

Abstract
The type I interferon pathway has been implicated in the pathogenesis of a number of rheumatic diseases, including systemic lupus erythematosus, Sjögren syndrome, myositis, systemic sclerosis, and rheumatoid arthritis. In normal immune responses, type I interferons have a critical role in the defence against viruses, yet in many rheumatic diseases, large subgroups of patients demonstrate persistent activation of the type I interferon pathway. Genetic variations in type I interferon-related genes are risk factors for some rheumatic diseases, and can explain some of the heterogeneity in type I interferon responses seen between patients within a given disease. Inappropriate activation of the immune response via Toll-like receptors and other nucleic acid sensors also contributes to the dysregulation of the type I interferon pathway in a number of rheumatic diseases. Theoretically, differences in type I interferon activity between patients might predict response to immune-based therapies, as has been demonstrated for rheumatoid arthritis. A number of type I interferon and type I interferon pathway blocking therapies are currently in clinical trials, the results of which are promising thus far. This Review provides an overview of the many ways in which the type I interferon system affects rheumatic diseases.
Overactivity of the type I interferon pathway has been observed in several rheumatic conditions, including both monogenic diseases (for example, Aicardi–Goutieres syndrome (AGS)1) and polygenic diseases (for example, systemic lupus erythematosus (SLE)2–4). Human genetic studies of rheumatic diseases have identified numerous disease-risk genes that function within the type I interferon pathway. In many cases, these genetic variations augment the function of type I interferons. Data suggest that heterogeneity in type I interferon pathway activation and genetic make-up contribute to the clinical heterogeneity observed in rheumatic diseases5. Given that the type I interferon pathway is deeply entwined with the pathogenesis of multiple rheumatic diseases, a robust effort is underway to determine whether type I interferon activity might be a predictor of treatment response, or whether the type I interferon pathway could be targeted by treatment. It is attractive to think that differences in type I interferon-related genes and pathway activation between patients might lead to rational selection of immunomodulatory therapy.
In this Review, we discuss the role of the type I interferon pathway in rheumatic diseases, focusing on the clinical implications. We briefly discuss type I interferon biology, followed by the interferon signature and other measurements of type I interferon in rheumatic diseases. We also examine genetic factors related to the type I interferon pathway, focusing on common gene variants associated with rheumatic disease, as well as the rare monogenic interferonopathies. We review current data regarding type I interferons in SLE, Sjögren syndrome, myositis, systemic sclerosis (SSc), and rheumatoid arthritis (RA). Finally, we consider treatments targeting type I interferons and the type I interferon pathway in rheumatic diseases.
Type I interferon biology
Interferons are functionally related cytokines that have important roles in infection, cancer, inflammation and autoimmunity. The antiviral properties of interferons were identified more than 50 years ago6, and the roles of interferons in cell survival, proliferation, differentiation and activation have since been highlighted and are reviewed elsewhere7. There are three major types of interferon: type I, type II and type III. Each type signals via a specific cell surface receptor complex. The type I interferons in humans include twelve interferon-α (IFNα) subtypes, IFNβ, IFNω, IFNκ and IFNε (reviewed elsewhere8). Each subtype is produced by particular cells in response to specific stimuli. In this Review, we will focus on IFNα and IFNβ, the most extensively studied type I interferons in rheumatic diseases.
Type I interferon induction
Many cells can produce type I interferons (FIG. 1); plasmacytoid dendritic cells (pDCs) are the predominant producers of IFNα, whereas many cell types (for example, fibroblasts, epithelial cells, dendritic cells, phagocytes and synoviocytes) produce IFNβ. Production of type I interferons depends on the cell type and the environmental context. For example, pDCs constitutively express high levels of interferon regulatory factor 7 (IRF7), which, in part, enables them to produce relatively high amounts of IFNα9,10, whereas other cell types must be ‘primed’ before high levels of type I interferons can be produced10. In a steady state, IFNβ is present at physiological levels, which seems to be important for priming cells for subsequent exposures11,12. Of note, over the past 5–10 years there has been strong interest in the microbiome and its effect on inflammation and rheumatic diseases. Interestingly, commensal intestinal flora influence this baseline production of IFNβ13.
Fig. 1 |. Major pathways of induction of type I interferon production in different cell lineages.
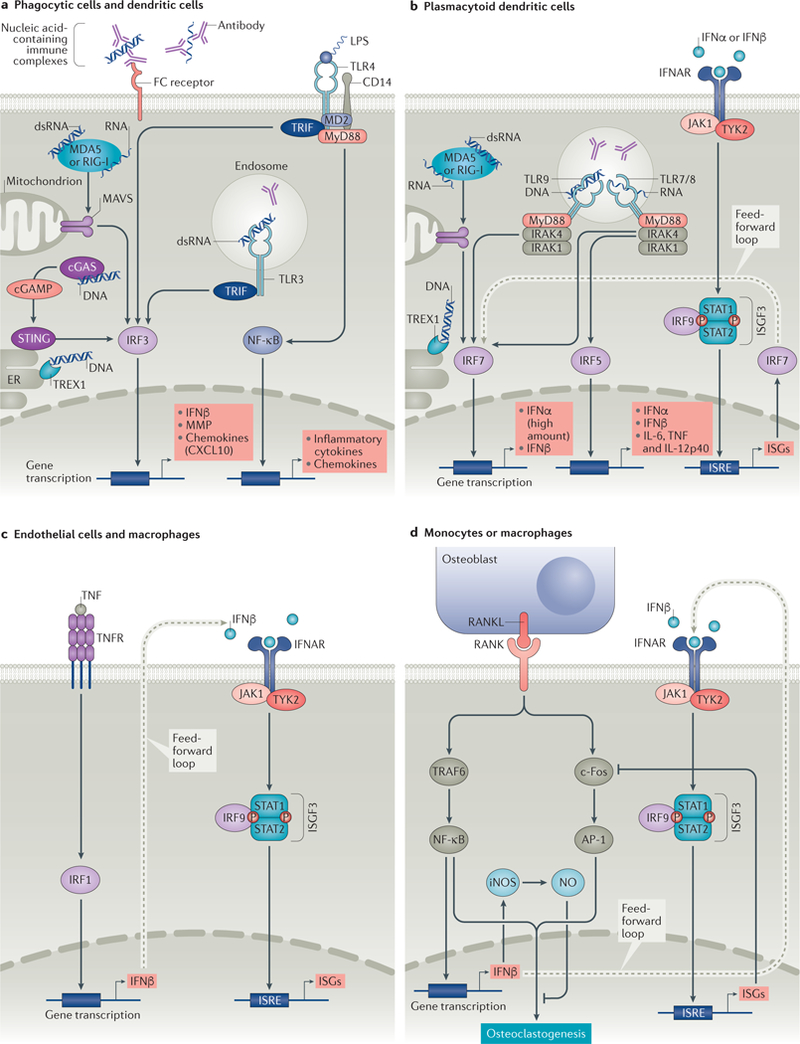
a | In phagocytes and dendritic cells, stimulation of surface Toll-likereceptor 4 (TLR4) by lipopolysaccharide (LPS) or endosomal TLR3 by double-stranded RNA (dsRNA) results in activation of interferon regulatory factor 3 (IRF3) via a TIR domain-containing adaptor molecule 1 (TICAM-1, also known as TRIF)-dependent pathway, and nuclear factor-κB (NF-κB) via myeloid differentiation primary response protein (MyD88). Activation of cytosolic nucleic acid sensors (melanoma differentiation-associated protein 5 (MDA5) or retinoic acid inducible gene 1(RIG-I) by RNA, or stimulator of interferon genes protein (STING) by DNA (via cyclic GMP-AMP synthase (cGAS)) also prompt activation of IRF3. IRF3 translocates to the nucleus and induces transcription of IFNβ. b | In plasmacytoid dendritic cells (pDCs), activation of endosomal TLR7 or TLR8 by RNA results in activation of IRF7 and/or IRF5. Activation of endosomal TLR9 by DNA or of cytosolic sensors MDA5 or RIG-I by RNA results in activation of IRF7. IRF7 translocates to the nucleus, where it induces transcription of type I interferons. Translocation of IRF5 to the nucleus culminates in transcription of type I interferons and pro-inflammatory cytokines. In pDCs, binding of type I interferon to the type I interferon receptor (IFNAR) results in activation of the canonical Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway that results in transcription of type I interferon stimulated genes (ISGs). ISGs include IRF7, which provides a feed-forward mechanism for production of more type I interferon. c | In macrophages and endothelial cells, TNF induces IFNβ via IRF1 and can induce an IFNβ autocrine loop that acts in synergy with canonical TNF signals to induce sustained expression of inflammatory genes and delayed expression of STAT1-dependent ISGs that prime cells for enhanced responses to subsequent challenge. d | Receptor activator of nuclear factor-κB (RANK)–RANK ligand (RANKL) interaction activates TNF receptor-associated factor 6 (TRAF6) and c-Fos pathways. TRAF6 activation results in induction of NFκB. c-Fos, together with activator protein 1 (AP-1) leads to a cascade that promotes osteoclastogenesis. NF-κB and c-Fos stimulate production of IFNβ. IFNβ promotes transcription of genes that inhibit c-Fos activity and results in the induction of nitric oxide (NO), which inhibits osteoclastogenesis. cGAMP, cyclic GMP-AMP; CXCL10, CXC-chemokine 10; ER, endoplasmic reticulum; iNOS, inducible nitric oxide synthase; IRAK, interleukin-1 receptor-associated kinase; ISGF3, interferon-stimulated gene factor 3; ISRE, interferon-sensitive response element; MAVS, mitochondrial antiviral-signalling protein; MD2, myeloid differentiation 2; MMP, matrix metalloproteinase; TNFR, TNF receptor; TYK2, tyrosine kinase 2; TREX1, three-prime repair exonuclease 1.
Type I interferon production can be induced fol-lowing the detection of microbial products by patternrecognition receptors (PRRs), such as Toll-like receptors (TLRs) or cytosolic nucleic acid sensors14,15; for example, lipopolysaccharide (LPS), a microbial cell wall component, is detected by surface TLR4; endosomal TLRs (TLR3, TLR7, TLR8, and TLR9) are ligated by nucleic acids delivered to the endosome via immune complexes; and nucleic acids in the cytosol are detected by sensors such as retinoic acid inducible gene 1 (RIG-I, also known as DDX58), melanoma differentiation-associated protein 5 (MDA5, also known as IFIH1) and stimulator of interferon genes protein (STING) (FIG. 1). In normal immune responses, these events occur after the sensing of pathogen-derived material. However, PRRs can also detect nucleic acids from endogenous sources (for example, nucleic acids within nucleic acid-containing antibody complexes, nucleic acids released as a result of defective nucleic acid metabolism16 or reactivity with endogenous transcripts that contain virus-like nuclear repeat elements (NREs)17) and might thereby contribute to the pathogenesis of rheumatic diseases. Interestingly, activation of the inflammasome can negatively regulate type I interferon production via the cyclic GMP-AMP synthase (cGAS)–STING pathway in the context of viral infection18. In normal conditions, cytosolic double-stranded DNA (dsDNA) triggers the synthesis of cyclic GMP-AMP (cGAMP) by cGAS, which activates STING, leading to type I interferon production. However, upon canonical and non-canonical inflammasome activation, caspase-1 cleaves cGAS and thereby dampens STING-mediated type I interferon production18.
Interferon regulatory factors (IRFs) are activated downstream of PRRs, and translocate to the nucleus, where they function as transcription factors19. In phagocytes and dendritic cells, stimulation of TLR3 or TLR4 leads to the activation of IRF3 via the adaptor TIR domain-containing adaptor molecule 1 (TICAM1, also known as TRIF)20. Activation of cytosolic nucleic acid sensors (MDA5 and RIG-I by RNA or STING by DNA) also upregulate the activation of IRF3 (REF. 21), which upregulates expression of IFNB1. The adaptor mitochondrial antiviral-signalling protein (MAVS) interacts with RIG-I and MDA5 to facilitate activation of IRF3 in phagocytic and dendritic cells, and of IRF7 in pDCs. In pDCs, recognition of nucleic acids by TLR7, TLR8 or TLR9 leads to recruitment of the adaptor protein MyD88, which in turn interacts with IL-1 receptor-associated kinase 1 (IRAK1) and IRAK4 (REF. 22) (FIG. 1b). This signalling complex results in the phosphorylation of IRFs, such as IRF5 and/or IRF7. The translocation of IRF5 to the nucleus culminates in the transcription of genes encoding type I interferons, pro-inflammatory cytokines (IL-6 and TNF) and IL-12p40, whereas IRF7 promotes expression of type I interferons23.
IFNβ production is also stimulated as a result of sig-nalling through TNF receptors (TNFRs), such as receptor activator of nuclear factor-κB (RANK) and TNFR2. In macrophages and endothelial cells, TNF induces IFNβ production via IRF1, and can also induce an IFNβ autocrine loop that functions in synergy with canonical TNF signals to induce sustained expression of inflammatory genes and delayed expression of signal transducer and activator of transcription 1 (STAT1)-dependent interferon stimulated genes (ISGs)24 (FIG. 1c). This synergy primes macrophages for increased responses to subsequent challenges24. In human endothelial cells, this cascade seems to depend on TNFR2 and results in the promotion of monocyte recruitment25. Interaction between RANK and RANK ligand (RANKL) activates pathways that include TNFR associated factor 6 (TRAF6) and c-Fos, which promote expression of IFNβ and can also promote osteoclastogenesis (FIG. 1d). IFNβ promotes the transcription of genes that inhibit c-Fos activity26 and induce nitric oxide, which inhibits osteoclastogenesis26.
Type I interferon signalling
Type I interferons bind to a shared cell surface receptor, the type I interferon receptor (IFNAR). IFNα and IFNβ induce different conformational changes in the cytosolic portion of the receptor, which enables differential signalling by the two cytokines through the same receptor28. Upon engagement, IFNAR activates kinases (for example, Janus kinase 1 (JAK1) and tyrosine kinase 2 (TYK2) in canonical type I interferon signalling), prompting phosphorylation, dimerization and nuclear translocation of STAT proteins. The resulting STAT complexes control distinct gene-expression programmes. For example, the interferon-stimulated gene factor 3 (ISGF3) complex (composed of STAT1, STAT2 and IRF9) activates classic antiviral genes. By contrast, STAT1 homodimers induce pro-inflammatory gene expression, and STAT3 homodimers suppress pro-inflammatory gene expression29.
IFNα signalling activates antigen-presenting cells, and increases the expression of CD86, as well as MHC class I and II molecules on these cells, which provide co-stimulatory signals and augment antigen presentation, respectively30. Thus, IFNα can bridge the innate and adaptive immune systems, demonstrating its importance in setting thresholds for self-reactivity and autoimmunity. IFNβ shares many downstream signalling properties with IFNα, but also has anti-inflammatory and antiproliferative properties. ISG expression is complex and seems to be cell and context dependent31 (reviewed elsewhere29). Signalling pathways that are also triggered by interferon receptor engagement (for example, mitogen-activated protein kinase (MAPK), nuclear factor-κB (NF-κB) and protein kinase B (RACα serine/threonine-protein kinase, also known as AKT) pathways) influence the transcription of ISGs and/or translation of ISG mRNA downstream of type I interferon-activated JAK–STAT pathways32,33. Physiological activation of the type I interferon pathway is even more complex, as IFNβ can function in synergy with TNF, which is produced at early stages following innate recognition of a pathogen34. Co-stimulation with IFNβ and TNF induces a synergy-dependent delayed antiviral response via an as yet uncharacterized pathway that is dependent on TYK2, STAT2 and IRF9, but is independent of STAT1 signalling34. Thus, various cytokine signalling pathways functioning through different receptors can affect the outcome of type I interferon signalling.
Measuring type I interferon in blood
Traditionally, the term ‘interferon signature’ has been used to describe the pattern of increased expression of >100 type I ISGs in studies comparing the expression of genes in peripheral blood cells from patients with SLE and controls2–4. In addition to being present in SLE, such a signature has been found in other rheumatic diseases, including Sjögren syndrome, myositis, SSc and RA35. An important caveat regarding the interferon signature is that genes that are type I interferon-induced can sometimes also be induced by other factors. For example, type II interferons can induce the expression of some of the same genes as type I interferons, and evidence is accumulating for a circulating type II interferon signature in SLE36.
Many studies of the interferon signature have examined gene expression in either whole blood or in peripheral blood mononuclear cells (PBMCs)2,37. In these approaches, multiple different cell types are mixed together. Different individuals typically have different proportions of immune cell types; thus, a difference in the amount of a measured transcript reflects a combination of the amount of transcript expressed by each cell type and the proportion of each cell type in the cellular mixture. This limitation can be partially addressed by enumerating the proportion of each immune cell type in the sample before study, although adjusting for these differences in cell numbers with covariates is also challenging and cannot account for every possibility in the data. To address this issue, individual immune cell populations can be sorted before lysing the cells and measuring gene expression31,38. Interestingly, such analysis has shown that different immune cell types from the same blood sample express different ISGs31. These data suggest a great diversity in the downstream type I interferon responses of different cell types, and highlight the fact that we are still just beginning to understand the varied consequences of chronic type I interferon stimulation in human cellular immunity.
To address the limitations of interferon signature studies, functional assays have also been used to assess type I interferon activity in large cohorts of patients39 (BOX 1). These functional assays are sensitive and utilize IFNAR and the downstream gene expression cascade to detect even very small amounts of type I interferons. To date, many commercial enzyme-linked immunosorbent assays (ELISAs) and multiplex assays that measure type I interferon protein levels have proven to be insufficiently sensitive or specific in detecting type I interferons in human samples40. However, a new method for detecting type I interferons was described by Wilson et al. in 2016 (REF. 41) that uses single-molecule array (Simoa) digital ELISA technology. This method reportedly detects attomolar (femtograms per milliliter) concentrations of IFNα protein in human samples41. This methodology is based on counting individual enzyme-labelled immune complexes captured on paramagnetic beads in single-molecule arrays41,42 and utilizes unique high-affinity anti-IFNα antibodies isolated from patients with autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APECED)43. Additional validation of this technology is anticipated in the future.
Box 1 |. Functional assays of type I interferon activity .
The most well-known functional assays for measuring type I interferon activity are the luciferase203 and WISH cell124 reporter assays. Both reporter assays rely on a cell line bearing the receptor for type I interferon
Luciferase reporter assay
The luciferase assay uses a cell line that is transfected with a plasmid carrying the luciferase gene under the control of a type I interferon inducible promoter. The cell line is exposed to samples containing type I interferon, and type I interferon activity is determined by measuring the luciferase expression203
Wish cell reporter assay
The WISH cell assay uses the WISH epithelial cell line to measure the ability of patient sera to promote type I interferon-induced gene expression. WISH cells are exquisitely sensitive to type I interferon, but do not produce type I interferons and lack other pattern recognition receptors such as TLRs204,205. Expression of the interferon stimulated genes (ISGs) MX1, IFIT1 and EIF2AK2 is measured using quantitative PCR (qPCR). The relative expression of each of these three genes is standardized to that generated with healthy donor sera and summed to generate a score reflecting the ability of sera to cause interferon-induced gene expression, which is referred to as type I interferon activity. The type I interferon activity is reflective of the amount of type I interferon protein present in the sample to ligate the type I interferon receptor. Additional aliquots from the same patients can be tested following pre-incubation with anti-IFNα or anti-IFNβ antibodies to determine how much of the total type I interferon activity is due to IFNβ activity, and how much is due to IFNα activity. IFNγ, TNF, IL-6, and IFNλ do not induce substantial expression of these three transcripts in the WISH cells (REF. 124 and unpublished data).
Type I interferons in SLE
Disease initiation
Type I interferons have been linked with SLE initiation. Some patients being treated with recombinant human IFNα for viral hepatitis and haematologic malignancy develop de novo SLE44,45. When IFNα therapy is stopped, the rheumatic symptoms usually improve, supporting a causal role for type I interferons in the initiation of SLE in some patients45. Circulating type I interferon activity is frequently high in unaffected relatives of patients with SLE and familial correlations in type I interferon activity have been observed46,47, suggesting that high levels of type I interferon in the circulation is a heritable risk factor for SLE. This heritability of high levels of type I interferon activity is shared across patients with SLE from all ancestral backgrounds46 and follows a polygenic inheritance pattern. In longitudinal studies of serum samples from patients before they were diagnosed with SLE, type I interferon activity increases precipitously in the year before disease onset48, also supporting the importance of type I interferon in disease initiation in SLE.
Genetic factors
Among the SLE-associated loci identified in case–control genetic studies, there is an over-representation of genes involved in type I interferon signalling, production and response49. In general, many of the SLE-associated variants in type I interferon pathway genes are associated with increased activity in the type I interferon pathway in humans (reviewed elsewhere49). Variants have also been associated with increased circulating type I interferon activity or increased ISG expression50,51. Genes that are overexpressed in peripheral blood cells (creating the interferon signature) are not necessarily the same as the genes that are implicated as genetic risk factors5.
Interferon response factors.
IRFs coordinate type I interferon and ISG expression in a cell-type specific manner (reviewed elsewhere23). IRF5 is involved in the production of both pro-inflammatory cytokines and type I interferons in innate immune cells52, and IRF5 also influences B cell responses downstream of TLR stimuli53. Human genetic variants in IRF5 have been identified as rheumatic disease susceptibility factors in SLE54,55, Sjögren syndrome56, SSc57, RA58 and juvenile idiopathic arthritis (JIA)59.
In SLE, risk variants of IRF5 are associated with increased circulating type I interferon activity in patients with SLE; this association is dependent upon the presence of anti-RNA-binding protein (RBP) or anti-dsDNA autoantibodies60,61. These data suggest that IRF5 risk variants might be an important factor in the ‘stimulated’ scenario, in which the autoantibody immune complexes provide a constant endogenous stimulus that synergizes with a hyperresponsive TLR system, resulting in chronic over-production of type I interferons. The SLE IRF5 risk haplotype is associated with the production of autoantibodies in otherwise healthy individuals62, suggesting a potential feed-forward loop. In such a loop, an IRF5 risk variant predisposes individuals to the production of autoantibodies, possibly via hyperactivity of the TLR pathway in B cells, and the autoantibodies produced can then form nucleic acid immune complexes that stimulate the overactive TLR system in innate immune cells. Genetic variants in both IRF7 and IRF8 have been associated with an increased risk of SLE55,63,64, and such variants are also associated with altered type I interferon responses in patients with SLE23,65,66.
Other interferon-related genes.
Beyond the IRF family, a number of other genes associated with risk of SLE (for example, STAT4, MAVS, IFIH1 (which encodes MDA5) and PTPN22) have also been demonstrated to alter type I interferon pathway function50,51,67–69. Variants in some of these genes have also been associated with other rheumatic diseases, for example, STAT4 is associated with Sjögren syndrome70, SSc71, RA72, psoriasis73 and, possibly, JIA74; IFIH1 is associated with late-onset psoriasis75; and PTPN22 is associated with RA and JIA76,77. Overall, type I interferon activity is clearly controlled to some degree by genetic factors and is a polygenic trait.
Interestingly, thus far, robust evidence demonstrating gene–gene interactions between interferon pathway genes (that is, the effect of one gene being modified by one or several interferon pathway genes) is lacking. Studies that have examined type I interferon in patients in the context of these risk variants have demonstrated additive effects without evidence for either synergy or redundancy to date50,51,65. In addition, other factors, such as epigenetic regulation78, probably influence the effect of these risk variants.
Tissue expression.
Most studies in SLE have examined circulating type I interferons, however, the action of type I interferons in the tissue is likely to be important and complex. Genetic polymorphisms in IFNK (encoding IFNκ) are implicated in the pathogenesis of cutaneous lupus erythematosus, and disease associated single-nucleotide polymorphisms in IFNK differ between male and female patients79. Interestingly, type I interferon activity was frequently increased in the circulation of female patients with such IFNK variants compared with that in healthy controls79. However, IFNκ was not a major contributor to the type I interferon activity observed in the circulation of these patients46,79. The IFNK variants could instead be influencing type I interferon production by pDCs in the affected skin, and thereby increasing type I interferon activity in the circulation79. Keratinocytes from the skin of patients with cutaneous lupus erythematosus produce more IL-6 in vitro than keratinocytes from healthy individuals after exposure to TLR agonists or ultraviolet B (UVB) radiation; this increased IL-6 production seems to be dependent on IFNκ80. Such cytokine production might also contribute to the skin inflammation observed in cutaneous lesions in SLE.
Heterogeneity in SLE
IFNα is the predominant circulating type I interferon in patients with SLE46. Serum IFNα activity varies widely between patients with SLE, and in 40–50% of patients, serum IFNα activity is normal46. Therefore, type I interferon is probably not an important pathogenic factor for all patients with SLE, contributing to the pathological heterogeneity of this disease. A high degree of functional circulating type I interferon activity is strongly correlated with the presence of anti-RBPs, such as antibodies to 52 kDa SSA/Ro antigen (Ro52, also known as TRIM21) and ribonucleoprotein (RNP), in patients with SLE39. These autoantibody titres frequently do not change considerably over time, supporting the idea of a stable subset of patients with SLE who have high levels of type I interferon activity. A study comparing gene expression in African American and European American patients with SLE demonstrated that patients from both ancestral backgrounds had a type I interferon signature, but in African Americans this signature was particularly dependent on the presence of anti-RBP autoantibodies81. This finding is interesting as these autoantibodies, particularly anti-RNP and anti-Sm antibodies, are more common in African American patients than in European American patients39, suggesting differences in the molecular pathogenesis of SLE between ancestral backgrounds.
Case–case genome-wide genetic studies, which com-pare patients with high levels of type I interferon activity to those with low levels of type I interferon activity, have implicated additional genes that modulate circulating IFNα activity in patients with SLE5,47. These studies identified a number of novel loci associated with risk of SLE and high degree of type I interferon activity that were not identified in case–control studies, including risk loci in PRKG1, PNP, and ANKS1A5,47. Further bioinformatic analyses suggested that these loci mediate functional effects in DCs and natural killer (NK) cells5. NK cells cooperate with DCs to induce IFNα production in SLE82. The PNP variant is a loss-of-function mutation in the gene encoding purine nucleoside phosphorylase (PNP), an enzyme involved in purine metabolism, that leads to cell-cycle abnormality (a block in S phase entry) and type I interferon pathway activation in human lymphocytes83. Interestingly, this block in S phase can be rescued in vitro by providing hypoxanthine and adenosine, supporting the notion that relative PNP deficiency is the cause of the S-phase block, and suggesting a potential for personalized therapeutics in patients with SLE who harbour this PNP variant83.
Clinical implications
In SLE, the peripheral blood type I interferon signature correlates with disease severity2. In a cross-sectional study, patients with a prominent peripheral type I interferon signature fulfilled a substantially higher number of SLE clinical diagnostic criteria and, upon retrospective review, more commonly had kidney, central nervous system (CNS) and/or haematologic involvement at some point during the course of their disease2. However, in longitudinal studies, the interferon signature in blood is relatively stable and cannot be used to predict SLE disease flares over time84,85. The expression of certain chemokines (CXC-chemokine ligand 10 (CXCL10), CC-chemokine 2 (CCL2), and CCL-chemokine ligand 19 (CCL19)) that are induced by interferons and other cytokines also correlate with disease activity and might predict risk of flares over time in SLE86, suggesting that other factors beyond type I interferon are involved in disease flares. Gene expression studies support this idea, showing that other non-type I interferon-induced gene signatures, such as the plasmablast signature, correlated more strongly with disease activity than the interferon signature37. Thus, type I interferons might be more important in disease initiation and in the early phases of disease than in disease flares.
Type I interferon in Sjögren syndrome
Some of the genes associated with increased type I interferon pathway activation in SLE (such as IRF5 and STAT4) are also associated with risk of Sjögren syndrome56, and a type I interferon signature has been reported in both the blood and tissues of patients with Sjögren syndrome87–89. In Sjögren syndrome, a peripheral blood type I interferon signature strongly correlates with the presence of anti-SSA/Ro antibodies88, which parallels the association observed between anti-RBP antibodies and the interferon signature in SLE. Thus, despite the many clinical differences that exist between Sjögren syndrome and SLE, parallels can be drawn between these two diseases regarding type I interferon pathway activation with respect to autoantibody associations and background genetics. Although anti-SSA/Ro antibodies are associated with increased type I interferon activity in patients with either SLE or Sjögren syndrome, asymptomatic individuals with high anti-Ro antibody titres do not have high levels of circulating type I interferon activity90. This finding suggests that other disease-associated factors must be present in addition to anti-SSA/Ro antibodies to cause a chronic increase of circulating type I interferon90.
In Sjögren syndrome, a type I interferon signature might help identify clinically meaningful subgroups of patients. A peripheral blood monocyte type I interferon signature identified a subgroup of patients with Sjögren syndrome who had high levels of clinical disease activity, autoantibodies and the expression of B-cell activating factor (BAFF, also known as TNFSF13)-encoding mRNA in their monocytes91. OAS1, one of the ISGs, is a Sjögren syndrome risk locus, and disease-associated variants of OAS1 result in alternate splicing of the gene transcript, leading in multiple alternative transcripts that result in a lack of translational response to type I interferon stimulation92. The 620W polymorphism in PTPN22 is also associated with Sjögren syndrome and with a low expression of ISGs, implying the presence of distinct genetic backgrounds among subsets of patients with Sjögren syndrome that can be defined by type I interferon activity93. Interestingly, investigators found that the pattern of expression of RNA-sensing receptors (TLR7, RIG-I and MDA5) in monocytes and pDCs from patients with Sjögren syndrome differed substantially between those who did and did not have a peripheral type I interferon signature94. This type of differentiation might help identify subsets of patients who will benefit from therapies targeting these pathways.
In contrast to the type I interferon signature that predominates in the blood in patients with Sjögren syndrome, a type II interferon signature predominates in minor salivary gland (MSG) biopsy samples from such patients95. Concomitant low expression of IFNα-encoding mRNA and high expression of IFNγ-encoding mRNA in MSG tissue is strongly associated with lymphomagenesis, suggesting that the ratio between these two mRNA species in MSG biopsy samples can serve as a biomarker for in situ Sjögren syndrome-related lymphoma95.
Type I interferon in myositis
In patients with either dermatomyositis or polymyositis, type I interferon levels are increased in the circulation and a type I interferon signature is detectable in muscle tissue35,96,97. Muscle tissue from patients with juvenile dermatomyositis has increased numbers of infiltrating pDCs and increased expression of the ISG MX198 compared with tissue from healthy controls. Multiple studies have shown an association between type I interferon in the circulation and disease activity in myositis96,99,100. These studies provide stronger evidence for an association between type I interferon activity and longitudinal disease activity than has been observed in SLE.
Although the genetic basis of inflammatory disease is currently less well described in myositis compared with SLE, a number of polymorphisms in several genes associated with increased type I interferon activity in patients with SLE (for example, OPN rs28357094G and TNFA-308A alleles) have been associated with high levels of type I interferon activity in patients with dermatomyositis101. Furthermore, type I interferon levels are higher in patients with dermatomyositis who have a family history of SLE compared with in those without a family history of SLE102, which supports the idea of a shared genetic basis for type I interferon pathway activation in various rheumatic diseases. The presence of anti-RBP antibodies in patients with myositis, such as anti-SSA/Ro and anti-Sm antibodies, is associated with high levels of circulating type I interferon activity103, paralleling that seen in other rheumatic diseases. Interestingly, the use of TNF inhibitors in patients with myositis104 or Sjögren syndrome105 results in increased type I interferon activity, which, in myositis, is associated with lack of improvement or worsening of disease104.
Circulating IFNα is an important contributor to the total functional type I interferon activity observed in dermatomyositis96; however, some studies support the idea that IFNβ also contributes to the interferon signature seen in PBMCs from patients with dermatomyositis106. TLR3 stimulation of cultured myoblasts induces the production of IFNβ when combined with IFNγ stimulation, and upregulates the expression of HLA class I molecules107. In muscle biopsy samples from patients with polymyositis or dermatomyositis, immature muscle precursor cells that overexpress HLA class I are a source of IFNβ107. Thus, IFNβ from immature muscle precursor cells might contribute to the type I interferon signature seen in muscle tissue in myositis. A 2015 study of muscle tissue from patients with dermatomyositis demonstrated that TLR3 and RIG-I are preferentially expressed in the perifascicular fibres, indicating that these type I interferon pathway components might be involved in the formation of perifascicular atrophy, a hallmark feature of dermatomyositis108. In the same study, the investigators found that expression of TLRs and RIG-I was upregulated in the muscle tissue of patients with dermatomyositis compared with controls (which included patients with polymyositis, facioscapulohumeral muscular dystrophy, and patients without neuromuscular disease) and that TLR4 and TLR9 were expressed mainly in inflammatory infiltrates108. The researchers concluded that endogenous production of type I interferon in dermatomyositis is generated by pDCs, mainly through the TLR9 pathway. However, the TLR4 pathway can also contribute to type I interferon induction20 (FIG. 1a), and as TLR4 was also expressed in the inflammatory infiltrates, it is conceivable that TLR4 might also contribute to the endogenous type I interferon found in the muscle of patients. Additionally, non-immune cells that produce IFNβ (such as endothelial cells, FIG. 1c) might also contribute to type I interferon production in myositis.
Type I interferon in SSc
A number of studies have documented increased type I interferon-induced gene expression in patients with SSc, in both circulating blood cells and in affected lung tissue109–112. Interestingly, patients with SSc who have antiSSB/Ro antibodies are more likely to have high levels of type I interferon than patients without these antibodies, resembling associations seen in myositis, Sjögren syndrome and SLE113. Other autoantibodies have also been associated with high circulating type I interferon expression in SSc, including anti-U1 RNP and antitopoisomerase autoantibodies109. This finding suggests that a similar process of immune complex-mediated type I interferon generation might contribute to the increased circulating type I interferon levels observed in many rheumatic diseases.
A number of variants in type I interferon pathway genes (for example, IRF5 (REF. 57), IRF7 (REF. 114), IRF8 (REF. 115), TREX1 (REF. 116), IRAK1 (REF. 114), and STAT4 (REF. 71)) are associated with SSc. pDCs are also implicated in SSc pathogenesis. In SSc, in addition to the role of pDCs in type I interferon production, there is a striking and disease-specific over-production of CXC-chemokine ligand 4 (CXCL4, also known as platelet factor 4) by pDCs, which corresponds with severe skin disease and lung fibrosis117. CXCL4, a potent antiangiogenic chemokine that also has profibrotic properties and stimulates the proliferation of regulatory T cells that have impaired function118–120, is suspected to have a major role in the vasculopathy of SSc and to influence fibrosis by downregulating FLI1 in endothelial cells and fibroblasts117. It is speculated that CXCL4 does not act in isolation117; hence other factors, such as alterations in the type I interferon pathway, could function together with CXCL4 to contribute to SSc pathogenesis. Intramuscular administration of recombinant IFNα showed some initial promise in improving or stabilizing skin scores in a pilot study of patients with diffuse cutaneous SSc121. However, in keeping with IFNα having a pathogenic role in SSc, a randomized, double-blind, placebo-controlled trial showed that recombinant IFNα therapy in SSc is ineffective and might in fact be harmful, as those who received the IFNα treatment showed less improvement in skin scores and greater deterioration of lung function than the placebo group122.
Type I interferon in RA
A type I interferon signature is detectable in the peripheral blood of patients with RA, and can be present in the preclinical phase of the disease123. The relative level of expression of ISGs in the circulation in RA is lower than that observed in SLE and other autoimmune connective tissue diseases35,124. However, some of the genes associated with increased type I interferon pathway activation in SLE are also associated with the risk of RA, such as IRF5 (REF. 58), IRAK1 (REF. 125), STAT4 (REF. 72) and PTPN22 (REF. 77). The finding that particular polymorphisms are associated with the risk of developing a number of rheumatic diseases supports the idea that there is a shared pathway in these diseases126.
The presence of pDCs and the expression of ISGs, IFNα and IFNβ have been documented in the synovium of patients with RA127–130. IFNα positively correlates with TLR3 and TLR7 in the lining and sub-lining of RA synovium. IFNα increases the expression of TLR3 and TLR7 and downstream production of IL-6 and TNF. Additionally, IFNα markedly potentiates TLR4-mediated production of IL-1β and IL-18 in synovial cells from patients with RA130. By contrast, IFNβ has an anti-inflammatory effect in inflammatory arthritis. In PMBCs, IFNβ can inhibit the production of IL-1β and TNF and can also increase the production of IL-1 receptor antagonist (IL1Ra) in a dosedependent manner131. IFNβ also dose-dependently increases IL1Ra secretion by synovial fibroblasts and enhances the secretion of IL1Ra induced by IL1β in synovial fibroblasts and chondrocytes132. Treatment with IFNβ is effective in alleviating arthritis in the collagen-induced arthritis mouse model of RA133,134. However, in a multicentre, randomized, double-blind,placebo-controlled phase II study, treatment with subcutaneous recombinant IFNβ resulted in no improvement in patients with active RA135.
In RA, type I interferon is potentially a predictive biomarker of response to biologic therapies. For example, the presence of a pretreatment type I interferon signature reportedly predicts response to the B cell-depleting therapy rituximab136. In another study, the ratio of pretreatment IFNβ activity to IFNα activity (IFNβ:IFNα activity ratio) could predict the response to anti-TNF therapy in RA137. A larger study from 2016 supported this idea, finding that the pretreatment serum IFNβ:IFNα activity ratio was strongly predictive of non-response to TNF inhibitors in both discovery and independent replication cohorts138. Although the reasons for the differences in the relative proportions of IFNα versus IFNβ in the circulation are unknown, other studies support the idea that these two type I interferons exist in different proportions in different rheumatic diseases139, with IFNα predominating in the circulation in SLE46,96, and IFNβ being relatively more abundant in RA138,139. Reason for the discrepancy between the finding that IFNβ was anti-inflammatory in early functional studies131–134,140 and the failure of the clinical trial of recombinant IFNβ treatment135, as well as the relatively increased IFNβ levels observed in the circulation of patients with RA who are unlikely to respond to anti-TNF therapy138, is unclear. Given the complexity of type I interferon signalling regulation (reviewed elsewhere29), the effects of IFNβ are probably influenced by the amount, duration and location (for example, the circulation or tissue) of IFNβ expression, and the environmental context.
Monogenic interferonopathies
The gene variants mentioned thus far moderately effect activation of the type I interferon pathway and/or susceptibility to complex polygenic rheumatic diseases, and it is likely that combinations of these genetic variations are probably required to predispose to disease. However, a number of monogenic diseases are characterized by interferon pathway activation. Interestingly, these diseases are considered to lie on an autoimmune– autoinflammation spectrum that depends on the driver of dysregulated type I interferon production141. On the basis of this spectrum, Kim et al.141 have proposed grouping these conditions into ‘autoinflammatory’ interferonopathies (those caused by a problem in the innate immune sensing system), and ‘autoimmune’ interferonopathies (those caused by immune complex stimulation of endosomal TLRs in B cells and pDCs). Dysregulation can occur from both processes in a given patient; however, the initial ‘driver’ of the interferonopathy is typically at one end of this spectrum. Monogenic forms of SLE (such as those caused by loss-of-function mutations in the genes encoding complement protein C1q, deoxyribonuclease 1 or deoxyribonuclease-γ) are considered to be autoimmune monogenic interferonopathies. Important examples among the autoinflammatory monogenic interferonopathies include AGS, chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE), and STING-associated vasculopathy with onset in infancy (SAVI).
AGS and related monogenic diseases
AGS is caused by gain-of-function mutations in TREX1 or IFIH1. The TREX1 gene encodes the major mammalian 3ʹ−5ʹ DNA exonuclease that degrades endogenous DNA in the cytoplasm142. In addition to AGS143,144, mutations in TREX1 have also been reported in SLE143,145, familial chilblain lupus144 and retinal vasculopathy with cerebral leukodystrophy (RVCL) (reviewed elsewhere146). AGS and RVCL are characterized clinically by CNS inflammation and high levels of type I interferon in the circulation and cerebrospinal fluid. Mutations in the gene encoding nucleic acid sensor RIG-I, DDX58, cause an atypical Singleton–Merten syndrome, which manifests with variable clinical presentations of glaucoma, aortic calcification and skeletal abnormalities, such as acro-osteolysis without dental anomalies147. Although there are no classical signs of apparent inflammation in patients with Singleton–Merten syndrome, the clinical manifestations are suspected to relate to chronic inflammation, at least in part conferred by constitutive activation of RIG-I resulting in increased type I interferon activity and ISG expression.
CANDLE
CANDLE is caused by mutations in protein subunits of the proteosome–immunoproteasome system. Disease can be the result of any of several recessive mutations in different protein subunits of the system, located either in one single subunit (monogenic, homozygous or compound heterozygous inheritance) or in two different subunits (digenic and compound heterozygous inheritance)148,149. Defects in the catalytic activity of the proteasome–immunoproteasome system result in a sustained production of type I interferon148,149 that is independent of STING and MAVS141.
SAVI
SAVI is caused by gain-of-function mutations in STING and is characterized by cutaneous vasculopathy and pulmonary inflammation150. In vitro studies indicate that these STING variants stimulate IFNB1 expression and other gene targets of STING150. Data from STING N153S knock-in mice demonstrates that at least some of the phenotype of SAVI occurs independently of IRF3, suggesting that the phenotype is not solely ISGrelated151. However, patients with SAVI have a strong type I interferon signature in their PMBCs, and JAK inhibitors reduce the constitutive upregulation of phosphorylated STAT1 in the lymphocytes of these patients in vitro, indicating that JAK inhibition could be a promising therapy for SAVI150.
Type I interferon pathway therapies
Insights gleaned from studies of the type I interferon pathway, including those identifying disease risk loci and functional studies of molecules involved in the type I interferon pathway, might help explain the heterogeneity in the molecular pathogenesis of rheumatic diseases. Such insights might explain some of the heterogeneity in treatment responses observed in these diseases, and type I interferon pathway studies could also reveal new targets. These insights should inform the development of new therapies and the design of clinical trials. Multiple antiIFNα, anti-IFNAR and anti-TLR strategies are currently in clinical development for the treatment of rheumatic diseases (TABLE 1).
Table 1 |. Biologic therapies in development for rheumatic diseases that target the type I interferon pathway.
| Treatment name | Type | Targeted rheumatic disease(s) | Phase of testing |
|---|---|---|---|
| Anti-IFNα therapies | |||
| Rontalizumab | Humanized anti-IFNα IgG1 mAb | Systemic lupus erythematosus | II |
| Sifalimumab | Humanized anti-IFNα IgG1 mAb | Systemic lupus erythematosus | II |
| Psoriasis | II | ||
| Dermatomyositis and polymyositis | I | ||
| AGS-009 | Humanized anti-IFNα IgG4 mAb | Systemic lupus erythematosus | I |
| IFNα Kinoid | IFNα vaccine | Systemic lupus erythematosus | II |
| Dermatomyositis | II | ||
| Anti-IFNAR therapy | |||
| Anifrolumab | Fully human anti-IFNAR mAb | Systemic lupus erythematosus | II/III |
| Systemic sclerosis | I | ||
| TLR inhibition | |||
| NI-0101 | Humanized anti-TLR4 mAb | Rheumatoid arthritis | II |
IFNAR, type I interferon receptor; mAb, monoclonal antibody; TLR, Toll-like receptor;
Anti-IFNα therapies
Anti-IFNα monoclonal antibodies (such as sifalimumab and rontalizumab) can inhibit the expression of the type I interferon signature in patients with SLE152–154, and phase II studies examining clinical responses to these antibodies in patients with SLE have had mixed results155–157. Rontalizumab did not meet the primary endpoint in one phase II trial, but did demonstrate some efficacy in a subset of patients with SLE and a low type I interferon signature metric (a set of 3 ISGs (HERC5, EPSTI and CMPK2) were used as a surrogate for the type I interferon signature)157. Treatment with sifalimumab did result in clinical improvement in various clinical end points in patients with SLE in another phase II study, and the effect was strongest in those patients with a high type I interferon signature score (based on a set of four ISGs: IFI27, IFI44, IFI44L and RSAD2)156. Although these phase II trial findings seem somewhat contradictory, it is interesting that in both trials the pretreatment type I interferon status of the patients affected the treatment response to anti-IFNα antibodies. It is possible that differences in the strength of interferon blockade between the two therapeutics or the dosing level could explain these differences in clinical efficacy.
Results from phase I and II studies investigating the induction of humoral polyclonal anti-IFNα responses by immunization with IFNα kinoid (a conjugate of an inactive form of human IFNα and a carrier protein, keyhole limpet haemocyanin) in patients with SLE have also shown some promise in improving control of the disease158. Furthermore, in a phase Ib trial, sifalimumab reduced the expression of a type I interferon signature observed in the blood of patients with dermatomyositis or polymyositis159.
Anti-IFNAR therapies
Anifrolumab is an antibody that binds to the IFNAR and blocks signals from both IFNα and IFNβ160. In a phase II study of patients with moderate to severe SLE161, anifrolumab treatment resulted in greater rates of improvement across a broad range of composite and organ-specific disease activity measures; a greater proportion of patients achieving and maintaining low disease activity or corticosteroid tapering as well as a trend toward a reduction in flare rate compared with placebo. Greater efficacy was seen in all end points in patients with a high baseline type I interferon signature compared with those with a low baseline interferon gene signature, suggesting that the former group represents a subpopulation of patients who are likely to benefit from anifrolumab treatment. However, the sample size of the low baseline type I interferon signature group was small, limiting interpretations of the data from this group; thus, further studies are warranted to determine efficacy in this subpopulation.
In early phase studies in patients with SSc, anifrolumab inhibited a type I interferon signature (as measured by a composite score from five ISGs: RSAD2, IFI44, IFI44L, IFI27 and IFI6), and this inhibition correlated with decreases in T cell-related transcripts and increases in collagen degradation-related transcripts in the skin162. Thus far there have not been overly concerning safety signals with regard to viral infection or malignancy risk with these anti-IFNα and anti-IFNAR therapies, although herpes zoster reactivation has occurred in some patients161. In lupus-prone mice, type I interferon-induced synapse loss and behavioural phenotypes are prevented by blocking signalling at IFNAR163, suggesting that anifrolumab might be helpful in treating neuropsychiatric lupus and should be considered for future clinical trials.
Hydroxychloroquine and TLR inhibition
Treatment with hydroxychloroquine impairs the ability of pDCs from patients with SLE to produce IFNα and TNF in response to stimulation with TLR9 and TLR7 agonists in vitro164. Unless contraindicated, hydroxychloroquine is advocated for use in all patients with SLE owing to its efficacy in reducing the number and intensity of flares, and in reducing damage accrual165,166. In the treatment of RA, hydroxychloroquine has also been used in combination with other drugs (for example, the widely use combination of methotrexate plus sulfasalazine and hydroxychloroquine, known as the ‘triple therapy’ regimen); however, hydroxychloroquine has limited efficacy in treating disease activity on its own. In a 2017 systematic review and meta-analysis of studies evaluating the effects of hydroxychloroquine on cardiovascular outcomes in patients with RA, hydroxychloroquine seemed to decrease insulin resistance and incidence of cardiovascular disease; however, the data were too few for meta-analysis167. Hydroxychloroquine is currently being tested in phase II trials for endothelial dysfunction in RA168, antiphospholipid antibody syndrome169, neonatal SLE170, incomplete SLE171 and in the prevention of clinically apparent RA in seropositive individuals172.
Several additional TLR-targeting strategies are in early development in SLE173, and a humanized anti-TLR4 monoclonal antibody is currently being tested in phase II of trials for the treatment of RA174. TLR4 inhibition could be interesting in RA if TLR4 activation contributes to the IFNβ levels observed in this disease, which is associated with non-response to anti-TNF therapies.
Kinase inhibition
The development of small molecule kinase inhibitors that target proteins in the type I interferon pathway has been a major area of drug development, and a number of clinical trials of these inhibitors in various rheumatic diseases are currently underway (TABLE 2).
Table 2 |. Kinase inhibitor therapies that target the type I interferon pathway and are in clinical development for rheumatic diseases.
| Treatment name | Type | Targeted rheumatic disease(s) | Phase of testing |
|---|---|---|---|
| Tofacitinib | JAK inhibitor (JAK1, JAK3) | Rheumatoid arthritis | IV (FDA approved) |
| • Juvenile idiopathic arthritis • Psoriatic arthritis • Psoriasis • Ulcerative colitis |
III | ||
| • Systemic sclerosis • Alopecia areata • Ankylosing spondylitis • Crohn’s disease |
II | ||
| Systemic lupus erythematosus | I/II | ||
| Dermatomyositis | I | ||
| Baricitinib | JAK inhibitor (JAK1, JAK2) | • Autoinflammatory syndromes (SAVI, AGS, CANDLE) • Juvenile dermatomyositis |
III (Compassionate use protocol) |
| Rheumatoid arthritis | III | ||
| • Giant cell arteritis • Psoriasis |
II | ||
| Solcitinib (GSK2586184) | JAK inhibitor (JAK1) | Rheumatoid arthritis | II/III |
| • Systemic lupus erythematosus • Psoriasis |
II | ||
| Decernotinib (VX-509) | JAK inhibitor (JAK3) | Rheumatoid arthritis | II/III |
| Ruxolitinib (INCB018424) | JAK inhibitor (JAK1, JAK2) | Secondary haemophagocytic syndrome (macrophage activation syndrome) | II |
| • Rheumatoid arthritis • Psoriasis |
I | ||
| PF-06651600 | JAK inhibitor (JAK3) | • Rheumatoid arthritis • Alopecia areata • Ulcerative colitis |
II |
| Lestaurtinib | JAK inhibitor (JAK2) | Psoriasis | II |
| INCB018424 (topical) | JAK inhibitor (JAK1, JAK2) | • Psoriasis • Vitiligo • Alopecia areata |
II |
| Filgotinib (GLPG0634) | JAK and TYK inhibitor (JAK1, JAK2, TYK2) | • Rheumatoid arthritis • Systemic lupus erythematosus (membranous lupus nephritis) • Non-infectious uveitis • Crohn’s disease |
II |
| PF-06700841 | JAK and TYK inhibitor (JAK1, TYK2) | • Alopecia areata • Psoriasis • Crohn’s disease • Ulcerative colitis |
II |
| PF-06263276 (topical) | JAK and TYK inhibitor (JAK1, JAK2, JAK3, TYK2) | Psoriasis | I |
| PF-06650833 | IRAK inhibitor (IRAK4) | Rheumatoid arthritis | II |
AGS, Aicardi– Goutieres syndrome; CANDLE, chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; IRAK, interleukin-1 receptor-associated kinase; JAK, Janus kinase; SAVI, STING-associated vasculopathy with onset in infancy; TYK, tyrosine kinase.
Tofacitinib, a JAK1 and JAK3 inhibitor, was approved by the FDA in 2012 for the treatment of patients with RA who have had an inadequate response or intolerance to methotrexate. Tofacitinib is now being tested in phase III trials for use in other rheumatic diseases (such as JIA175–177 and psoriatic arthritis178–180), and is being investigated in earlier phases studies for use in SLE181–183 and dermatomyositis184. Baricitinib, a JAK1 and JAK2 inhibitor, is being evaluated in stage III trials for the treatment of RA185–190, in addition to being used as part of a compassionate use protocol for the treatment of autoinflammatory syndromes marked by high type I interferon (for example, SAVI, AGS and CANDLE)191. JAK inhibitors that also inhibit TYK2 are in phase II trials for the treatment of RA192–197, psoriatic arthritis198,199 and membranous lupus nephritis200. Finally, an IRAK4 inhibitor is currently in phase II trials for the treatment of RA201. Kinase inhibitors are also being tested in additional chronic autoimmune or autoinflammatory diseases not covered in this Review (TABLE 2).
Clinical implications
Differences in type I interferon levels explain some of the heterogeneity in the clinical phenotypes and treatment responses across various rheumatic diseases. Thus, it would be reasonable to divide patients with a given disease (for example, Sjögren syndrome, SLE or RA) into subsets by their type I interferon pathway activity in clinical trials. Such a strategy has already been tested in trials of therapies targeting type I interferons in SLE156,157, but this same strategy might also yield informative results in the treatment of other rheumatic diseases with either therapies that target the type I interferon pathway or other drugs.
Stratifying patients by type I interferon pathway activity might reveal important differences in particular subgroups of patients that would otherwise be missed and might also enable the prediction of a patient’s treatment response to particular therapies, such as that observed with anti-TNF therapy138. Monitoring type I interferons during treatment might also be desirable in some patients. For example, caspase inhibitors are an attractive therapy for use in autoinflammatory disorders that result in increased inflammasome activation, such as NLRC4-related macrophage activation syndrome (NLRC4-MAS, also known as syndrome of enterocolitis and autoinflammation associated with mutation in NLRC4 (SCAN4)). However, blocking caspase-1 activity also pathologically increases type I interferon production in some patients (particularly those who have relatively high levels of type I interferon activity at baseline)18. Regular assessment of type I interferon pathway activation could enable better monitoring for possible unwanted consequences in this scenario.
Interestingly, many rheumatic diseases are more frequent in females than in males. In a 2017 transcriptome analysis of human skin samples, the genes that were overexpressed in female healthy skin (compared with male healthy skin) were frequently genes that are associated with autoimmune diseases such as SLE, SSc and Sjögren syndrome202. The presence of sex hormones, such as oestradiol or testosterone, did not affect the expression of these genes in cultured keratinocytes. Some of these overexpressed genes were regulated by the transcription cofactor vestigial-like protein 3 (VGLL3), the expression of which also has a strong female bias. ISGs (LY6E, OAS1, MX1 and IFI44) were among the genes that were targeted by VGLL3. In monocytes, maximal induction of the ISGs identified required the expression of VGLL3, suggesting that VGLL3 might promote inflammation by supporting type I interferon responses202. Thus, as we move towards precision medicine, we will need to carefully consider whether it is best to also subset patients with sex-discordant rheumatic diseases by sex in clinical trials.
Conclusions
The type I interferon pathway is central in both immunity and tolerance, and alterations in this pathway underlie the pathogenesis of different rheumatic conditions. Rheumatic diseases such as SLE, SSc, myositis and RA are heterogeneous and some of the differences observed between patients with rheumatic diseases could be explained by variations in the expression of interferon-related genes or activation of the type I interferon pathway. Hence, certain genetic factors and/or pathogenic pathways might explain particular disease phenotypes, and these underlying factors and/or pathways will not be shared between all patients who have the same rheumatic disease. We suspect variation in the type I interferon pathway is a major factor in the currently unexplained heritability of rheumatic disease.
Studies that compare patient subgroups based on their type I interferon signature or type I interferon activity have furthered our understanding of the molecular mechanisms underpinning the heterogeneity of these diseases and treatment responses. Additional molecular phenotyping should help to further advance our understanding of the pathogenesis of disease subtypes, and help to guide therapy. For example, medications that have seemingly failed in clinical trials of a complex rheumatic disease might still be helpful for treating a subgroup of patients with this disease. Thus, the study of individual samples from clinical trials is important, and insights gleaned from such studies should inform the next steps in an iterative fashion, including the subgrouping of patients by molecular phenotype in subsequent trials.
Functional studies of causal allelic variants should advance our ability to translate genetic associations into clinical applications. A delicate balance exists between the autoimmune and/or autoinflammatory effects and the antipathogen and anticancer effects of type I interferon. Increasing our understanding of the regulation of this pathway in humans will have important therapeutic and safety implications. By understanding the genetic regulation and molecular underpinnings of type I interferon in rheumatic diseases, we might be able to intervene therapeutically in a more personalized fashion, on the basis of the molecular dysregulation present in a given individual.
Key points.
Type I interferon has a pathogenic role in many rheumatic conditions, including systemic lupus erythematosus, Sjögren syndrome, myositis and systemic sclerosis.
Many genetic risk factors for rheumatic diseases lie within the type I interferon pathway as gain-of-function polymorphisms, and both polygenic and monogenic influences have been described.
Stratifying patients by type I interferon activity levels will inform us about both disease pathogenesis and treatment response in rheumatic diseases.
A number of therapeutics that target type I interferons, the type I interferon receptor, or the type I interferon pathway are currently in various stages of development.
Acknowledgements
The work of T.L.W.M. is supported by grants from the Central Society for Clinical and Translational Research. The work of T.B.N. is supported by grants from the NIH (AR060861, AR057781and AR065964), the Rheumatology Research Foundation, CureJM Foundation, the Myositis Association, the Lupus Research Alliance and the Colton Center for Autoimmunity.
Competing interests
T.B.N. declares that he has received research grants from EMD Serono and Janssen. T.L.W.N declares no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Rice G et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am. J. Hum. Genet 81, 713–725 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baechler EC et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl Acad. Sci. USA 100, 2610–2615 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett L et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J. Exp. Med 197, 711–723 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crow MK & Wohlgemuth J Microarray analysis of gene expression in lupus. Arthritis Res. Ther 5, 279–287 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kariuki SN et al. Genetic analysis of the pathogenic molecular sub-phenotype interferon-α identifies multiple novel loci involved in systemic lupus erythematosus. Genes Immun 16, 15–23 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Isaacs A & Lindenmann J Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci 147, 258–267 (1957). [PubMed] [Google Scholar]
- 7.Pestka S, Krause CD & Walter MR Interferons, interferon-like cytokines, and their receptors. Immunol. Rev 202, 8–32 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Pestka S Purification and cloning of interferon α. Curr. Top. Microbiol. Immunol 316, 23–37 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Ito T, Kanzler H, Duramad O, Cao W & Liu YJ Specialization, kinetics, and repertoire of type I interferon responses by human plasmacytoid predendritic cells. Blood 107, 2423–2431 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Prakash A, Smith E, Lee CK & Levy DE Tissuespecific positive feedback requirements for production of type I interferon following virus infection. J. Biol. Chem 280, 18651–18657 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Severa M et al. Sensitization to TLR7 agonist in IFNβ-preactivated dendritic cells. J. Immunol 178, 6208–6216 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Nir U, Maroteaux L, Cohen B & Mory I Priming affects the transcription rate of human interferon-β1 gene. J. Biol. Chem 260, 14242–14247 (1985). [PubMed] [Google Scholar]
- 13.Weiss G et al. MyD88 drives the IFN-β response to Lactobacillus acidophilus in dendritic cells through a mechanism involving IRF1, IRF3, and IRF7. J. Immunol 189, 2860–2868 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Kawai T & Akira S Innate immune recognition of viral infection. Nat. Immunol 7, 131–137 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Oliveira L, Sinicato NA, Postal M, Appenzeller S & Niewold TB Dysregulation of antiviral helicase pathways in systemic lupus erythematosus. Front. Genet 5, 418 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crow YJ et al. Mutations in the gene encoding the 3ʹ−5ʹ DNA exonuclease TREX1 cause AicardiGoutieres syndrome at the AGS1 locus. Nat. Genet 38, 917–920 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Mavragani CP et al. Expression of long interspersed nuclear element 1 retroelements and induction of type I interferon in patients with systemic autoimmune disease. Arthritis Rheumatol 68, 2686–2696 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y et al. Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity 46, 393–404 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Tamura T, Yanai H, Savitsky D & Taniguchi T The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol 26, 535–584 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Akira S, Uematsu S & Takeuchi O Pathogen recognition and innate immunity. Cell 124, 783–801 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Shrivastav M & Niewold TB Nucleic acid sensors and type I interferon production in systemic lupus erythematosus. Front. Immunol 4, 319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blasius AL & Beutler B Intracellular toll-like receptors. Immunity 32, 305–315 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Jensen MA & Niewold TB Interferon regulatory factors: critical mediators of human lupus. Transl Res 165, 283–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yarilina A, Park-Min KH, Antoniv T, Hu X & Ivashkiv LB TNF activates an IRF1-dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I interferonresponse genes. Nat. Immunol 9, 378–387 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Venkatesh D et al. Endothelial TNF receptor 2 induces IRF1 transcription factor-dependent interferon-β autocrine signaling to promote monocyte recruitment. Immunity 38, 1025–1037 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takayanagi H et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature 416, 744–749 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Xiong Q, Zhang L, Ge W & Tang P The roles of interferons in osteoclasts and osteoclastogenesis. Joint Bone Spine 83, 276–281 (2016). [DOI] [PubMed] [Google Scholar]
- 28.de Weerd NA et al. Structural basis of a unique interferon-beta signaling axis mediated via the receptor IFNAR1. Nat. Immunol 14, 901–907 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Ivashkiv LB & Donlin LT Regulation of type I interferon responses. Nat. Rev. Immunol 14, 36–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hervas-Stubbs S et al. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res 17, 2619–2627 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Sharma S et al. Widely divergent transcriptional patterns between SLE patients of different ancestral backgrounds in sorted immune cell populations. J. Autoimmun 60, 51–58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goh KC, Haque SJ & Williams BR p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J 18, 5601–5608 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaur S et al. Role of the Akt pathway in mRNA translation of interferon-stimulated genes. Proc. Natl Acad. Sci. USA 105, 4808–4813 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fink K et al. IFNβ/TNFα synergism induces a noncanonical STAT2/IRF9-dependent pathway triggering a novel DUOX2 NADPH oxidase-mediated airway antiviral response. Cell Res 23, 673–690 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Higgs BW et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann. Rheum. Dis 70, 2029–2036 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Karonitsch T et al. Activation of the interferongamma signaling pathway in systemic lupus erythematosus peripheral blood mononuclear cells. Arthritis Rheum 60, 1463–1471 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Banchereau R et al. Personalized immunomonitoring uncovers molecular networks that stratify lupus patients. Cell 165, 1548–1550 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Becker AM et al. SLE peripheral blood B cell, T cell and myeloid cell transcriptomes display unique profiles and each subset contributes to the interferon signature. PLoS ONE 8, e67003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weckerle CE et al. Network analysis of associations between serum interferon-α activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum 63, 1044–1053 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jabs WJ, Hennig C, Zawatzky R & Kirchner H Failure to detect antiviral activity in serum and plasma of healthy individuals displaying high activity in ELISA for IFN-α and IFN-β. J. Interferon Cytokine Res 19, 463–469 (1999). [DOI] [PubMed] [Google Scholar]
- 41.Wilson DR et al. The Simoa HD-I analyzer: a novel fully automated digital immunoassay analyzer with single-molecule sensitivity and multiplexing. J. Lab. Autom 21, 533–547 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Rissin DM et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol 28, 595–599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meyer S et al. AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies. Cell 166, 582–595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ronnblom LE, Alm GV & Oberg KE Possible induction of systemic lupus erythematosus by interferon-α treatment in a patient with a malignant carcinoid tumour. J. Intern. Med 227, 207–210 (1990). [DOI] [PubMed] [Google Scholar]
- 45.Niewold TB & Swedler WI Systemic lupus erythematosus arising during interferon-α therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin. Rheumatol 24, 178–181 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Niewold TB, Hua J, Lehman TJ, Harley JB & Crow MK High serum IFN-α activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun 8, 492–502 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kariuki SN et al. Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res. Ther 12, R151 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Munroe ME et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann. Rheum. Dis 75, 2014–2021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghodke-Puranik Y & Niewold TB Genetics of the type I interferon pathway in systemic lupus erythematosus. Int. J. Clin. Rheumtol 10.2217/ijr.13.58 (2013). [DOI] [PMC free article] [PubMed]
- 50.Kariuki SN et al. Cutting edge: Autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-α in lupus patients in vivo. J. Immunol 182, 34–38 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson T et al. Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-α and serologic autoimmunity in lupus patients. J. Immunol 187, 1298–1303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barnes BJ, Moore PA & Pitha PM Virusspecific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon α genes. J. Biol. Chem 276,23382–23390 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Yasuda K et al. Interferon regulatory factor-5 deficiency ameliorates disease severity in the MRL/lpr mouse model of lupus in the absence of a mutation in DOCK2. PLoS ONE 9, e103478 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Graham RR et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc. Natl Acad. Sci. USA 104, 6758–6763 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harley JB et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat. Genet 40, 204–210 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lessard CJ et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat. Genet 45, 1284–1292 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Radstake TR et al. Genome-wide association study of systemic sclerosis identifies CD247 as a new susceptibility locus. Nat. Genet 42, 426–429 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dieguez-Gonzalez R et al. Association of interferon regulatory factor 5 haplotypes, similar to that found in systemic lupus erythematosus, in a large subgroup of patients with rheumatoid arthritis. Arthritis Rheum 58, 1264–1274 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Nordang GB et al. Interferon regulatory factor 5 gene polymorphism confers risk to several rheumatic diseases and correlates with expression of alternative thymic transcripts. Rheumatology (Oxford) 51, 619–626 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Niewold TB et al. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum 58, 2481–2487 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Niewold TB et al. IRF5 haplotypes demonstrate diverse serological associations which predict serum interferon alpha activity and explain the majority of the genetic association with systemic lupus erythematosus. Ann. Rheum. Dis 71, 463–468 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cherian TS et al. Brief Report: IRF5 systemic lupus erythematosus risk haplotype is associated with asymptomatic serologic autoimmunity and progression to clinical autoimmunity in mothers of children with neonatal lupus. Arthritis Rheum 64, 3383–3387 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu Q et al. Association of a functional IRF7 variant with systemic lupus erythematosus. Arthritis Rheum 63, 749–754 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lessard CJ et al. Identification of IRF8, TMEM39A, and IKZF3-ZPBP2 as susceptibility loci for systemic lupus erythematosus in a large-scale multiracial replication study. Am. J. Hum. Genet 90, 648–660 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Salloum R et al. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-α activity in lupus patients. Arthritis Rheum 62, 553–561 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chrabot BS et al. Genetic variation near IRF8 is associated with serologic and cytokine profiles in systemic lupus erythematosus and multiple sclerosis. Genes Immun 14, 471–478 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pothlichet J et al. A loss-of-function variant of the antiviral molecule MAVS is associated with a subset of systemic lupus patients. EMBO Mol. Med 3, 142–152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kariuki SN, Crow MK & Niewold TB The PTPN22 C1858T polymorphism is associated with skewing of cytokine profiles toward high interferon-α activity and low tumor necrosis factor α levels in patients with lupus. Arthritis Rheum 58, 2818–2823 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y et al. The autoimmunity-associated gene PTPN22 potentiates toll-like receptor-driven, type I interferon-dependent immunity. Immunity 39, 111–122 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gestermann N et al. STAT4 is a confirmed genetic risk factor for Sjögren’s syndrome and could be involved in type I interferon pathway signaling. Genes Immun 11, 432–438 (2010). [DOI] [PubMed] [Google Scholar]
- 71.Dieude P et al. STAT4 is a genetic risk factor for systemic sclerosis having additive effects with IRF5 on disease susceptibility and related pulmonary fibrosis. Arthritis Rheum 60, 2472–2479 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Remmers EF et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N. Engl.J. Med 357, 977–986 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zervou MI, Goulielmos GN, Castro-Giner F, Tosca AD & Krueger-Krasagakis S STAT4 gene polymorphism is associated with psoriasis in the genetically homogeneous population of Crete, Greece. Hum. Immunol 70, 738–741 (2009). [DOI] [PubMed] [Google Scholar]
- 74.Liang YL et al. Association of STAT4 rs7574865 polymorphism with autoimmune diseases: a metaanalysis. Mol. Biol. Rep 39, 8873–8882 (2012). [DOI] [PubMed] [Google Scholar]
- 75.Hebert HL et al. Identification of loci associated with late-onset psoriasis using dense genotyping of immunerelated regions. Br. J. Dermatol 172, 933–939 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Lee YH et al. The PTPN22 C1858T functional polymorphism and autoimmune diseases — a metaanalysis. Rheumatology (Oxford) 46, 49–56 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Carlton VE et al. PTPN22 genetic variation: evidence for multiple variants associated with rheumatoid arthritis. Am. J. Hum. Genet 77, 567–581 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mok A et al. Genome-wide profiling identifies associations between lupus nephritis and differential methylation of genes regulating tissue hypoxia and type I interferon responses. Lupus Sci. Med 3, e000183 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harley ITW et al. The role of genetic variation near interferon-κ in systemic lupus erythematosus. J. Biomed. Biotechnol 2010, 706825 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stannard JN et al. Lupus skin is primed for IL-6 inflammatory responses through a keratinocytemediated autocrine type I interferon loop. J. Invest. Dermatol 137, 115–122 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ko K, Koldobskaya Y, Rosenzweig E & Niewold TB Activation of the interferon pathway is dependent upon autoantibodies in African-American SLE patients, but not in European-American SLE patients. Frontiers Immunol 4, 309 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hagberg N et al. IFN-α production by plasmacytoid dendritic cells stimulated with RNA-containing immune complexes is promoted by NK cells via MIP-1β and LFA-1. J. Immunol 186, 5085–5094 (2011). [DOI] [PubMed] [Google Scholar]
- 83.Ghodke-Puranik Y et al. Lupus-associated functional polymorphism in PNP causes cell cycle abnormalities and interferon pathway activation in human immune cells. Arthritis Rheumatol 69, 2328–2337 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Landolt-Marticorena C et al. Lack of association between the interferon-α signature and longitudinal changes in disease activity in systemic lupus erythematosus. Ann. Rheum. Dis 68, 1440–1446 (2009). [DOI] [PubMed] [Google Scholar]
- 85.Petri M et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus 18, 980–989 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bauer JW et al. Interferon-regulated chemokines as biomarkers of systemic lupus erythematosus disease activity: a validation study. Arthritis Rheum 60, 3098–3107 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hjelmervik TO, Petersen K, Jonassen I, Jonsson R & Bolstad AI Gene expression profiling of minor salivary glands clearly distinguishes primary Sjögren’s syndrome patients from healthy control subjects. Arthritis Rheum 52, 1534–1544 (2005). [DOI] [PubMed] [Google Scholar]
- 88.Emamian ES et al. Peripheral blood gene expression profiling in Sjögren’s syndrome. Genes Immun 10, 285–296 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gottenberg JE et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjögren’s syndrome. Proc. Natl Acad. Sci. USA 103, 2770–2775 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Niewold TB, Rivera TL, Buyon JP & Crow MK Serum type I interferon activity is dependent on maternal diagnosis in anti-SSA/Ro-positive mothers of children with neonatal lupus. Arthritis Rheum 58, 541–546 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brkic Z et al. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjögren’s syndrome and association with disease activity and BAFF gene expression. Ann. Rheum. Dis 72, 728–735 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li H et al. Identification of a Sjögren’s syndrome susceptibility locus at OAS1 that influences isoform switching, protein expression, and responsiveness to type I interferons. PLoS Genet 13, e1006820 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vlachogiannis NI et al. Increased frequency of the PTPN22W* variant in primary Sjögren’s syndrome: association with low type I IFN scores. Clin. Immunol 173, 157–160 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Maria NI et al. Contrasting expression pattern of RNA-sensing receptors TLR7, RIG-I and MDA5 in interferon-positive and interferon-negative patients with primary Sjögren’s syndrome. Ann. Rheum. Dis 76, 721–730 (2017). [DOI] [PubMed] [Google Scholar]
- 95.Nezos A et al. Type I and II interferon signatures in Sjögren’s syndrome pathogenesis: contributions in distinct clinical phenotypes and Sjögren’s related lymphomagenesis. J. Autoimmun 63, 47–58 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Niewold TB, Kariuki SN, Morgan GA, Shrestha S & Pachman LM Elevated serum interferon-α activity in juvenile dermatomyositis: associations with disease activity at diagnosis and after thirty-six months of therapy. Arthritis Rheum 60, 1815–1824 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.O’Connor KA, Abbott KA, Sabin B, Kuroda M & Pachman LM MxA gene expression in juvenile dermatomyositis peripheral blood mononuclear cells: association with muscle involvement. Clin. Immunol 120, 319–325 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shrestha S et al. Lesional and non lesional skin from untreated juvenile dermatomyositis (JDM) displays increased mast cells and mature plasmacytoid dendritic cells. Arthritis Rheum 62, 2813–2822 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reed AM et al. Changes in novel biomarkers of disease activity in juvenile and adult dermatomyositis are sensitive biomarkers of disease course. Arthritis Rheum 64, 4078–4086 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Greenberg SA et al. Relationship between disease activity and type I interferonand other cytokineinducible gene expression in blood in dermatomyositis and polymyositis. Genes Immun 13, 207–213 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Niewold TB, Kariuki SN, Morgan GA, Shrestha S & Pachman LM Gene-gene-sex interaction in cytokine gene polymorphisms revealed by serum interferon α phenotype in juvenile dermatomyositis. J. Pediatr 157, 653–657 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Niewold TB, Wu SC, Smith M, Morgan GA & Pachman LM Familial aggregation of autoimmune disease in juvenile dermatomyositis. Pediatrics 127, e1239–e1246 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Balboni I et al. Detection of anti-Ro, La, Smith and RNP autoantibodies by autoantigen microarray analysis and interferon-α induction in juvenile dermatomyositis. Arthritis Rheum 2424–2429 (2013). [DOI] [PMC free article] [PubMed]
- 104.Dastmalchi M et al. A high incidence of disease flares in an open pilot study of infliximab in patients with refractory inflammatory myopathies. Ann. Rheum. Dis 67, 1670–1677 (2008). [DOI] [PubMed] [Google Scholar]
- 105.Mavragani CP et al. Augmented interferon-α pathway activation in patients with Sjögren’s syndrome treated with etanercept. Arthritis Rheum 56, 3995–4004 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liao AP et al. Interferon β is associated with type I interferon-inducible gene expression in dermatomyositis. Ann. Rheum. Dis 70, 831–836 (2011). [DOI] [PubMed] [Google Scholar]
- 107.Tournadre A, Lenief V, Eljaafari A & Miossec P Immature muscle precursors are a source of interferon-β in myositis: role of Toll-like receptor 3 activation and contribution to HLA class I up-regulation. Arthritis Rheum 64, 533–541 (2012). [DOI] [PubMed] [Google Scholar]
- 108.Li L et al. Role of Toll-like receptors and retinoic acid inducible gene I in endogenous production of type I interferon in dermatomyositis. J. Neuroimmunol 285, 161–168 (2015). [DOI] [PubMed] [Google Scholar]
- 109.Assassi S et al. Systemic sclerosis and lupus: points in an interferon-mediated continuum. Arthritis Rheum 62, 589–598 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brkic Z et al. The interferon type I signature is present in systemic sclerosis before overt fibrosis and might contribute to its pathogenesis through high BAFF gene expression and high collagen synthesis. Ann. Rheum. Dis 75, 1567–1573 (2016). [DOI] [PubMed] [Google Scholar]
- 111.York MR et al. A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and toll-like receptor agonists. Arthritis Rheum 56, 1010–1020 (2007). [DOI] [PubMed] [Google Scholar]
- 112.Christmann RB et al. Association of Interferonand transforming growth factor β-regulated genes and macrophage activation with systemic sclerosis-related progressive lung fibrosis. Arthritis Rheumatol 66, 714–725 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wuttge DM et al. Increased serum type I interferon activity in early systemic sclerosis patients is associated with antibodies against Sjögren’s syndrome antigens and nuclear ribonucleoprotein antigens. Scand. J. Rheumatol 42, 235–240 (2013). [DOI] [PubMed] [Google Scholar]
- 114.Mahoney JM et al. Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms. PLoS Comput. Biol 11, e1004005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gorlova O et al. Identification of novel genetic markers associated with clinical phenotypes of systemic sclerosis through a genome-wide association strategy. PLoS Genet 7, e1002178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Barizzone N et al. Rare variants in the TREX1 gene and susceptibility to autoimmune diseases. Biomed. Res. Int 2013, 471703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van Bon L et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N. Engl. J. Med 370, 433–443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Aidoudi S, Bujakowska K, Kieffer N & Bikfalvi A The CXC-chemokine CXCL4 interacts with integrins implicated in angiogenesis. PLoS ONE 3, e2657 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Romagnani P et al. CXCR3-mediated opposite effects of CXCL10 and CXCL4 on T(H)1 or T-H cytokine production. J. Allergy Clin. Immunol 116, 1372–1379 (2005). [DOI] [PubMed] [Google Scholar]
- 120.Liu CY et al. Platelet factor 4 differentially modulates CD4(+)CD25(+) (regulatory) versus CD4(+)CD25(−) (nonregulatory) T cells. J. Immunol 174, 2680–2686 (2005). [DOI] [PubMed] [Google Scholar]
- 121.Stevens W, Vancheeswaran R & Black CM Alpha interferon-2a (Roferon-A) in the treatment of diffuse cutaneous systemic sclerosis: a pilot study. Br. J. Rheumatol 31, 683–689 (1992). [DOI] [PubMed] [Google Scholar]
- 122.Black CM et al. Interferon-α does not improve outcome at one year in patients with diffuse cutaneous scleroderma: results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 42, 299–305 (1999). [DOI] [PubMed] [Google Scholar]
- 123.Lubbers J et al. The type I IFN signature as a biomarker of preclinical rheumatoid arthritis. Ann. Rheum. Dis 72, 776–780 (2013). [DOI] [PubMed] [Google Scholar]
- 124.Hua J, Kirou K, Lee C & Crow MK Functional assay of type I interferon in systemic lupus erythematosus plasma and association with anti-RNA binding protein autoantibodies. Arthritis Rheum 54, 1906–1916 (2006). [DOI] [PubMed] [Google Scholar]
- 125.Han TU et al. Association of an activity-enhancing variant of IRAK1 and an MECP2-IRAK1 haplotype with increased susceptibility to rheumatoid arthritis. Arthritis Rheum 65, 590–598 (2013). [DOI] [PubMed] [Google Scholar]
- 126.Orozco G et al. Association of a functional singlenucleotide polymorphism of PTPN22, encoding lymphoid protein phosphatase, with rheumatoid arthritis and systemic lupus erythematosus. Arthritis Rheum 52, 219–224 (2005). [DOI] [PubMed] [Google Scholar]
- 127.Lande R et al. Characterization and recruitment of plasmacytoid dendritic cells in synovial fluid and tissue of patients with chronic inflammatory arthritis. J. Immunol 173, 2815–2824 (2004). [DOI] [PubMed] [Google Scholar]
- 128.Cavanagh LL et al. Rheumatoid arthritis synovium contains plasmacytoid dendritic cells. Arthritis Res. Ther 7, R230–240 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.van Holten J, Smeets TJ, Blankert P & Tak PP Expression of interferon β in synovial tissue from patients with rheumatoid arthritis: comparison with patients with osteoarthritis and reactive arthritis. Ann. Rheum. Dis 64, 1780–1782 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Roelofs MF et al. Type I interferons might form the link between Toll-like receptor (TLR) 3/7 and TLR4-mediated synovial inflammation in rheumatoid arthritis (RA). Ann. Rheum. Dis 68, 1486–1493 (2009). [DOI] [PubMed] [Google Scholar]
- 131.Coclet-Ninin J, Dayer JM & Burger D Interferon-β not only inhibits interleukin-1β and tumor necrosis factor-α but stimulates interleukin-1 receptor antagonist production in human peripheral blood mononuclear cells. Eur. Cytokine Netw 8, 345–349 (1997). [PubMed] [Google Scholar]
- 132.Palmer G et al. Interferon β stimulates interleukin 1 receptor antagonist production in human articular chondrocytes and synovial fibroblasts. Ann. Rheum.Dis 63, 43–49 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Triantaphyllopoulos KA, Williams RO, Tailor H & Chernajovsky Y Amelioration of collagen-induced arthritis and suppression of interferon-γ, interleukin-12, and tumor necrosis factor alpha production by interferon-β gene therapy. Arthritis Rheum 42, 90–99 (1999). [DOI] [PubMed] [Google Scholar]
- 134.van Holten J et al. Treatment with recombinant interferon-β reduces inflammation and slows cartilage destruction in the collagen-induced arthritis model of rheumatoid arthritis. Arthritis Res. Ther 6, R239–R249 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.van Holten J et al. A multicentre, randomised, double blind, placebo controlled phase II study of subcutaneous interferon β−1a in the treatment of patients with active rheumatoid arthritis. Ann. Rheum.Dis 64, 64–69 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Raterman HG et al. The interferon type I signature towards prediction of non-response to rituximab in rheumatoid arthritis patients. Arthritis Res. Ther 14, R95 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Mavragani CP, La DT, Stohl W & Crow MK Association of the response to tumor necrosis factor antagonists with plasma type I interferon activity and interferon-β/α ratios in rheumatoid arthritis patients: a post hoc analysis of a predominantly Hispanic cohort. Arthritis Rheum 62, 392–401 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wampler Muskardin T et al. Increased pretreatment serum IFN-β/α ratio predicts non-response to tumour necrosis factor α inhibition in rheumatoid arthritis. Ann. Rheum. Dis 75, 1757–1762 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.de Jong TD et al. Physiological evidence for diversification of IFNαand IFNβ-mediated response programs in different autoimmune diseases. Arthritis Res. Ther 18, 49 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Coclet-Ninin J, Dayer JM & Burger D Interferon-β not only inhibits interleukin-1 β and tumor necrosis factor-α but stimulates interleukin-1 receptor antagonist production in human peripheral blood mononuclear cells. Eur. Cytokine Netw 8, 345–349 (1997). [PubMed] [Google Scholar]
- 141.Kim H, Sanchez GA & Goldbach-Mansky R Insights from Mendelian interferonopathies: comparison of CANDLE, SAVI with AGS, monogenic lupus. J. Mol. Med 94, 1111–1127 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Paludan SR & Bowie AG Immune sensing of DNA. Immunity 38, 870–880 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Namjou B et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun 12, 270–279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rice G et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am. J. Hum. Genet 80, 811–815 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lee-Kirsch MA et al. Mutations in the gene encoding the 3ʹ−5ʹ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet 39, 1065–1067 (2007). [DOI] [PubMed] [Google Scholar]
- 146.Rice GI, Rodero MP & Crow YJ Human disease phenotypes associated with mutations in TREX1. J. Clin. Immunol 35, 235–243 (2015). [DOI] [PubMed] [Google Scholar]
- 147.Jang MA et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet 96, 266–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Torrelo A CANDLE syndrome as a paradigm of proteasome-related autoinflammation. Front. Immunol 8, 927 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Brehm A et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J. Clin. Invest 125, 4196–4211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Liu Y et al. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med 371, 507–518 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Warner JD et al. STING-associated vasculopathy develops independently of IRF3 in mice. J. Exp. Med 10.1084/jem.20171351 (2017). [DOI] [PMC free article] [PubMed]
- 152.Merrill JT et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon alpha monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann. Rheum. Dis 70, 1905–1913 (2011). [DOI] [PubMed] [Google Scholar]
- 153.McBride JM et al. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: results of a phase I, placebocontrolled, double-blind, dose-escalation study. Arthritis Rheum 64, 3666–3676 (2012). [DOI] [PubMed] [Google Scholar]
- 154.Petri M et al. Sifalimumab, a human antiinterferon-α monoclonal antibody, in systemic lupus erythematosus: a phase I randomized, controlled, dose-escalation study. Arthritis Rheum 65, 1011–1021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Niewold TB Connective tissue diseases: Targeting type I interferon in systemic lupus erythematosus. Nat. Rev. Rheumatol 12, 377–378 (2016). [DOI] [PubMed] [Google Scholar]
- 156.Khamashta M et al. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis 75, 1909–1916 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kalunian KC et al. A phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-alpha) in patients with systemic lupus erythematosus (ROSE). Ann. Rheum. Dis 75, 196–202 (2016). [DOI] [PubMed] [Google Scholar]
- 158.Lauwerys BR et al. Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon α-kinoid. Arthritis Rheum 65, 447–456 (2013). [DOI] [PubMed] [Google Scholar]
- 159.Higgs BW et al. A phase Ib clinical trial evaluating sifalimumab, an anti-IFN-α monoclonal antibody, shows target neutralisation of a type I IFN signature in blood of dermatomyositis and polymyositis patients. Ann. Rheum. Dis 73, 256–262 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Peng L, Oganesyan V, Wu H, Dall’Acqua WF & Damschroder MM Molecular basis for antagonistic activity of anifrolumab, an anti-interferon-α receptor 1 antibody. mAbs 7, 428–439 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Furie R et al. Anifrolumab, an anti-interferon-α receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol 69, 376–386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Guo X et al. Suppression of T cell activation and collagen accumulation by an anti-IFNAR1 mAb, anifrolumab, in adult patients with systemic sclerosis. J. Invest. Dermatol 135, 2402–2409 (2015). [DOI] [PubMed] [Google Scholar]
- 163.Bialas AR et al. Microglia-dependent synapse loss in type I interferon-mediated lupus. Nature 546, 539–543 (2017). [DOI] [PubMed] [Google Scholar]
- 164.Sacre K, Criswell LA & McCune JM Hydroxychloroquine is associated with impaired interferon-α and tumor necrosis factor-α production by plasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res. Ther 14, R155 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Alarcon GS et al. Effect of hydroxychloroquine on the survival of patients with systemic lupus erythematosus: data from LUMINA, a multiethnic US cohort (LUMINA L). Ann. Rheum. Dis 66, 1168–1172 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Bruce IN et al. Factors associated with damage accrual in patients with systemic lupus erythematosus: results from the Systemic Lupus International Collaborating Clinics (SLICC) Inception Cohort. Ann. Rheum. Dis 74, 1706–1713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rempenault C et al. Metabolic and cardiovascular benefits of hydroxychloroquine in patients with rheumatoid arthritis: a systematic review and metaanalysis. Ann. Rheum. Dis 10.1136/annrheumdis-2017-211836 (2017). [DOI] [PubMed]
- 168.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT03085940 (2017). [DOI] [PubMed]
- 169.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT02595346 (2016). [DOI] [PubMed]
- 170.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01379573 (2017). [DOI] [PubMed]
- 171.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT03030118 (2018). [DOI] [PubMed]
- 172.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT02603146 (2017). [DOI] [PubMed]
- 173.Postal M, Sinicato NA, Appenzeller S & Niewold TB Drugs in early clinical development for systemic lupus erythematosus. Expert Opin. Invest. Drugs 25, 573–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT03241108 (2017). [DOI] [PubMed]
- 175.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT02592434 (2018). [DOI] [PubMed]
- 176.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01500551 (2018). [DOI] [PubMed]
- 177.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT03000439 (2018). [DOI] [PubMed]
- 178.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01976364 (2018). [DOI] [PubMed]
- 179.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01882439 (2017). [DOI] [PubMed]
- 180.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01877668 (2017). [DOI] [PubMed]
- 181.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT03159936 (2017). [DOI] [PubMed]
- 182.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT02535689 (2018). [DOI] [PubMed]
- 183.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT03288324 (2018). [DOI] [PubMed]
- 184.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT03002649 (2017). [DOI] [PubMed]
- 185.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT02265705 (2017). [DOI] [PubMed]
- 186.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01721044 (2018). [DOI] [PubMed]
- 187.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01885078 (2018). [DOI] [PubMed]
- 188.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01721057 (2017). [DOI] [PubMed]
- 189.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01710358 (2017). [DOI] [PubMed]
- 190.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01711359 (2017). [DOI] [PubMed]
- 191.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01724580 (2017). [DOI] [PubMed]
- 192.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT02065700 (2017). [DOI] [PubMed]
- 193.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01894516 (2016). [DOI] [PubMed]
- 194.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01888874 (2016). [DOI] [PubMed]
- 195.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01668641 (2013). [DOI] [PubMed]
- 196.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT01384422 (2012). [DOI] [PubMed]
- 197.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT02885181 (2017). [DOI] [PubMed]
- 198.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT03320876 (2018). [DOI] [PubMed]
- 199.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT03101670 (2018). [DOI] [PubMed]
- 200.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT03285711 (2018). [DOI] [PubMed]
- 201.US National Library of Medicine. ClinicalTrials.govhttps://clinicaltrials.gov/ct2/show/NCT02996500 (2018). [DOI] [PubMed]
- 202.Liang Y et al. A gene network regulated by the transcription factor VGLL3 as a promoter of sexbiased autoimmune diseases. Nat. Immunol 18, 152–160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Canosi U et al. A highly precise reporter gene bioassay for type I interferon. J. Immunol. Methods 199, 69–76 (1996). [DOI] [PubMed] [Google Scholar]
- 204.Khabar KS et al. Expressed gene clusters associated with cellular sensitivity and resistance towards anti-viral and anti-proliferative actions of interferon. J. Mol. Biol 342, 833–846 (2004). [DOI] [PubMed] [Google Scholar]
- 205.Niewold TB, Clark DN, Salloum R & Poole BD Interferon α in systemic lupus erythematosus. J. Biomed. Biotechnol 2010, 948364 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]