

Abstract
Factors governing the development of liver fibrosis in nonalcoholic steatohepatitis (NASH) are only partially understood. We recently identified adipocyte enhancer binding protein 1 (AEBP1) as a member of a core set of dysregulated fibrosis-specific genes in human NASH. Here we sought to investigate the relationship between AEBP1 and hepatic fibrosis. We confirmed that hepatic AEBP1 expression is elevated in fibrosis compared to lobular inflammation, steatosis, and normal liver, and increases with worsening fibrosis in NASH patients. AEBP1 expression was upregulated 5.8-fold in activated hepatic stellate cells and downregulated during chemical and contact induction of biological quiescence. In LX-2 and HepG2 cells treated with high glucose (25 mM), AEBP1 expression increased over 7-fold compared to normal glucose conditions. In response to treatment with either fructose or palmitate, AEBP1 expression in primary human hepatocytes increased 2.4-fold or 9.6-fold, but was upregulated 55.8-fold in the presence of fructose and palmitate together. AEBP1 knockdown resulted in decreased expression of nine genes previously identified to be part of a predicted AEBP1-associated NASH co-regulatory network and confirmed to be upregulated in fibrotic tissue. We identified binding sites for two miRNAs known to be downregulated in NASH fibrosis, miR-372-3p and miR-373-3p in the AEBP1 3’ untranslated region. Both miRNAs functionally interacted with AEBP1 to regulate its expression. These findings indicate a novel AEBP1-mediated pathway in the pathogenesis of hepatic fibrosis in NASH.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a chronic, frequently progressive condition resulting from excessive accumulation of fat in hepatocytes. Nonalcoholic steatohepatitis (NASH) is a clinically advanced form of NAFLD characterized by hepatic inflammation with or without scarring that is associated with increased liver-related morbidity and mortality [1]. Severe liver fibrosis represents the end-stage pathology evolving from a number of pathogenic mechanisms [2] and is considered a major risk factor for the development of hepatocellular carcinoma [3]. Although oxidative stress [4], pro-inflammatory cytokines [5, 6], and immune response [7, 8] are associated with inflammation and fibrosis in NASH, the molecular mechanisms by which fibrosis develops and progresses in these patients remain only partially understood.
In the course of our studies on differential gene expression in NASH patients with severe fibrosis, we observed upregulation of adipocyte enhancer binding protein 1 (AEBP1), also known as aortic carboxypeptidase (ACLP) compared to NASH patients with no histological evidence of fibrosis [9]. These findings were consistent with those showing increased hepatic ACLP/AEBP1 protein expression with NAFLD progression in humans and mice [10]. AEBP1 is a multifunctional protein implicated in a wide range of biological processes including adipogenesis [11–13], cell differentiation [14–16], and macrophage cholesterol homeostasis [17], although a role for this protein in forming collagen-rich tissues and in the development of tissue fibrosis is also emerging. Mutations in the AEBP1 gene can cause an inherited connective tissue disorder [18]. In patients with idiopathic pulmonary fibrosis and mice with bleomycin-induced pulmonary fibrosis, AEBP1 expression is upregulated in lung tissue [19], where it controls myofibroblast differentiation through a mechanism involving activation of transforming growth factor beta receptors, stimulation of SMAD3, enhanced expression of smooth muscle actin, and subsequent collagen production [19]. AEBP1 was recently found to be expressed in hepatic stellate cells (HSCs) and shown to complex with frizzled-8 and low-density lipoprotein-related receptor 6 to activate canonical WNT signaling, resulting in activation of HSCs in mouse models of NASH [10]. In parallel, AEBP1 was identified as a key control gene of a NASH co-regulatory network constructed using publicly available microarray data [20]. The same group also showed that in the presence of a high fat/high cholesterol diet, ApoE-/- mice with NASH showed higher hepatic AEBP1 expression relative to animals with NAFLD [20]. These aggregate findings suggest a role for AEBP1 in the development of fibrosis within the pathological context of NASH.
Here we sought to further investigate the relationship between AEBP1 and hepatic fibrosis in NASH. AEBP1 expression paralleled worsening severity of fibrosis in NASH patients, was upregulated in human hepatic stellate cell activation, and modulated by glucose, fructose, and palmitate. We found that AEBP1 regulates the expression of genes involved in extracellular matrix (ECM) maintenance, as well as a number of predicted target genes that were differentially expressed in NASH fibrosis. We identified and confirmed functional interactions between miR-372-3p and miR-373-3p and the AEBP1 3’-untranslated region and demonstrated that both miRNAs regulate AEBP1 expression in liver cells. These results implicate AEBP1 in a novel pathway associated with the development of hepatic fibrosis in NASH.
Materials and methods
RNA sequencing data analysis
Details of patient samples, RNA sequencing experiments, and raw data processing are provided elsewhere [9]. Previously generated RNA-sequencing data are freely available in the NCBI Bioproject database (https://www.ncbi.nlm.nih.gov/bioproject/512027). In our analyses here, we filtered the raw count table data to include only genes with an average count greater than five across all samples. We normalized raw counts using the voom algorithm [21] and adjusted for batch effects using the ComBat algorithm [22]. Following removal of outliers, the final sample size across the four histological grades was normal (n = 36), steatosis (n = 50), inflammation (n = 52), and fibrosis (n = 53). We performed a pairwise comparison of AEBP1 expression in severe fibrosis versus the other histological classes (i.e., normal, steatosis, or inflammation) using bivariate logistic regression. To investigate the relationship between AEBP1 gene expression and histological grade of fibrosis, we modeled an ordinal logistic regression considering grade as a dependent variable, adjusting for sex and age. P values were adjusted for multiple comparisons using the Bonferroni method (Padj). All analyses were conducted using R.
RNA extraction from liver wedge biopsies
Liver wedge biopsies were obtained from individuals enrolled in the Bariatric Surgery Program at the Geisinger Clinic Center for Nutrition and Weight Management [23]. Details of the study population can be found elsewhere [24–26] and in S1 Table. All study participants provided written informed consent for research, which was conducted according to The Code of Ethics of the World Medical Association (Declaration of Helsinki). The Institutional Review Boards of Geisinger Health System, Translational Genomics Research Institute, and Temple University School of Medicine approved the research protocol. Approximately 5–10 mg of biopsied liver tissue was homogenized in lysis buffer (Qiagen; Germantown, MD). Total RNA was extracted from homogenized lysate using the RNeasy Mini Kit (Qiagen) and quantified using the NanoDrop One spectrophotometer (Applied Biosystems; Foster City, CA).
Cell culture
LX-2 cells (Merck Millipore; Billerica, MA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific; Waltham, MA) supplemented with 2% fetal bovine serum (FBS) and 1% Pen/Strep (Omega Scientific; Tarzana, CA). Approximately 1 x 106 cells were thawed in T-75 flasks (Corning Life Sciences; Corning, NY) containing 12 mL cell culture medium and placed at 37°C in a Hera Cell 5% CO2 incubator (Thermo Fisher Scientific). Culture medium was replaced the first day after thawing, and then every 72 hours until 80% confluent. HepG2 and HEK293 cells (ATCC; Manassas, VA) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS) and 1% Pen/Strep (Omega Scientific). Culture medium was replaced the first day after thawing, and then every 48 hours until 80% confluent. Primary human hepatocytes (Thermo Fisher Scientific) were thawed in 50 mL Cryopreserved Hepatocyte Recovery Medium (CHRM) and plated in 500 υL William's E Medium supplemented with Hepatocyte Plating Supplement Pack on collagen-coated 24-well plates (Thermo Fisher Scientific). Culture medium was replaced the first day after thawing with William's E Medium supplemented with Hepatocyte Maintenance Supplement Pack.
Cell treatments
LX-2 cells were seeded at 0.5 x 106 cell/well on 6-well culture dishes (VWR International; Radnor, PA) and serum-starved overnight. Cell culture medium was aspirated and replaced with DMEM, 10% FBS, 0.5 mM isobutylmethylxanthine, 1 μM dexamethasone, and 167 nM insulin [i.e., MDI solution (Sigma-Aldrich; St. Louis, MO)] for 72 hours to induce a state resembling biological quiescence [27, 28].
Primary human hepatocytes were serum-starved overnight, and then treated with 1 mM palmitate (Sigma-Aldrich) conjugated with bovine serum albumin (BSA; [Omega Scientific]), 20 mM fructose, or a combination of the two for 48 hours. Treatment with 1% BSA was included as a negative control. Oil Red O staining was used to assess lipid uptake.
Total RNA extraction and quantification from cells
Total RNA was extracted from cells using the RNeasy mini kit (Qiagen) according to the manufacturer’s protocol. RNA quality and concentration were determined by absorbance at 260 nm using the NanoDrop One spectrophotometer (Thermo Fisher Scientific). MicroRNA was extracted from cells using the miRNeasy mini kit (Qiagen) following the product protocol. The miRNA quantity and quality were assessed using the 2100 Bioanalyzer System (Agilent Technologies; Santa Cruz, CA).
Quantitative real-time PCR (qPCR)
The TaqMan RNA-to-Ct 1-Step kit (Thermo Fisher Scientific) in conjunction with the QuantStudio 6 Flex Real-Time PCR system (Thermo Fisher Scientific) and TaqMan commercial primers were used to measure transcript levels. MiRNA was processed using the Taqman MicroRNA Reverse Transcription Kit (Applied Biosystems), followed by amplification using commercial probes (Applied Biosystems). Cycle threshold (Ct) levels were generated using QuantStudio Real-Time PCR Software 1.0. Messenger RNA and miRNA data were normalized using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and RNU6B (both of which showed invariant expression levels), respectively. The– ΔΔCt method was used to determine fold-change of gene expression.
Protein extraction and quantification
Cells were lysed in 350 uL RIPA buffer and lysate was transferred to 1.5 ml tubes and centrifuged at 5000 x g for 5 minutes at 4°C. Total protein concentrations for each sample were determined using the Pierce BCA Protein Assay kit (ThermoFisher Scientific). Levels of AEBP1 and GAPDH were detected using western blot analysis. An equal amount of protein was loaded on a NuPage pre-cast Bis-Tris protein gel with a 4–12% polyacrylamide gradient (Life Technologies), and then transferred from gels to nitrocellulose membranes (Life Technologies). Membranes were placed in Pierce Protein-Free (PBS) Blocking Buffer (ThermoFisher Scientific) for ninety minutes at room temperature, and then incubated overnight at 4°C with primary antibodies directed against ACLP/AEBP1 (anti-mouse 1:1000 dilution; Santa Cruz; Dallas, TX; catalog number: sc-271374) or GAPDH (anti-rabbit 1:1000; Cell Signaling Technology; Danvers, MA). Membranes were washed for eight minutes in 1X TBS-Tween buffer, and then incubated with secondary antibodies, either anti-mouse or anti-rabbit, labeled with horseradish peroxidase (1:3000; Cell Signaling Technology) for one hour at room temperature. Membranes were incubated with Clarity Western ECL Blotting Substrate (Bio-Rad; Hercules, CA) and protein bands were imaged using the Odyssey FC imaging system (LI-COR Biotechnology; Lincoln, NE).
Transfection with AEBP1 siRNA
Approximately 2x105 LX-2 cells/well were seeded in a 24-well plate with 6 uL Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific) containing either 50 nM or 100 nM Silencer Select Pre-Designed siRNA directed against AEBP1 or Silencer Select Negative Control Number 1 siRNA (both Thermo Fisher Scientific) and incubated at 37°C. Cells were harvested after 48 or 72 hours. Total RNA was extracted and expression levels of the genes of interest were assessed as described.
Prediction of miRNAs targeting the 3’ untranslated region (3’UTR) of AEBP1
Potential AEBP1-targeting miRNAs were identified using four prediction algorithms (miRWalk, miRanda, RNA22, and Targetscan) as implemented in miRWalk 2.0 (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/). The miRmap tool [29] was used to predict miRNA target repression strength. MiRNA conservation was determined using the phyloP (“phylogenetic P-values”) program (http://compgen.bscb.cornell.edu/phast).
3’UTR dual-luciferase reporter assay
The 3’ untranslated region (3’UTR) of AEBP1 and a scrambled sequence insert were generated by PCR and cloned into the multiple cloning site (MCS) of the pmirGLO vector (Promega Corp; Madison, WI) by the Emory Integrated Genomics Core (Atlanta, GA). The resulting (pGLO-AEBP1) and (pGLO-Scramble) reporter plasmids were sequence-verified. Approximately 2x105 HEK293 cells/well were seeded in a 24-well plate, co-transfected with 2 μg of the indicated vector and 10 pM miR-372-3p mimic, miR-373-3p mimic, or negative control mimic using 4 ul Lipofectamine 2000 per well (Thermo Fisher Scientific), and then incubated at 37°C for 48 hours. Luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega). Luminescence was detected using the EnVision 2105 Multimode Plate Reader (PerkinElmer; Waltham, MA). The ratio of firefly luciferase activity to Renilla luciferase activity was used to calculate normalized luciferase activity. The effect of miR-372-3p and miR-373-3p on gene expression was determined by setting the normalized luciferase activity of the negative mimic control + pGLO-AEBP1 3’UTR vector to 100% and showing the normalized luciferase activity of miR-372-3p mimic or miR-373-3p mimic + AEBP1 3’UTR vector as a percentage. For the control conditions, relative luciferase activity was determined by showing the normalized luciferase activity of miR-372-3p mimic or miR-373-3p mimic + pGLO-Scramble vector as a percentage of the negative mimic control + vector control. The Mann-Whitney U test was used to assess differences between conditions.
Transfection with miRNA mimics and inhibitors
Approximately 2x105 LX-2 cells/well were seeded in a 24-well plate with 6 uL Lipofectamine RNAiMAX transfection reagent (Thermo Fisher Scientific) containing either 75 nM mirVana miR-372-3p/miR-373-3p mimic, 75 nM anti-miR-372-3p/miR-373-3p inhibitor, or 75 nM scrambled sequence control (mirVana Negative Control #1 [Thermo Fisher Scientific]) and incubated for 48 hours at 37°C. Total RNA was extracted using the miRNeasy kit (Qiagen; Germantown, MD) and miRNA over-expression and knockdown was verified using qPCR.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 8 (GraphPad Software; La Jolla, CA). Data were analyzed as the mean ± standard deviation (SD) from at least three independent assays. The Kruskal-Wallis one-way analysis of variance test with a Dunn post-hoc test was used to assess differences between conditions, unless otherwise indicated. A P-value <0.05 was considered statistically significant.
Results
AEBP1 expression is upregulated in NASH fibrosis
We previously reported upregulated hepatic AEBP1 expression in NASH patients with fibrosis compared to those with NASH but no histological evidence for fibrosis identified using massively parallel RNA-sequencing [9]. These results are concordant with those of Teratani et al [10], who recently reported enhanced AEBP1 expression in liver tissue from NASH patients (N = 44) compared to individuals with NAFLD (N = 16) and metastatic liver cancer patients with no evidence of steatosis, inflammation, or fibrosis (N = 14). Here we assessed AEBP1 expression in NASH fibrosis versus other NAFLD histological classes (S1 Table) using pairwise comparisons. We observed significant differences in hepatic AEBP1 expression between fibrosis and normal (Padj = 1.5E-05), steatosis (Padj <0.001), and inflammation (Padj <0.001) (Fig 1A). Analysis of AEBP1 expression using quantitative RT-PCR in a subset of NAFLD patients validated genome-wide results (Fig 1B). We then examined the relationship across fibrosis stage using an ordinal logistic regression (adjusting for sex and age) in an analysis of the RNA-sequencing data. We observed a significant trend of increasing AEBP1 levels with increasing severity of fibrosis with advanced fibrosis (i.e., cirrhosis) > incomplete cirrhosis > bridging fibrosis (beta = 0.906; P = 0.028). The median expression level increased from 4.69 (bridging fibrosis) to 4.97 (incomplete cirrhosis) to 5.37 (cirrhosis). (Fig 1C). The partial regression P values for bridging fibrosis vs incomplete fibrosis, and incomplete cirrhosis vs cirrhosis were statistically significant (P < 0.01).
Fig 1. Hepatic AEBP1 expression in NAFLD patients.
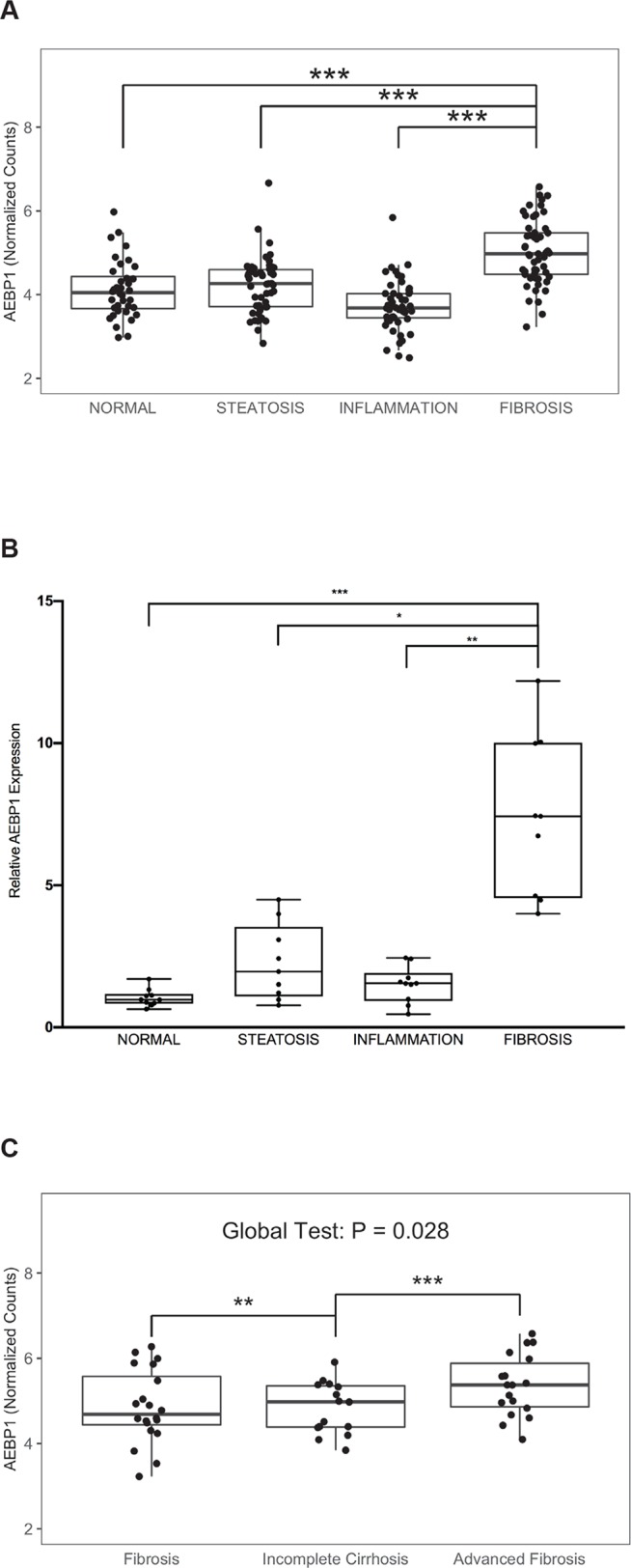
A) Using RNA-sequencing data [9], we determined average AEBP1 transcript abundance (normalized counts) in NAFLD samples across a spectrum of liver histology (36 normal, 50 steatosis, 52 inflammation, and 53 fibrosis). The ordinal logistic regression showed a significant positive trend (β = 0.244; P = 4.8E-4), whereas the pairwise comparisons conducted with logistic regression showed a significant difference between normal and fibrosis histological grades (Padj = 1.3E-03). B) AEBP1 gene expression was measured in a subset of NAFLD patients with steatosis (n = 10), lobular inflammation (n = 10), cirrhosis (n = 10), and normal liver histology (n = 10). Real time qPCR was performed using Taqman gene expression assays for AEBP1 and GAPDH. The Kruskal-Wallis one-way analysis of variance test with a Dunn post-hoc test was used to assess differences in expression between fibrosis and the other histological grades. *P≤0.05, **P≤0.001, and *** P≤ 0.0001. C) Using RNA-sequencing data (9), we determined AEBP1 transcript abundance (normalized counts) in human NAFLD samples with bridging fibrosis (n = 20), incomplete cirrhosis (n = 15), and advanced fibrosis (n = 18). The ordinal logistic regression adjusted for sex and age showed a global significant positive trend (beta = 0.906; P = 0.028), with the median expression level across different stages of fibrosis.
AEBP1 expression is upregulated in activated hepatic stellate cells
Findings of increased hepatic AEBP1 expression in NASH patients with fibrosis led us to ask whether the gene was differentially regulated during myofibroblastic transformation of hepatic stellate cells (HSCs), the key fibrogenic cells of the liver. We compared AEBP1 expression levels in HSCs (LX-2 cells) grown on plastic to achieve a myofibroblast phenotype or treated with MDI solution to induce a state resembling biological quiescence [30]. Increased ACTA2/αSMA expression was indicative of LX-2 cell activation (Fig 2A and 2B). In untreated LX-2 cells, AEBP1 transcript and protein expression was 5.8-fold and 44% higher, respectively, compared to those treated with MDI solution (Figs 2A and 2B and S1). The form of the ACLP/AEBP1 protein (~185 kDA) detected was the same as that reported earlier [10]. We also observed that AEBP1 expression decreased over time in response to MDI treatment, showing a 2.1-, 3.1-, and 3.6-fold (log2) reduction at 24, 48, and 72 hours, respectively (Fig 2C). In LX-2 cells grown on Matrigel-coated plates, AEBP1 levels decreased in a manner similar to that seen in MDI-treated cells (S2 Fig).
Fig 2. AEBP1 expression in untreated and MDI-treated LX-2 cells.
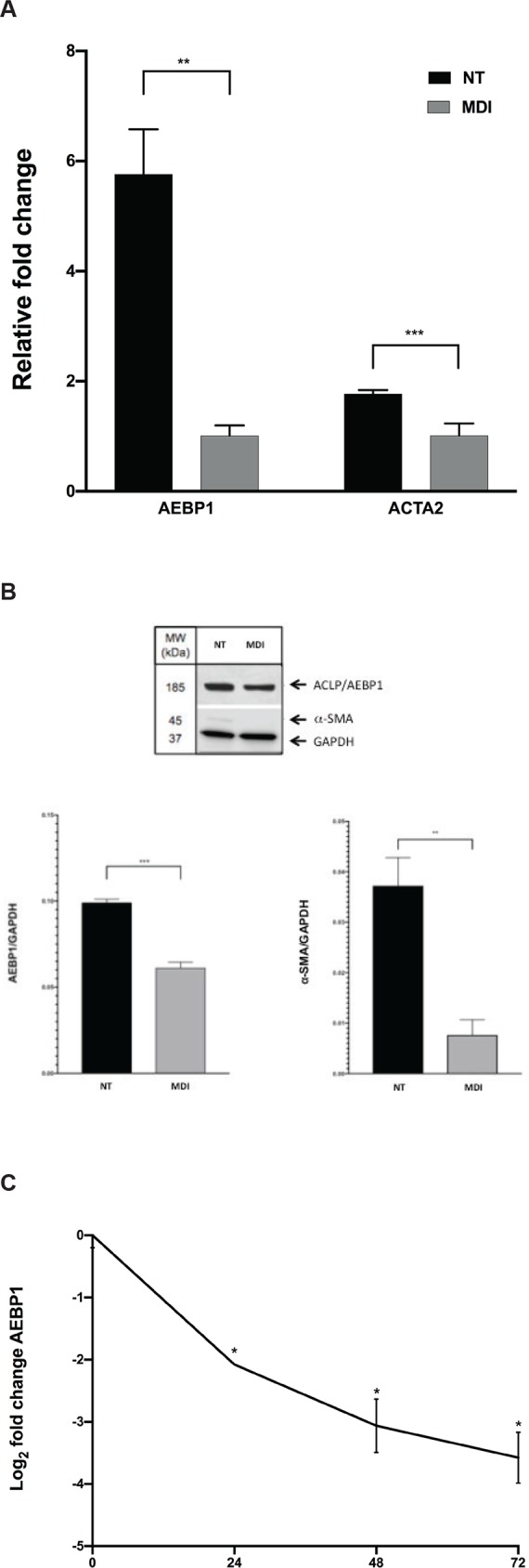
A) AEBP1 and ACTA2 mRNA levels were measured by qPCR in no treatment controls (NT; i.e., activated) and MDI-treated (i.e., quiescent) LX-2 cells. AEBP1 and ACTA2 levels were normalized to GAPDH for RNA and protein expression analysis. B) Protein levels of AEBP1, αSMA, and GAPDH were assayed using western blot. Imaging was performed and used for densitometry. C) Time course of AEBP1 expression over 72 hours of MDI treatment. Transcript levels of AEBP1 expression were normalized to GAPDH. All experiments were performed in triplicate. *P≤0.05.
Effect of glucose, palmitate, and fructose on AEBP1 expression in liver cells
Because of the close association between type 2 diabetes and NAFLD, we investigated the effects of glucose in LX-2 cells and HepG2 cells, both of which are widely used for in vitro studies of NASH. In both cell types, AEBP1 expression increased >7.0—fold over 24 hours and remained upregulated up to 48 hours under high glucose (HG: 25 mM) compared to normal glucose (NG: 5 mM) conditions (Fig 3A). We have previously observed effects of lipid loading on target gene expression in activated HSCs [31] and in primary cultured murine HSCs, palmitate was observed to additively increase AEBP1 expression [10]. To extend these studies to hepatocytes, we measured the effect of lipid loading on AEBP1 expression in primary human hepatocytes treated with 1 mM palmitate. In these cells, AEBP1 expression increased 9.6-fold compared to vehicle (Fig 3B). We next sought to investigate the effects of fructose on AEBP1 expression given the relationship between high fructose consumption and severity of NAFLD [32] and found that AEBP1 expression increased 2.4-fold compared to the control cells (Fig 3B). Interestingly, in response to treatment with a mixture of 1 mM palmitate and 20 mM fructose, we saw a large, non-additive effect (>55.8-fold increase) on AEBP1 expression.
Fig 3. Effect of glucose, palmitate, and fructose on AEBP1 expression.
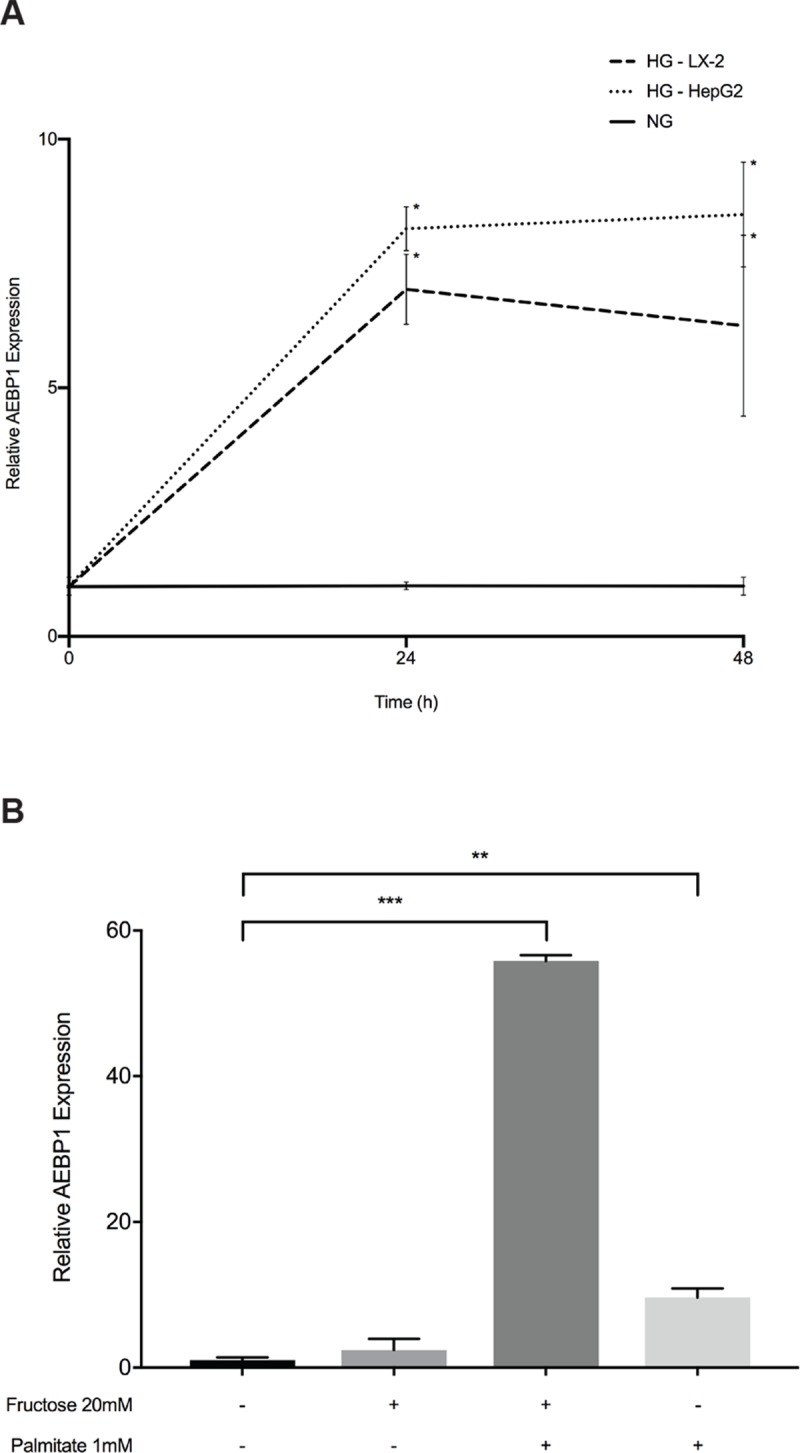
LX-2 and HepG2 cells were serum-starved for 24 hours to arrest and synchronize cell growth. After this time, cells were grown in medium supplemented with 5% FBS, containing either normal glucose (NG: 5.5 mM) or high glucose (HG: 25 mM) for 24 or 48 hours. A) Relative quantification of AEBP1 transcript levels was performed using quantitative PCR. AEBP1 expression levels were normalized using GADPH, setting values to one under NG conditions at each time point. Data are presented as mRNA fold-increase incubated under HG or NG conditions. Results represent average of three independent experiments. Data are mean ± SD.*P<0.05. B) Primary human hepatocytes were treated with 1 mM BSA-conjugated palmitate, 20 mM fructose, or a combination of the two for 48 hours. The black bar represents treatment with BSA-only. All experiments were performed in triplicate. Data are mean ± SD. Differences between conditions were assessed using one-way ANOVA test with a Dunnett’s multiple comparison post-test. **P<0.005; ***P<0.0001.
AEBP1 regulates predicted target genes uniquely associated with NASH fibrosis
In a recent study, we identified 34 transcripts that were differentially expressed in NASH patients with or without fibrosis [9], nine of which overlapped with predicted AEBP1 target genes identified by bioinformatic analysis [20]. We first confirmed increased expression of these genes in an independent sample of NASH patients with fibrosis relative to normal histology (Fig 4A). To establish an interaction between AEBP1 and these predicted targets, we knocked down AEBP1 expression and measured levels of the nine genes in LX-2 cells. Knockdown efficiency of AEBP1 was verified through qPCR analysis (S3 Fig). As shown in Fig 4B, in the presence of AEBP1 siRNA, we observed a reduction of aldo-keto reductase family 1 member 10 (AKR1B10; 7.8-fold), EGF containing fibulin extracellular matrix protein 1 (EFEMP1; 2.3-fold), integrin beta-like protein 1 (ITGBL1; 2.7-fold), and secreted phosphoprotein 1 (SPP1; 3.4-fold). Expression levels of dermatopontin (DPT), laminin subunit gamma-3 (LAMC3), and stathmin 2 (STMN2) were not detectable in LX-2 cells.
Fig 4. Expression of predicted AEBP1 target genes.
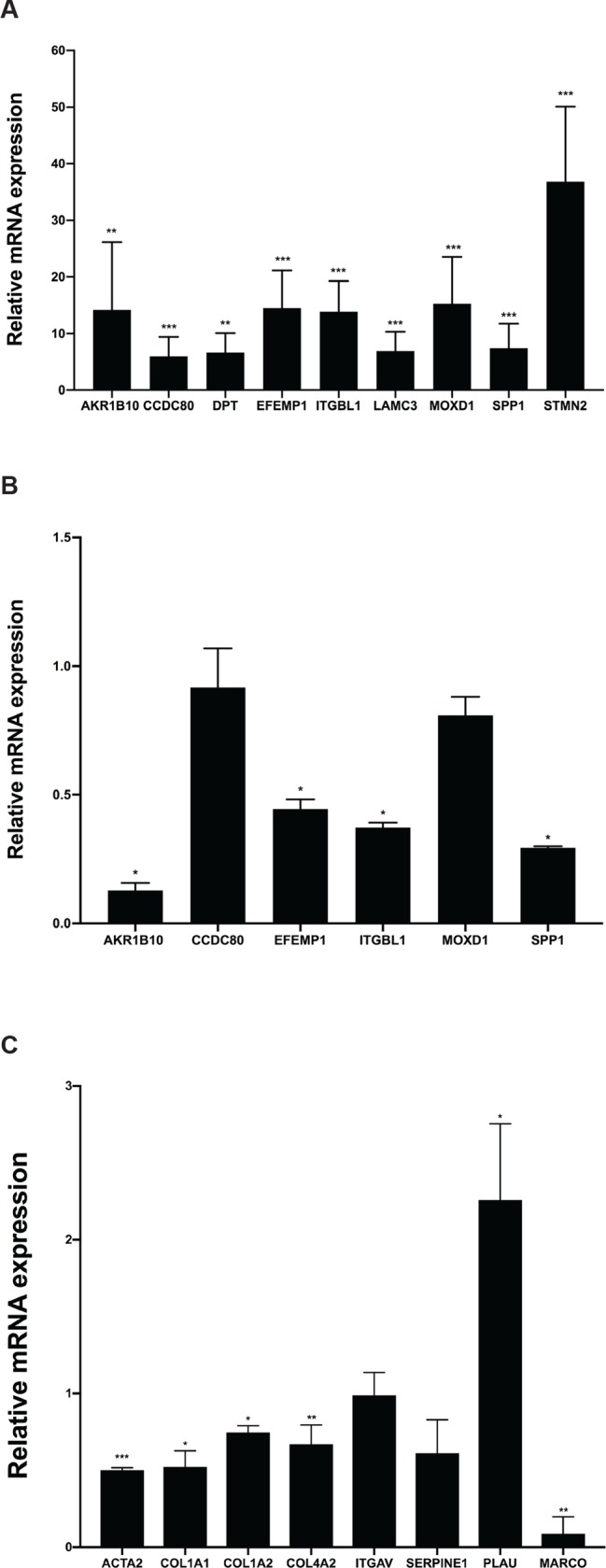
A) We measured levels of the genes of interest using qPCR and hepatic RNA from NAFLD patients with normal liver histology and severe fibrosis. Transcript levels were normalized to GAPDH, which showed invariant expression. Effects of AEBP1 knockdown on B) fibrosis-specific predicted target genes and C) predicted target genes involved with ECM maintenance. Data are presented as relative fold-change. A Mann-Whitney U test was performed to assess statistical significance. *P≤0.05, **P≤0.001, and *** P≤ 0.0001.
To determine the role of AEBP1 in the accumulation of ECM proteins, we reduced AEBP1 expression using siRNA and quantified levels of selected genes involved in ECM structure and maintenance by qPCR. We also analyzed levels of integrin subunit alpha V (ITGAV), collagen type IV alpha 2 chain (COL4A2), and macrophage receptor with collagenous structure (MARCO), which encode known cell adhesion proteins and are predicted AEBP1 target genes [20], although these genes were not part of the set of fibrosis-specific genes identified in our previous work [9]. As shown in Fig 4C, levels of COL4A2 and MARCO were decreased 1.5-fold and 11.3-fold, respectively, compared to levels obtained in cells transfected with the negative control siRNA. Given the upregulation of AEBP1 observed in activated HSCs, we also investigated effects of AEBP1 depletion on expression of selected genes known to be affected by myofibroblastic transition [30]. We found that knockdown of AEBP1 expression resulted in 2.0-fold, 1.9-fold, 1.3-fold, and 1.6-fold (log2) decrease in levels of actin alpha 2 smooth muscle (ACTA2), collagen type 1 alpha 1 chain (COL1A1), collagen type 1 alpha 2 chain (COL1A2), and serpin family E member 1 (SERPINE1) expression, respectively, and a 2.3-fold increase in urokinase plasminogen activator (PLAU) expression (Fig 4C).
AEBP1 is regulated by miRNAs downregulated in NASH fibrosis
In colorectal cancer cells, AEBP1 expression is negatively regulated by miR-214 [33]. To explore potential mechanisms of miRNA-mediated regulation of AEBP1 in NASH fibrosis, we analyzed the AEBP1 3’UTR to identify putative miRNA binding sites using a comparative platform comprised of four different algorithms (see Methods section). Seven miRNA binding site predictions were identified using all four algorithms (S2 Table). Two of these, miR-372-3p and miR-373-3p were found to be downregulated in NAFLD patients with fibrosis compared to those with normal histology: miR-372-3p (log2 fold-change = -1.7; Padj = 0.031) and miR-373-3p (log2 fold-change = -2.4; Padj = 0.001) [24]. Levels of miR-372-3p were 2.1-fold higher, while those of miR-373-3p were 2.9-fold lower, in quiescent versus activated LX-2 cells, respectively (S4 Fig). According to miRmap [29], miR-372-3p and miR-373-3p share overlapping binding sites at positions 3829–3835 bp (NM_001129) in the AEBP1 3’UTR (Fig 5A). To validate this interaction, we used a dual luciferase reporter assay to measure direct binding between the two miRNAs and the AEBP1 3’UTR. Co-transfection of a construct containing the AEBP1 3’UTR and exogenous miR-372-3p or miR-373-3p resulted in a 26% and 46.7% reduction in relative luciferase activity compared to control conditions, respectively (Fig 5B).
Fig 5. miR-372-3p and miR-373-3p functionally interact with AEBP1 to regulate its expression.

A) The target site for miR-372-3p and miR-373-3p was predicted to align to positions 3829–3835 in the AEBP1 3’UTR (GenBank Accession NM_001129.4). Vertical lines indicate paired alignment between the miRNA seed region and AEBP1 3’UTR sequence. B) Approximately 20,000 human embryonic kidney 293 (HEK293) cells were co-transfected with the pGLO-AEBP1 3’UTR vector or scrambled control vector (pGLO-Scramble) and miR-372-3p mimic, miR-373-3p mimic, or scrambled sequence control miRNA (negative control [NC] mimic), and luciferase activity was measured 48 hours post-transfection using the Dual-Glo Luciferase Assay System (Promega). The ratio of firefly luciferase activity to renilla luciferase activity was determined for each sample and the firefly/renilla relative luminometer units (RLU) in HEK293 cells co-transfected with miRNA mimic and the pGLO-AEBP1 3’UTR vector were compared with those from cells co-transfected with pGLO-Scramble and NC mimic. The Mann-Whitney U test was used to assess differences between conditions; differences between the negative controls were not statistically significant. For functional studies, activated LX-2 cells were transfected with 75 nM mirVana miRNA-372-3p/miR-373-3p mimic, 75 nM anti-miR-372-3p/miR-373-3p inhibitor, or 75 nM scrambled sequence control. After 48 hours cells were harvested and total RNA was extracted. The TaqMan RNA-to-Ct 1-Step kit (Thermo Fisher Scientific) and TaqMan commercial primers were used to measure C) miR-372-3p and AEBP1 levels and D) miR-373-3p and AEBP1 levels in the presence of the respective mimic and siRNA. Cycle threshold levels were generated using QuantStudio Real-Time PCR Software 1.0. miRNA and mRNA data were normalized using RNU6B + RNU48 and GAPDH, respectively. Black bars represent the scrambled miRNA negative control, while the gray bars represent treatment conditions. All experiments were performed in triplicate. Data are means ±SD. A paired t-test was used to compare the treatment group to the no treatment control group (NTC) *P<0.05; **P<0.001; ***P<0.0001.
We next transfected LX-2 cells with mimics or siRNAs to determine possible functional consequences of miR-372-3p and miR-373-3p on AEBP1 expression. As expected, transfection with miRNA mimic or inhibitor corresponded with an increase or reduction, respectively, in miR-372-3p (Fig 5C) and miR-373-3p (Fig 5D). In the presence of miR-372-3p mimic, AEBP1 expression was reduced 1.8-fold (p<0.05). miR-372-3p inhibitor increased AEBP1 expression, but the change was not statistically significant (Fig 5C). Treatment of LX-2 cells with miR-373-3p mimic reduced AEBP1 levels 1.5-fold (P = 0.0573), although the presence of miR-373-3p inhibitor did not significantly alter AEBP1 expression.
Discussion
Given the strong link between fibrosis and risk of liver-related mortality in NAFLD patients [34, 35], efforts to identify and characterize the specific mechanisms contributing to NAFLD progression are critical for the development of effective therapeutic and preventive strategies. We previously identified 34 genes that were upregulated in NASH fibrosis, relative to non-fibrotic NASH. One of these genes, AEBP1, was recently reported to be a potential central regulator driving the transition of NASH, possibly through the modulation of target genes [20]. One of the major findings of the current work demonstrated that AEBP1 regulates the expression of nine algorithm-predicted target genes that overlapped with our set of fibrosis-specific genes, including AKR1B10, CCDC80, DPT, EFEMP1, ITGBL1, LAMC3, MOXD1, SPP1, and STMN2. We also showed that AEBP1 regulates expression of genes known to play a role in ECM production and maintenance, concordant with recent findings from a comprehensive analysis of AEBP1 in mouse models of NAFLD [10]
We found that hepatic AEBP1 levels were elevated in fibrotic tissue compared to other non-fibrotic histologic grades. We also observed a significant trend of increasing AEBP1 expression concomitant with worsening severity of fibrosis with the highest hepatic levels found in patients with advanced fibrosis. Teratani et al [10] found increased hepatic AEBP1 staining in NASH patients compared to NAFLD patients. In that study, however, AEBP1 expression relative to fibrosis was not investigated; thus, the current results extend these findings to indicate that AEBP1 expression in the liver parallels the onset of fibrosis in NASH and suggest that AEBP1 may represent a specific therapeutic target to prevent the development of NASH fibrosis.
The presence of increasing AEBP1 levels in parallel with worsening fibrosis is consistent with changes in AEBP1 expression occurring with transdifferentiation of LX-2 cells, an immortalized human hepatic stellate cell line that retains important features of primary hepatic stellate cells [30]. We observed that AEBP1 levels decreased in a time-dependent manner in LX-2 cells treated with MDI solution or grown on Matrigel to induce quiescence compared to cells grown on plastic that develop a myofibroblastic phenotype. These findings are in contrast to those showing more abundant AEBP1 expression in quiescent rat aortic smooth muscle cells compared to actively proliferating cells [15]. However, a recent study demonstrated that recombinant AEBP1 enhanced myofibroblast differentiation and promoted fibrogenesis [14], consistent with our results in LX-2 cells. Further, AEBP1 expression was elevated in HSCs concomitant with NAFLD progression [10], indicating that increased, not decreased, expression occurs with myofibroblastic activation. Importantly, in that work HSC-specific AEBP1-knockout mice developed NASH following a high fat/high cholesterol diet for 24 weeks, but exhibited significantly less fibrosis compared to AEBP1-floxed animals under the same conditions. Repression of AEBP1 expression also inhibited HSC activation and significantly decreased expression of genes involved with ECM [10]. Together, these findings support a role for AEBP1 in HSC activation and initiation of fibrosis, a process that may involve increased STAT3 signaling in response to elevated free fatty acids, leptin, and IL-6 [10].
Previous work reported that AEBP1 expression was restricted to stellate cells and not observed in hepatocytes [10] using immunohistochemistry to distinguish cell specificity. In the current work, we detected AEBP1 expression in primary human hepatocytes and in the HepG2 hepatoma cell line. These discrepant findings may be attributed to differences in measurement: immunohistochemistry to visualize protein expression is less sensitive than PCR or sequencing to detect mRNA. In addition, AEBP1 is a secreted protein, thus intracellular levels may be low relative to its location in the extracellular matrix.
To extend AEBP1 expression findings from human liver biopsy tissue, we used in vitro models of diabetes and NAFLD by assaying the effects of glucose, fructose, and palmitate treatments on human stellate and liver cells. There is a strong association between diabetes and liver fibrosis, with a much higher risk of progression of NAFLD in patients with diabetes [36]; thus, glucose metabolism appears to play a role in the development of steatohepatitis and fibrosis. Like glucose, fructose has been implicated in NAFLD. Daily consumption of fructose was associated with a higher stage of fibrosis and a lower grade of steatosis in patients with NASH [32]. In mice, fructose, combined with a diet high in fat and cholesterol, can induce NASH and hepatic fibrosis within four to six months [37]. Our findings that glucose, fructose, and palmitate upregulate AEBP1 expression are consistent with a role for AEBP1 in the underlying mechanism (s) of these risk factors. However, the mechanism (s) by which these factors upregulate AEBP1 expression is not yet known. In primary human hepatocytes, palmitate and palmitate plus high glucose were both found to induce expression of the fatty acid binding protein (FABP4) gene [38]. AEBP1 binds to the proximal promoter of FABP4 [39], suggesting it may have a role in the response to palmitate and glucose. AEBP1 also interacts with tumor suppressor phosphatase and tensin homolog deleted on chromosome ten (PTEN), which is a negative regulator of insulin signaling and insulin sensitivity in adipose tissue [40]. Saturated fatty acids increase PTEN levels [41], which may be another pathway by which palmitate influences AEBP1 expression. Why fructose and palmitate together caused a dramatically higher level of upregulation than either treatment alone warrants further exploration; however, it is possible that AEBP1 may be a common regulatory point for gene expression changes induced by both treatments.
Another common regulatory point may be WNT signaling. Interestingly, AEBP1 has been shown to be associated with activation of the canonical WNT pathway in HSCs [10]. Activation of this pathway causes a methylation-dependent downregulation of PPARγ, which is involved in HSC activation [42]. Increased AEBP1 expression from exogenous sugars or fats could induce canonical WNT pathway signaling that would reduce PPARγ promoter activity and subsequent expression in HSCs, leading to decreased PPARγ expression and resultant HSC activation. This is consistent with the observed synergistic effect of a high fat diet with fructose on decreasing PPARγ expression in mice [43]. However, palmitic acid was also observed to increase PPARγ expression in HepG2 cells, suggesting that stellate cells may possess a response to increased fatty acid exposure that is distinct from hepatocytes.
We used a combination of four predictive algorithms to identify miR-372-3p and miR-373-3p as potential AEPB1-targeting miRNAs and found that both miRNAs negatively regulated AEBP1 expression. Both miR-372-3p and miR-373-3p were significantly downregulated in NAFLD patients with fibrosis compared to those with normal histology [24], which would be consistent with upregulated AEBP1 expression. Other studies have reported involvement of both miRNAs in human malignant tumors through the targeted downregulation of a number of genes including fibroblast growth factor 9 [44], dickkopf WNT signaling pathway inhibitor 1 [45], and others [46–48], and miR-372-3p was shown to promote epithelial mesenchymal transition in breast carcinoma through WNT pathway activation [46]. miR-372-3p and miR-373-3p share overlapping binding sites at position 3829–3835 bp in the AEBP1 3’UTR. This position is proximal to a binding site for miR-214 (3842–3848 bp), which was found to directly target and downregulate AEBP1 expression in colorectal cancer (HT-29) cells [33]. The presence of clustered functional miRNA binding sites indicates that this region may be important for the regulation of AEBP1. More detailed studies involving mutagenesis of the predicted sites will be needed to further characterize the post-transcriptional miRNA regulation of AEBP1.
The current findings add to our understanding of the role of AEBP1 in hepatic fibrosis within the context of NASH in several ways. First, AEBP1-mediated regulation of fibrosis-specific genes, as well as those involved in ECM production and maintenance, suggest that this protein contributes to hepatic fibrosis through modulation of a gene network that may be specific to HSCs. Second, obesity-related factors that have also been linked to NAFLD, including glucose, fructose, and palmitate, increase AEBP1 expression, thereby exacerbating expression of these genes. Finally, miR-372-3p and miR-373-3p, which may function to downregulate AEBP1 and are known to regulate canonical WNT signaling [45, 46], are reduced in NASH patients with advanced fibrosis. Together, these results provide a potential mechanism by which AEBP1 may contribute to NASH-related fibrosis (Fig 6).
Fig 6. Potential mechanism by which AEBP1 affects the development of fibrosis in NASH.

Hyperglycemia, lipid loading, and fructose treatment cause an increase in AEBP1 expression in liver cells. miR-372-3p and miR-373-3p, which are reduced in NAFLD patients with advanced fibrosis, functionally interact with AEBP1 to downregulate its expression. Increased expression of AEBP1 is associated with altered expression of key components of the ECM and algorithmically predicted target genes belong to a core set of fibrosis-specific genes previously identified by the authors.
The multicellular nature of biopsied tissue limits the interpretation of differential gene expression using RNA from intact liver. We were able to demonstrate patterns of AEBP1 expression similar to human liver in human stellate, HepG2, and primary human hepatocyte cell cultures, but we cannot exclude the possibility that other resident hepatic cell types may also contribute to its differential regulation with fibrosis. AEBP1 is expressed by perivascular and vascular cells, as well as the stromal-vascular fraction of white adipose tissue [14]. Whether these or other cell types in the liver express AEBP1 will require in situ hybridization or single cell analysis. We also chose to focus our studies on human tissue and cells. Despite the apparent phenotypic aspects of various mouse models that can be observed to parallel human NASH, there appears to be little mechanistic overlap at the level of gene expression [49]. Human studies of NAFLD and NASH are unfortunately limited by the difficulties in obtaining tissue for analyses, thus other models such as organoids [50], may be required to more faithfully recapitulate NAFLD and NASH for gene expression studies.
In summary, we found that AEBP1 expression was increased in human liver biopsies from patients with NASH fibrosis, in activated human stellate cells, and in human liver cells treated with glucose, fructose, and palmitate. AEBP1 regulated the expression of nine fibrosis-specific genes that were also members of an algorithm-predicted AEBP1 target gene network in NASH. We also found that AEBP1 functionally interacted with two miRNAs. Taken together, these findings support the idea that AEBP1 may be a central regulator of a complex fibrosis gene expression network in human liver.
Supporting information
(TIF)
LX-2 cells were seeded at 1 x 105 cell/well on 6-well plates coated with 350 μL of (1mg/mL) Matrigel Growth Factor Reduced (GFR) basement membrane matrix (Corning Inc; Corning, NY) and cultured for 72 hours at 37°C to induce a state resembling quiescence. Transcript levels of AEBP1 expression were normalized to GAPDH. All experiments were performed in triplicate. *P≤0.05.
(TIF)
AEBP1 expression was reduced and gene expression measured as described in the Methods section. A t-test was performed to assess statistical significance. ***P = 0.0001.
(TIF)
RT-qPCR was performed and miRNAs levels analyzed as described in the Methods section. Data were analyzed using a two-tailed t-test. *P<0.05.
(TIF)
(DOCX)
(DOCX)
Acknowledgments
We appreciate the vector cloning services provided by Dr. Oskar Laur at the Emory Integrated Genomics Core (EIGC) at Emory University School of Medicine.
Data Availability
The data are available in the NCBI public repository (https://www.ncbi.nlm.nih.gov/bioproject/512027).
Funding Statement
This study was funded in part by the National Institute of Diabetes and Digestive and Kidney Diseases (http://www.niddk.nih.gov) to JKD (DK088231). The NIDDK did not play any role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clinics in liver disease. 2004;8(3):521–33, viii. Epub 2004/08/28. 10.1016/j.cld.2004.04.004 . [DOI] [PubMed] [Google Scholar]
- 2.Altamirano-Barrera A, Barranco-Fragoso B, Mendez-Sanchez N. Management strategies for liver fibrosis. Ann Hepatol. 2017;16(1):48–56. 10.5604/16652681.1226814 . [DOI] [PubMed] [Google Scholar]
- 3.Ghouri YA, Mian I, Rowe JH. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J Carcinog. 2017;16:1 10.4103/jcar.JCar_9_16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koek GH, Liedorp PR, Bast A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin Chim Acta. 2011;412(15–16):1297–305. 10.1016/j.cca.2011.04.013 . [DOI] [PubMed] [Google Scholar]
- 5.Jorge AS, Andrade JM, Paraiso AF, Jorge GC, Silveira CM, de Souza LR, et al. Body mass index and the visceral adipose tissue expression of IL-6 and TNF-alpha are associated with the morphological severity of non-alcoholic fatty liver disease in individuals with class III obesity. Obes Res Clin Pract. 2016. 10.1016/j.orcp.2016.03.009 . [DOI] [PubMed] [Google Scholar]
- 6.Liu W, Baker RD, Bhatia T, Zhu L, Baker SS. Pathogenesis of nonalcoholic steatohepatitis. Cell Mol Life Sci. 2016;73(10):1969–87. 10.1007/s00018-016-2161-x . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate Immunity and Inflammation in NAFLD/NASH. Dig Dis Sci. 2016. 10.1007/s10620-016-4049-x . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heymann F, Tacke F. Immunology in the liver—from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13(2):88–110. 10.1038/nrgastro.2015.200 . [DOI] [PubMed] [Google Scholar]
- 9.Gerhard GS, Legendre C, Still CD, Chu X, Petrick A, DiStefano JK. Transcriptomic Profiling of Obesity-Related Nonalcoholic Steatohepatitis Reveals a Core Set of Fibrosis-Specific Genes. J Endocr Soc. 2018;2(7):710–26. Epub 2018/07/07. 10.1210/js.2018-00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teratani T, Tomita K, Suzuki T, Furuhashi H, Irie R, Nishikawa M, et al. Aortic carboxypeptidase-like protein, a WNT ligand, exacerbates nonalcoholic steatohepatitis. J Clin Invest. 2018;128(4):1581–96. Epub 2018/03/20. 10.1172/JCI92863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He GP, Muise A, Li AW, Ro HS. A eukaryotic transcriptional repressor with carboxypeptidase activity. Nature. 1995;378(6552):92–6. Epub 1995/11/02. 10.1038/378092a0 . [DOI] [PubMed] [Google Scholar]
- 12.Kim SW, Muise AM, Lyons PJ, Ro HS. Regulation of adipogenesis by a transcriptional repressor that modulates MAPK activation. The Journal of biological chemistry. 2001;276(13):10199–206. Epub 2001/01/21. 10.1074/jbc.M010640200 . [DOI] [PubMed] [Google Scholar]
- 13.Park JG, Muise A, He GP, Kim SW, Ro HS. Transcriptional regulation by the gamma5 subunit of a heterotrimeric G protein during adipogenesis. EMBO J. 1999;18(14):4004–12. Epub 1999/07/16. 10.1093/emboj/18.14.4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jager M, Lee MJ, Li C, Farmer SR, Fried SK, Layne MD. Aortic carboxypeptidase-like protein enhances adipose tissue stromal progenitor differentiation into myofibroblasts and is upregulated in fibrotic white adipose tissue. PLoS One. 2018;13(5):e0197777 Epub 2018/05/26. 10.1371/journal.pone.0197777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Layne MD, Endege WO, Jain MK, Yet SF, Hsieh CM, Chin MT, et al. Aortic carboxypeptidase-like protein, a novel protein with discoidin and carboxypeptidase-like domains, is up-regulated during vascular smooth muscle cell differentiation. The Journal of biological chemistry. 1998;273(25):15654–60. Epub 1998/06/23. 10.1074/jbc.273.25.15654 . [DOI] [PubMed] [Google Scholar]
- 16.Tumelty KE, Smith BD, Nugent MA, Layne MD. Aortic carboxypeptidase-like protein (ACLP) enhances lung myofibroblast differentiation through transforming growth factor beta receptor-dependent and -independent pathways. The Journal of biological chemistry. 2014;289(5):2526–36. Epub 2013/12/18. 10.1074/jbc.M113.502617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majdalawieh A, Zhang L, Fuki IV, Rader DJ, Ro HS. Adipocyte enhancer-binding protein 1 is a potential novel atherogenic factor involved in macrophage cholesterol homeostasis and inflammation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(7):2346–51. Epub 2006/02/08. 10.1073/pnas.0508139103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blackburn PR, Xu Z, Tumelty KE, Zhao RW, Monis WJ, Harris KG, et al. Bi-allelic Alterations in AEBP1 Lead to Defective Collagen Assembly and Connective Tissue Structure Resulting in a Variant of Ehlers-Danlos Syndrome. Am J Hum Genet. 2018;102(4):696–705. Epub 2018/04/03. 10.1016/j.ajhg.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schissel SL, Dunsmore SE, Liu X, Shine RW, Perrella MA, Layne MD. Aortic carboxypeptidase-like protein is expressed in fibrotic human lung and its absence protects against bleomycin-induced lung fibrosis. Am J Pathol. 2009;174(3):818–28. 10.2353/ajpath.2009.080856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lou Y, Chen YD, Sun FR, Shi JP, Song Y, Yang J. Potential Regulators Driving the Transition in Nonalcoholic Fatty Liver Disease: a Stage-Based View. Cell Physiol Biochem. 2017;41(1):239–51. Epub 2017/02/20. 10.1159/000456061 . [DOI] [PubMed] [Google Scholar]
- 21.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome biology. 2014;15(2):R29 Epub 2014/02/04. 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28(6):882–3. Epub 2012/01/20. 10.1093/bioinformatics/bts034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wood GC, Chu X, Manney C, Strodel W, Petrick A, Gabrielsen J, et al. An electronic health record-enabled obesity database. BMC medical informatics and decision making. 2012;12(1):45 Epub 2012/05/30. 10.1186/1472-6947-12-45 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leti F, Malenica I, Doshi M, Courtright A, Van Keuren-Jensen K, Legendre C, et al. High-throughput sequencing reveals altered expression of hepatic microRNAs in nonalcoholic fatty liver disease-related fibrosis. Translational research: the journal of laboratory and clinical medicine. 2015. 10.1016/j.trsl.2015.04.014 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DiStefano JK, Kingsley C, Craig Wood G, Chu X, Argyropoulos G, Still CD, et al. Genome-wide analysis of hepatic lipid content in extreme obesity. Acta diabetologica. 2015;52(2):373–82. 10.1007/s00592-014-0654-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gerhard GS, Benotti P, Wood GC, Chu X, Argyropoulos G, Petrick A, et al. Identification of novel clinical factors associated with hepatic fat accumulation in extreme obesity. Journal of obesity. 2014;2014:368210 10.1155/2014/368210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.She H, Xiong S, Hazra S, Tsukamoto H. Adipogenic transcriptional regulation of hepatic stellate cells. The Journal of biological chemistry. 2005;280(6):4959–67. Epub 2004/11/13. 10.1074/jbc.M410078200 . [DOI] [PubMed] [Google Scholar]
- 28.Wu Y, Liu X, Zhou Q, Huang C, Meng X, Xu F, et al. Silent information regulator 1 (SIRT1) ameliorates liver fibrosis via promoting activated stellate cell apoptosis and reversion. Toxicol Appl Pharmacol. 2015;289(2):163–76. 10.1016/j.taap.2015.09.028 . [DOI] [PubMed] [Google Scholar]
- 29.Vejnar CE, Zdobnov EM. MiRmap: comprehensive prediction of microRNA target repression strength. Nucleic Acids Res. 2012;40(22):11673–83. Epub 2012/10/05. 10.1093/nar/gks901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, Blaner WS, et al. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut. 2005;54(1):142–51. Epub 2004/12/14. 10.1136/gut.2004.042127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu X, Jin Q, Chen H, Wood GC, Petrick A, Strodel W, et al. CCL20 is up-regulated in non-alcoholic fatty liver disease fibrosis and is produced by hepatic stellate cells in response to fatty acid loading. J Transl Med. 2018;16(1):108 Epub 2018/04/25. 10.1186/s12967-018-1490-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdelmalek MF, Suzuki A, Guy C, Unalp-Arida A, Colvin R, Johnson RJ, et al. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51(6):1961–71. Epub 2010/03/20. 10.1002/hep.23535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S, Li C, Fang Z. MicroRNA 214 inhibits adipocyte enhancer-binding protein 1 activity and increases the sensitivity of chemotherapy in colorectal cancer. Oncol Lett. 2019;17(1):55–62. Epub 2019/01/19. 10.3892/ol.2018.9623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver Fibrosis, but No Other Histologic Features, Is Associated With Long-term Outcomes of Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology. 2015;149(2):389–97 e10. 10.1053/j.gastro.2015.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ekstedt M, Hagstrom H, Nasr P, Fredrikson M, Stal P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61(5):1547–54. 10.1002/hep.27368 . [DOI] [PubMed] [Google Scholar]
- 36.Lonardo A, Lugari S, Ballestri S, Nascimbeni F, Baldelli E, Maurantonio M. A round trip from nonalcoholic fatty liver disease to diabetes: molecular targets to the rescue? Acta diabetologica. 2018. Epub 2018/12/07. 10.1007/s00592-018-1266-0 . [DOI] [PubMed] [Google Scholar]
- 37.Jahn D, Kircher S, Hermanns HM, Geier A. Animal models of NAFLD from a hepatologist's point of view. Biochim Biophys Acta Mol Basis Dis. 2018. Epub 2018/07/11. 10.1016/j.bbadis.2018.06.023 . [DOI] [PubMed] [Google Scholar]
- 38.Latorre J, Moreno-Navarrete JM, Mercader JM, Sabater M, Rovira O, Girones J, et al. Decreased lipid metabolism but increased FA biosynthesis are coupled with changes in liver microRNAs in obese subjects with NAFLD. Int J Obes (Lond). 2017;41(4):620–30. Epub 2017/01/26. 10.1038/ijo.2017.21 . [DOI] [PubMed] [Google Scholar]
- 39.Ladha J, Sinha S, Bhat V, Donakonda S, Rao SM. Identification of genomic targets of transcription factor AEBP1 and its role in survival of glioma cells. Mol Cancer Res. 2012;10(8):1039–51. Epub 2012/06/23. 10.1158/1541-7786.MCR-11-0488 . [DOI] [PubMed] [Google Scholar]
- 40.Ro HS, Zhang L, Majdalawieh A, Kim SW, Wu X, Lyons PJ, et al. Adipocyte enhancer-binding protein 1 modulates adiposity and energy homeostasis. Obesity (Silver Spring). 2007;15(2):288–302. Epub 2007/02/15. 10.1038/oby.2007.569 . [DOI] [PubMed] [Google Scholar]
- 41.Wang D, Wei Y, Frye M, Gentile CL, Pagliassotti MJ. Saturated Fatty Acid-induced cytotoxicity in liver cells does not involve phosphatase and tensin homologue deleted on chromosome 10. J Nutr Metab. 2013;2013:514206 Epub 2013/05/22. 10.1155/2013/514206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhu NL, Wang J, Tsukamoto H. The Necdin-Wnt pathway causes epigenetic peroxisome proliferator-activated receptor gamma repression in hepatic stellate cells. The Journal of biological chemistry. 2010;285(40):30463–71. Epub 2010/07/29. 10.1074/jbc.M110.156703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bartolini D, Torquato P, Barola C, Russo A, Rychlicki C, Giusepponi D, et al. Nonalcoholic fatty liver disease impairs the cytochrome P-450-dependent metabolism of alpha-tocopherol (vitamin E). J Nutr Biochem. 2017;47:120–31. Epub 2017/06/20. 10.1016/j.jnutbio.2017.06.003 . [DOI] [PubMed] [Google Scholar]
- 44.Wang Q, Liu S, Zhao X, Wang Y, Tian D, Jiang W. MiR-372-3p promotes cell growth and metastasis by targeting FGF9 in lung squamous cell carcinoma. Cancer Med. 2017;6(6):1323–30. Epub 2017/04/26. 10.1002/cam4.1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weng J, Zhang H, Wang C, Liang J, Chen G, Li W, et al. miR-373-3p Targets DKK1 to Promote EMT-Induced Metastasis via the Wnt/beta-Catenin Pathway in Tongue Squamous Cell Carcinoma. Biomed Res Int. 2017;2017:6010926 Epub 2017/03/25. 10.1155/2017/6010926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fan X, Xu S, Yang C. miR-373-3p promotes lung adenocarcinoma cell proliferation via APP. Oncol Lett. 2018;15(1):1046–50. Epub 2018/02/02. 10.3892/ol.2017.7372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiu X, Zhu J, Sun Y, Fan K, Yang DR, Li G, et al. TR4 nuclear receptor increases prostate cancer invasion via decreasing the miR-373-3p expression to alter TGFbetaR2/p-Smad3 signals. Oncotarget. 2015;6(17):15397–409. Epub 2015/05/20. 10.18632/oncotarget.3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu SY, Xu PF, Gao TT. MiR-372-3p inhibits the growth and metastasis of osteosarcoma cells by targeting FXYD6. Eur Rev Med Pharmacol Sci. 2018;22(1):62–9. Epub 2018/01/25. 10.26355/eurrev_201801_14101 . [DOI] [PubMed] [Google Scholar]
- 49.Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, et al. Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues From Patients. Gastroenterology. 2016;151(3):513–25 e0. 10.1053/j.gastro.2016.05.051 . [DOI] [PubMed] [Google Scholar]
- 50.Sendi H, Mead I, Wan M, Mehrab-Mohseni M, Koch K, Atala A, et al. miR-122 inhibition in a human liver organoid model leads to liver inflammation, necrosis, steatofibrosis and dysregulated insulin signaling. PLoS One. 2018;13(7):e0200847 Epub 2018/07/20. 10.1371/journal.pone.0200847 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
LX-2 cells were seeded at 1 x 105 cell/well on 6-well plates coated with 350 μL of (1mg/mL) Matrigel Growth Factor Reduced (GFR) basement membrane matrix (Corning Inc; Corning, NY) and cultured for 72 hours at 37°C to induce a state resembling quiescence. Transcript levels of AEBP1 expression were normalized to GAPDH. All experiments were performed in triplicate. *P≤0.05.
(TIF)
AEBP1 expression was reduced and gene expression measured as described in the Methods section. A t-test was performed to assess statistical significance. ***P = 0.0001.
(TIF)
RT-qPCR was performed and miRNAs levels analyzed as described in the Methods section. Data were analyzed using a two-tailed t-test. *P<0.05.
(TIF)
(DOCX)
(DOCX)
Data Availability Statement
The data are available in the NCBI public repository (https://www.ncbi.nlm.nih.gov/bioproject/512027).