or enteric nervous system) could play a key role in influencing the type of α-synuclein pathology that propagates throughout the nervous system, and with which speed it will progress (Fig. 3).
Potential Cellular Mechanisms of propagation
There remain many outstanding questions as to how Lewy pathology propagates across the nervous system. In this section, we discuss the cellular mechanisms that might underlie cell-to-cell transfer of α-synuclein oligomers and fibrils. The pathogenic α-synuclein conformers must be released from the neuron, taken up by neighboring neurons, and escape into the cytosol where they seed conversion of endogenous α-synuclein to an abnormal aggregate-prone form. Recent data suggest how α-synuclein aggregates might be taken up by neurons. Fibrillar α-synuclein binds to heparin sulfate proteoglycans (HSPGs) and stimulates uptake by macropinocytosis in a manner independent of dynamin (Holmes et al., 2013). Heparin which blocks binding to HSPGs, and chlorate which blocks sulfation, inhibit the uptake of α-synuclein fibrils. The receptor, lymphocyte-activation gene 3 (LAG3), a member of the immunoglobulin superfamily, selectively binds α-synuclein fibrils and mediates their uptake (Mao et al., 2016). Antibodies to LAG3 or gene knockout of LAG3 prevent α-synuclein fibril-induced seeding of inclusions and protects against dopamine neuron loss when fibrils are injected into the striatum. It will be of great interest to learn whether neurexin or amyloid precursor-like protein (APLP), which also selectively bind fibrillar α-synuclein and are highly enriched in the brain, also facilitate propagation of α-synuclein pathology. The realization that some of the molecular pathways that mediate uptake of pathogenic α-synuclein assemblies exhibit a degree of specificity opens up for the exciting possibility that the uptake process might be targeted therapeutically with small molecules.
How α-synuclein aggregates are released from the neuron remains a partial mystery. As mentioned before, α-synuclein is found in the cerebrospinal fluid (El-Agnaf et al., 2003). Both monomeric and aggregated α-synuclein can be released from neurons (Alvarez-Erviti et al., 2011; Danzer et al., 2012). α-Synuclein has been detected in exosomes and in non-exosome, soluble fractions from the conditioned media of “neuronotypic” cells, although the amount of α-synuclein associated with the exosomes appears to be relatively low (Danzer et al., 2012). Aggregates have been detected in exosomes and association of α-synuclein with cell-derived exosomes or artificial exosomes (containing the key lipids, but lacking exosomal proteins) may even accelerate pathogenic aggregation (Grey et al., 2015). Monomeric and oligomeric α-synuclein associates with exosomes, but to date, no fibrillar α-synuclein has been detected on or inside exosomes (Brahic et al., 2016). Exosomes are small vesicles that reside inside multivesicular bodies (MVBs) that are released when the MVBs fuse with the plasma membrane. Given that α-synuclein aggregates in the neuron are cytosolic, they would have to somehow bind the limiting membrane of the MVBs and be internalized into the internal vesicles. Thus, α-synuclein aggregates would be inside the internal vesicles and would remain inside the vesicles when they are released into the extracellular space (Danzer et al., 2012). Because peripheral administration of antibodies to α-synuclein can prevent propagation of pathology in experimental models (Masliah et al., 2005; Tran et al., 2014; Spencer et al., 2017) this means the antibodies have to interact with and bind α-synuclein extracellularly, implying that exosomal release may not be the only mechanism for the extracellular spread of α-synuclein aggregates. Recently, tunneling nanotubes were shown to be a mechanism by which neurons share aggregates α-synuclein (Abounit et al., 2016). A final, as yet to be demonstrated mechanism, is that the α-synuclein fibrils permeabilize the plasma membrane directly to gain access to the extracellular space.
Another mystery is related to how imported α-synuclein gains access to the endogenous α-synuclein of a recipient cell. After endocytosis, either by a receptor-mediated mechanism or by a general macropinocytosis mechanism, fibrillar α-synuclein must penetrate the endosomal membrane to gain access to the cytoplasm where it can seed conversion of endogenous α-synuclein. The most compelling findings are that upon entry into cells, α-synuclein aggregates can rupture endosomes and lysosomes, and thereby directly enter the cytosol in a manner similar to the mechanism used by some viruses (Freeman et al., 2013; Flavin et al., 2017).
Potential for therapeutics that target the prion-like process
The prion-like hypothesis for α-synucleinopathies has led to the development of novel therapeutic strategies for preventing disease progression. Because α-synuclein is released into the extracellular space under certain conditions, it can be targeted by antibodies. The interest in developing immunotherapy to target α-synuclein has been around for over a decade (Masliah et al., 2005), but this research field really took off dramatically in the wake of studies demonstrating cell-to-cell transfer of the pathogenic protein. Experiments performed in rodent models of α-synucleinopathy support that antibodies targeting α-synuclein can prevent the progression of disease pathology and reduce associated behavioral phenotypes (Masliah et al., 2005; Games et al., 2014; Tran et al., 2014; Christiansen et al., 2016; Spencer et al., 2017). Immunotherapy can be divided into two different strategies, passive immunotherapy or active immunotherapy. With passive immunotherapy, antibodies to α-synuclein are generated in the laboratory and then administered to the patient. One of these antibodies, which targets the C-terminus of α-synuclein, PRX002/RG7935, was recently demonstrated by the Prothena Corporation to have an acceptable safety and tolerability profile in a Phase 1b double-blind placebo controlled dose study (Schenk et al., 2017). Biogen also initiated a Phase I clinical trial on an antibody, BIIB054, which targets human α-synuclein and was isolated from a B cell library from elderly healthy subjects based on findings that α-synuclein antibodies or auto-antibodies exist in healthy individuals and those with PD (Papachroni et al., 2007; Neff et al., 2008). By contrast, AFFiRiS has utilized an active immunotherapy approach in which patients are immunized with “AFFITOPES” or small peptides that mimic α-synuclein, in order to generate a B-cell response and antibodies to α-synuclein. AFFiRiS has completed a Phase 1b trial showing that the vaccinations are safe and tolerable.
For all immunotherapies targeting α-synuclein, a potential drawback of the approach is that it might be detrimental to deplete normal, α-helical α-synuclein which has been shown to regulate presynaptic vesicle endo/exocytosis. In addition, because α-synuclein is a cytosolic protein, there may be cell intrinsic mechanisms that cause α-synuclein to convert to an abnormal conformation, and extracellular antibodies may not be able to access these molecules and prevent this conversion. That said, the realization that pathological α-synuclein can be found in the extracellular space means that the idea of generating antibodies that only access this compartment is still viable. The specific epitopes that the antibodies are directed against, and the type of conformations that allow these epitopes to be exposed, are likely to be crucial factors. For example, antibodies that selectively recognize beta-sheet, fibrillar α-synuclein may be more effective with fewer potential side effects than antibodies against the monomeric protein. If fact, AbbVie and Bioarctic Neuroscience also recently joined to develop a novel antibody, BAN0805, which targets a “toxic” form of α-synuclein (http://www.bioarctic.se/news/-bioarctic-enters-into-collaboration-with-abbvie-for-parkinsons-disease-research). The emerging concept of different strains of α-synuclein aggregates (see detailed discussion above), however, might mean that there will be a need to have tailor-made, patient-specific antibodies depending on the type of α-synuclein fibrils that dominate in the brain in that particular patient. As we learn more about α-synuclein aggregates and the effects of immunotherapies in experimental models and from clinical trials, it will become apparent if this therapeutic approach will require a personalized “precision medicine”strategy.
Treatments to prevent α-synuclein aggregation or disrupt aggregates are in clinical trials as well. Neuropore has completed a Phase I clinical trial of its compound, NPT200–11, which may prevent accumulation of oligomeric α-synuclein within membranes (Wrasidlo et al., 2016). Proclara Biosciences has a compound, NPT088, in a Phase 1b clinical trial for Alzheimer’s disease. This compound utilizes a general amyloid interaction motif to target multiple amyloid domains. The goal is to inhibit aggregation of multiple proteins implicated in neurodegenerative disease including amyloid β, tau, prion, transthyretin and α-synuclein. However, recent studies of the NACore domain of α-synuclein using microelectron diffraction and nanocrystals has revealed crucial differences in the α-synuclein amyloid domain from amyloid fibrils formed by other proteins, such as the 11-amino acid width of the zipper, which is larger, and the inclusion of water molecules within the interface (Rodriguez et al., 2015). Thus, it is possible that not all amyloid domains are created equal and a generic amyloid targeting compound may not be effective for all neurodegenerative diseases.
Antisense oligonucleotides (ASOs) to reduce levels of α-synuclein may provide an alternate strategy prevent pathologic templating of α-synuclein amyloid fibrils. ASOs have recently emerged as promising, strategies to treat neurodegenerative diseases (Ottesen, 2017). ASOs are single stranded synthetic nucleic acids that bind target mRNA via Watson-Crick base pairing, resulting in degradation of target mRNA by RNAse H, a ubiquitously expressed mammalian enzyme. Phosphorothioate-modified deoxynucleotide (DNA) and 2’-O-methoxyethyl (2’-MOE) sugar modifications enable ASOs to be water soluble, resistant to exonucleases, diffusible, and exhibit dose-dependent activity in vitro and in vivo (Bennett and Swayze, 2010). Importantly, ASOs can be targeted directly to the brain by intracerebroventricular injections and thus bypass adverse effects in the systemic organs. As mentioned above, slightly increased cytoplasmic levels of α-synuclein may facilitate the conversion of α-synuclein from a disordered protein to form beta-sheets and amyloid. Thus, slight reductions in α-synuclein may prevent it from forming inclusions. However, it is important to point out that absence of the SNCA gene or RNAi to knock down levels of α-synuclein in the rodent brain cause a reductions in total dopamine levels in the striatum which could cause adverse events in PD patients who already have low levels of dopamine (Zharikov et al., 2015). Thus, the concentrations of the ASOs would have to be carefully titrated.
The findings that cell surface receptors such as LAG3 can selectively bind to fibrillar but not monomeric α-synuclein provides a potentially novel therapeutic strategy (Mao et al., 2016). Inhibitors of the LAG3 and α-synuclein interactions could also be designed that could inhibit uptake and seeding of pathologic α-synuclein fibrils. Likely other receptors that mediate the uptake and spread of α-synuclein pathology will be identified that can also be targeted. Another potentially interesting strategy is inferring with the binding of α-synuclein with heparin sulfate glucosaminoglycans to prevent uptake of seeds (Holmes et al., 2013).
Recently, combination therapeutic approaches to treating neurodegenerative diseases have received attention. As mentioned above, inflammation may contribute to the aggregation of α-synuclein, neuron death and progression of the disease. In turn, α-synuclein aggregates may stimulate pro-inflammatory response, causing a vicious cycle that amplifies the disease process. Neuroinflammation is widespread in brains of PD (McGeer et al., 1988; Brochard et al., 2009). In addition, the HLA-DR (MHC-II) gene locus is one of the top genetic links to PD in genome wide association studies, suggesting that the immune system plays a role in PD (Hamza et al., 2010). Extracellular α-synuclein binds to Toll-like receptor 2 on microglia and activates inflammatory responses (Kim et al., 2013). Activated monocytes in the brain may internalize and process released α-synuclein, and influence its spread throughout the nervous system (Lee et al., 2008). Targeting inflammation can be neuroprotective in mouse models of PD (Harms et al., 2013; Qin et al., 2016). Thus, both preventing α-synuclein aggregation and propagation in addition to inhibiting pro-inflammatory cascades may be the most effective strategy for modifying disease progression. AstraZeneca has completed a Phase 2 clinical trial on AZD3241 in patients with PD (Jucaite et al., 2015). AD3241 inhibits myeloperoxidase which is a reactive oxygen generating enzyme expressed by microglia.
Concluding remarks
The goal of current PD therapeutics is to prevent the progression of the disease. The evidence that pathogenic α-synuclein spreads throughout the nervous system in a prion-like manner has opened up wide avenues for novel therapeutic strategies. Current strategies already in preclinical development include preventing the abnormal conversion of α-synuclein, or immunotherapy to block the spread of extracellular pathogenic seeds. Emerging basic science research is honing in on the molecular and cellular mechanisms that increase the likelihood of α-synuclein to convert to an abnormal, aggregation prone conformation, and on how these aggregates can propagate within the cell, to neighboring neurons and from one brain region to another. This research will be critical for identifying novel therapeutic targets and to understand if halting propagation of pathologic α-synuclein is sufficient to stop disease progression. Other factors may also contribute to disease progression such as inflammation and metabolic failure in remaining neurons. Perhaps combination therapies that target both misfolded α-synuclein and impaired metabolic pathways will prove to be the most beneficial (Ghosh et al., 2016; Valera and Masliah, 2016).
Acknowledgements
We thank Dr Nolwen Rey for providing images for Figure 2, and Dr Nolwen Rey, Dr Martha Galvis Escobar, Dr Jennifer Steiner and Dr Sonia George for input and feedback on Figure 3. L Volpicelli-Daley reports grants from the Department of Defense, Neurotoxin Exposure Treatment Parkinson’s Disease, and the Michael J. Fox Foundation. P. Brundin acknowledges support from the Van Andel Research Institute. P. Brundin also reports grants from The Michael J. Fox Foundation, National Institutes of Health, Cure Parkinson’s Trust.
References
- Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L, Olivo-Marin JC, Melki R, Zurzolo C (2016) Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J 35:2120–2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegre-Abarrategui J, Christian H, Lufino MM, Mutihac R, Venda LL, Ansorge O, Wade-Martins R (2009) LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Human molecular genetics 18:4022–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM (2011) Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis 42:360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry-Watson J, Sasse J, Boyer S, Shirohi S, Brooks R, Eschbacher J, White CL 3rd, Akiyama H, Caviness J, Shill HA, Connor DJ, Sabbagh MN, Walker DG, Arizona Parkinson’s Disease C (2009) Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatman EL, Massey A, Shives KD, Burrack KS, Chamanian M, Morrison TE, Beckham JD (2015) Alpha-Synuclein Expression Restricts RNA Viral Infections in the Brain. Journal of virology 90:2767–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedarf JR, Hildebrand F, Coelho LP, Sunagawa S, Bahram M, Goeser F, Bork P, Wullner U (2017) Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naive Parkinson’s disease patients. Genome medicine 9:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CF, Swayze EE (2010) RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annual review of pharmacology and toxicology 50:259–293. [DOI] [PubMed] [Google Scholar]
- Bousset L, Pieri L, Ruiz-Arlandis G, Gath J, Jensen PH, Habenstein B, Madiona K, Olieric V, Bockmann A, Meier BH, Melki R (2013) Structural and functional characterization of two alpha-synuclein strains. Nat Commun 4:2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Rub U, Gai WP, Del Tredici K (2003a) Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. Journal of neural transmission 110:517–536. [DOI] [PubMed] [Google Scholar]
- Braak H, Rub U, Gai WP, Del Tredici K (2003b) Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 110:517–536. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003c) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- Brahic M, Bousset L, Bieri G, Melki R, Gitler AD (2016) Axonal transport and secretion of fibrillar forms of alpha-synuclein, Abeta42 peptide and HTTExon 1. Acta Neuropathol 131:539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM, Duyckaerts C, Flavell RA, Hirsch EC, Hunot S (2009) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. The Journal of clinical investigation 119:182–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundin P, Li JY, Holton JL, Lindvall O, Revesz T (2008) Research in motion: the enigma of Parkinson’s disease pathology spread. Nature reviews Neuroscience 9:741–745. [DOI] [PubMed] [Google Scholar]
- Burre J, Sharma M, Sudhof TC (2015) Definition of a molecular pathway mediating alpha-synuclein neurotoxicity. J Neurosci 35:5221–5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen JR, Olesen MN, Otzen DE, Romero-Ramos M, Sanchez-Guajardo V (2016) alpha-Synuclein vaccination modulates regulatory T cell activation and microglia in the absence of brain pathology. Journal of neuroinflammation 13:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y, Kordower JH (2007) Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol Dis 25:134–149. [DOI] [PubMed] [Google Scholar]
- Chu Y, Dodiya H, Aebischer P, Olanow CW, Kordower JH (2009) Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol Dis 35:385–398. [DOI] [PubMed] [Google Scholar]
- Collinge J, Clarke AR (2007) A general model of prion strains and their pathogenicity. Science 318:930–936. [DOI] [PubMed] [Google Scholar]
- Danzer KM, Kranich LR, Ruf WP, Cagsal-Getkin O, Winslow AR, Zhu L, Vanderburg CR, McLean PJ (2012) Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener 7:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E (2012) Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 109:9611–9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 106:13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, Selkoe D (2015) Parkinson-causing alpha-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun 6:7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman JW, Schapira AH, Gwinn K, Hardy J, Lewis PA, Kunath T (2011) Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun 2:440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, Coron E, Bruley des Varannes S, Naveilhan P, Nguyen JM, Neunlist M, Derkinderen P (2013) Colonic inflammation in Parkinson’s disease. Neurobiol Dis 50:42–48. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Braak H, Duda JE, Duyckaerts C, Gasser T, Halliday GM, Hardy J, Leverenz JB, Del Tredici K, Wszolek ZK, Litvan I (2009) Neuropathological assessment of Parkinson’s disease: refining the diagnostic criteria. Lancet Neurol 8:1150–1157. [DOI] [PubMed] [Google Scholar]
- Djelloul M, Holmqvist S, Boza-Serrano A, Azevedo C, Yeung MS, Goldwurm S, Frisen J, Deierborg T, Roybon L (2015) Alpha-Synuclein Expression in the Oligodendrocyte Lineage: an In Vitro and In Vivo Study Using Rodent and Human Models. Stem cell reports 5:174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Mabon ME, Miller DC, Golbe LI, Lee VM, Trojanowski JQ (2002) Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol 104:7–11. [DOI] [PubMed] [Google Scholar]
- Edwards TL, Scott WK, Almonte C, Burt A, Powell EH, Beecham GW, Wang L, Zuchner S, Konidari I, Wang G, Singer C, Nahab F, Scott B, Stajich JM, Pericak-Vance M, Haines J, Vance JM, Martin ER (2010) Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 74:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, Gibson MJ, Curran MD, Court JA, Mann DM, Ikeda S, Cookson MR, Hardy J, Allsop D (2003) Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 17:1945–1947. [DOI] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW (2004) Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 55:174–179. [DOI] [PubMed] [Google Scholar]
- Flavin WP, Bousset L, Green ZC, Chu Y, Skarpathiotis S, Chaney MJ, Kordower JH, Melki R, Campbell EM (2017) Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. [DOI] [PubMed] [Google Scholar]
- Freeman D, Cedillos R, Choyke S, Lukic Z, McGuire K, Marvin S, Burrage AM, Sudholt S, Rana A, O’Connor C, Wiethoff CM, Campbell EM (2013) Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8:e62143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujishiro H, Imamura AY, Lin WL, Uchikado H, Mark MH, Golbe LI, Markopoulou K, Wszolek ZK, Dickson DW (2013) Diversity of pathological features other than Lewy bodies in familial Parkinson’s disease due to SNCA mutations. American journal of neurodegenerative disease 2:266–275. [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T (2002) alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4:160–164. [DOI] [PubMed] [Google Scholar]
- Galvagnion C (In Press) The role of lipids interacting with a-synuclein in the pathogenesis of Parkinson’s disease. Journal of Parkinson’s Disease. [DOI] [PubMed] [Google Scholar]
- Galvagnion C, Buell AK, Meisl G, Michaels TC, Vendruscolo M, Knowles TP, Dobson CM (2015) Lipid vesicles trigger alpha-synuclein aggregation by stimulating primary nucleation. Nature chemical biology 11:229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Games D, Valera E, Spencer B, Rockenstein E, Mante M, Adame A, Patrick C, Ubhi K, Nuber S, Sacayon P, Zago W, Seubert P, Barbour R, Schenk D, Masliah E (2014) Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci 34:9441–9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Tyson T, George S, Hildebrandt EN, Steiner JA, Madaj Z, Schulz E, Machiela E, McDonald WG, Escobar Galvis ML, Kordower JH, Van Raamsdonk JM, Colca JR, Brundin P (2016) Mitochondrial pyruvate carrier regulates autophagy, inflammation, and neurodegeneration in experimental models of Parkinson’s disease. Science translational medicine 8:368ra174. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM (2000) Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 290:985–989. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI (2006) Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 311:1471–1474. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Spillantini MG (2017) The Synucleinopathies: Twenty Years On. J Parkinsons Dis 7:S53–S71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenamyre JT, Higgins DS, Eller RV (1992) Quantitative autoradiography of dihydrorotenone binding to complex I of the electron transport chain. J Neurochem 59:746–749. [DOI] [PubMed] [Google Scholar]
- Grey M, Dunning CJ, Gaspar R, Grey C, Brundin P, Sparr E, Linse S (2015) Acceleration of alpha-synuclein aggregation by exosomes. J Biol Chem 290:2969–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VM (2013) Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton RL (2000) Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain pathology 10:378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, Kusel VI, Collura R, Roberts J, Griffith A, Samii A, Scott WK, Nutt J, Factor SA, Payami H (2010) Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 42:781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen C, Angot E, Bergstrom AL, Steiner JA, Pieri L, Paul G, Outeiro TF, Melki R, Kallunki P, Fog K, Li JY, Brundin P (2011) alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. The Journal of clinical investigation 121:715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms AS, Cao S, Rowse AL, Thome AD, Li X, Mangieri LR, Cron RQ, Shacka JJ, Raman C, Standaert DG (2013) MHCII is required for alpha-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci 33:9592–9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI (2013) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A 110:E3138–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser MC, Tansey MG (2017) The gut-brain axis: is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? npj Parkinson’s Disease 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, Van Deerlin V, Lee VM, Leverenz JB, Montine TJ, Duda JE, Hurtig HI, Trojanowski JQ (2012) Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 72:587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ et al. (2017) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 16:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizawa T, Mattila P, Davies P, Wang D, Dickson DW (2003) Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol 62:389–397. [DOI] [PubMed] [Google Scholar]
- Jucaite A, Svenningsson P, Rinne JO, Cselenyi Z, Varnas K, Johnstrom P, Amini N, Kirjavainen A, Helin S, Minkwitz M, Kugler AR, Posener JA, Budd S, Halldin C, Varrone A, Farde L (2015) Effect of the myeloperoxidase inhibitor AZD3241 on microglia: a PET study in Parkinson’s disease. Brain 138:2687–2700. [DOI] [PubMed] [Google Scholar]
- Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, Proukakis C, Quinn N, Lees AJ, Hardy J, Revesz T, Houlden H, Holton JL (2013) alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol 125:753–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ (2013) Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4:1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YM, Jang WH, Quezado MM, Oh Y, Chung KC, Junn E, Mouradian MM (2011) Proteasome inhibition induces alpha-synuclein SUMOylation and aggregate formation. J Neurol Sci 307:157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW (2008a) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14:504–506. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Olanow CW, Freeman TB (2008b) Transplanted dopaminergic neurons develop PD pathologic changes: a second case report. Mov Disord 23:2303–2306. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Dodiya HB, Kordower AM, Terpstra B, Paumier K, Madhavan L, Sortwell C, Steece-Collier K, Collier TJ (2011) Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol Dis 43:552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka K (1978) Lewy bodies in cerebral cortex, report of three cases. Acta Neuropathol 42:127–134. [DOI] [PubMed] [Google Scholar]
- Kramer ML, Schulz-Schaeffer WJ (2007) Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci 27:1405–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumova P, Meulmeester E, Garrido M, Tirard M, Hsiao HH, Bossis G, Urlaub H, Zweckstetter M, Kugler S, Melchior F, Bahr M, Weishaupt JH (2011) Sumoylation inhibits alpha-synuclein aggregation and toxicity. J Cell Biol 194:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurowska Z, Englund E, Widner H, Lindvall O, Li JY, Brundin P (2011) Signs of degeneration in 12–22-year old grafts of mesencephalic dopamine neurons in patients with Parkinson’s disease. J Parkinsons Dis 1:83–92. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Patel S, Lee SJ (2005) Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 25:6016–6024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Bae EJ, Lee SJ (2008) Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochemical and biophysical research communications 372:423–428. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ (2010) Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 285:9262–9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Englund E, Widner H, Rehncrona S, Bjorklund A, Lindvall O, Brundin P (2010) Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson’s disease. Mov Disord 25:1091–1096. [DOI] [PubMed] [Google Scholar]
- Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Bjorklund A, Widner H, Revesz T, Lindvall O, Brundin P (2008) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14:501–503. [DOI] [PubMed] [Google Scholar]
- Liu B, Fang F, Pedersen NL, Tillander A, Ludvigsson JF, Ekbom A, Svenningsson P, Chen H, Wirdefeldt K (2017) Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012a) Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. The Journal of experimental medicine 209:975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM (2012b) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Song C, O’Brien P, Stieber A, Branch JR, Brunden KR, Trojanowski JQ, Lee VM (2009) Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 106:20051–20056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Covell DJ, Kehm VM, Zhang B, Song IY, Byrne MD, Pitkin RM, Decker SC, Trojanowski JQ, Lee VM (2016) Molecular and Biological Compatibility with Host Alpha-Synuclein Influences Fibril Pathogenicity. Cell Rep 16:3373–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A (2013) RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 77:425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X et al. (2016) Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D (2005) Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron 46:857–868. [DOI] [PubMed] [Google Scholar]
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H, Hasegawa M (2014) Pathological alpha-synuclein propagates through neural networks. Acta neuropathologica communications 2:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M (2013) Prion-like spreading of pathological alpha-synuclein in brain. Brain 136:1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D (2011) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1291. [DOI] [PubMed] [Google Scholar]
- Miller DW, Hague SM, Clarimon J, Baptista M, Gwinn-Hardy K, Cookson MR, Singleton AB (2004) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62:1835–1838. [DOI] [PubMed] [Google Scholar]
- Neff F, Wei X, Nolker C, Bacher M, Du Y, Dodel R (2008) Immunotherapy and naturally occurring autoantibodies in neurodegenerative disorders. Autoimmunity reviews 7:501–507. [DOI] [PubMed] [Google Scholar]
- Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M (2010) Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem 285:34885–34898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM (2013) Interplay of LRRK2 with chaperone-mediated autophagy. Nature neuroscience 16:394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottesen EW (2017) ISS-N1 makes the First FDA-approved Drug for Spinal Muscular Atrophy. Translational neuroscience 8:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakpoor J, Noyce A, Goldacre R, Selkihova M, Mullin S, Schrag A, Lees A, Goldacre M (2017) Viral hepatitis and Parkinson disease: A national record-linkage study. Neurology. [DOI] [PubMed] [Google Scholar]
- Papachroni KK, Ninkina N, Papapanagiotou A, Hadjigeorgiou GM, Xiromerisiou G, Papadimitriou A, Kalofoutis A, Buchman VL (2007) Autoantibodies to alpha-synuclein in inherited Parkinson’s disease. J Neurochem 101:749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp MI, Kahn JE, Lantos PL (1989) Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 94:79–100. [DOI] [PubMed] [Google Scholar]
- Paul KC, Sinsheimer JS, Rhodes SL, Cockburn M, Bronstein J, Ritz B (2016) Organophosphate Pesticide Exposures, Nitric Oxide Synthase Gene Variants, and Gene-Pesticide Interactions in a Case-Control Study of Parkinson’s Disease, California (USA). Environmental health perspectives 124:570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, Sandoval IM, Fleming S, Dirr E, Polinski NK, Trojanowski JQ, Lee VM, Sortwell CE (2015) Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 82:185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V (2015) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522:340–344. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, Woerman AL, Mordes DA, Watts JC, Rampersaud R, Berry DB, Patel S, Oehler A, Lowe JK, Kravitz SN, Geschwind DH, Glidden DV, Halliday GM, Middleton LT, Gentleman SM, Grinberg LT, Giles K (2015) Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 112:E5308–5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H, Buckley JA, Li X, Liu Y, Fox TH 3rd, Meares GP, Yu H, Yan Z, Harms AS, Li Y, Standaert DG, Benveniste EN (2016) Inhibition of the JAK/STAT Pathway Protects Against alpha-Synuclein-Induced Neuroinflammation and Dopaminergic Neurodegeneration. J Neurosci 36:5144–5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recasens A, Dehay B, Bove J, Carballo-Carbajal I, Dovero S, Perez-Villalba A, Fernagut PO, Blesa J, Parent A, Perier C, Farinas I, Obeso JA, Bezard E, Vila M (2014) Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75:351–362. [DOI] [PubMed] [Google Scholar]
- Rey NL, Wesson DW, Brundin P (2016a) The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, Petit GH, Bousset L, Melki R, Brundin P (2013) Transfer of human alpha-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126:555–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, Steiner JA, Maroof N, Luk KC, Madaj Z, Trojanowski JQ, Lee VM, Brundin P (2016b) Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. The Journal of experimental medicine 213:1759–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez JA et al. (2015) Structure of the toxic core of alpha-synuclein from invisible crystals. Nature 525:486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacino AN, Brooks M, Thomas MA, McKinney AB, Lee S, Regenhardt RW, McGarvey NH, Ayers JI, Notterpek L, Borchelt DR, Golde TE, Giasson BI (2014) Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci U S A 111:10732–10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, Chesselet MF, Keshavarzian A, Shannon KM, Krajmalnik-Brown R, Wittung-Stafshede P, Knight R, Mazmanian SK (2016) Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 167:1469–1480 e1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake W et al. (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41:1303–1307. [DOI] [PubMed] [Google Scholar]
- Schenk DB, Koller M, Ness DK, Griffith SG, Grundman M, Zago W, Soto J, Atiee G, Ostrowitzki S, Kinney GG (2017) First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov Disord 32:211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, Haapaniemi E, Kaakkola S, Eerola-Rautio J, Pohja M, Kinnunen E, Murros K, Auvinen P (2015) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30:350–358. [DOI] [PubMed] [Google Scholar]
- Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH (2012) Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord 27:716–719. [DOI] [PubMed] [Google Scholar]
- Sharon G, Sampson TR, Geschwind DH, Mazmanian SK (2016) The Central Nervous System and the Gut Microbiome. Cell 167:915–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimozawa A, Ono M, Takahara D, Tarutani A, Imura S, Masuda-Suzukake M, Higuchi M, Yanai K, Hisanaga SI, Hasegawa M (2017) Propagation of pathological alpha-synuclein in marmoset brain. Acta neuropathologica communications 5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Sanchez J et al. (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41:1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldner F, Stelzer Y, Shivalila CS, Abraham BJ, Latourelle JC, Barrasa MI, Goldmann J, Myers RH, Young RA, Jaenisch R (2016) Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 533:95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza JM, Giasson BI, Chen Q, Lee VM, Ischiropoulos H (2000) Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J Biol Chem 275:18344–18349. [DOI] [PubMed] [Google Scholar]
- Spencer B, Valera E, Rockenstein E, Overk C, Mante M, Adame A, Zago W, Seubert P, Barbour R, Schenk D, Games D, Rissman RA, Masliah E (2017) Anti-alpha-synuclein immunotherapy reduces alpha-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta neuropathologica communications 5:7. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840. [DOI] [PubMed] [Google Scholar]
- Stokholm MG, Danielsen EH, Hamilton-Dutoit SJ, Borghammer P (2016) Pathological alpha-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann Neurol 79:940–949. [DOI] [PubMed] [Google Scholar]
- Svensson E, Horvath-Puho E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, Sorensen HT (2015) Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 78:522–529. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Razzaq A, Ghetti B, Lilley KS, Spillantini MG (2003) Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J Biol Chem 278:44405–44411. [DOI] [PubMed] [Google Scholar]
- Tomlinson JJ, Shutinoski B, Dong L, Meng F, Elleithy D, Lengacher NA, Nguyen AP, Cron GO, Jiang Q, Roberson ED, Nussbaum RL, Majbour NK, El-Agnaf OM, Bennett SA, Lagace DC, Woulfe JM, Sad S, Brown EG, Schlossmacher MG (2017) Holocranohistochemistry enables the visualization of alpha-synuclein expression in the murine olfactory system and discovery of its systemic anti-microbial effects. Journal of neural transmission. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran HT, Chung CH, Iba M, Zhang B, Trojanowski JQ, Luk KC, Lee VM (2014) Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell Rep 7:2054–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenovic M, Tresse E, Mazzulli JR, Taylor JP, Krainc D (2012) Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci 32:4240–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera E, Masliah E (2016) Combination therapies: The next logical Step for the treatment of synucleinopathies? Mov Disord 31:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbaan D, Marinus J, Visser M, van Rooden SM, Stiggelbout AM, van Hilten JJ (2007) Patient-reported autonomic symptoms in Parkinson disease. Neurology 69:333–341. [DOI] [PubMed] [Google Scholar]
- Vicente Miranda H et al. (2017) Glycation potentiates alpha-synuclein-associated neurodegeneration in synucleinopathies. Brain. [DOI] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM (2011) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Abdelmotilib H, Liu Z, Stoyka L, Daher JP, Milnerwood AJ, Unni VK, Hirst WD, Yue Z, Zhao HT, Fraser K, Kennedy RE, West AB (2016) G2019S-LRRK2 Expression Augments alpha-Synuclein Sequestration into Inclusions in Neurons. J Neurosci 36:7415–7427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts JC, Giles K, Oehler A, Middleton L, Dexter DT, Gentleman SM, DeArmond SJ, Prusiner SB (2013) Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A 110:19555–19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrasidlo W et al. (2016) A de novo compound targeting alpha-synuclein improves deficits in models of Parkinson’s disease. Brain 139:3217–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ysselstein D, Joshi M, Mishra V, Griggs AM, Asiago JM, McCabe GP, Stanciu LA, Post CB, Rochet JC (2015) Effects of impaired membrane interactions on alpha-synuclein aggregation and neurotoxicity. Neurobiol Dis 79:150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zharikov AD, Cannon JR, Tapias V, Bai Q, Horowitz MP, Shah V, El Ayadi A, Hastings TG, Greenamyre JT, Burton EA (2015) shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. The Journal of clinical investigation 125:2721–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]